Abstract
Neurodegenerative diseases (NDs) are among the most feared of the disorders that afflict humankind for the lack of specific diagnostic tests and effective treatments. Understanding the molecular, cellular, biochemical changes of NDs may hold therapeutic promise against debilitating central nerve system (CNS) disorders. In the present review, we summarized the clinical presentations and biology backgrounds of NDs, including Parkinson’s disease (PD), Huntington’s disease (HD), and Alzheimer’s disease (AD) and explored the role of molecular mechanisms, including dys-regulation of epigenetic control mechanisms, Ataxia-telangiectasia-mutated protein kinase (ATM), and neuroinflammation in the pathogenesis of NDs. Targeting these mechanisms may hold therapeutic promise against these devastating diseases.
1. Introduction
Neurodegeneration occurs when structures or functions of neurons are progressively lost. Acute neurodegenerative diseases (NDs), such as stroke [] or traumatic brain injury [] may result neurons partial or entire loss; chronic NDs, including Parkinson’s disease (PD) [], Huntington’s disease (HD) [], Alzheimer’s disease (AD) [], etc., are among the most feared of the disorders that afflict humankind because of the lack of specific diagnostic tests and effective treatments. The World Health Organization (WHO) predicts that NDs are going to overtake cancer in the rank of top causes of death by 2050 []. The estimated figures, severity and chronicity of these diseases, and a vast economic and emotional burden on individuals, communities and governments generate an urgent need to better understand pathophysiology, improve early diagnosis and develop effective treatments of NDs.
Early studies suggested that misfolding proteins or polyglutamine-dependent pathogenesis resulted in an excessive amount or abnormal structural aggregation-prone proteins accumulation, causing several distinct NDs []. However, numerous theories, such as impaired ubiquitin-proteasome and/or autophagy-lysosomal pathways [], mitochondrial dys-function [], programmed cell death [], glutamatergic activity, reactive oxygen species (ROS), etc., suggest the complexity of NDs. Insights into the cellular and molecular pathogenesis of NDs may broaden our understanding of the underlying mechanisms and hold therapeutic promise against debilitating CNS disorders. Hence, this review will discuss the clinical manifestations of three distinct NDs: PD, HD, and AD through the associated molecular machineries, including epigenetic misregulation, Ataxia-telangiectasia-mutated protein kinase (ATM), and neuroinflammation. Greater understanding of the diseases may aid development of better therapeutics.
2. Molecular Mechanisms of NDs
2.1. Epigenetic Misregulation
Normally, DNA is tightly coiled into dense chromatin and not available for reading and transcription in the nucleus of mammals. Relaxation of chromatin allows DNA active transcription. Nucleosomes, the smallest structural unit of chromatin, are composed of eight histone core molecules, including doublets of histone 2A, 2B, 3, and 4, with two loops of 147 bp DNA. The process of coiling and uncoiling the genome is mainly modulated via post-translational modifications. Although there may appear to be a bewildering array of histone modifications, there are at least methylation, acetylation, phosphorylation, ubiquitination, sumoylation, etc. []. These epigenetic changes involve the covalent chemical reactions of histones by DNA methyltransferases (DNMTs), histone acetyltransferases (HATs) and histone deacetylases (HDACs), the polycomb repressive complex 1 (PRC1) and PRC2, ubiquitination- and sumoylation-related proteins to regulate activation or inactivation of gene expression (Figure 1).
