RIP1 Is a Novel Component of γ-ionizing Radiation-Induced Invasion of Non-Small Cell Lung Cancer Cells

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
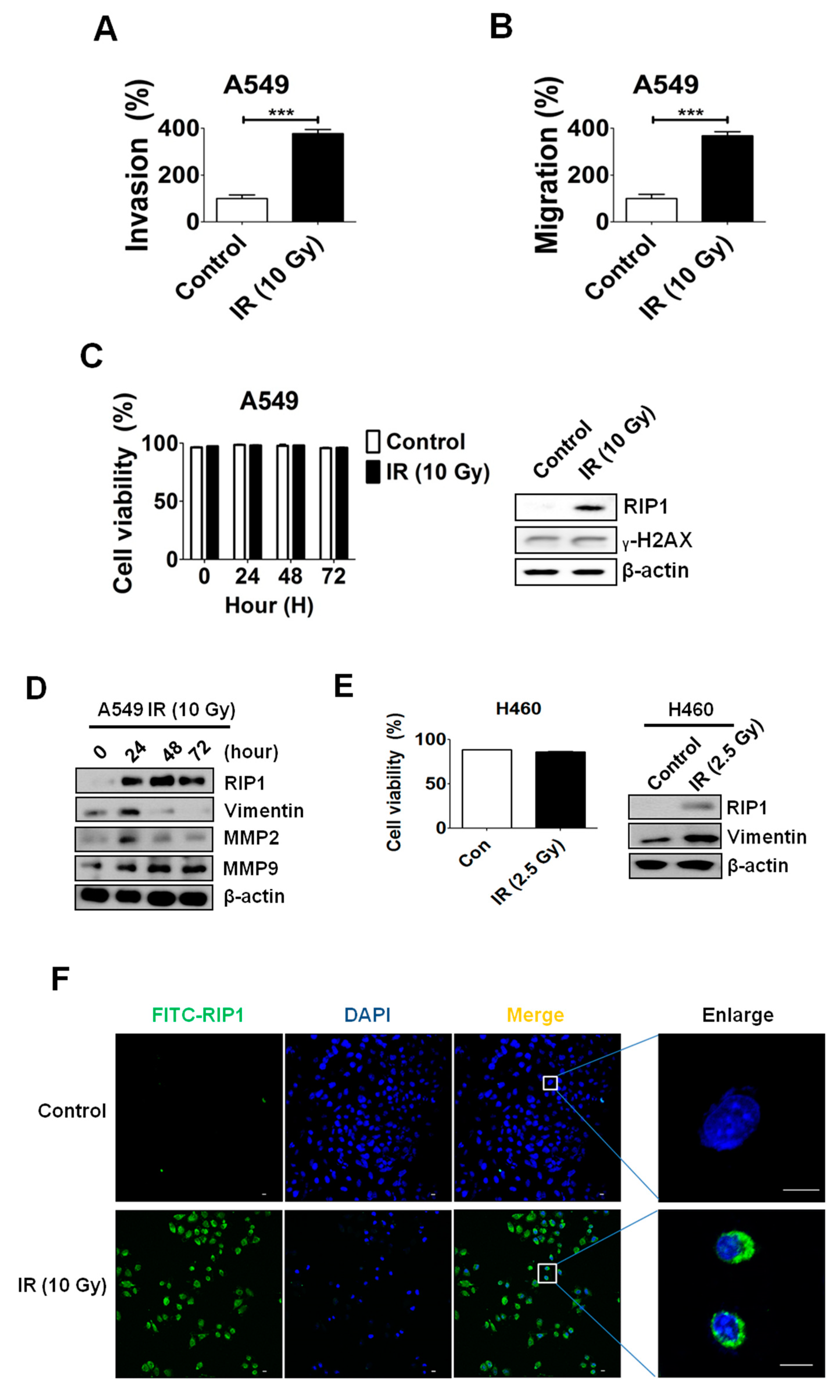
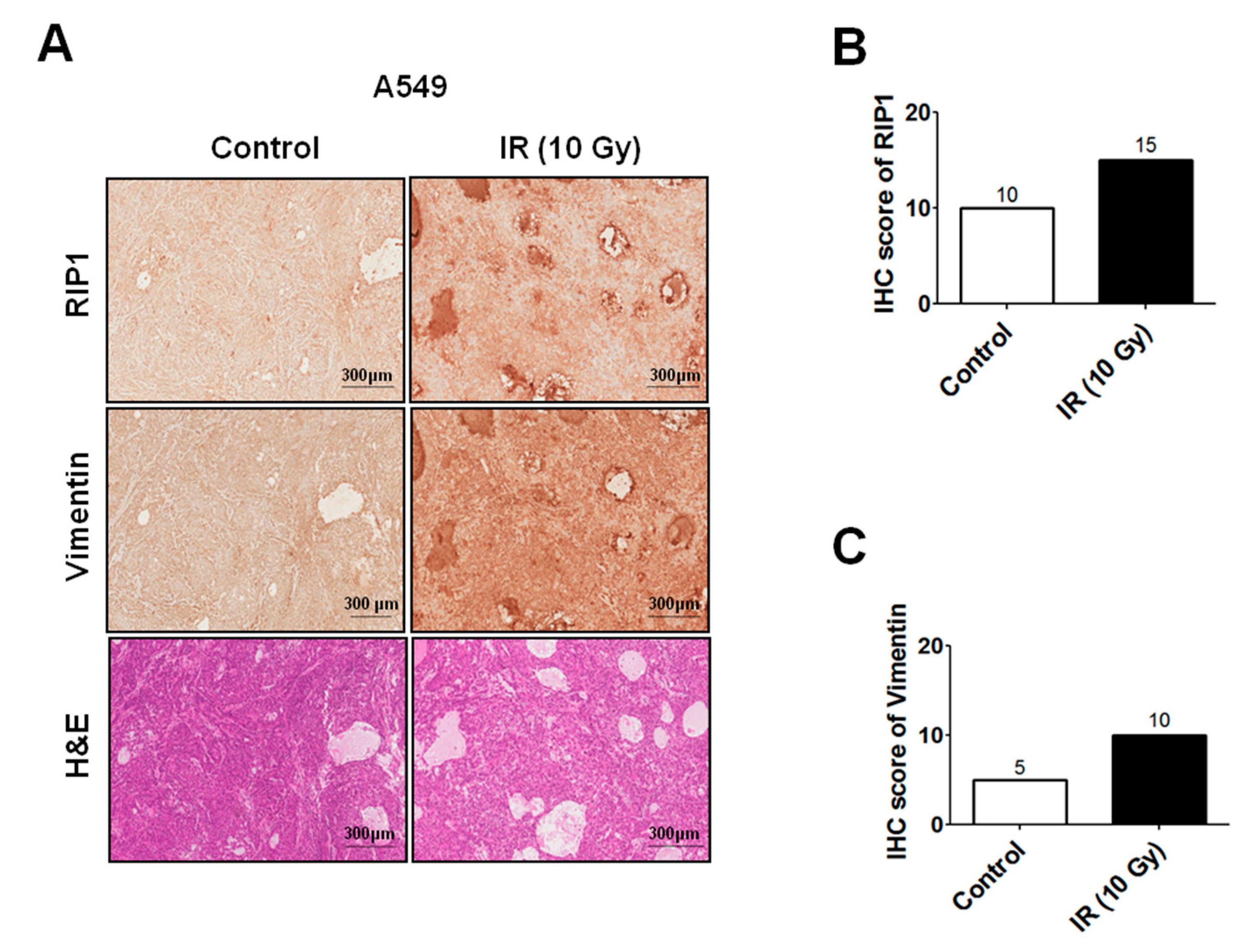
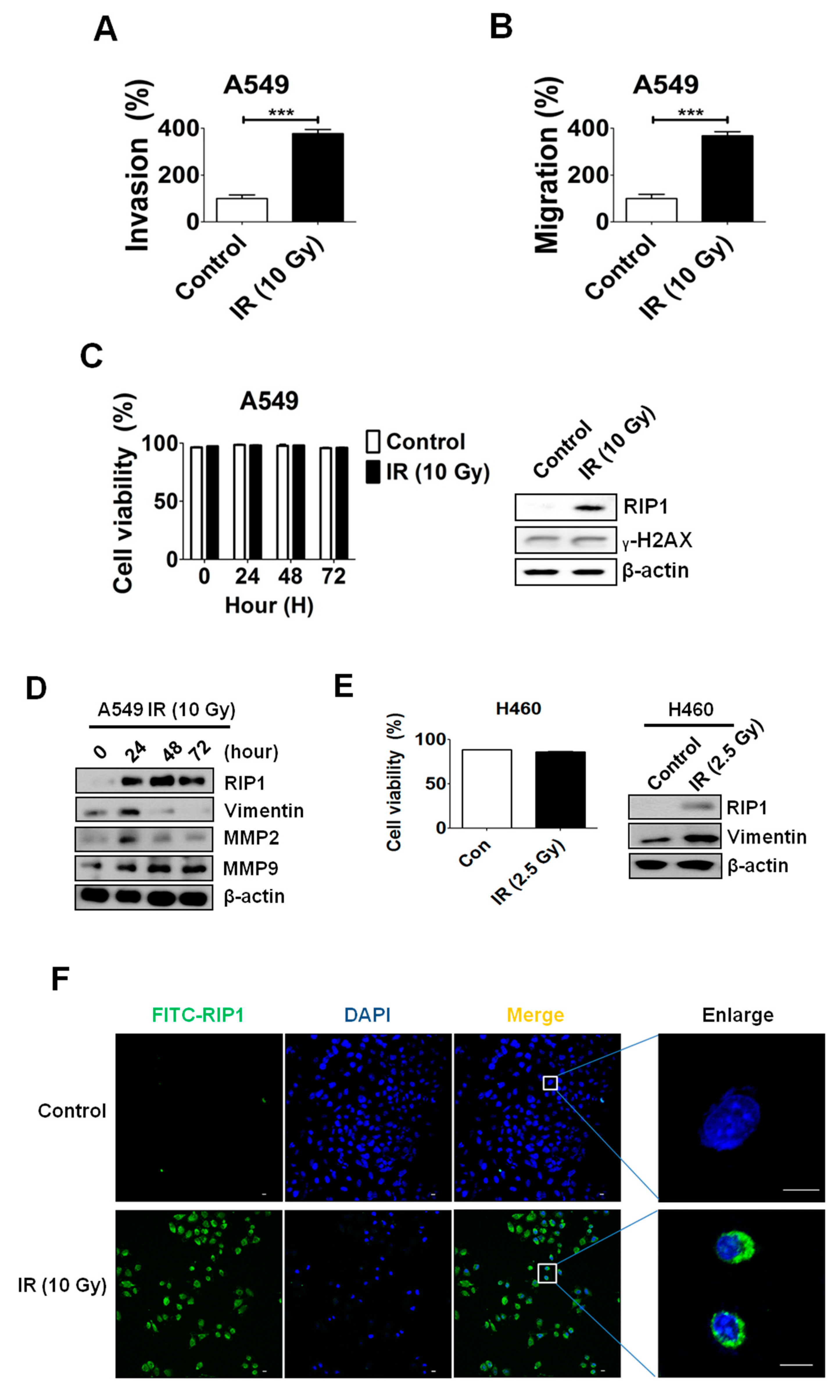
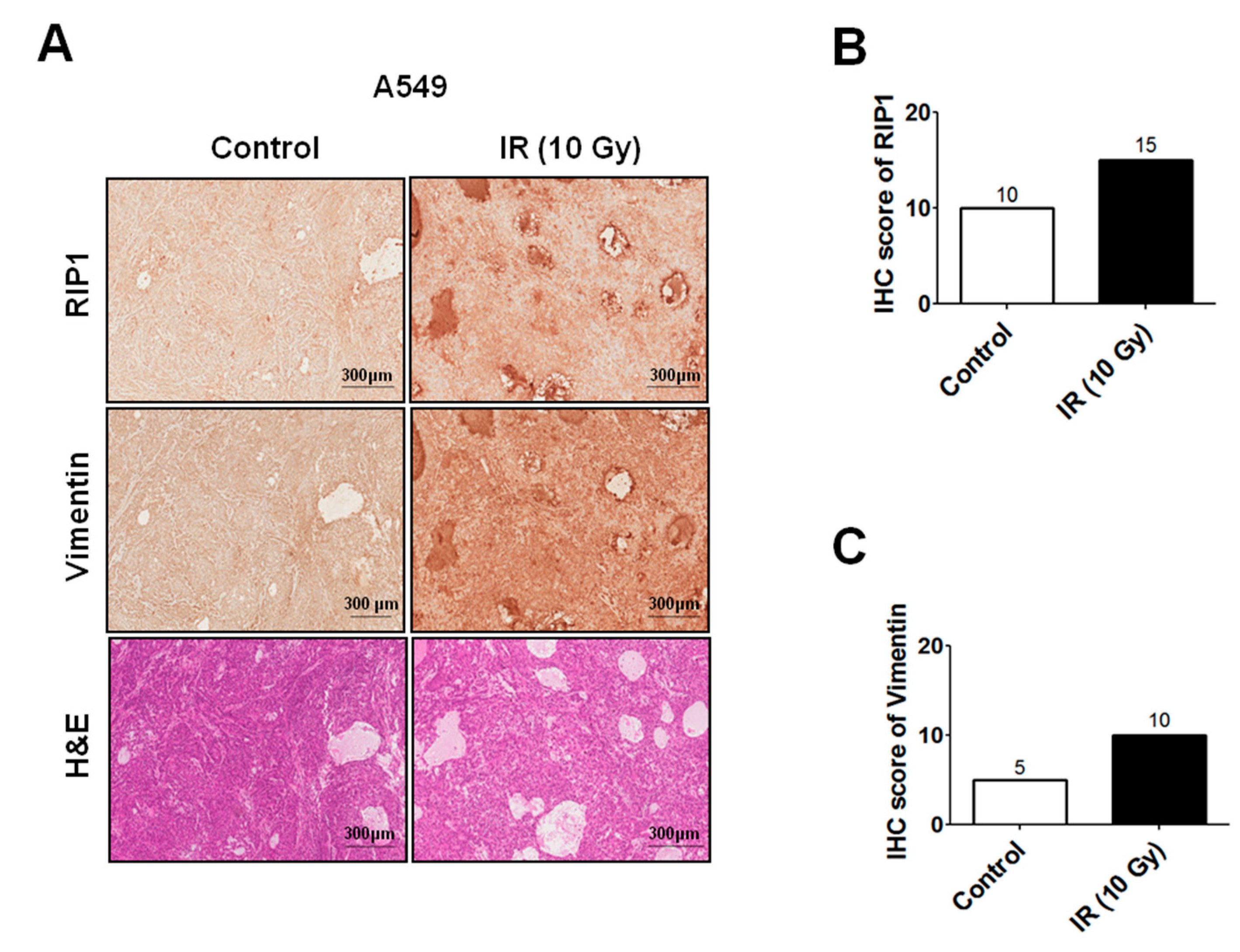
2.1. IR Induces Invasion/Migration via Upregulation of EMT and RIP1
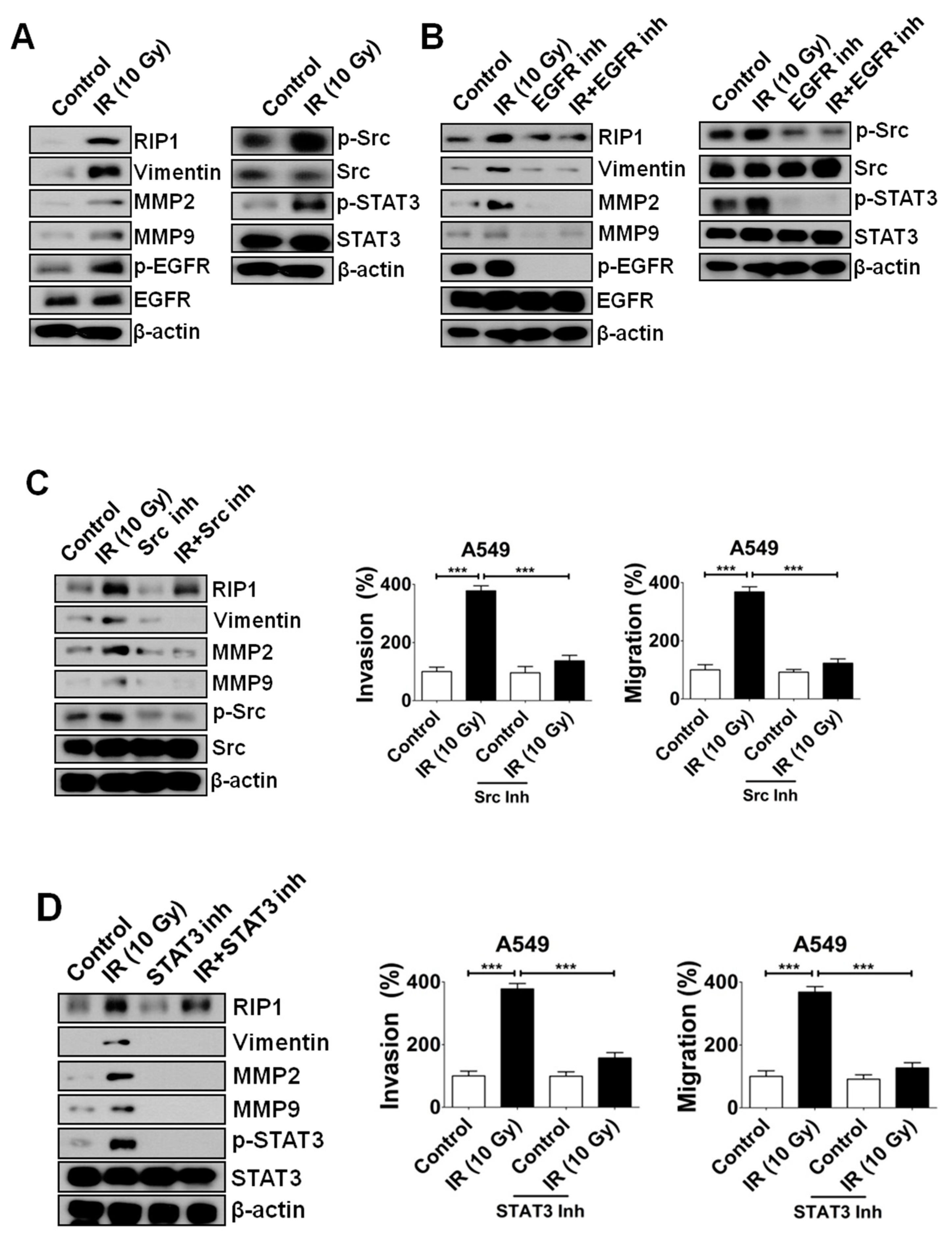
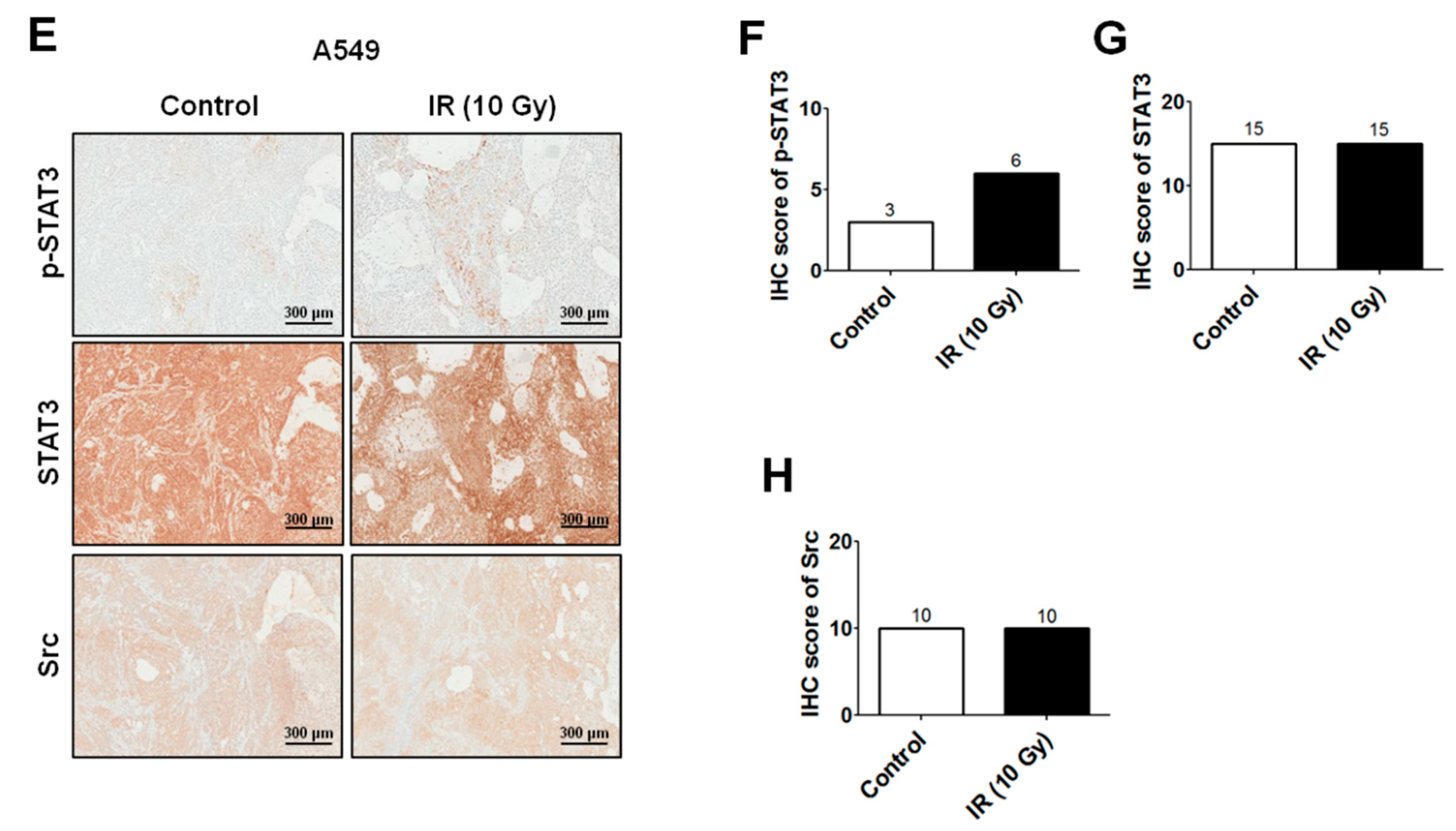
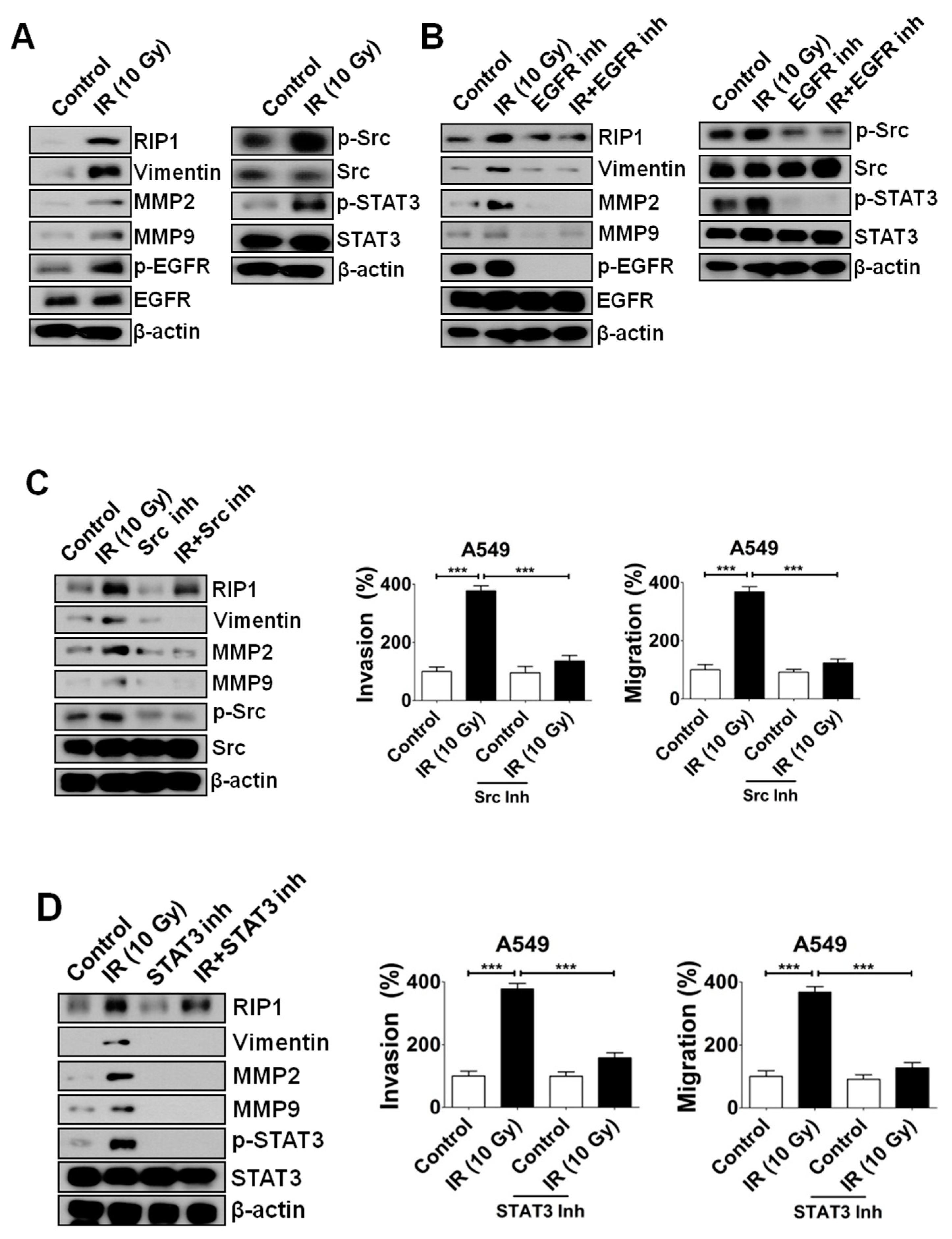
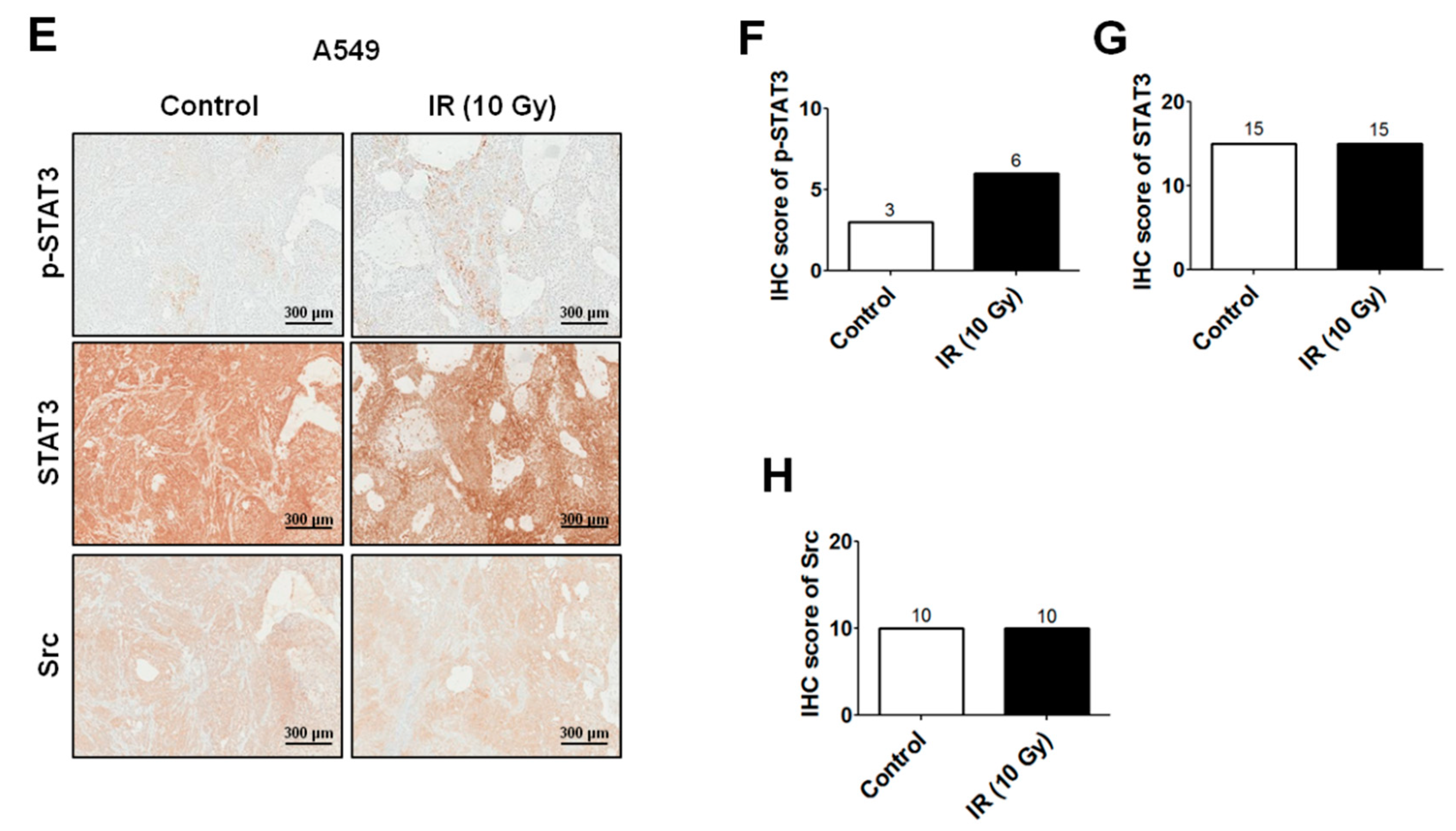
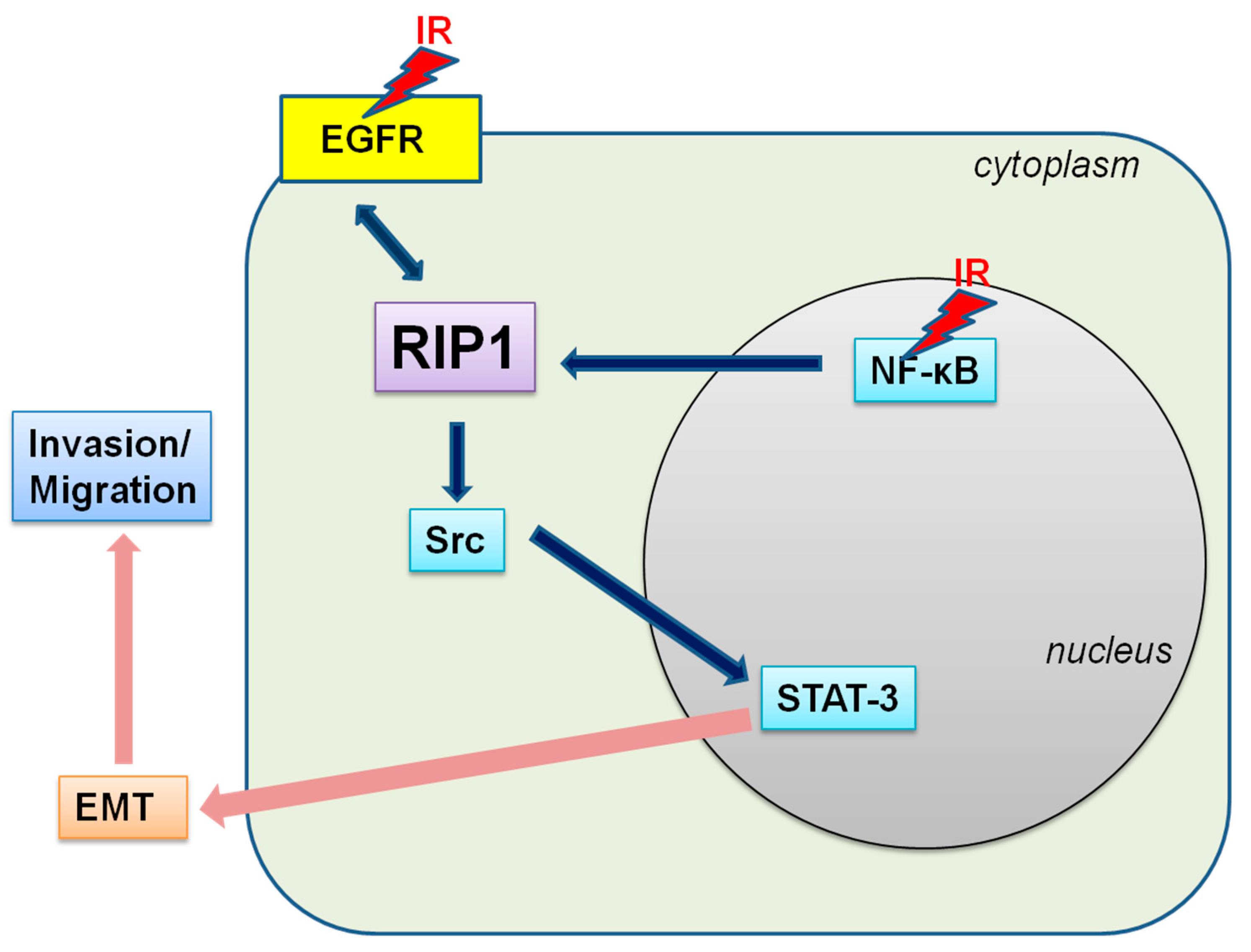
2.2. IR-Induced Invasion/Migration Is Mediated by the EGFR/Src/STAT3 Pathway
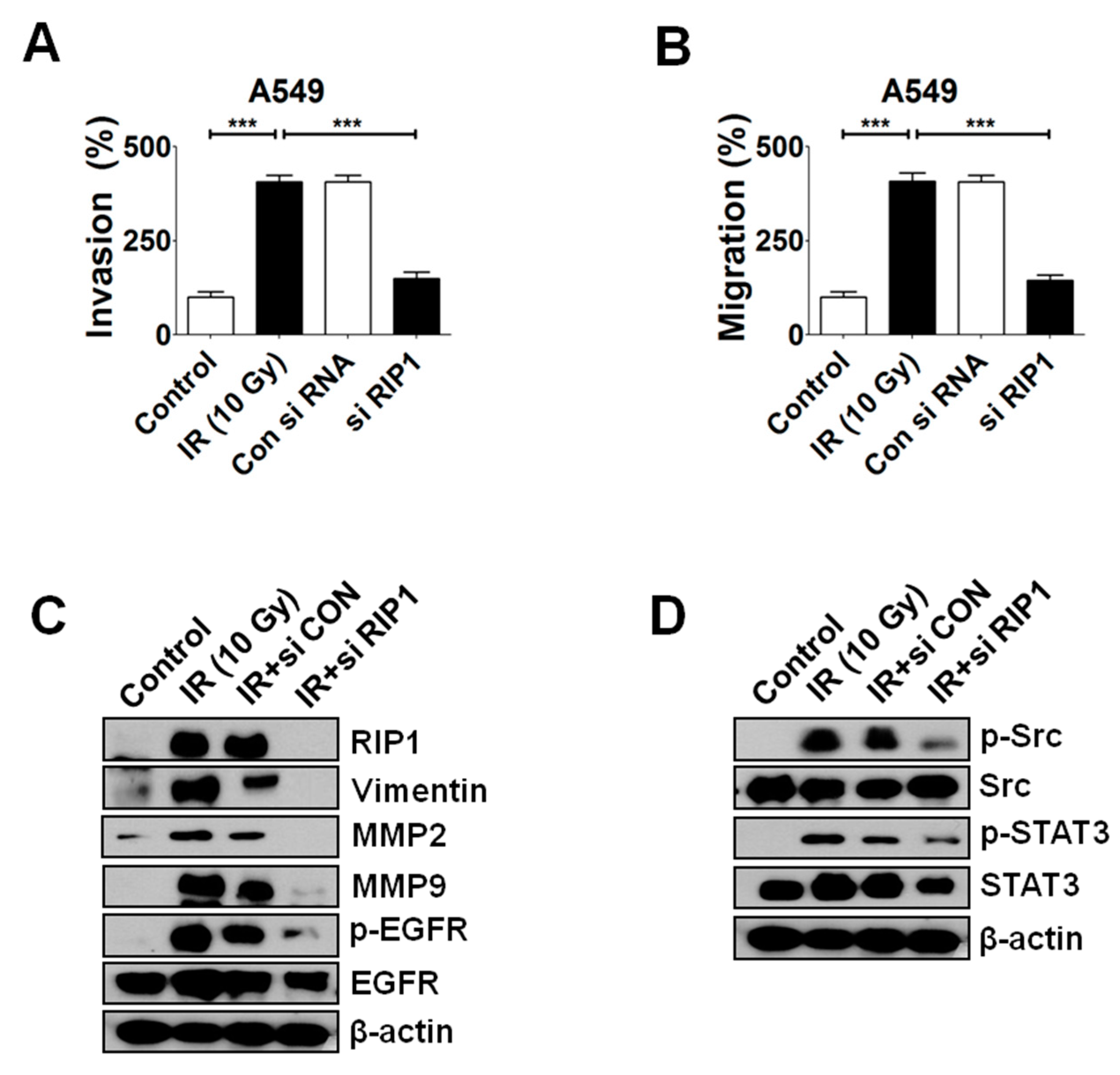
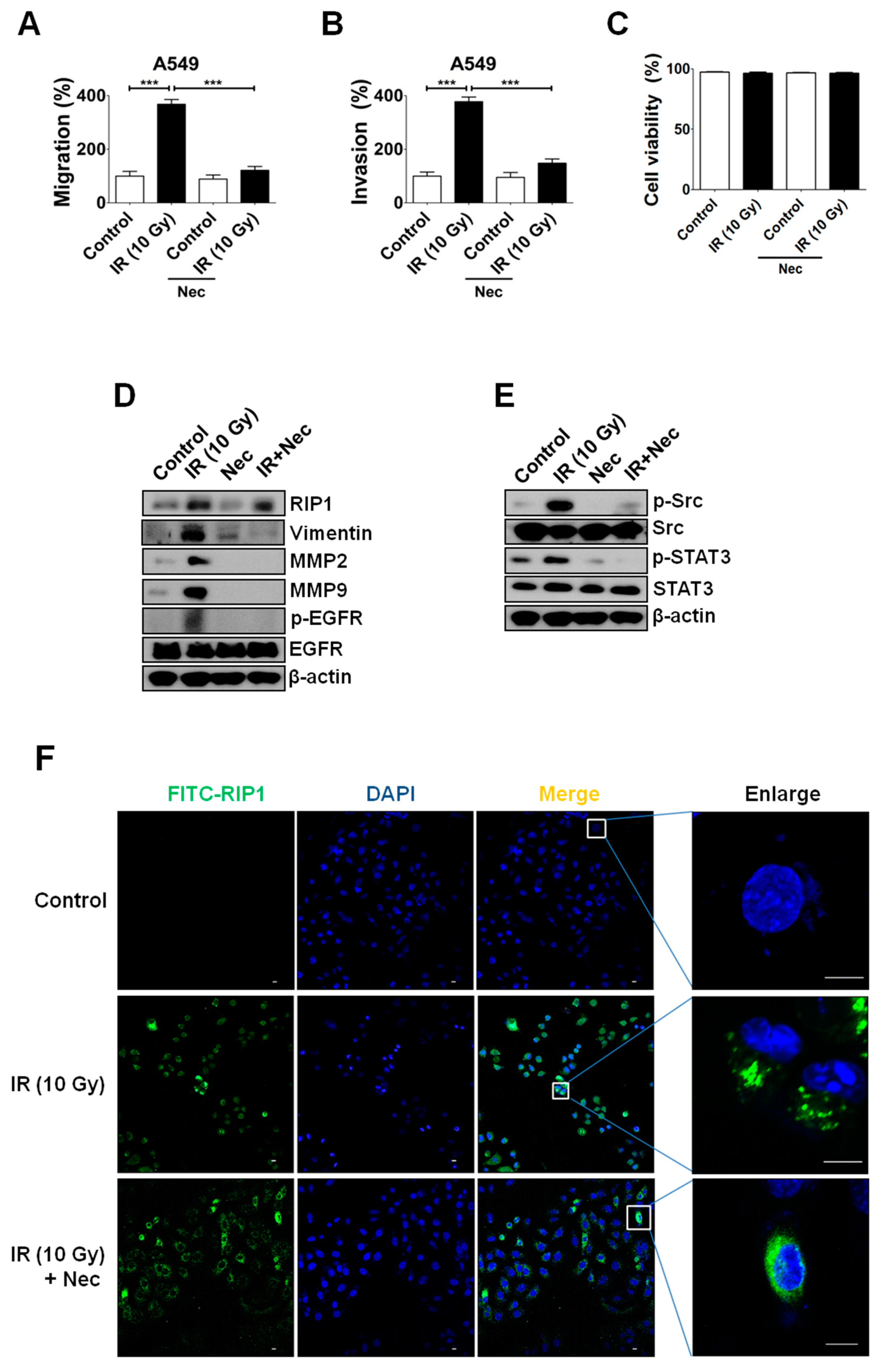
2.3. Inhibition of RIP1 Protein Suppresses Invasion/Migration in IR-Treated A549 Cells
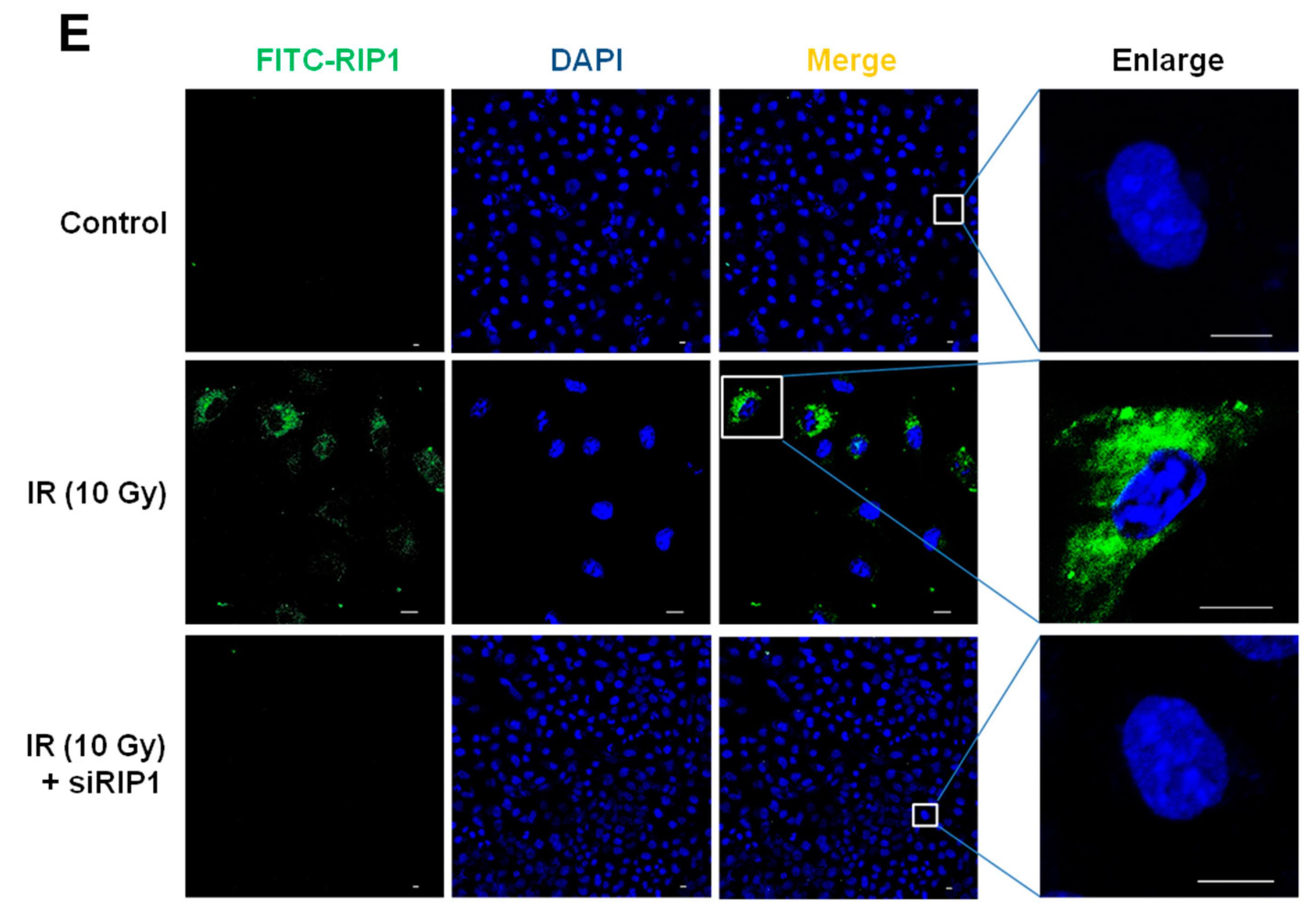
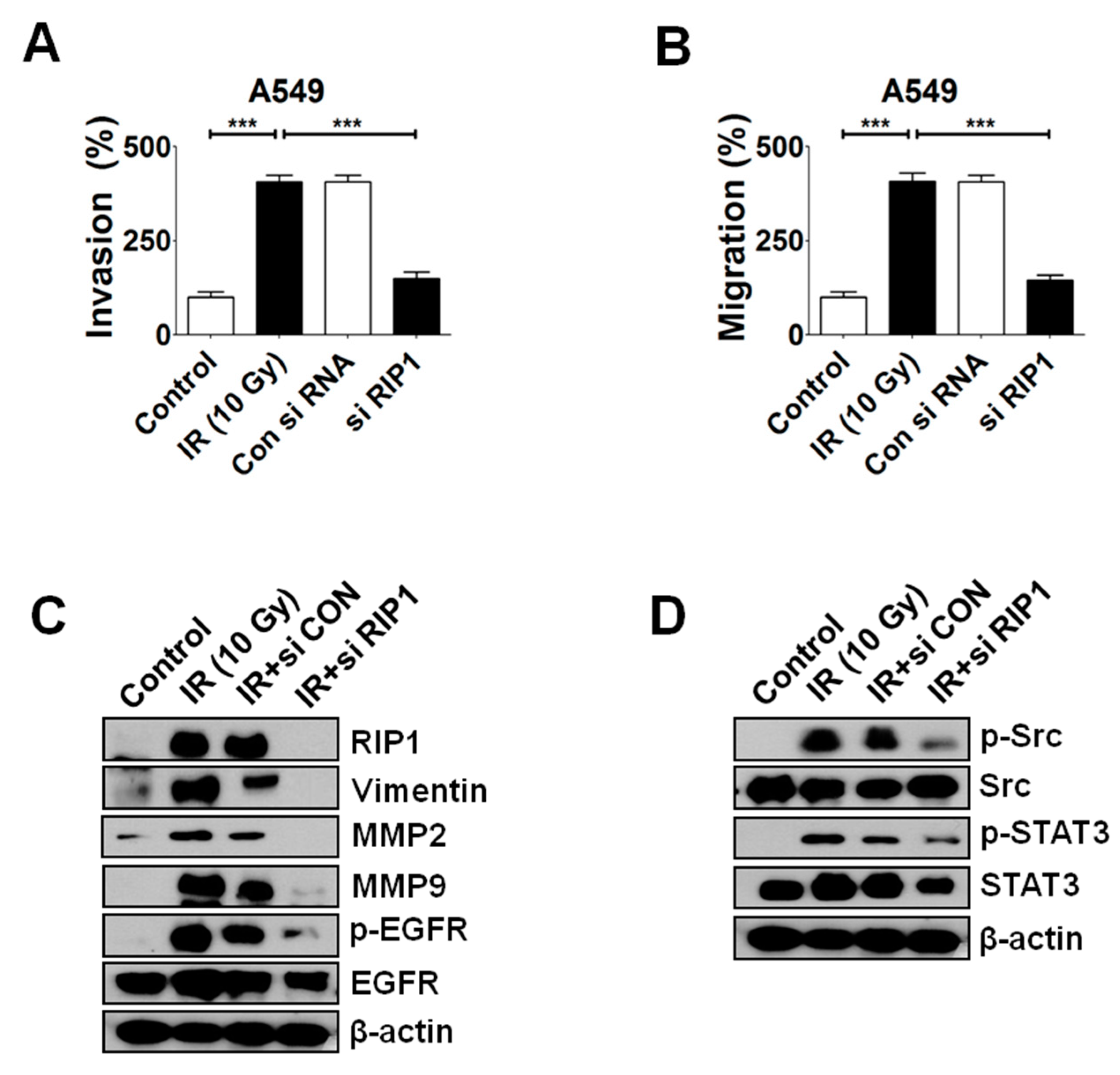
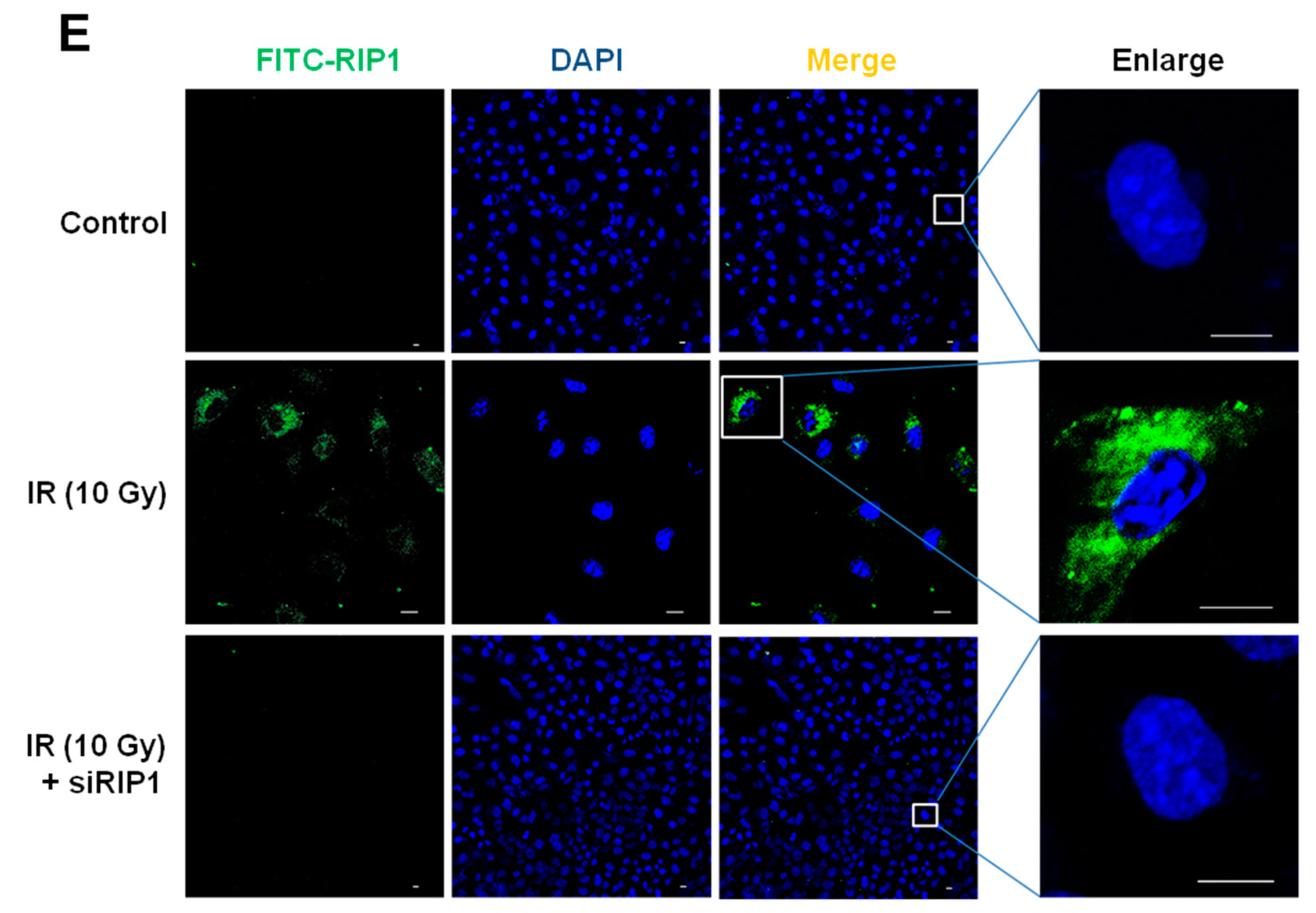
2.4. Knockdown of RIP1 Suppresses Invasion/Migration of IR-Treated A549 Cells
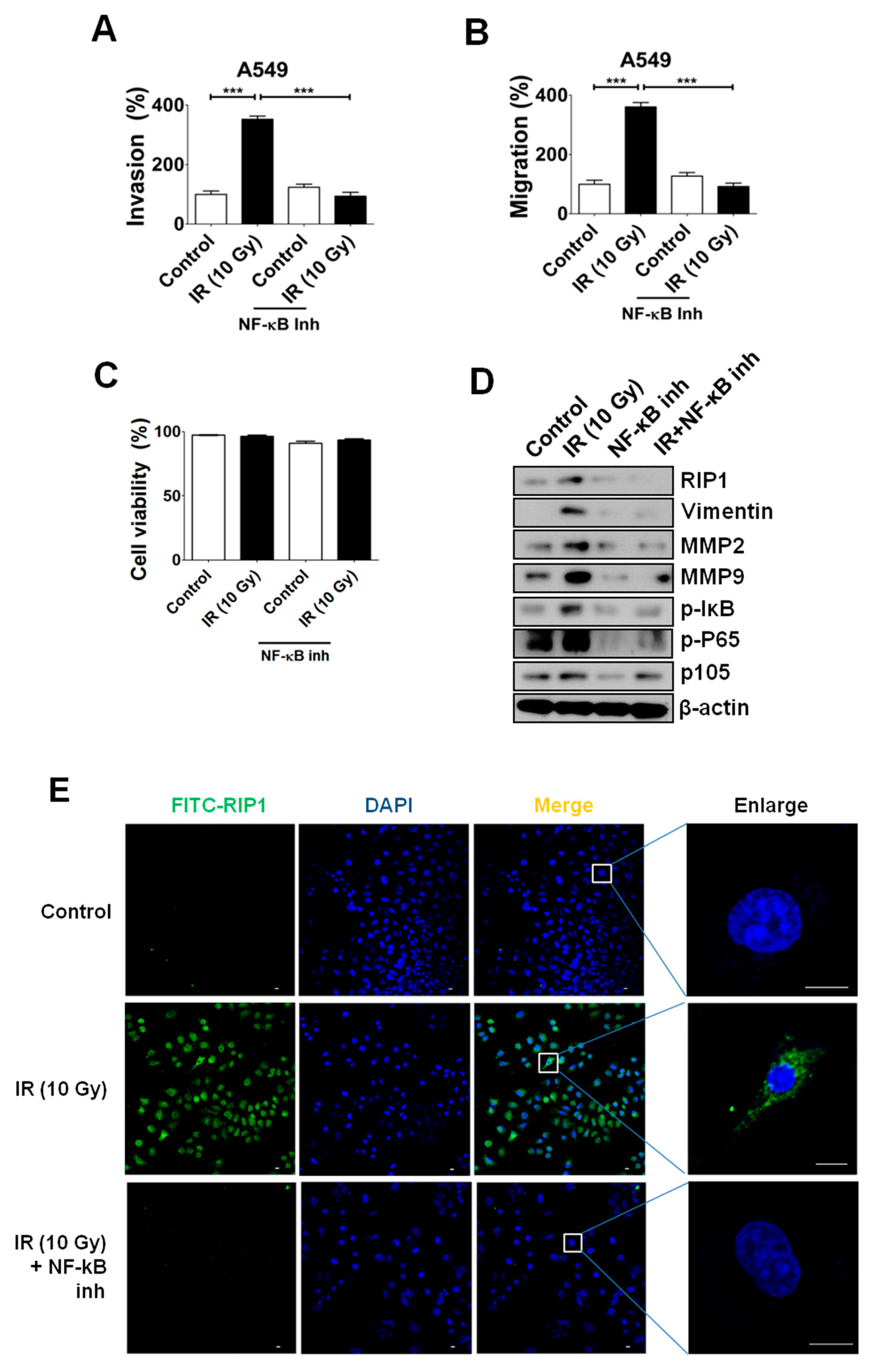
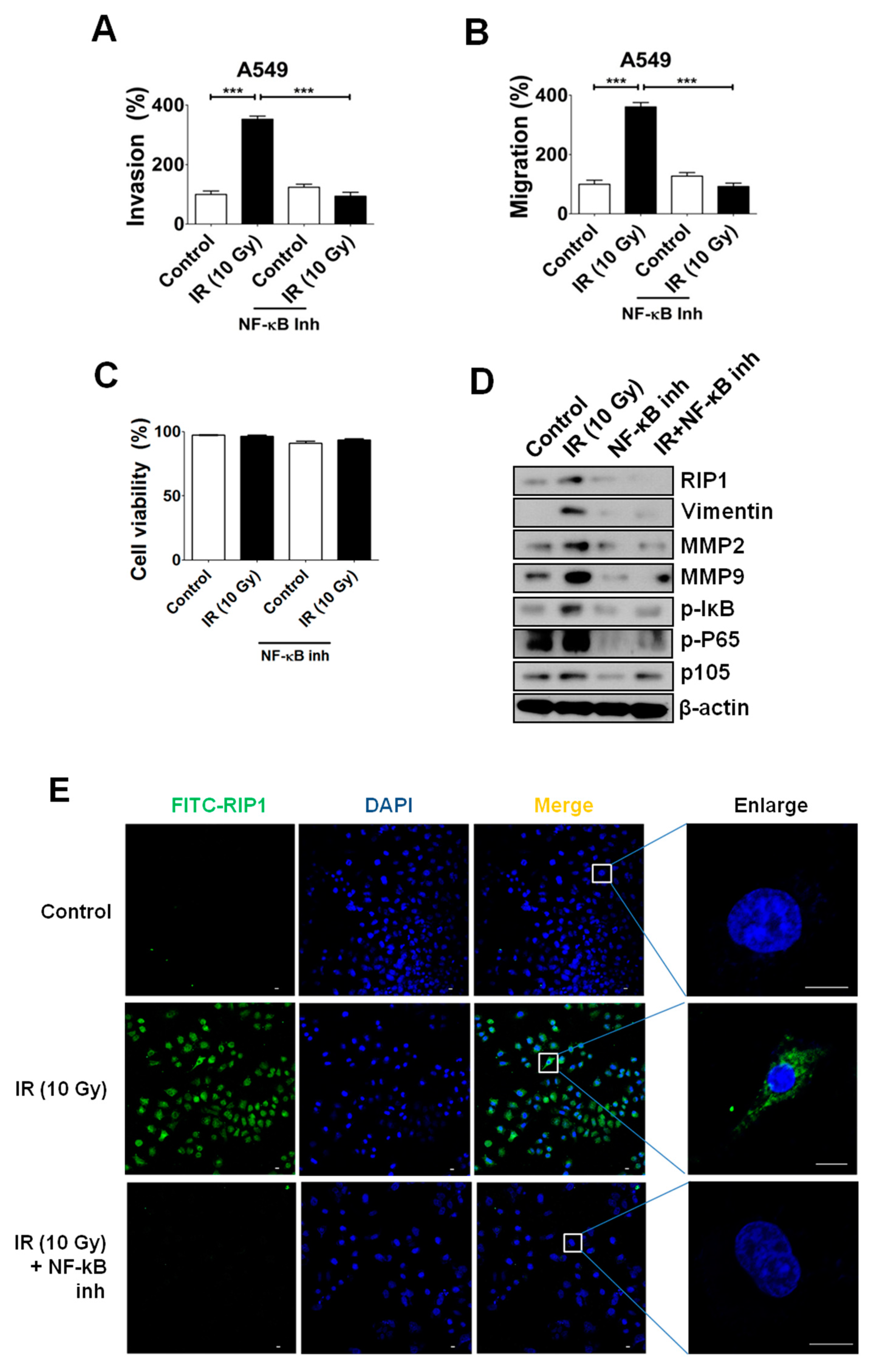
2.5. NF-κB Is Associated with RIP1 Expression and EMT in IR-Treated A549 Cells
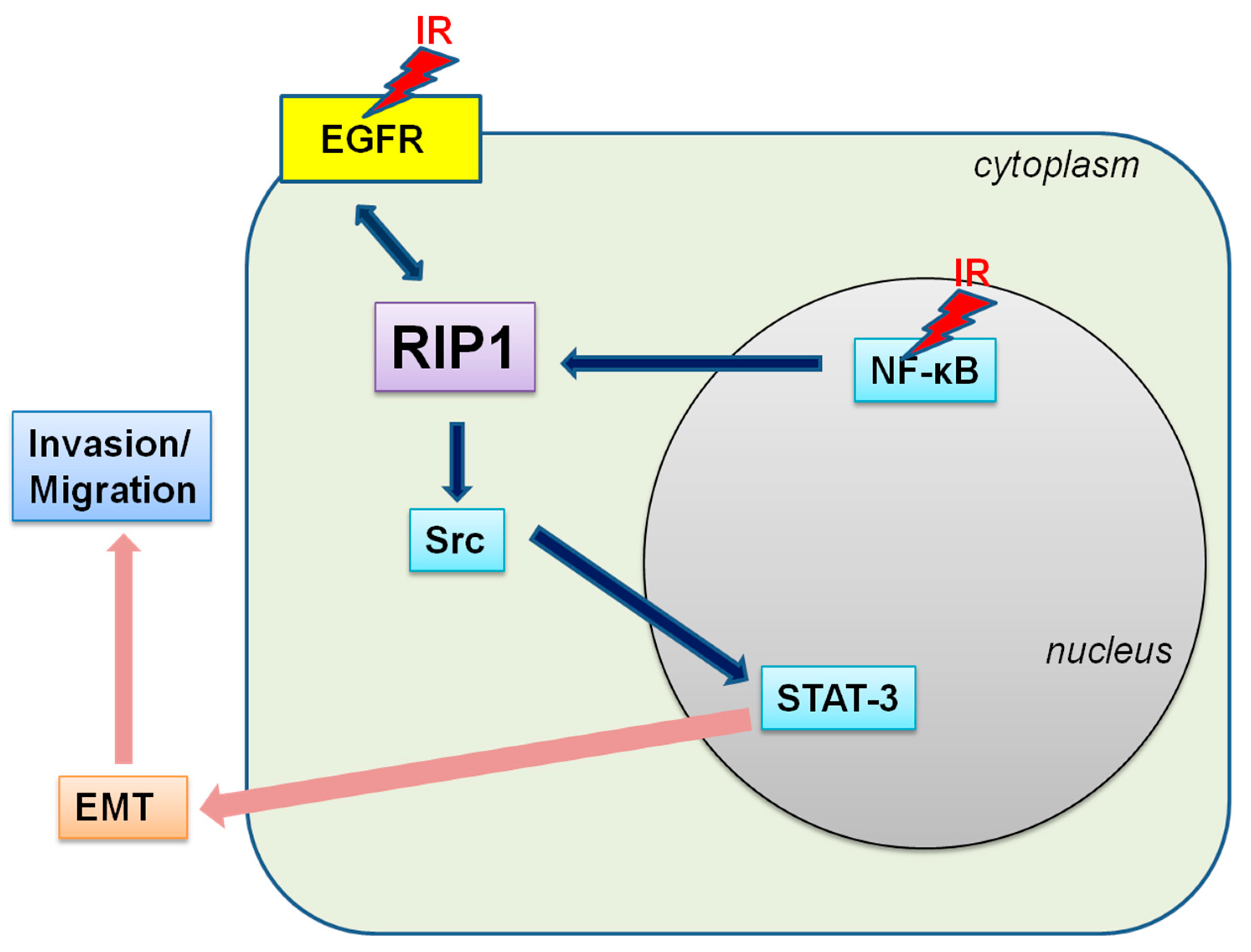
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Chemical Reagents
4.2. Treatment with γ-IR
4.3. Propidium Iodide (PI) Uptake Assay
4.4. Migration and Invasion Assays
4.5. Immunoblot Analysis
4.6. Immunofluorescence (IF) Staining
4.7. Small Interference RNA and Transfection
4.8. Xenograft Tumorigenicity Assays and Immunohistochemistry
