The Inescapable Effects of Ribosomes on In-Cell NMR Spectroscopy and the Implications for Regulation of Biological Activity

Abstract
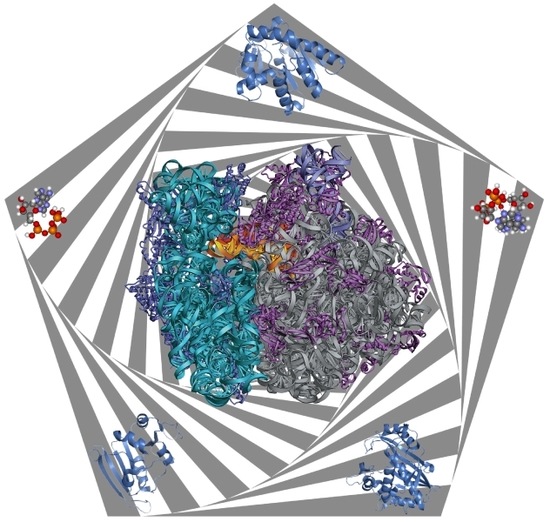
:
1. Introduction
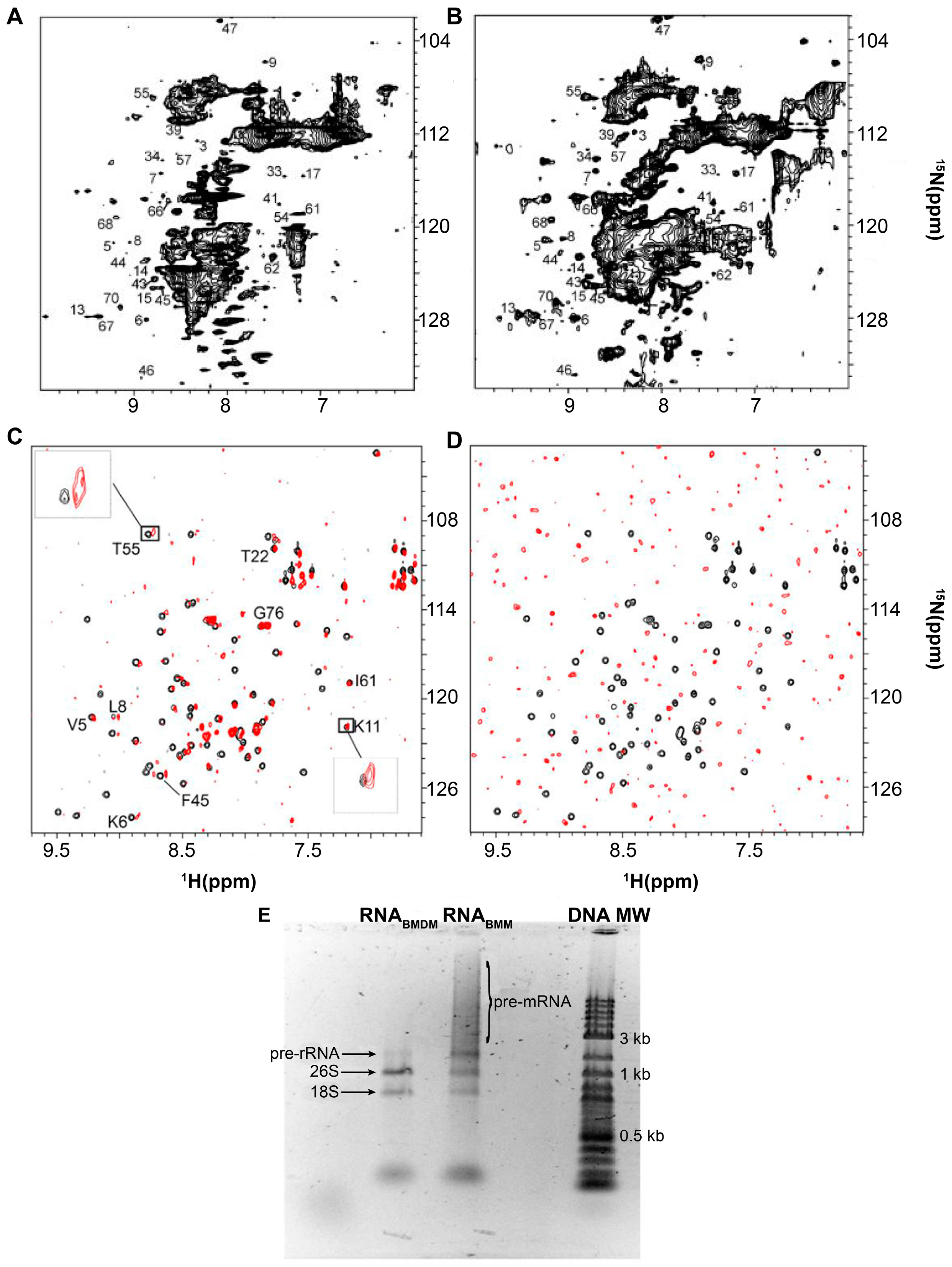
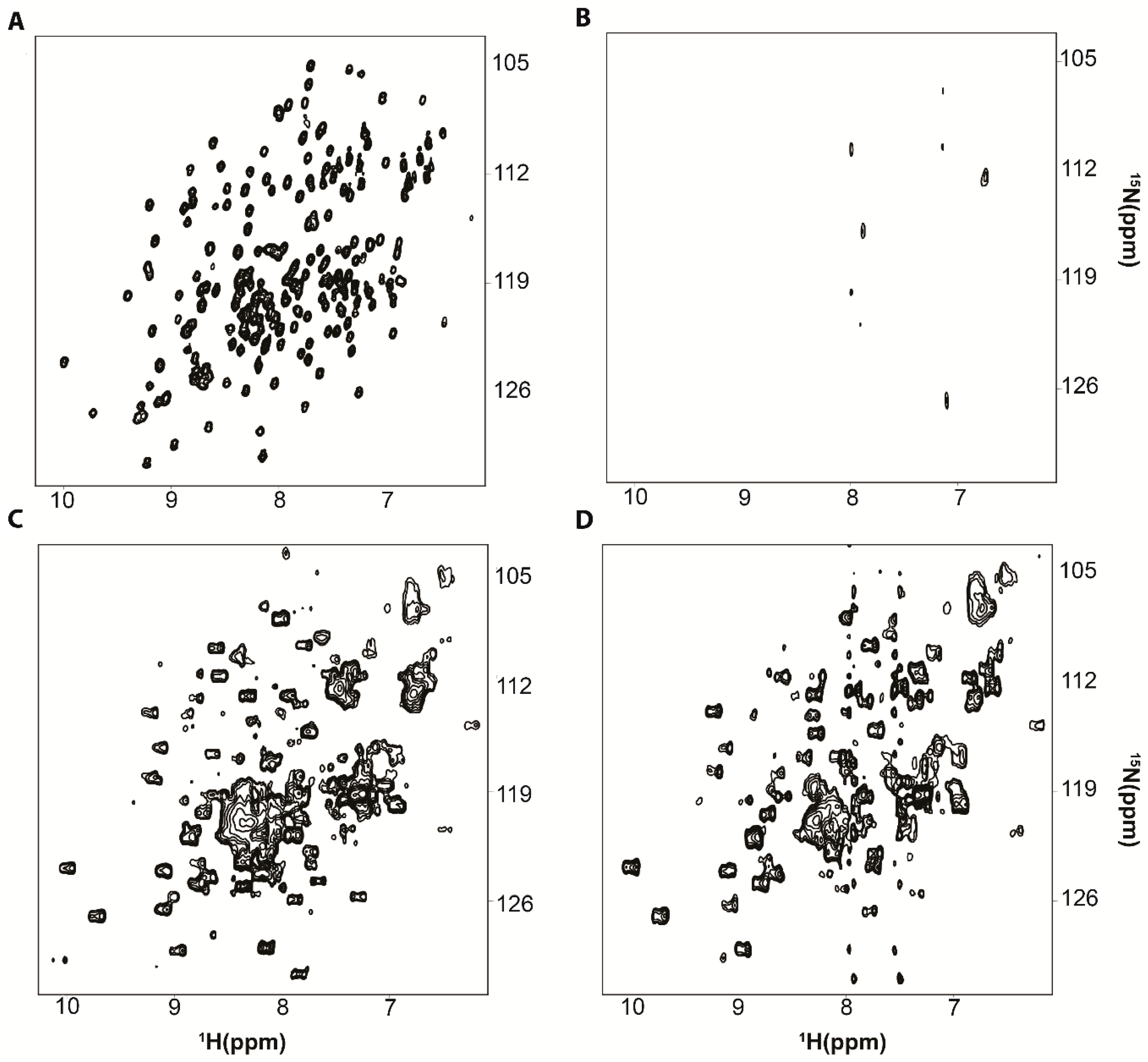
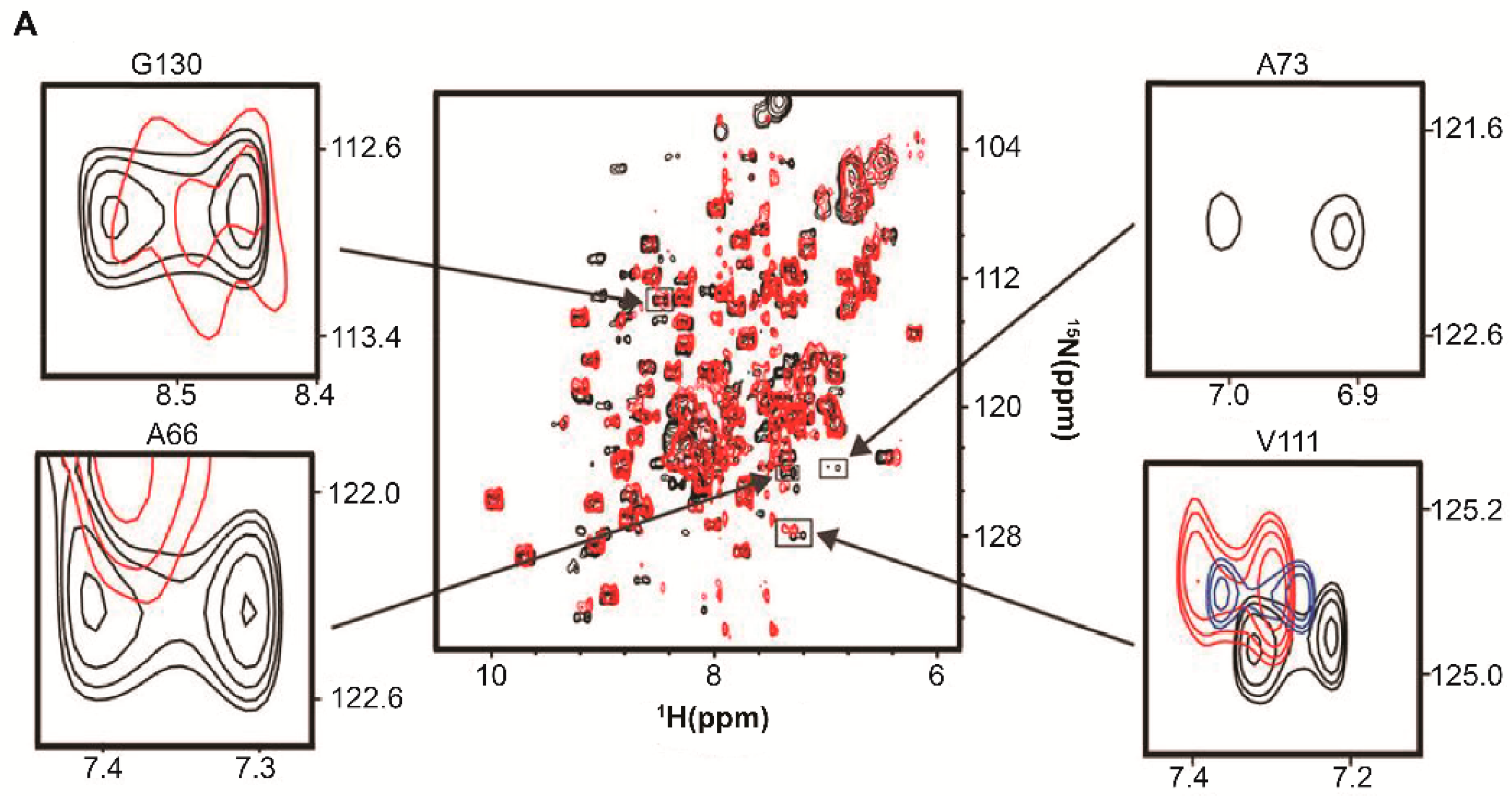
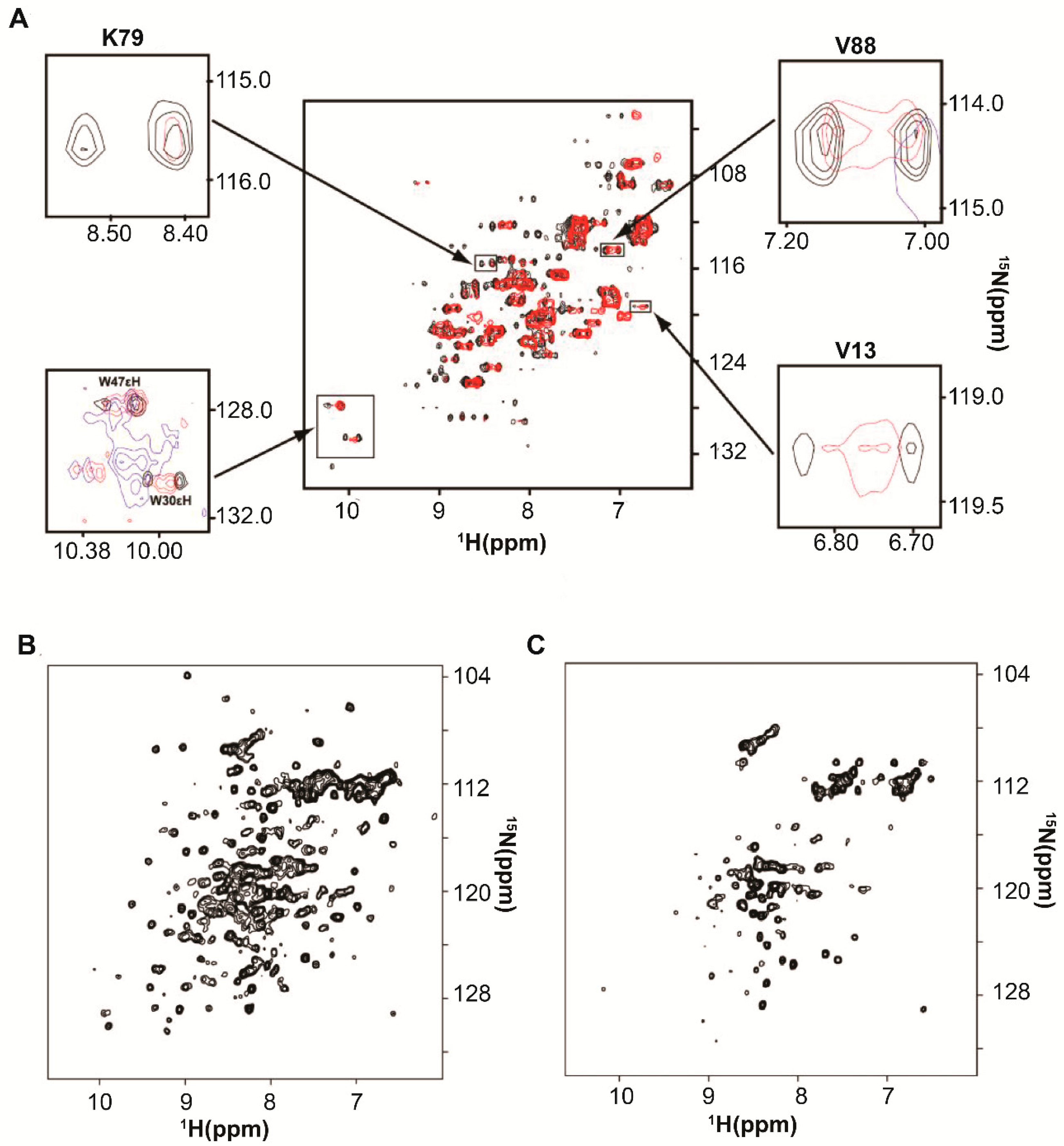
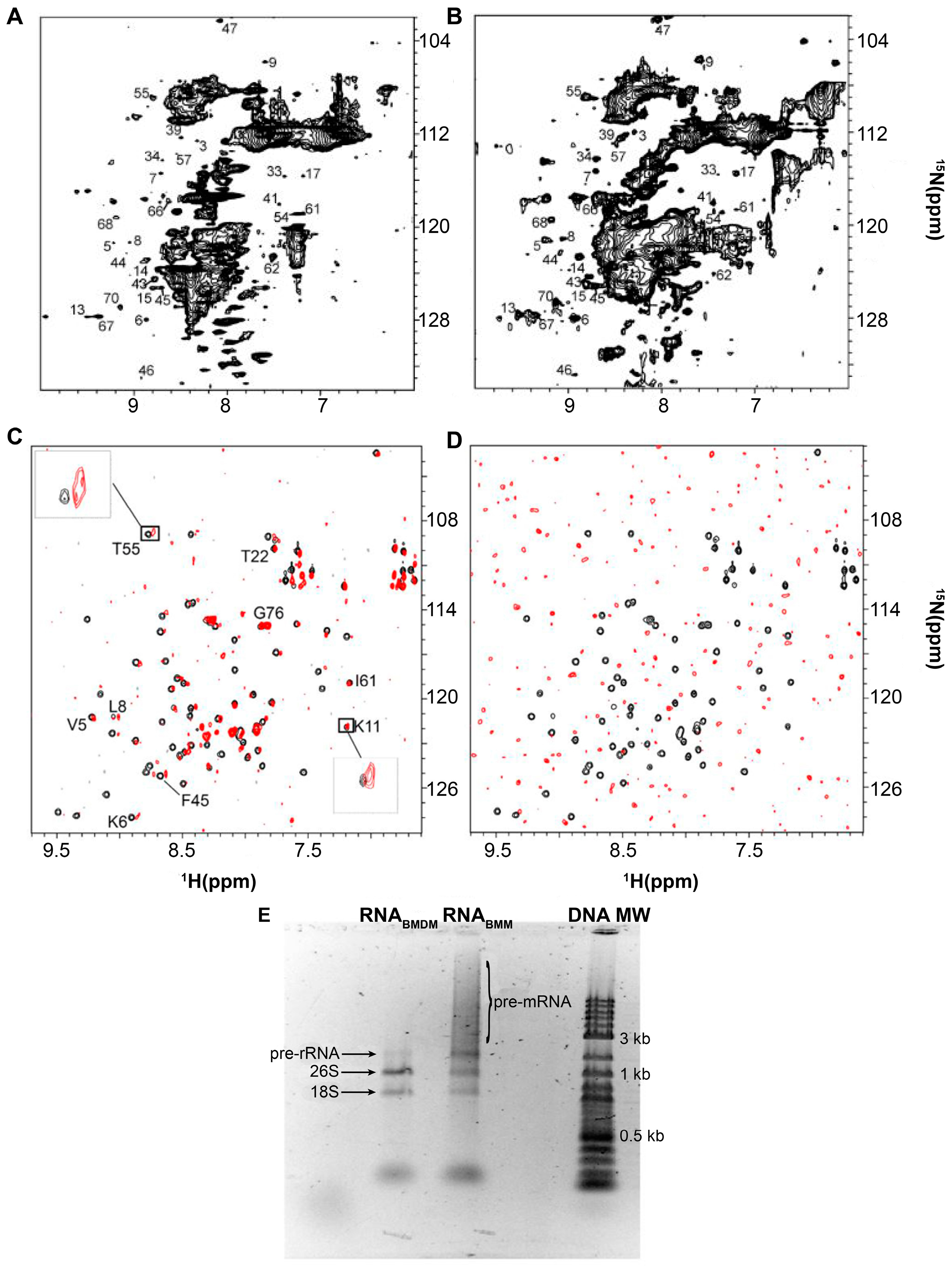
2. Protein-RNA Interactions Broaden Target Protein NMR Spectra
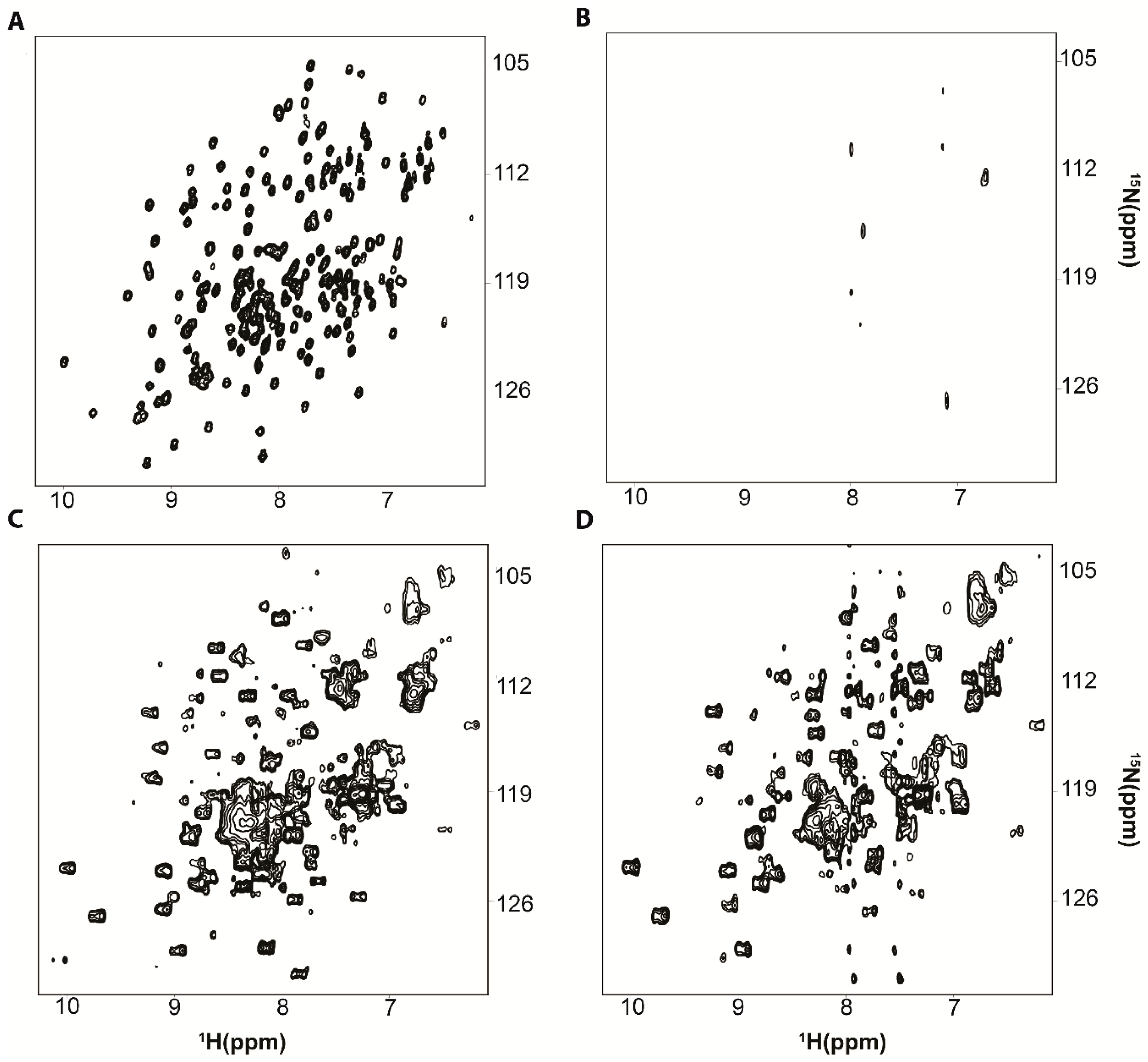
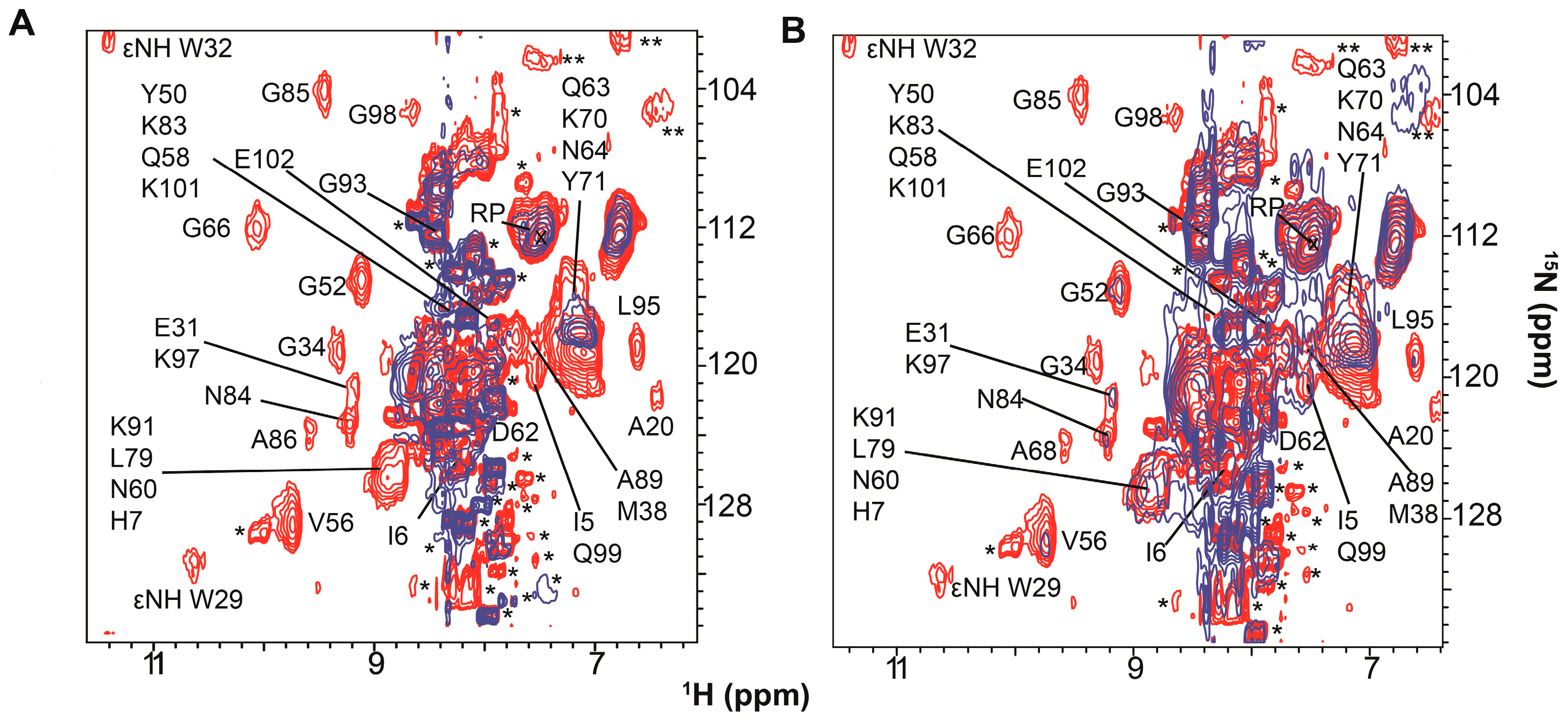
3. Resolving Target Protein Bound to RNA
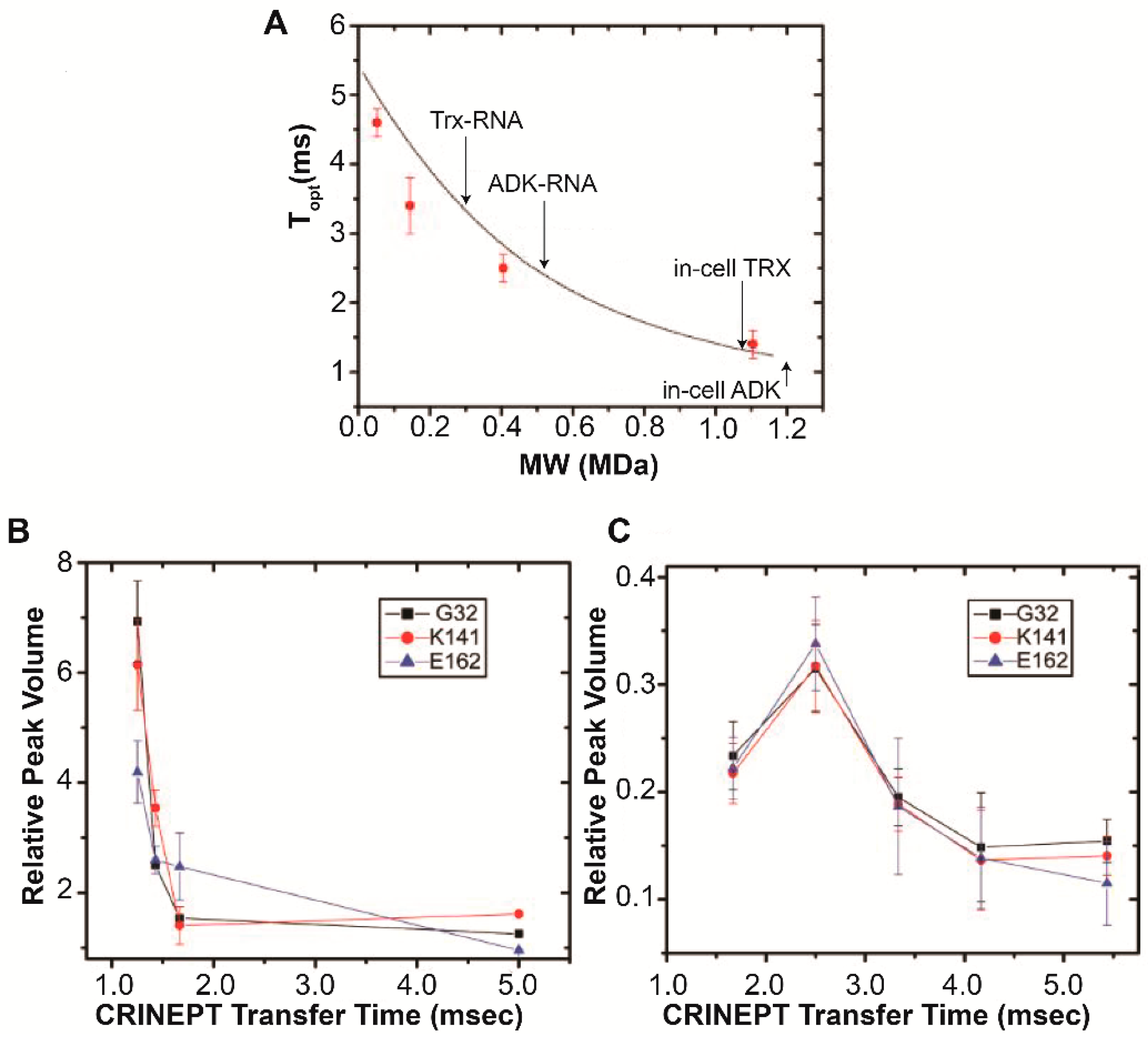
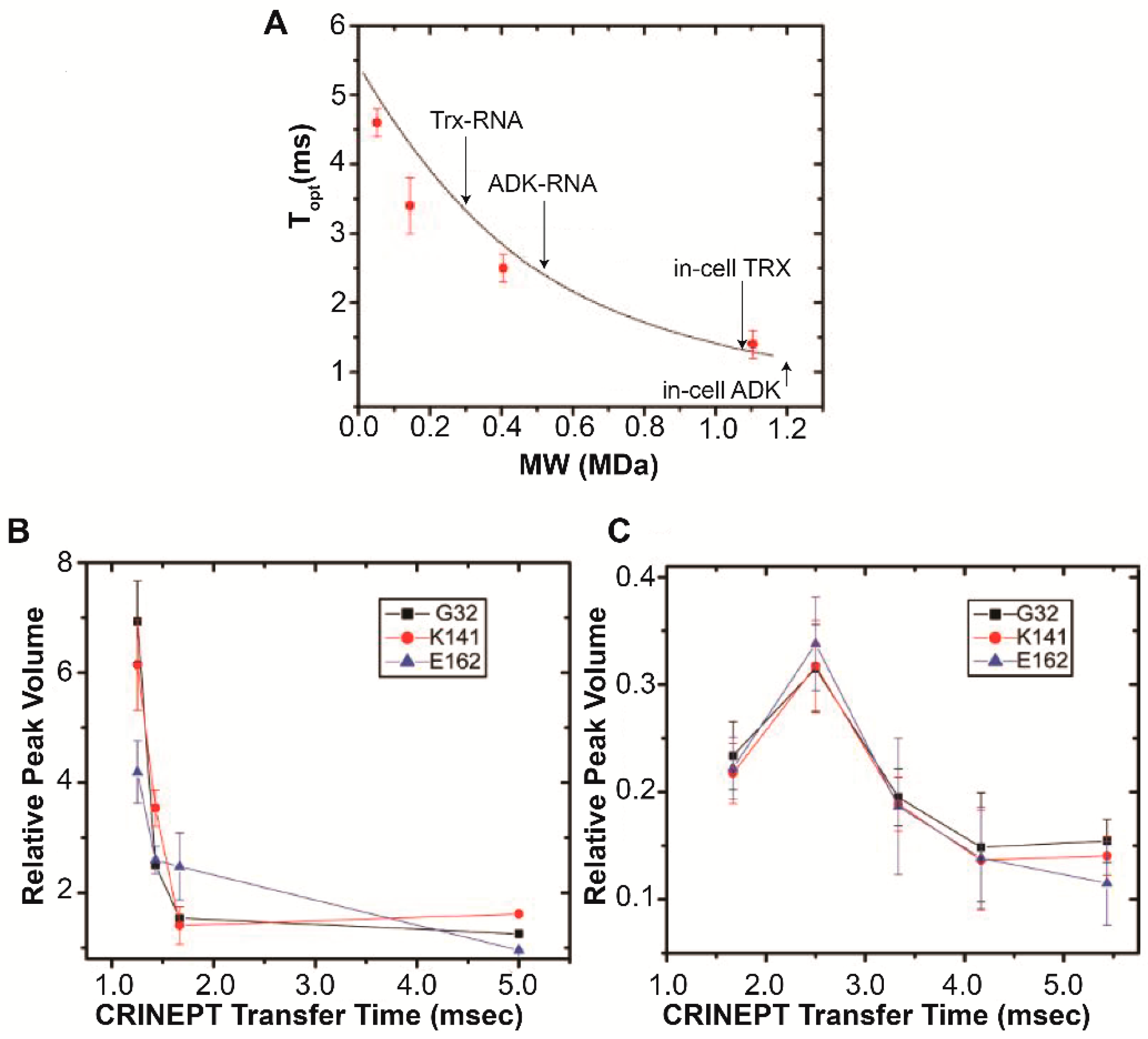
4. Target Protein-RNA Complexes Exhibit Megadalton Apparent Molecular Masses
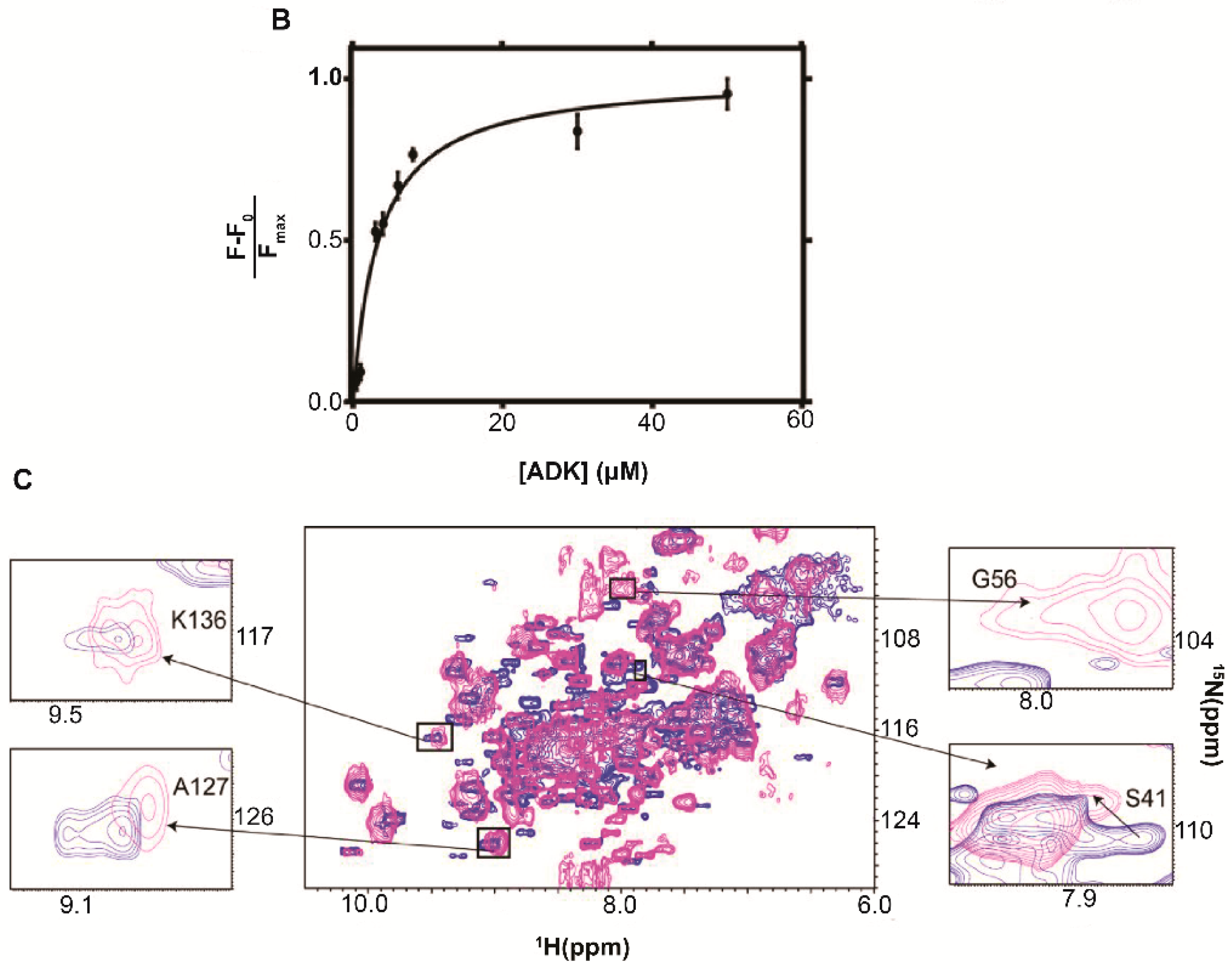
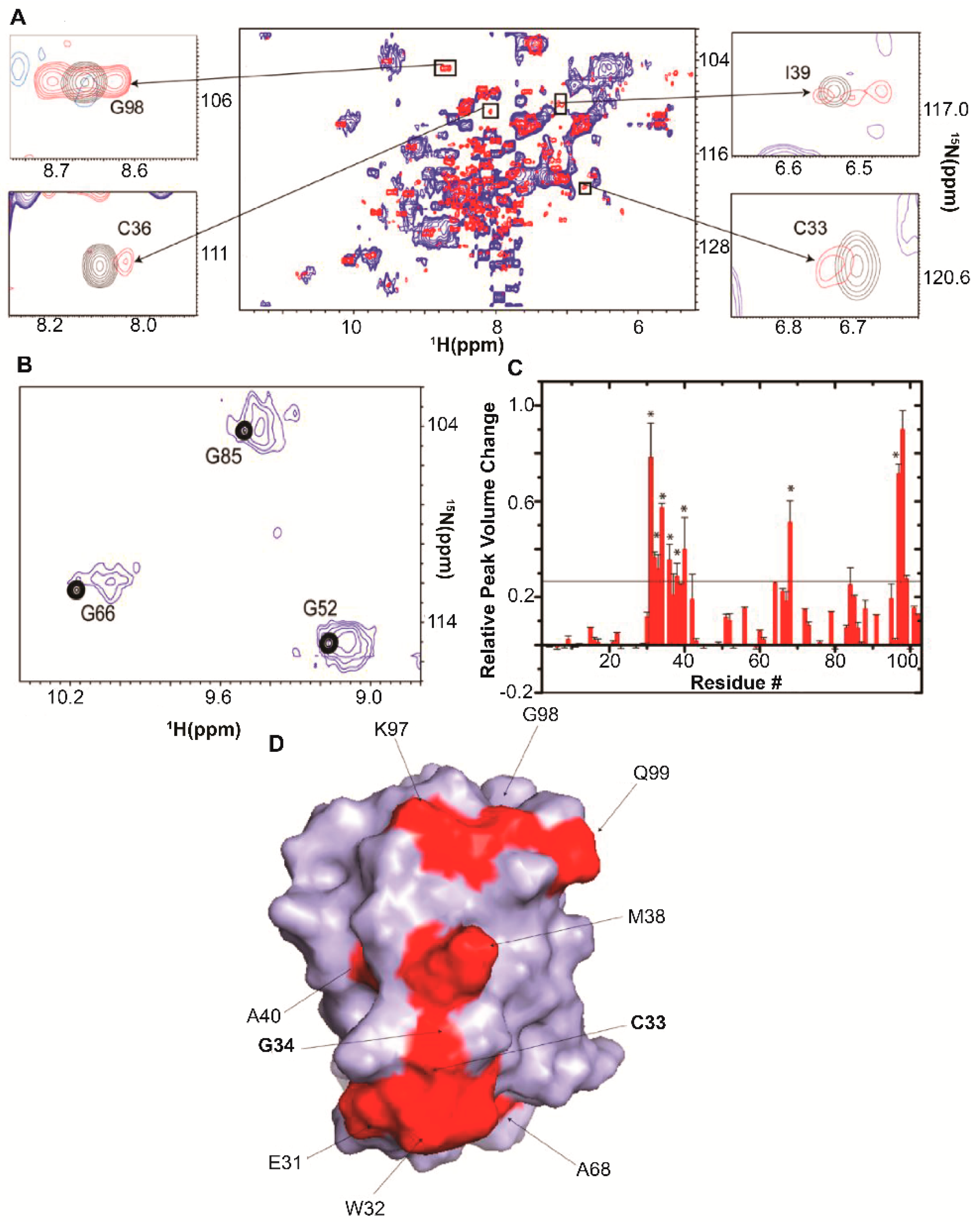
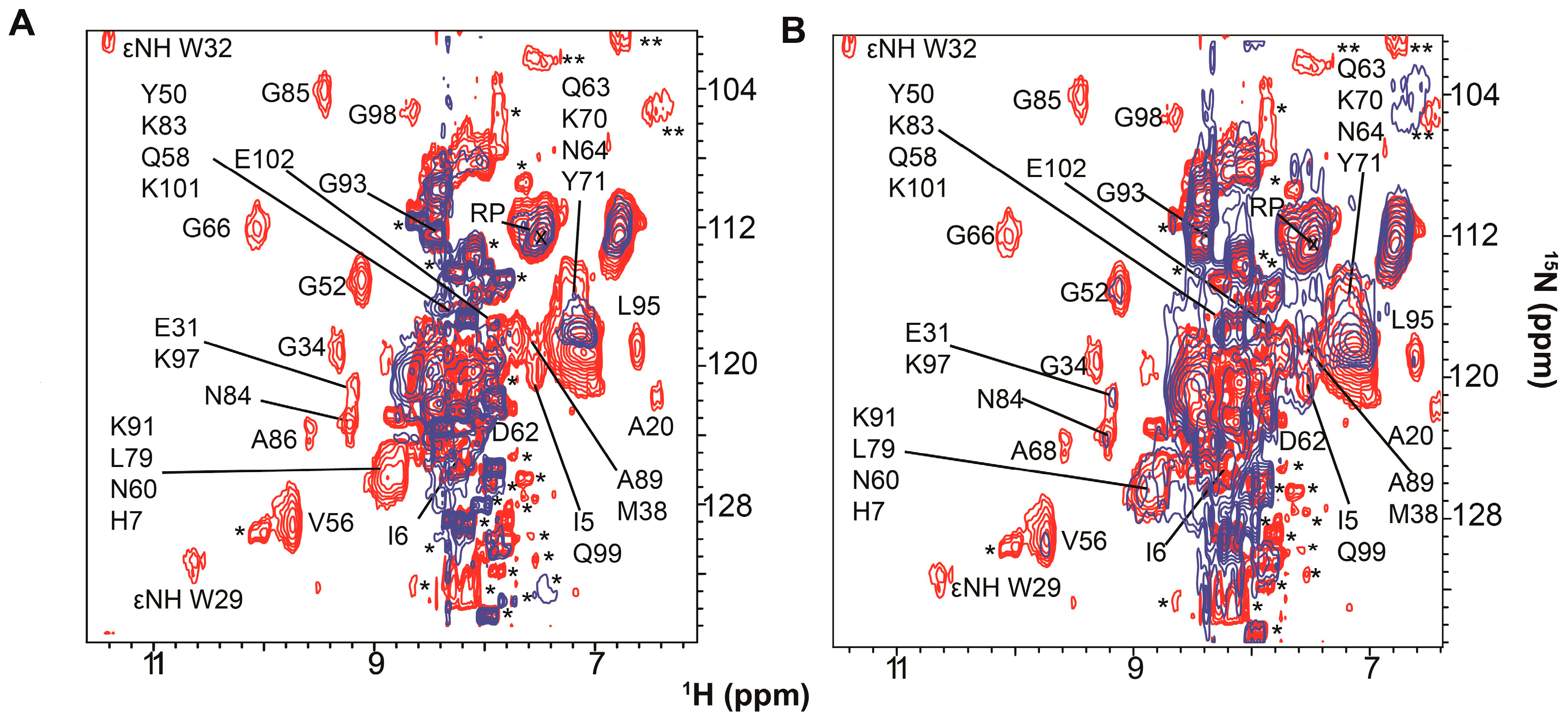
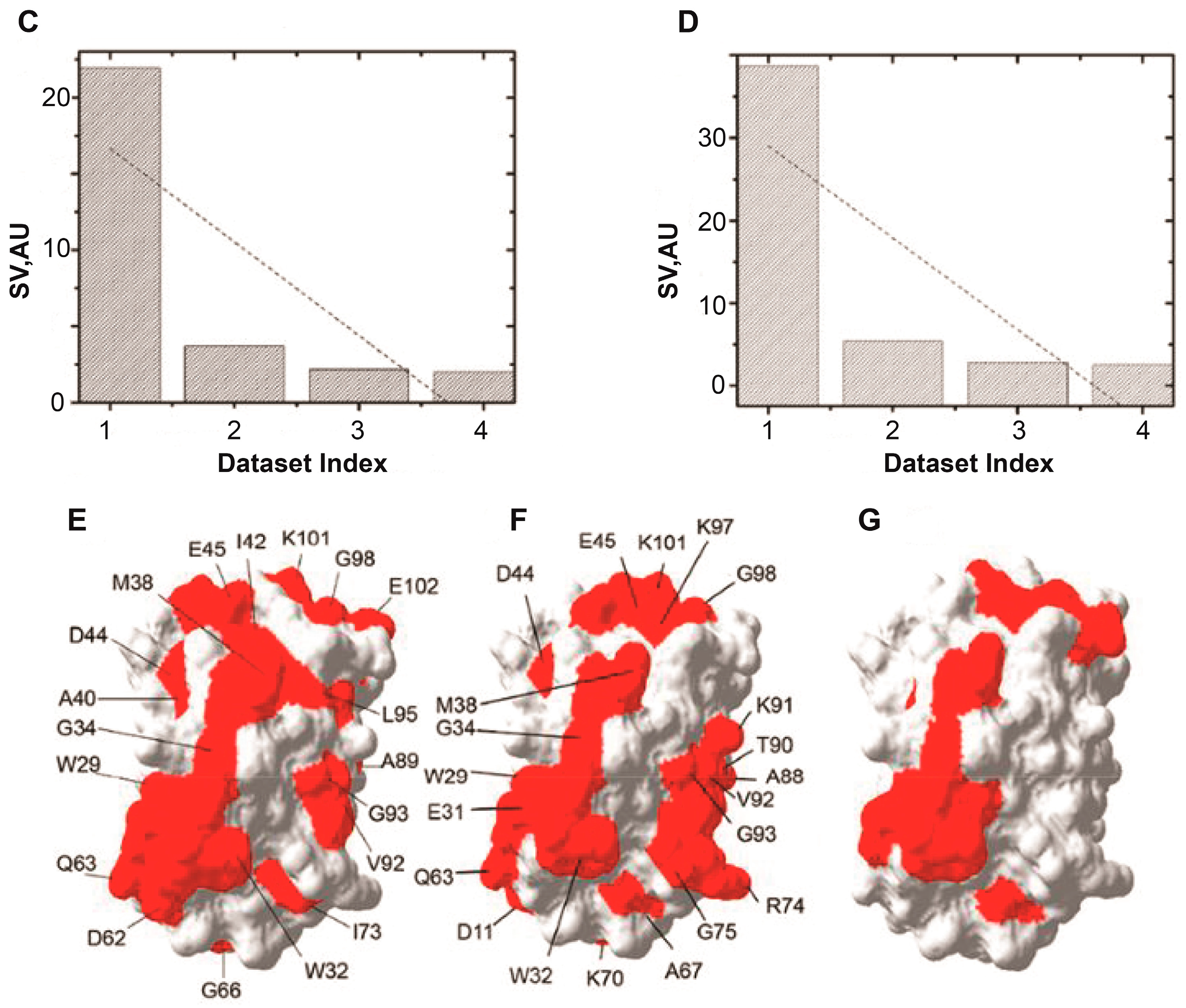
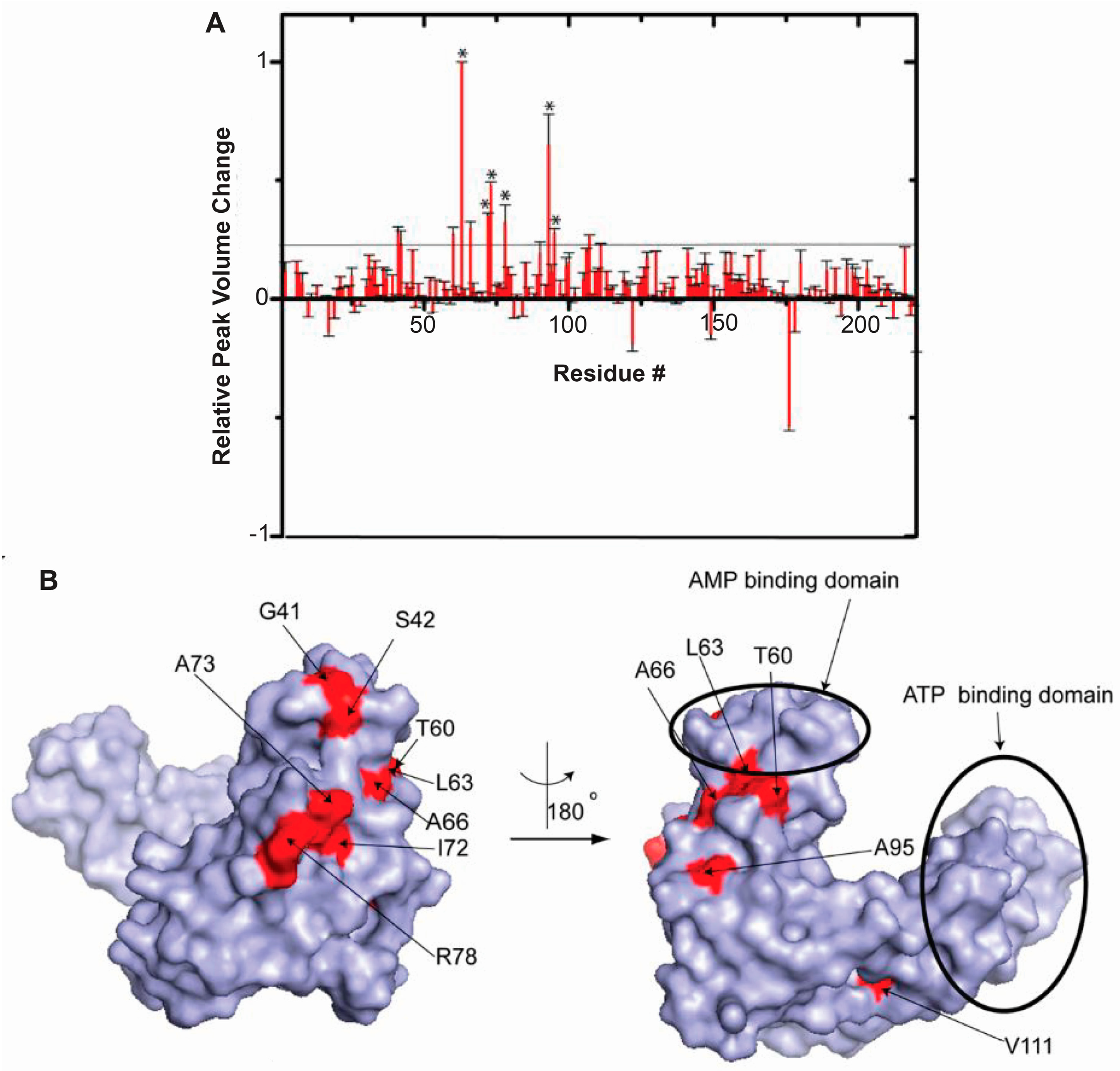
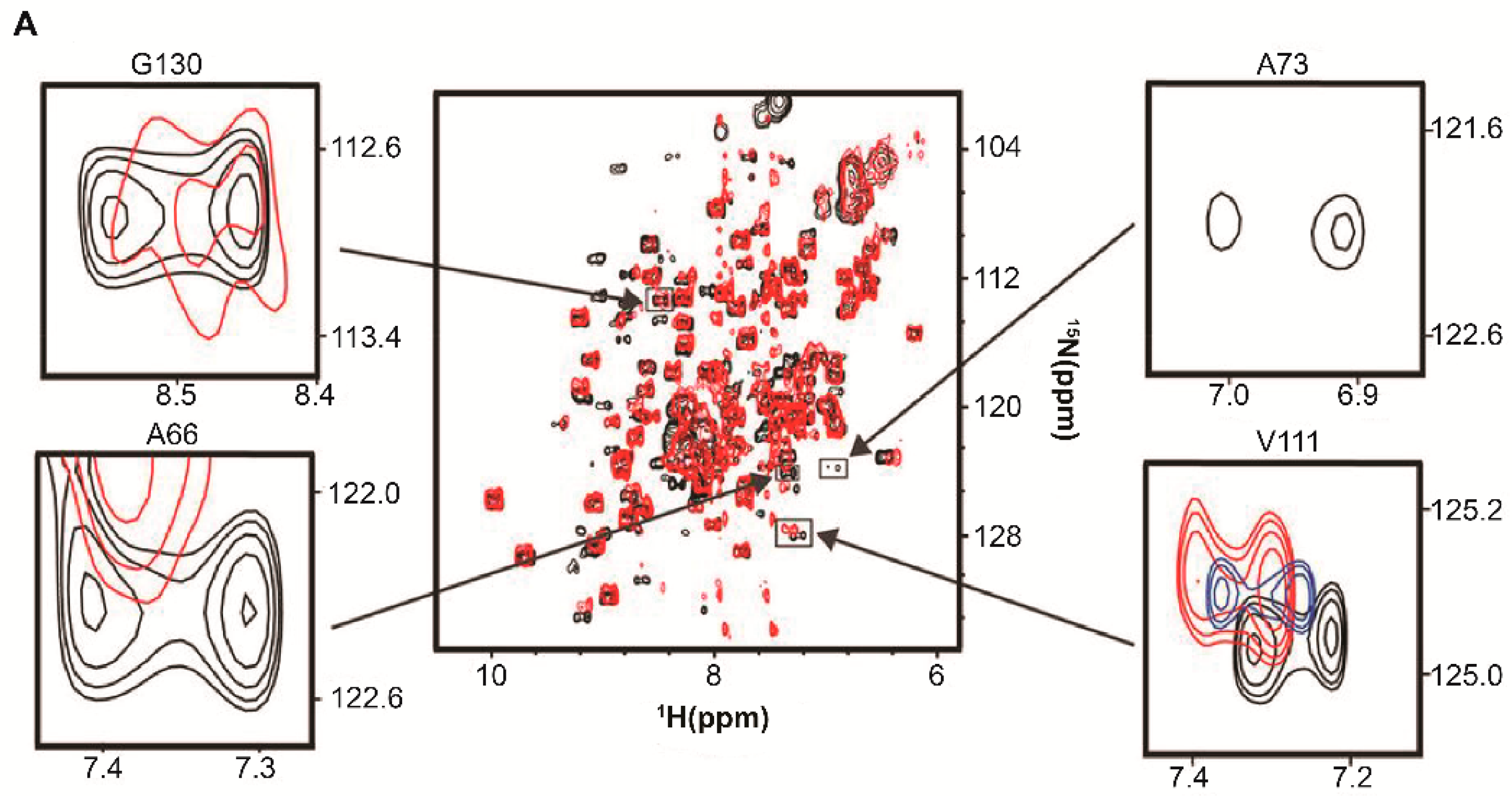
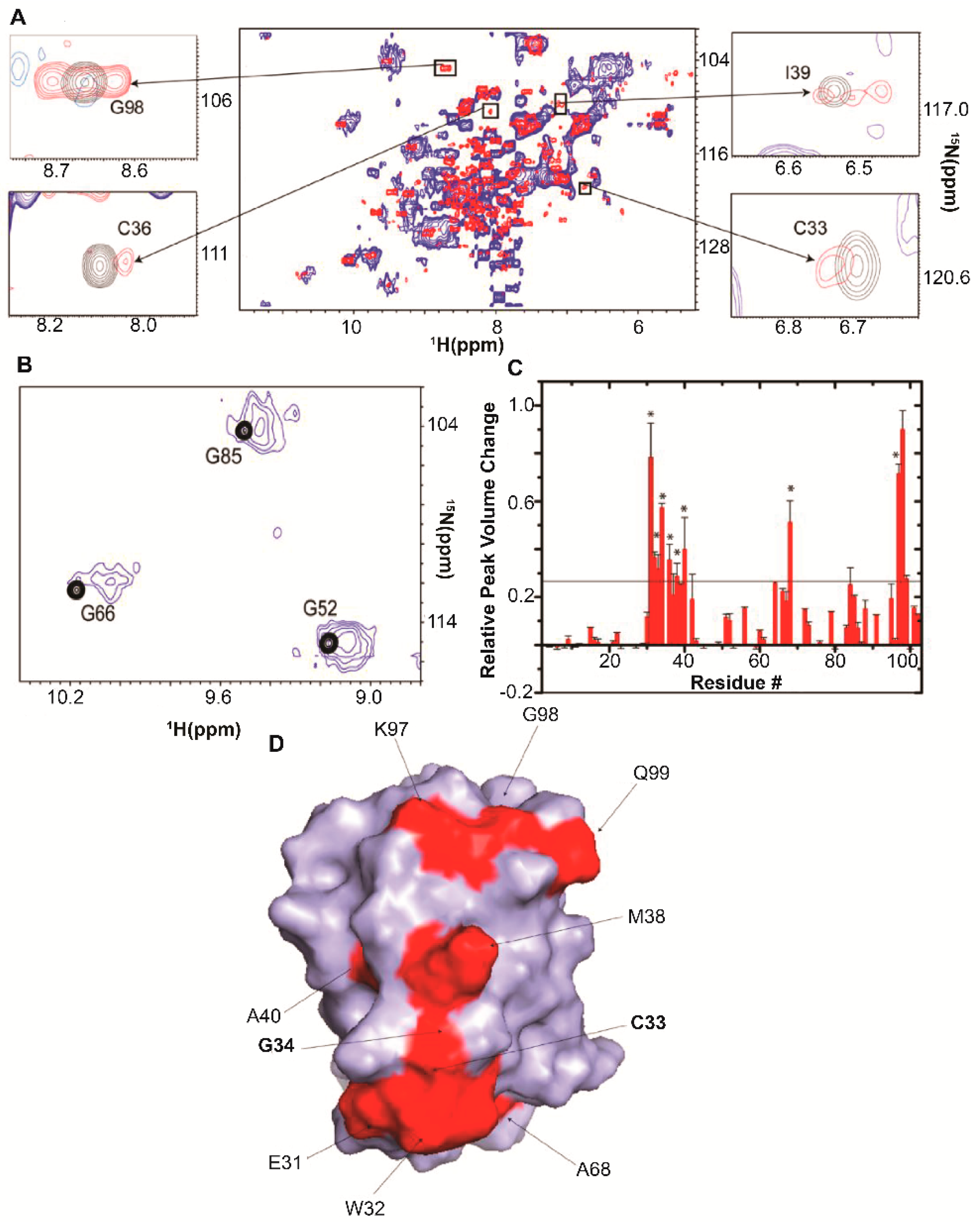
5. RNA-Mediated Quinary Interaction Surfaces
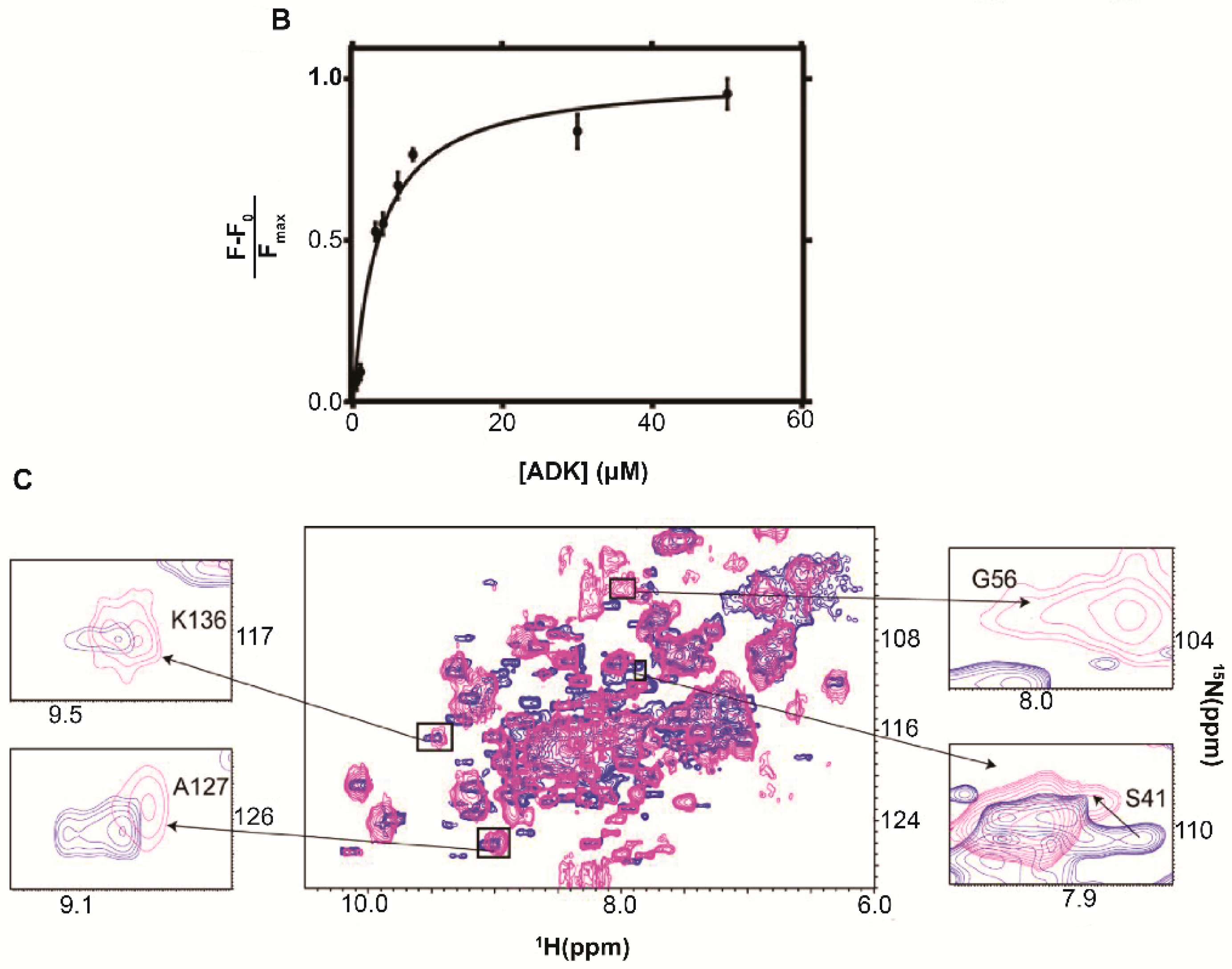
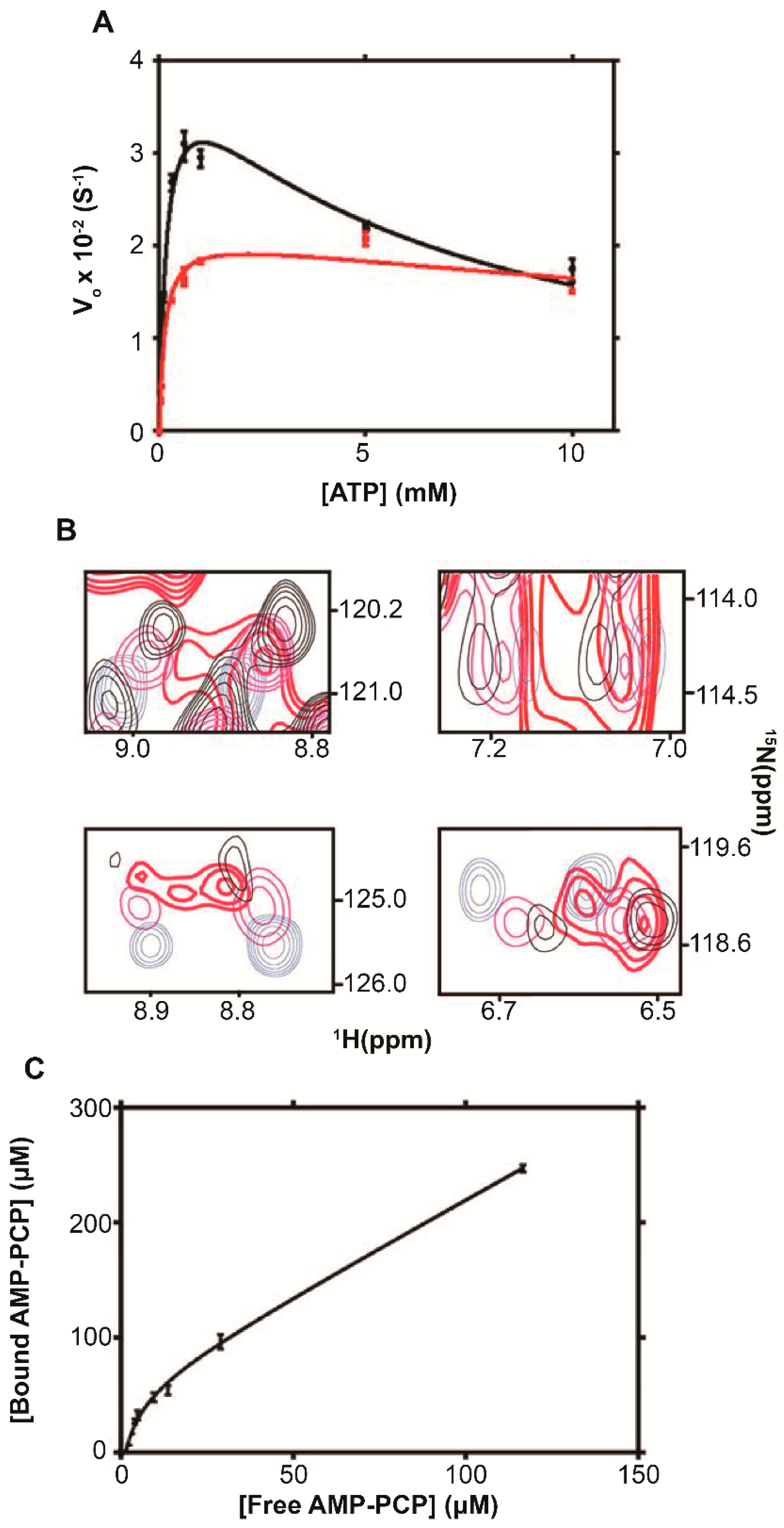
5.1. Adenylate Kinase
5.2. Thioredoxin
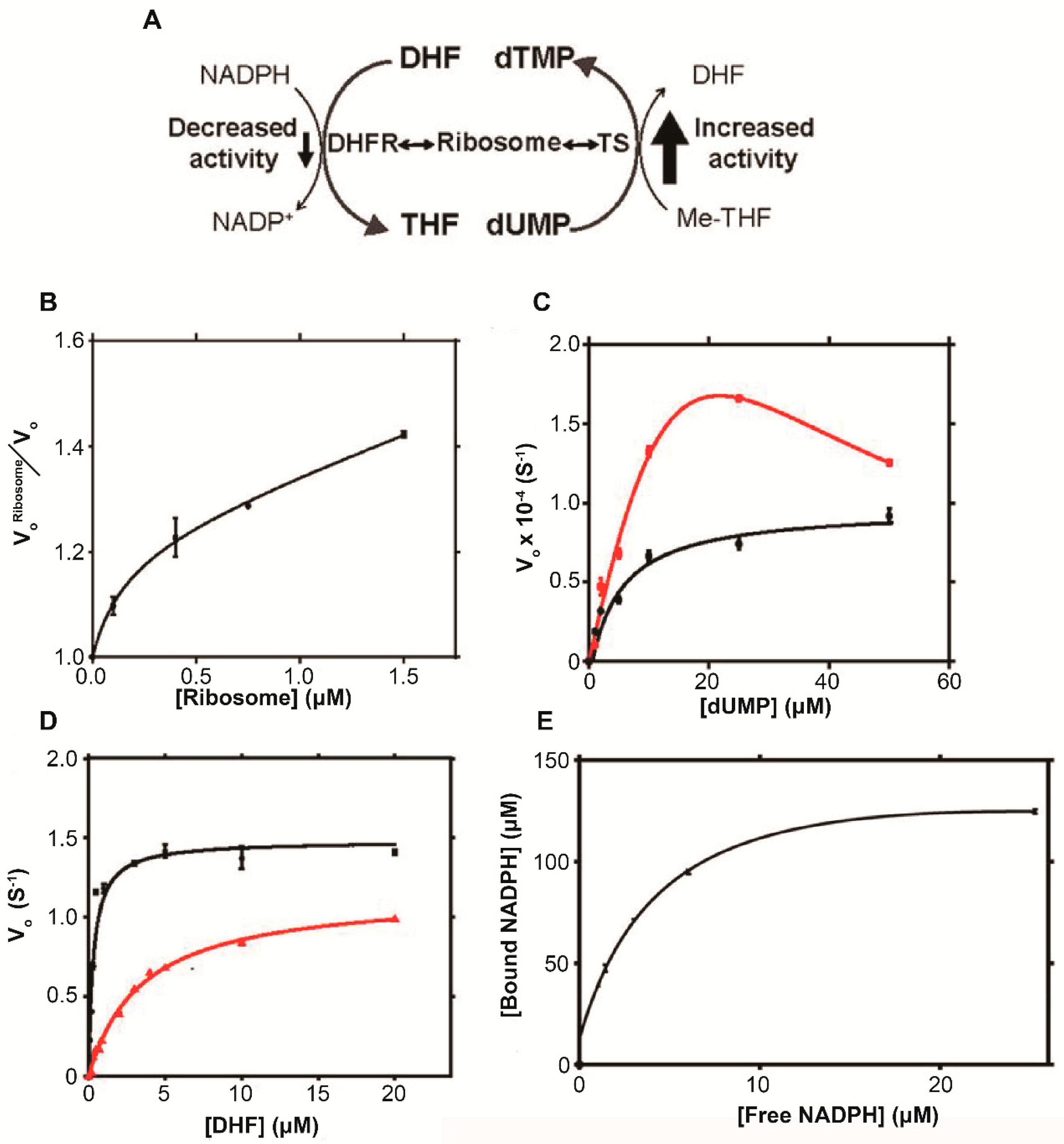
5.3. Dihydrofolate reductase and Thymidylate synthase
6. Ribosome-Mediated Regulation of Biological Activity
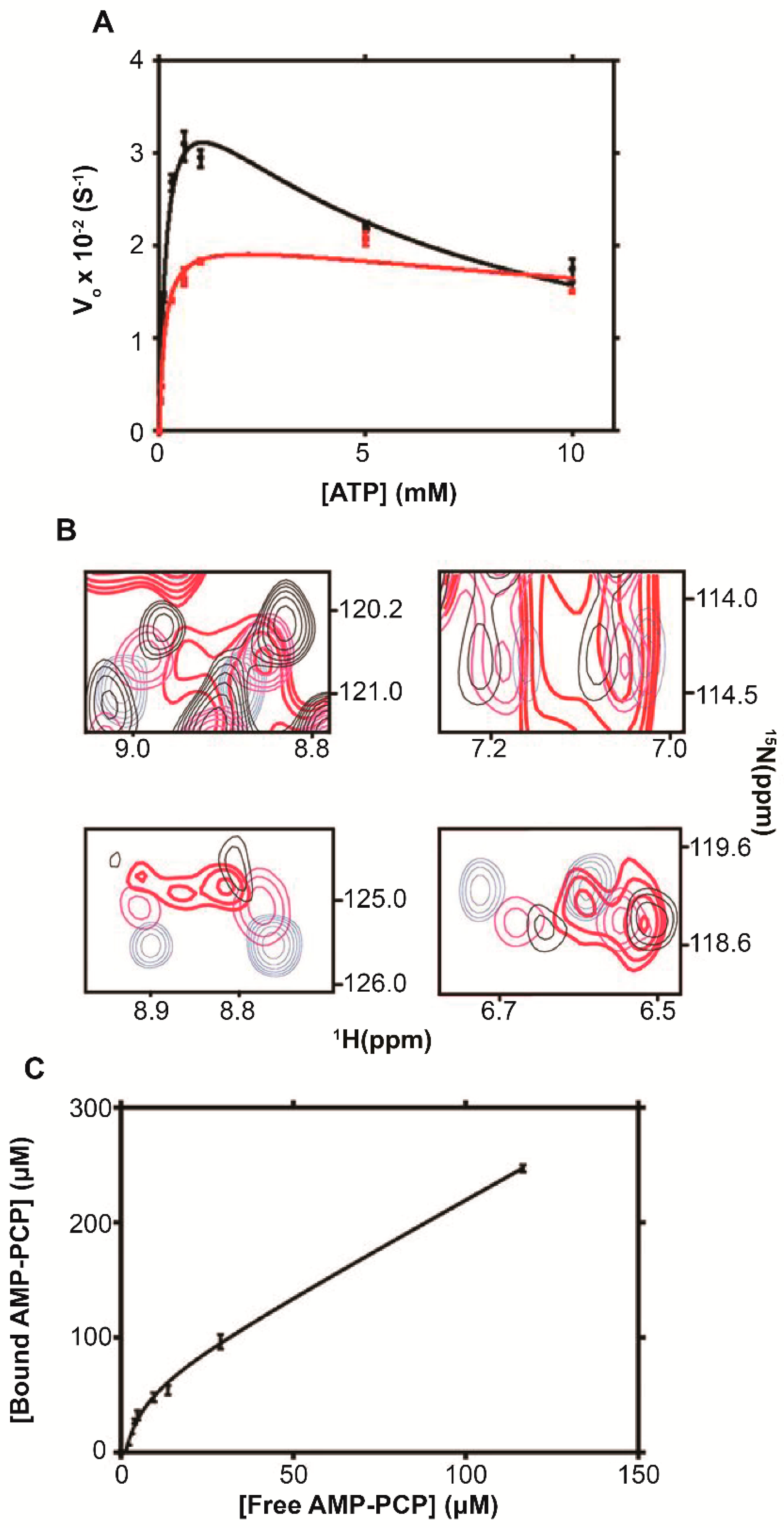
6.1. Adenylate Kinase
6.2. Dihydrofolate Reductase and Thymidylate Synthase
7. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Serber, Z.; Dotsch, V. In-cell NMR spectroscopy. Biochemistry 2001, 40, 14317–14323. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, D.; Sasaki, A.; Ikeya, T.; Hamatsu, J.; Hanashima, T.; Mishima, M.; Yoshimasu, M.; Hayashi, N.; Mikawa, T.; Walchli, M.; et al. Protein structure determination in living cells by in-cell NMR spectroscopy. Nature 2009, 458, 102–105. [Google Scholar] [CrossRef]
- Inomata, K.; Ohno, A.; Tochio, H.; Isogai, S.; Tenno, T.; Nakase, I.; Takeuchi, T.; Futaki, S.; Ito, Y.; Hiroaki, H.; et al. High-resolution multi-dimensional NMR spectroscopy of proteins in human cells. Nature 2009, 458, 106–109. [Google Scholar] [CrossRef]
- Burz, D.S.; Dutta, K.; Cowburn, D.; Shekhtman, A. Mapping structural interactions using in-cell NMR spectroscopy (STINT-NMR). Nat. Methods 2006, 3, 91–93. [Google Scholar] [CrossRef] [Green Version]
- Theillet, F.X.; Binolfi, A.; Bekei, B.; Martorana, A.; Rose, H.M.; Stuiver, M.; Verzini, S.; Lorenz, D.; van Rossum, M.; Goldfarb, D.; et al. Structural disorder of monomeric alpha-synuclein persists in mammalian cells. Nature 2016, 530, 45–50. [Google Scholar] [CrossRef]