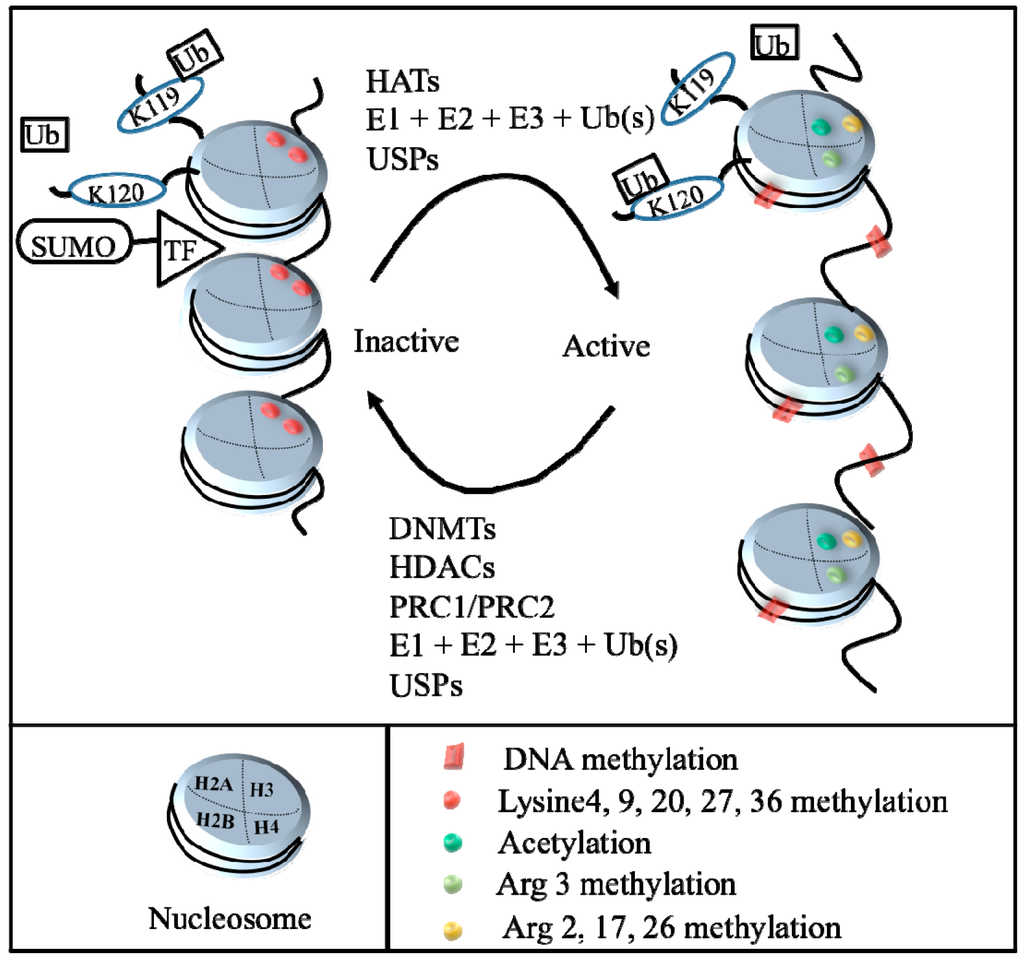
Figure 1.
Illustration of epigenetic mechanisms. The process of DNA condensation and relaxation is controlled principally through histone post-translational modifications, such as methylation, acetylation, phosphorylation, ubiquitination, sumoylation, etc. Histone acetyltransferases (HATs), mono- and poly-ubiquitination, and ubiquitin-specific proteases (USPs) may cause uncoiling chromatin (euchromatin) and allow transcriptional factor access to the DNA (right); whereas DNA methyltransferases (DNMTs), histone deacetylases (HDACs), mono- and poly-ubiquitination (Ub), ubiquitin-specific proteases (USPs), Polycomb repressive complex 1(PRC1)/Polycomb repressive complex 2 (PRC2), and Small ubiquitin-related modifier (SUMO) modification may result in coiling chromatin (heterochromatin) and prevent transcription factor access to DNA, leading to transcriptional repression. TF: transcription factor.
DNMTs contain at least four subtypes, including DNMT1, DNMT2, DNMT3a and DNMT3b. After DNA replication, the parent strand preserves methylated but the newly synthesized strand dose not. DNMT1 binds to these hemi-methylated CpG sites and methylates the cytosine on the newly synthesized stand to maintain established CpG methylation patterns through mitosis. DNMT2 may have a very low DNA-cysteine methylation activity though DNMT2 may possess a protective function []. DNMT3a and DNMT3b are essential for methylation and early development, and the loss of either is lethal [].