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| IR | γ-ionizing radiation |
| RIP | Receptor-interacting protein |
| MMP | Matrix metalloprotease |
| EMT | Epithelial-mesenchymal transition |
| EGFR | Epidermal growth factor receptor |
| PI | Propidium iodide |
| IF | Immunofluorescence |
| SD | Standard deviation |
| Nec | Necrostatin |
| TME | Tumor microenvironment |
| ECM | Extracellular matrix |
References
- Siegel, R.; Ma, J.; Zou, Z.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2014, 64, 929. [Google Scholar]
- Anglim, P.P.; Alonzo, T.A.; Laird-Offringa, I.A. DNA methylation-based biomarkers for early detection of non-small cell lung cancer: An update. Mol. Cancer 2008, 7, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazzari, C.; Spitaleri, G.; Catania, C.; Barberis, M.; Noberasco, C.; Santarpia, M.; Delmonte, A.; Toffalorio, F.; Conforti, F.; De Pas, T.M. Targeting ALK in patients with advanced Non Small Cell Lung Cancer: Biology, diagnostic and therapeutic options. Crit. Rev. Oncol. Hematol. 2014, 89, 358–365. [Google Scholar] [CrossRef]
- Begg, A.C.; Stewart, F.A.; Vens, C. Strategies to improve radiotherapy with targeted drugs. Nat. Rev. Cancer 2011, 11, 239–253. [Google Scholar] [CrossRef]
- Park, C.M.; Park, M.J.; Kwak, H.J.; Lee, H.C.; Kim, M.S.; Lee, S.H.; Park, I.C.; Rhee, C.H.; Hong, S.I. Ionizing radiation enhances matrix metalloproteinase2 secretion and invasion of glioma cells through Src/epidermal growth factor receptor mediated p38/Akt and phosphatidylinositol 3kinase/Akt signaling pathways. Cancer Res. 2006, 66, 8511–8519. [Google Scholar] [CrossRef] [Green Version]