- Banci, L.; Barbieri, L.; Bertini, I.; Luchinat, E.; Secci, E.; Zhao, Y.; Aricescu, A.R. Atomic-resolution monitoring of protein maturation in live human cells by NMR. Nat. Chem. Biol. 2013, 9, 297–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogino, S.; Kubo, S.; Umemoto, R.; Huang, S.; Nishida, N.; Shimada, I. Observation of NMR Signals from Proteins Introduced into Living Mammalian Cells by Reversible Membrane Permeabilization Using a Pore-Forming Toxin, Streptolysin O. J. Am. Chem. Soc. 2009, 131, 10834–10835. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, A.Y.; Burz, D.S.; Reverdatto, S.; Shekhtman, A. Fate of Pup inside the Mycobacterium proteasome studied by in-cell NMR. PLoS ONE 2013, 8, e74576. [Google Scholar] [CrossRef]
- Hough, L.E.; Dutta, K.; Sparks, S.; Temel, D.B.; Kamal, A.; Tetenbaum-Novatt, J.; Rout, M.P.; Cowburn, D. The molecular mechanism of nuclear transport revealed by atomic-scale measurements. Elife 2015, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wall, K.P.; Hough, L.E. In-Cell NMR within Budding Yeast Reveals Cytoplasmic Masking of Hydrophobic Residues of FG Repeats. Biophys. J. 2018, 115, 1690–1695. [Google Scholar] [CrossRef]
- Selenko, P.; Serber, Z.; Gadea, B.; Ruderman, J.; Wagner, G. Quantitative NMR analysis of the protein G B1 domain in Xenopus laevis egg extracts and intact oocytes. Proc. Natl. Acad. Sci. USA 2006, 103, 11904–11909. [Google Scholar] [CrossRef] [Green Version]
- Luchinat, E.; Gianoncelli, A.; Mello, T.; Galli, A.; Banci, L. Combining in-cell NMR and X-ray fluorescence microscopy to reveal the intracellular maturation states of human superoxide dismutase 1. Chem. Commun. 2015, 51, 584–587. [Google Scholar] [CrossRef]
- Maldonado, A.Y.; Burz, D.S.; Shekhtman, A. In-cell NMR spectroscopy. Prog. Nucl. Magn. Reson. Spectrosc. 2011, 59, 197–212. [Google Scholar] [CrossRef]
- Majumder, S.; Xue, J.; DeMott, C.M.; Reverdatto, S.; Burz, D.S.; Shekhtman, A. Probing protein quinary interactions by in-cell nuclear magnetic resonance spectroscopy. Biochemistry 2015, 54, 2727–2738. [Google Scholar] [CrossRef]
- Ye, Y.; Wu, Q.; Zheng, W.; Jiang, B.; Pielak, G.J.; Liu, M.; Li, C. Quantification of size effect on protein rotational mobility in cells by 19F NMR spectroscopy. Anal. Bioanal. Chem. 2018, 410, 869–874. [Google Scholar] [CrossRef]
- Ye, Y.; Liu, X.; Zhang, Z.; Wu, Q.; Jiang, B.; Jiang, L.; Zhang, X.; Liu, M.; Pielak, G.J.; Li, C. 19F NMR spectroscopy as a probe of cytoplasmic viscosity and weak protein interactions in living cells. Chemistry 2013, 19, 12705–12710. [Google Scholar] [CrossRef]
- Crowley, P.B.; Chow, E.; Papkovskaia, T. Protein interactions in the Escherichia coli cytosol: An impediment to in-cell NMR spectroscopy. Chembiochem 2011, 12, 1043–1048. [Google Scholar] [CrossRef]
- McConkey, E.H. Molecular evolution, intracellular organization, and the quinary structure of proteins. Proc. Natl. Acad. Sci. USA 1982, 79, 3236–3240. [Google Scholar] [CrossRef]