HATs adopted an acetyl group from acetyl-coenzyme A to counterbalance the positive charge of the lysine residues on the N-terminal tails of H2A, H2B, H3 and H4 [] to uncoil chromatin for transcriptional factors (TF)s to access and initiate transcription. This reaction is reversed by HDACs, which take away the acetyl groups from the lysine to coil chromatin and inhibit TF approaching. HDAC isoforms are extensively expressed in the brain and are at least 18 isoforms, which have been characterized and phylogenetically categorized into four main classes: Class I HDACs include HDACs 1, 2, 3, and 8. Class II HDACs is divided into class IIa, consisting of HDACs 4, 5, 7 and 9, and class IIb consisting of HDACs 6 and 10. Class III, the NAD+ dependent class, comprises of Sirtuin 1, 2, 3, 4, 5, 6, and 7. Class IV consists of HDAC 11 []. HDACI is multifunctional, including abolishing aberrant epigenetic modifications and abnormal transcriptional imbalance, modulating cytoskeletal and immune functions, and enhancing protein degradation. Pharmacological interventions using HDAC inhibitors (HDACI) are promising in the treatment of several diseases, including cancers, metabolic diseases, neuropsychiatric diseases, and NDs [,,].
Methylation is one of modifications of histone to regulate transcriptional expression and orchestrate numerous genes. Methylation on arginine or lysine residues of H3 or H4 can trigger a transcriptional cascades []. DNMTs transfer a methyl group, which is from S-adenosyl methionine (SAM), to target molecules. Moreover, most CpG islands are associated with functional genes and may contain promoters. Methylated CpG islands or gene promoter regions may cause gene silencing or activation. CpG methylation may partially rely on the ratio between SAM and S-adenosyl-homocysteine (SAH) []. After methylation, the methyl group is then back to SAH after cleavage by histone demethylases (HDMs). Furthermore, multiple methylation valences, including mono-methylated, di-methylated or trimethylated lysine and arginine residues on H3 and H4 are noted. For instance, activation signals include methylation at H3-Lys(K)4, K36, K79; Arg2, Arg17, Arg26 and H4-Arg3, usually link to uncoiling chromatin structure; inhibition signals include H3-Lys9, K27, K36, and H4-K20, Arg8 and Arg3 [,].
There are at least two subtypes of Polycomb (PcG) proteins: PRC1, and PRC2. PRC1, which catalyzes the mono-ubiquitination of histone H2A and plays a role in silencing maintenance, contains Bmi1/MEL18, polyhomeotic (PH), Ring1a, Ring1b and CBX/HPC PcG proteins. PRC2 is associated with transcriptional repression. PRC2 is composed of embryonic ectoderm development (EED), suppressor of zeste 12 (Suz12), zeste homolog 2 (EZH2) and RBAP48/46. PRC2 initially attached to chromatin and EZH2 and trimethylated H3K27 []. PRC1 recognized the methylated markers. E3 adhered RING1/2 and then mono-ubiquitinated H2AK119, making chromatin coiled and causing transcriptional repression []. EZH2 trimethylated H3K27, causing repression of promoters []. Excessive-expression EZH2 inhibits BRCA1 phosphorylation and thereby facilitates cell proliferation in breast cancer []. The histone methylation inhibitor 3-deazaneplanocin A (DZNep), a well-known EZH2 inhibitor, is promising in the treatment of cancers and other diseases [].
Ubiquitin is a highly conserved protein. Four genes, including UBB, UBC, UBA52 and RPS27A produce ubiquitin in the human genome []. Ubiquitination, is an enzymatic process, containing ubiquitin-activating enzymes (E1), which can activate the ubiquitin; ubiquitin conjugating enzymes (E2), which is a linker between the ubiquitin and E1; ubiquitin ligases (E3), which connects the glycine 76 of the ubiquitin to a lysine on the substrate protein through an isopeptide bond. In general, substrates, which were mono-ubiquitinated, represented as a signal carrying specific information. Poly-ubiquitinated substrate were for degradation []. H2A and H2B can be mono- and poly-ubiquitinated [,]. The same ubiquitin binding to H2A or H2B may cause distinct outcomes, because ubiquitinated histone H2A (uH2A) antagonizes transcription []. uH2B activates transcription []. The ubiquitination site of H2A is lysine 119 (K119). The site of H2B is K120. The reaction of mono-ubiquitination is reversible by ubiquitin-specific proteases (USPs) []. PRC1 is as ubiquitin ligase for H2A; PRC2 installs the H3K27me3 marks for PRC1 to recognize. After being ubiquitinated, uH2A repressed gene expression. Rad6 enhanced mono- and poly-ubiquitination on H2A and H2B []. Notably, in vitro studies showed that BRCA1 assisted ubiquitination on H2A and H2B, but the interactions in vivo remain unclear [].