- Ebos, J.M.L. Prodding the Beast: Assessing the Impact of Treatment-Induced Metastasis. Cancer Res. 2015, 75, 3427–3435. [Google Scholar] [CrossRef] [Green Version]
- Biard, D.S.; Martin, M.; Rhun, Y.L.; Duthu, A.; Lefaix, J.L.; May, E.; May, P. Concomitant p53 gene mutation and increased radiosensitivity in rat lung embryo epithelial cells during neoplastic development. Cancer Res. 1994, 54, 3361–3364. [Google Scholar]
- Lee, J.-U.; Hosotani, R.; Wada, M.; Doi, R.; Kosiba, T.; Fujimoto, K.; Miyamoto, Y.; Tsuji, S.; Nakajima, S.; Nishimura, Y.; et al. Role of Bcl-2 family proteins (Bax, Bcl-2 and Bcl-X) on cellular susceptibility to radiation in pancreatic cancer cells. Eur. J. Cancer 1999, 35, 1374–1380. [Google Scholar] [CrossRef]
- Vivanco, I.; Sawyers, C.L. The phosphatidylinositol 3-Kinase–AKT pathway in human cancer. Nat. Rev. Cancer 2002, 2, 489–501. [Google Scholar] [CrossRef]
- Cheng, J.C.; Chou, C.H.; Kuo, M.L.; Hsieh, C.J. Radiation enhanced hepatocellular carcinoma cell invasion with MMP9 expression through PI3K/Akt/NFkappaB signal transduction pathway. Oncogene 2006, 25, 7009–7018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, J.N.; Kang, G.Y.; Lee, S.S.; Kim, J.; Bae, I.H.; Hwang, S.G.; Um, H.D. Bcl-XL and STAT3 mediate malignant actions of gamma irradiation in lung cancer cells. Cancer Sci. 2010, 101, 1417–1423. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, K.M.; Li, J.J. NF-κB-mediated adaptive resistance to ionizing radiation. Free Radic. Biol. Med. 2007, 44, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piva, R.; Belardo, G.; Santoro, M.G. NF-κB: A Stress-Regulated Switch for Cell Survival. Antioxid. Redox Signal. 