- Wirth, A.J.; Gruebele, M. Quinary protein structure and the consequences of crowding in living cells: Leaving the test-tube behind. Bioessays 2013, 35, 984–993. [Google Scholar] [CrossRef]
- Cohen, R.D.; Pielak, G.J. A cell is more than the sum of its (dilute) parts: A brief history of quinary structure. Protein Sci. 2017, 26, 403–413. [Google Scholar] [CrossRef] [Green Version]
- Theillet, F.X.; Binolfi, A.; Frembgen-Kesner, T.; Hingorani, K.; Sarkar, M.; Kyne, C.; Li, C.; Crowley, P.B.; Gierasch, L.; Pielak, G.J.; et al. Physicochemical properties of cells and their effects on intrinsically disordered proteins (IDPs). Chem. Rev. 2014, 114, 6661–6714. [Google Scholar] [CrossRef]
- Luchinat, E.; Banci, L. A Unique Tool for Cellular Structural Biology: In-cell NMR. J. Biol. Chem. 2016, 291, 3776–3784. [Google Scholar] [CrossRef]
- Serber, Z.; Corsini, L.; Durst, F.; Dotsch, V. In-cell NMR spectroscopy. Methods Enzymol. 2005, 394, 17–41. [Google Scholar]
- Milo, R.; Philips, R. Cell Biology by the Numbers, 1st ed.; Garland Science: New York, NY, USA, 2015; p. 393. [Google Scholar]
- Milo, R.; Jorgensen, P.; Moran, U.; Weber, G.; Springer, M. BioNumbers—The database of key numbers in molecular and cell biology. Nucleic Acids Res. 2010, 38, D750–D753. [Google Scholar] [CrossRef]
- DeMott, C.M.; Majumder, S.; Burz, D.S.; Reverdatto, S.; Shekhtman, A. Ribosome mediated quinary interactions modulate in-cell protein activities. Biochemistry 2017, 56, 4117–4126. [Google Scholar] [CrossRef]
- Castello, A.; Fischer, B.; Frese, C.K.; Horos, R.; Alleaume, A.M.; Foehr, S.; Curk, T.; Krijgsveld, J.; Hentze, M.W. Comprehensive Identification of RNA-Binding Domains in Human Cells. Mol. Cell 2016, 63, 696–710. [Google Scholar] [CrossRef]
- Castello, A.; Fischer, B.; Eichelbaum, K.; Horos, R.; Beckmann, B.M.; Strein, C.; Davey, N.E.; Humphreys, D.T.; Preiss, T.; Steinmetz, L.M.; et al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell 2012, 149, 1393–1406. [Google Scholar] [CrossRef]
- Simsek, D.; Tiu, G.C.; Flynn, R.A.; Byeon, G.W.; Leppek, K.; Xu, A.F.; Chang, H.Y.; Barna, M. The Mammalian Ribo-interactome Reveals Ribosome Functional Diversity and Heterogeneity. Cell 2017, 169, 1051–1065. [Google Scholar] [CrossRef]
- Beckmann, B.M.; Horos, R.; Fischer, B.; Castello, A.; Eichelbaum, K.; Alleaume, A.M.; Schwarzl, T.; Curk, T.; Foehr, S.; Huber, W.; et al. The RNA-binding proteomes from yeast to man harbour conserved enigmRBPs. Nat. Commun. 2015, 6, 10127. [Google Scholar] [CrossRef] [Green Version]
- Feig, M.; Yu, I.; Wang, P.H.; Nawrocki, G.; Sugita, Y. Crowding in Cellular Environments at an Atomistic Level from Computer Simulations. J. Phys. Chem. B 2017, 121, 8009–8025. [Google Scholar] [CrossRef]
- Yu, I.; Mori, T.; Ando, T.; Harada, R.; Jung, J.; Sugita, Y.; Feig, M. Biomolecular interactions modulate macromolecular structure and dynamics in atomistic model of a bacterial cytoplasm. Elife 2016, 5. [Google Scholar] [CrossRef]