Small ubiquitin-related modifier (SUMO) modification (sumoylation) occurs on histones and results in transcriptional repression []. All four histones are sumoylated in S. cerevisiae [], whereas only H4 has been identified to be modified in mammalian cells []. H4 can connect E2 and be sumoylated in an E1- and E2-dependent pattern. Moreover, several molecules, including the histone demethylase LSD1, the histone methyltransferase SETDB1, chromatin-associated proteins HP1, L3MBTL1 and L3MBTL2, the nucleosome remodeling ATPase Mi-2, and deacetylase HDAC2 were recruited when SUMO proteins were covalently attached to a histone, leading to gene silencing through modulating the chromatin structure dynamics []. Because the structure of SUMO proteins is similar to that of ubiquitin [], their functions may also share similarities. Additionally, sumoylation can affect the distribution of proteins, initiate functions of enzymes, degrade or preserve target proteins, repress transcriptional factors, etc. [].
2.2. Ataxia-Telangiectasia and Ataxia-Telangiectasia Mutation (ATM)
Ataxia-telangiectasia (AT), also called Louis-Bar syndrome, is a rare and inherited human disease. A-T is characterized by predisposition to cancer, immunodeficiency and a significant loss of neurons causing neurological conditions [,]. The mutated ATM gene produced A-T phenotypes. ATM is a member of the PI3-kinase family and ubiquitously expressed throughout development. ATM involves the DNA repair system and maintains the integrity of its genome by controlling cell cycle checkpoints. When DNA is damaged by UV light, ionizing radiation, or ROS to cause lesions including DNA hydrolysis, DNA oxidation, DNA single-strand beaks (SSBs), and other damages []. If damaged DNA is left unrepaired, irreparable and toxic DNA double-strand breaks (DSBs) may be produced []. Functionally, ATM is activated by DSBs []. At very early step, activated ATM by DSBs can immediately phosphorylate histone H2AX at the site of the break []. ATM and Rad3 related (ATR) mutually works with ATR-interacting protein (ATRIP) to recognize SSB, which is fastened by replication protein A (RPA) binding. In response to DNA damage, ATR and ATM stimulated checkpoint kinases CHK1 and CHK2, respectively via P53 dependent and independent signaling pathways []. p53 dependent pathways: phosphorylated p53 initiated p21, which inhibited CDK1/cyclin B to regulate cell cycle []. p53 independent pathway: CHK1 and CHK2 phosphorylated CDC25, which then down-regulated CDC25A/B/C activity [], leading to the inhibition of CDK1/cyclin B []. CHK1 and CHK2 activated Wee 1 through phosphorylation. Phosphorylated CDC25 and Wee 1 arrested cell cycle at G2/M phase []. ATM and ATR sense and transduce damaged DNA signals to initiate DNA repair, apoptosis, rest and repair [,,] (Figure 2). For example, BRCA1 and P53, well-known tumor suppression genes (TSG), are regulated by ATM []. Mutated BRCA1 and P53 may be involved in the development and progression of cancers, and in the pathogenesis of NDs. Moreover, a study showed that ATM-mediated phosphorylation of EZH2 reduced protein stability [,]. Knockdown EZH2 improved the histological degeneration of Purkinje cells and mitigated behavioral impairment in ATM KO mice [], suggesting that epigenetics modulate ATM mediated NDs.