2006, 8, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.-C.; Hung, M.-C. Induction of Akt Activity by Chemotherapy Confers Acquired Resistance. J. Formos. Med. Assoc. 2009, 108, 180–194. [Google Scholar] [CrossRef] [Green Version]
- Kraus, A.C.; Ferber, I.; Bachmann, S.O.; Specht, H.; Wimmel, A.; Gross, M.W.; Schlegel, J.; Suske, G.; Schuermann, M. In vitro chemo- and radio-resistance in small cell lung cancer correlates with cell adhesion and constitutive activation of AKT and MAP kinase pathways. Oncogene 2002, 21, 8683–8695. [Google Scholar] [CrossRef] [Green Version]
- Criswell, T.; Leskov, K.; Miyamoto, S.; Luo, G.; Boothman, D.A. Transcription factors activated in mammalian cells after clinically relevant doses of ionizing radiation. Oncogene 2003, 22, 5813–5827. [Google Scholar] [CrossRef] [Green Version]
- Cho, J.H.; Hong, W.G.; Jung, Y.J.; Lee, J.; Lee, E.; Hwang, S.G.; Um, H.D.; Park, J.K. Γ-Ionizing radiation induced activation of the EGFR/p38/ERKSTAT3/CREB1EMT pathway promotes the migration/invasion of non small cell lung cancer cells and is inhibited by podophyllotoxin acetate. Tumor Biol. 2016, 37, 7315–7325. [Google Scholar] [CrossRef]
- Festjens, N.; Vanden Berghe, T.V.; Cornelis, S.; Vandenabeele, P. RIP1, a kinase on the crossroads of a cell’s decision to live or die. Cell Death Differ. 2007, 14, 400–410. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Lin, J.; Han, J. Receptor-interacting protein (RIP) kinase family. Cell. Mol. Immunol. 2010, 7, 243–249. [Google Scholar] [CrossRef] [Green Version]