- Schavemaker, P.E.; Smigiel, W.M.; Poolman, B. Ribosome surface properties may impose limits on the nature of the cytoplasmic proteome. Elife 2017, 6. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Poolman, B.; Boersma, A.J. Ionic Strength Sensing in Living Cells. ACS Chem. Biol. 2017, 12, 2510–2514. [Google Scholar] [CrossRef]
- Majumder, S.; DeMott, C.M.; Reverdatto, S.; Burz, D.S.; Shekhtman, A. Total Cellular RNA Modulates Protein Activity. Biochemistry 2016, 55, 4568–4573. [Google Scholar] [CrossRef] [Green Version]
- Breindel, L.; DeMott, C.; Burz, D.S.; Shekhtman, A. Real-Time In-Cell Nuclear Magnetic Resonance: Ribosome-Targeted Antibiotics Modulate Quinary Protein Interactions. Biochemistry 2018. [Google Scholar] [CrossRef]
- Ercikan-Abali, E.A.; Banerjee, D.; Waltham, M.C.; Skacel, N.; Scotto, K.W.; Bertino, J.R. Dihydrofolate reductase protein inhibits its own translation by binding to dihydrofolate reductase mRNA sequences within the coding region. Biochemistry 1997, 36, 12317–12322. [Google Scholar] [CrossRef]
- Chu, E.; Takimoto, C.H.; Voeller, D.; Grem, J.L.; Allegra, C.J. Specific binding of human dihydrofolate reductase protein to dihydrofolate reductase messenger RNA in vitro. Biochemistry 1993, 32, 4756–4760. [Google Scholar] [CrossRef]
- Brunn, N.D.; Dibrov, S.M.; Kao, M.B.; Ghassemian, M.; Hermann, T. Analysis of mRNA recognition by human thymidylate synthase. Biosci. Rep. 2014, 34, e00168. [Google Scholar] [CrossRef]
- Bertrand, K.; Reverdatto, S.; Burz, D.S.; Zitomer, R.; Shekhtman, A. Structure of proteins in eukaryotic compartments. J. Am. Chem. Soc. 2012, 134, 12798–12806. [Google Scholar] [CrossRef]
- Li, C.; Wang, G.F.; Wang, Y.; Creager-Allen, R.; Lutz, E.A.; Scronce, H.; Slade, K.M.; Ruf, R.A.; Mehl, R.A.; Pielak, G.J. Protein 19F NMR in Escherichia coli. J. Am. Chem. Soc. 2010, 132, 321–327. [Google Scholar] [CrossRef]
- Danielsson, J.; Inomata, K.; Murayama, S.; Tochio, H.; Lang, L.; Shirakawa, M.; Oliveberg, M. Pruning the ALS-associated protein SOD1 for in-cell NMR. J. Am. Chem. Soc. 2013, 135, 10266–10269. [Google Scholar] [CrossRef]
- Barnes, C.O.; Pielak, G.J. In-cell protein NMR and protein leakage. Proteins 2011, 79, 347–351. [Google Scholar] [CrossRef]
- Wuthrich, K. NMR of Proteins and Nucleic Acids; John Wiley&Sons: New York, NY, USA, 1986; p. 293. [Google Scholar]
- Cavanagh, J.F.; Fairbrother, W.J.; Palmer, A.G.; Rance, M.; Skelton, N.J. Protein NMR Spectroscopy; Academic Press: New York, NY, USA, 2007. [Google Scholar]
- Li, C.; Charlton, L.M.; Lakkavaram, A.; Seagle, C.; Wang, G.; Young, G.B.; Macdonald, J.M.; Pielak, G.J. Differential dynamical effects of macromolecular crowding on an intrinsically disordered protein and a globular protein: Implications for in-cell NMR spectroscopy. J. Am. Chem. Soc. 2008, 130, 6310–6311. [Google Scholar] [CrossRef]
- Pielak, G.J.; Li, C.; Miklos, A.C.; Schlesinger, A.P.; Slade, K.M.; Wang, G.F.; Zigoneanu, I.G. Protein nuclear magnetic resonance under physiological conditions. Biochemistry 2009, 48, 226–234. [Google Scholar] [CrossRef]
- Serber, Z.; Keatinge-Clay, A.T.; Ledwidge, R.; Kelly, A.E.; Miller, S.M.; Dotsch, V. High-resolution macromolecular NMR spectroscopy inside living cells. J. Am. Chem. Soc. 2001, 123, 2446–2447. [Google Scholar] [CrossRef]
- Riek, R.; Pervushin, K.; Wuthrich, K. TROSY and CRINEPT: NMR with large molecular and supramolecular structures in solution. Trends Biochem. Sci. 2000, 25, 462–468. [Google Scholar] [CrossRef]
- Pervushin, K.V.; Wider, G.; Wuthrich, K. Single Transition-to-single Transition Polarization Transfer (ST2-PT) in 15N-1H TROSY. J. Biomol. NMR 1998, 12, 345–348. [Google Scholar] [CrossRef]
- Riek, R.; Wider, G.; Pervushin, K.; Wuthrich, K. Polarization transfer by cross-correlated relaxation in solution NMR with very large molecules. Proc. Natl. Acad. Sci. USA 1999, 96, 4918–4923. [Google Scholar] [CrossRef] [Green Version]
- Goldman, M. Interference effects in the relaxation of a pair of unlike 1/2 nuclei. J. Magn. Reson. 1984, 60, 437–452. [Google Scholar] [CrossRef]
- Shekhtman, A.; Ghose, R.; Goger, M.; Cowburn, D. NMR structure determination and investigation using a reduced proton (REDPRO) labeling strategy for proteins. FEBS Lett. 2002, 524, 177–182. [Google Scholar] [CrossRef] [Green Version]
- Riek, R.; Fiaux, J.; Bertelsen, E.B.; Horwich, A.L.; Wuthrich, K. Solution NMR techniques for large molecular and supramolecular structures. J. Am. Chem. Soc. 2002, 124, 12144–12153. [Google Scholar] [CrossRef]
- Schimmel, P.R.; Cantor, C.R. Biophysical Chemistry: Part II: Techniques for the Study of Biologycal Structure and Function; W.H. Freeman: New York, NY, USA, 1980; p. 650. [Google Scholar]
- Segur, J.; Oberstar, H. Viscosity of Glycerol and its Aqueous Solutions. Ind. Eng. Chem. 1951, 43, 2117–2120. [Google Scholar] [CrossRef]
- Burz, D.S.; Dutta, K.; Cowburn, D.; Shekhtman, A. In-cell NMR for protein-protein interactions (STINT-NMR). Nat. Protoc. 2006, 1, 146–152. [Google Scholar] [CrossRef] [Green Version]
- Burz, D.S.; DeMott, C.M.; Aldousary, A.; Dansereau, S.; Shekhtman, A. Quantitative Determination of Interacting Protein Surfaces in Prokaryotes and Eukaryotes by Using In-Cell NMR Spectroscopy. Methods Mol. Biol. 2018, 1688, 423–444. [Google Scholar] [CrossRef]
- Burz, D.S.; Shekhtman, A. The STINT-NMR method for studying in-cell protein-protein interactions. Curr. Protoc. Protein Sci. 2010, 61, 17. [Google Scholar] [CrossRef]
- Majumder, S.; DeMott, C.M.; Burz, D.S.; Shekhtman, A. Using singular value decomposition to characterize protein-protein interactions by in-cell NMR spectroscopy. ChemBioChem 2014, 15, 929–933. [Google Scholar] [CrossRef]