- Humphries, F.; Yang, S.; Wang, B.; Moynagh, F.N. RIP kinases: Key decision makers in cell death and innate immunity. Cell Death Differ. 2014, 22, 225–236. [Google Scholar] [CrossRef] [Green Version]
- Silke, J.; Rickard, J.A.; Gerlic, M.; Gerlic, M. The diverse role of RIP kinases in necroptosis and inflammation. Nat. Immunol. 2015, 16, 689–697. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Chen, X.; Wang, X.; Li, X.; Du, Q.; Hong, H.; Tang, N.; She, F.; Chen, Y. Expression of the RIP-1 Gene and its Role in Growth and Invasion of Human Gallbladder Carcinoma. Cell. Physiol. Biochem. 2014, 34, 1152–1165. [Google Scholar] [CrossRef] [PubMed]
- Ofengeim, D.; Yuan, J. Regulation of RIP1 kinase signalling at the crossroads of inflammation and cell death. Nat. Rev. Mol. Cell Biol. 2013, 14, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Azijli, K.; Yuvaraj, S.; Peppelenbosch, M.P.; Würdinger, T.; Dekker, H.; Joore, J.; van Dijk, E.; Quax, W.J.; Peters, S.; de Jong, G.J.; et al. Kinome profiling of noncanonical TRAIL signaling reveals RIP1/Src/STAT3 dependent invasion in resistant non small cell lung cancer cells. J. Cell Sci. 2012, 125, 4651–4661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molavipordanjani, S.M.; Hosseinimehr, S.J. The Role of NF-κB Inhibitors in Cell Response to Radiation. Curr. Med. Chem. 2016, 23, 3951–3963. [Google Scholar] [CrossRef]
- Veuger, S.J.; Hunter, J.E.; Durkacz, B.W. Ionizing radiation-induced NF-κB activation requires PARP-1 function to confer radioresistance. Oncogene 2008, 28, 832–842. [Google Scholar] [CrossRef] [Green Version]