- Beis, I.; Newsholme, E.A. The contents of adenine nucleotides, phosphagens and some glycolytic intermediates in resting muscles from vertebrates and invertebrates. Biochem. J. 1975, 152, 23–32. [Google Scholar] [CrossRef] [Green Version]
- Henzler-Wildman, K.A.; Thai, V.; Lei, M.; Ott, M.; Wolf-Watz, M.; Fenn, T.; Pozharski, E.; Wilson, M.A.; Petsko, G.A.; Karplus, M.; et al. Intrinsic motions along an enzymatic reaction trajectory. Nature 2007, 450, 838–844. [Google Scholar] [CrossRef]
- Aden, J.; Verma, A.; Schug, A.; Wolf-Watz, M. Modulation of a pre-existing conformational equilibrium tunes adenylate kinase activity. J. Am. Chem. Soc. 2012, 134, 16562–16570. [Google Scholar] [CrossRef]
- Schneider, D.A.; Gourse, R.L. Relationship between growth rate and ATP concentration in Escherichia coli: A bioassay for available cellular ATP. J. Biol. Chem. 2004, 279, 8262–8268. [Google Scholar] [CrossRef]
- Freeberg, M.A.; Han, T.; Moresco, J.J.; Kong, A.; Yang, Y.C.; Lu, Z.J.; Yates, J.R.; Kim, J.K. Pervasive and dynamic protein binding sites of the mRNA transcriptome in Saccharomyces cerevisiae. Genome Biol. 2013, 14, R13. [Google Scholar] [CrossRef] [PubMed]
- Castello, A.; Hentze, M.W.; Preiss, T. Metabolic Enzymes Enjoying New Partnerships as RNA-Binding Proteins. Trends Endocrinol. Metab. 2015, 26, 746–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baltz, A.G.; Munschauer, M.; Schwanhausser, B.; Vasile, A.; Murakawa, Y.; Schueler, M.; Youngs, N.; Penfold-Brown, D.; Drew, K.; Milek, M.; et al. The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol. Cell 2012, 46, 674–690. [Google Scholar] [CrossRef]
- Vazquez-Laslop, N.; Mankin, A.S. Context-Specific Action of Ribosomal Antibiotics. Annu. Rev. Microbiol. 2018, 72, 185–207. [Google Scholar] [CrossRef]
- Futterman, S. Enzymatic reduction of folic acid and dihydrofolic acid to tetrahydrofolic acid. J. Biol. Chem. 1957, 228, 1031–1038. [Google Scholar]
- Schnell, J.R.; Dyson, H.J.; Wright, P.E. Structure, dynamics, and catalytic function of dihydrofolate reductase. Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 119–140. [Google Scholar] [CrossRef]
- Carreras, C.W.; Santi, D.V. The catalytic mechanism and structure of thymidylate synthase. Annu. Rev. Biochem. 1995, 64, 721–762. [Google Scholar] [CrossRef]
- Sinev, M.A.; Sineva, E.V.; Ittah, V.; Haas, E. Towards a mechanism of AMP-substrate inhibition in adenylate kinase from Escherichia coli. FEBS Lett. 1996, 397, 273–276. [Google Scholar] [CrossRef] [Green Version]
- Cleland, W.W. Substrate inhibition. Methods Enzymol. 1979, 63, 500–513. [Google Scholar]
- Kovermann, M.; Grundstrom, C.; Sauer-Eriksson, A.E.; Sauer, U.H.; Wolf-Watz, M. Structural basis for ligand binding to an enzyme by a conformational selection pathway. Proc. Natl. Acad. Sci. USA 2017, 114, 6298–6303. [Google Scholar] [CrossRef]