- Vandenabeele, P.; Declercq, W.; Van Herreweghe, F.; Vanden Berghe, T.V. The Role of the Kinases RIP1 and RIP3 in TNF-Induced Necrosis. Sci. Signal. 2010, 3, re4. [Google Scholar] [CrossRef]
- Kesanakurti, D.; Chetty, C.; Maddirela, D.R.; Gujrati, M.; Rao, J.S. Essential role of cooperative NF-κB and Stat3 recruitment to ICAM-1 intronic consensus elements in the regulation of radiation-induced invasion and migration in glioma. Oncogene 2012, 32, 5144–5155. [Google Scholar] [CrossRef] [Green Version]
- Alexander, S.; Friedl, P. Cancer invasion and resistance: Interconnected processes of disease progression and therapy failure. Trends Mol. Med. 2012, 18, 13–26. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egeblad, M.; Nakasone, E.S.; Werb, Z. Tumors as Organs: Complex Tissues that Interface with the Entire Organism. Dev. Cell 2010, 18, 884–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMillin, D.U.W.; Negri, J.M.; Mitsiades, C.S. The role of tumour–stromal interactions in modifying drug response: Challenges and opportunities. Nat. Rev. Drug Discov. 2013, 12, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Correia, A.L.; Bissell, M.J. The tumor microenvironment is a dominant force in multidrug resistance. Drug Resist. Updates 2012, 15, 39–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, R.F.; Maity, A. Radiotherapy and the Tumor Microenvironment: Mutual Influence and Clinical Implications. Adv. Exp. Med. Biol. 2013, 772, 147–165. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Kuonen, F.; Secondini, C.; Rüegg, C. Molecular Pathways: Emerging Pathways Mediating Growth, Invasion, and Metastasis of Tumors Progressing in an Irradiated Microenvironment. Clin. Cancer Res. 2012, 18, 5196–5202. [Google Scholar] [CrossRef] [Green Version]
- Rüegg, C.; Monnier, Y.; Kuonen, F.; Imaizumi, N. Radiation-induced modifications of the tumor microenvironment promote metastasis. Bull Cancer 2011, 98, 47–57. [Google Scholar] [CrossRef] [Green Version]
- Park, J.K.; Jang, S.J.; Kang, S.W.; Park, S.; Hwang, S.-G.; Kim, W.-J.; Kang, J.H.; Um, H.-D. Establishment of animal model for the analysis of cancer cell metastasis during radiotherapy. Radiat. Oncol. 2012, 7, 153. [Google Scholar] [CrossRef] [Green Version]
- Von Essen, C.F. Radiation enhancement of metastasis: A review. Clin. Exp. Metastasis 1991, 9, 77–104. [Google Scholar] [CrossRef]
- Ngwa, W.; Irabor, O.C.; Schoenfeld, J.D.; Hesser, J.; DeMaria, S.; Formenti, S.C. Using immunotherapy to boost the abscopal effect. Nat. Rev. Cancer 2018, 18, 313–322. [Google Scholar] [CrossRef]
- Declercq, W.; Berghe, T.V.; Vandenabeele, P. RIP kinases at the crossroads of cell death and survival. Cell 2009, 138, 229–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pao, W.; Chmielecki, J. Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat. Rev. Cancer 2010, 10, 760–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Normanno, N.; De Luca, A.; Bianco, C.; Strizzi, L.; Mancino, M.; Maiello, M.R.; Carotenuto, A.; De Feo, G.; Caponigro, F.; Salomon, D.S. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 2006, 366, 2–16. [Google Scholar] [CrossRef]
- Irwin, M.E.; Bohin, N.; Boerner, J.L. Src family kinases mediate epidermal growth factor receptor signaling from lipid rafts in breast cancer cells. Cancer Biol. Ther. 2011, 12, 718–726. [Google Scholar] [CrossRef] [Green Version]