- Fox, J.T.; Stover, P.J. Folate-mediated one-carbon metabolism. Vitam. Horm. 2008, 79, 1–44. [Google Scholar] [CrossRef]
- Goodsell, D. The Machinery of Life; Springer: New York, NY, USA, 2009; p. 167. [Google Scholar]
- Benedek, G.B.; Villars, F.M.H. Physics with Illustrative Examples from Medicine and Biology: Electricity and Magnetism; Springer: New York, NY, USA, 2000. [Google Scholar]
- Danielsson, J.; Mu, X.; Lang, L.; Wang, H.; Binolfi, A.; Theillet, F.X.; Bekei, B.; Logan, D.T.; Selenko, P.; Wennerstrom, H.; et al. Thermodynamics of protein destabilization in live cells. Proc. Natl. Acad. Sci. USA 2015, 112, 12402–12407. [Google Scholar] [CrossRef] [Green Version]
- Monteith, W.B.; Cohen, R.D.; Smith, A.E.; Guzman-Cisneros, E.; Pielak, G.J. Quinary structure modulates protein stability in cells. Proc. Natl. Acad. Sci. USA 2015, 112, 1739–1742. [Google Scholar] [CrossRef]
- Kyne, C.; Ruhle, B.; Gautier, V.W.; Crowley, P.B. Specific ion effects on macromolecular interactions in Escherichia coli extracts. Protein Sci. 2015, 24, 310–318. [Google Scholar] [CrossRef]
- Barbieri, L.; Luchinat, E.; Banci, L. Protein interaction patterns in different cellular environments are revealed by in-cell NMR. Sci. Rep. 2015, 5, 14456. [Google Scholar] [CrossRef] [Green Version]
- Kyne, C.; Crowley, P.B. Short Arginine Motifs Drive Protein Stickiness in the Escherichia coli Cytoplasm. Biochemistry 2017, 56, 5026–5032. [Google Scholar] [CrossRef]
- Li, C.; Pielak, G.J. Using NMR to distinguish viscosity effects from nonspecific protein binding under crowded conditions. J. Am. Chem. Soc. 2009, 131, 1368–1369. [Google Scholar] [CrossRef]
- Li, C.; Wang, Y.; Pielak, G.J. Translational and rotational diffusion of a small globular protein under crowded conditions. J. Phys. Chem. B 2009, 113, 13390–13392. [Google Scholar] [CrossRef]
- Jensen, K.F.; Pedersen, S. Metabolic growth rate control in Escherichia coli may be a consequence of subsaturation of the macromolecular biosynthetic apparatus with substrates and catalytic components. Microbiol. Rev. 1990, 54, 89–100. [Google Scholar]
- Gourse, R.L.; Gaal, T.; Bartlett, M.S.; Appleman, J.A.; Ross, W. rRNA transcription and growth rate-dependent regulation of ribosome synthesis in Escherichia coli. Annu. Rev. Microbiol. 1996, 50, 645–677. [Google Scholar] [CrossRef]
- Srere, P.A. Complexes of sequential metabolic enzymes. Annu. Rev. Biochem. 1987, 56, 89–124. [Google Scholar] [CrossRef]
- Sanamrad, A.; Persson, F.; Lundius, E.G.; Fange, D.; Gynna, A.H.; Elf, J. Single-particle tracking reveals that free ribosomal subunits are not excluded from the Escherichia coli nucleoid. Proc. Natl. Acad. Sci. USA 2014, 111, 11413–11418. [Google Scholar] [CrossRef] [Green Version]