- Karin, M.; Cao, Y.; Greten, F.R.; Li, Z.-W. NF-κB in cancer: From innocent bystander to major culprit. Nat. Rev. Cancer 2002, 2, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Shen, S.; Verma, I.M. NF-κB, an active player in human cancers. Cancer Immunol. Res. 2014, 2, 823–830. [Google Scholar] [CrossRef] [Green Version]
- Karin, M. NF-κB as a Critical Link Between Inflammation and Cancer. Cold Spring Harb. Perspect. Biol. 2009, 1, a000141. [Google Scholar] [CrossRef]
- Hoesel, B.; Schmid, J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [Green Version]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, A.-R.; Cho, J.H.; Lee, N.-G.; Song, J.-Y.; Hwang, S.-G.; Lee, D.-H.; Um, H.-D.; Park, J.K. RIP1 Is a Novel Component of γ-ionizing Radiation-Induced Invasion of Non-Small Cell Lung Cancer Cells. Int. J. Mol. Sci. 2020, 21, 4584. https://doi.org/10.3390/ijms21134584
Kang A-R, Cho JH, Lee N-G, Song J-Y, Hwang S-G, Lee D-H, Um H-D, Park JK. RIP1 Is a Novel Component of γ-ionizing Radiation-Induced Invasion of Non-Small Cell Lung Cancer Cells. International Journal of Molecular Sciences. 2020; 21(13):4584. https://doi.org/10.3390/ijms21134584
Chicago/Turabian StyleKang, A-Ram, Jeong Hyun Cho, Na-Gyeong Lee, Jie-Young Song, Sang-Gu Hwang, Dae-Hee Lee, Hong-Duck Um, and Jong Kuk Park. 2020. "RIP1 Is a Novel Component of γ-ionizing Radiation-Induced Invasion of Non-Small Cell Lung Cancer Cells" International Journal of Molecular Sciences 21, no. 13: 4584. https://doi.org/10.3390/ijms21134584