- Bakshi, S.; Siryaporn, A.; Goulian, M.; Weisshaar, J.C. Superresolution imaging of ribosomes and RNA polymerase in live Escherichia coli cells. Mol. Microbiol. 2012, 85, 21–38. [Google Scholar] [CrossRef]
- Fisher, J.K.; Bourniquel, A.; Witz, G.; Weiner, B.; Prentiss, M.; Kleckner, N. Four-dimensional imaging of E. coli nucleoid organization and dynamics in living cells. Cell 2013, 153, 882–895. [Google Scholar] [CrossRef]
- Conway, J.H.; Sloane, N.J.A. Sphere Packings, Latticies, and Groups; Springer: Berlin, Germany, 1999; p. 681. [Google Scholar]
- Ortiz, J.O.; Forster, F.; Kurner, J.; Linaroudis, A.A.; Baumeister, W. Mapping 70S ribosomes in intact cells by cryoelectron tomography and pattern recognition. J. Struct. Biol. 2006, 156, 334–341. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Binds | Effect of RNA-Binding |
|---|---|---|
| Ubiquitin (Ubq) | Total RNA [14,35] mRNA [28] | Blocks polyubiquitination sites, increases apparent MW [14] |
| Thioredoxin (Trx) | Total RNA [14], mRNA [28] | Increases apparent MW [14] Antibiotic binding to ribosome alters quinary structure [36] |
| Adenylate kinase (ADK) | Total RNA [14] mRNA [28] Ribosome [26,29] | Increases apparent MW [14] Noncompetitive kinetic inhibitor [26] |
| Dihydrofolate reductase (DHFR) | mRNA [37,38] Ribosome [26] | Competitive kinetic inhibitor [26] |
| Thymidylate synthase (TS) | Total RNA [26] mRNA [28,39] Ribosome [26,29] | Uncompetitive kinetic activator [26] |
| Enzyme | [ribosome] (μM) | Vmax (S−1) | VmaxRibosome/Vomax a | KM (μM) b | KI (mM) b | R2 |
|---|---|---|---|---|---|---|
| ADK | 0 | (4.2 ± 0.2) × 10−2 | ∼0.5 | 180 ± 20 | 6.1 ± 0.8 | 0.98 |
| 1 | (2.1 ± 0.1) × 10−2 | 130 ± 30 | 35 ± 2 | 0.96 | ||
| DHFR | 0 | (1.48 ± 0.04) × 10−4 | ∼0.8 | 0.32 ± 0.04 | 0.95 | |
| 0.5 | (1.16 ± 0.02) × 10−4 | 3.5 ± 0.1 | 0.99 | |||
| TS | 0 | (9.7 ± 0.4) × 10−5 | ∼20 | 5.4 ± 0.7 | 0.97 | |
| 0.5 | (2.0 ± 0.2) × 10−3 | 120 ± 20 | (3.9 ± 0.5) × 10−3 | 0.99 | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burz, D.S.; Breindel, L.; Shekhtman, A. The Inescapable Effects of Ribosomes on In-Cell NMR Spectroscopy and the Implications for Regulation of Biological Activity. Int. J. Mol. Sci. 2019, 20, 1297. https://doi.org/10.3390/ijms20061297
Burz DS, Breindel L, Shekhtman A. The Inescapable Effects of Ribosomes on In-Cell NMR Spectroscopy and the Implications for Regulation of Biological Activity. International Journal of Molecular Sciences. 2019; 20(6):1297. https://doi.org/10.3390/ijms20061297
Chicago/Turabian StyleBurz, David S., Leonard Breindel, and Alexander Shekhtman. 2019. "The Inescapable Effects of Ribosomes on In-Cell NMR Spectroscopy and the Implications for Regulation of Biological Activity" International Journal of Molecular Sciences 20, no. 6: 1297. https://doi.org/10.3390/ijms20061297