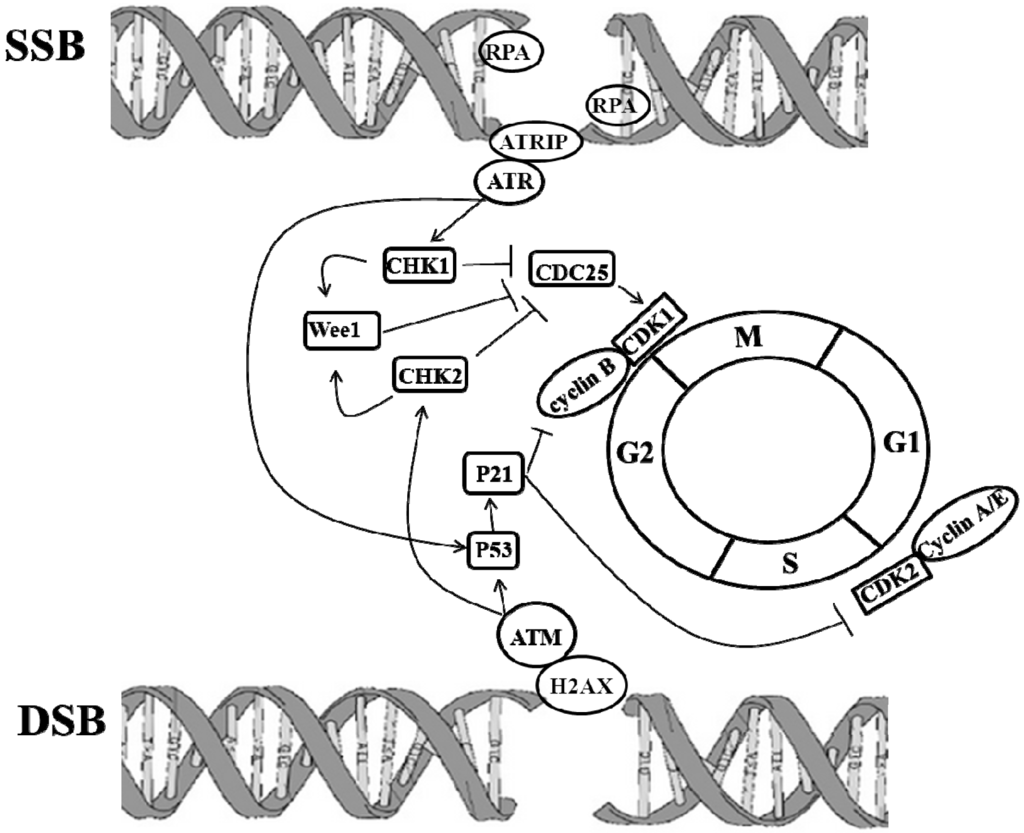
Figure 2.
Molecular cascades of DNA damages initiating repair systems and cell cycle progression. In response to DNA damage, including single-strand beaks (SSBs) and double-stranded breaks (DSB), the Ataxia-telangiectasia-mutated protein kinase (ATM)/ Ataxia-telangiectasia and Rad3 related protein (ATR) signaling pathways are activated, leading to the phosphorylation and activation of CHK1 and CHK2 and to the subsequent phosphorylation of CDC25. Phosphorylated CDC25 inhibits activation of cyclin B/CDK1, resulting in G2 arrest. Activated ATM/ATR pathways also activate p53-dependent signaling to arrest G2 through the activation of P21, which inhibits cyclin B/CDK1 complexes. Lines with arrow heads indicate activation, while lines with bar heads indicate inhibition.
2.3. Neuroinflammation: Pro-Inflammatory and Anti-Inflammatory Cytokines, and Irregular Tryptophan (TRP) Metabolism
2.3.1. Pro-Inflammatory and Anti-Inflammatory Cytokines
Neuroinflammation was originally defined as inflammation of the CNS, which may be triggered by viral and bacterial infection, ischemic stroke, toxic metabolites, HIV encephalopathy, and autoimmunity [,]. Immune responses can be triggered by these causes and exposed self-antigens in damaged CNS, causing autoimmune reactions that commonly follow these curs, because circulating peripheral immune cells can surpass a compromised blood-brain barrier (BBB) and activate the immune response to protect the CNS. Also, microglia, the resident innate immune cells in the CNS, act as scavengers to eradicate microbial pathogens, modulate immune responses and generate neurotrophic or toxic substances to trigger diseases []. The CNS is typically an immunologically privileged site because peripheral immune cells are blocked by the BBB. However, the widespread inflammation in the CNS attracted further migration of leukocytes infiltration, leakage, and disruption of the BBB, leading to neurodegeneration [].
TNF and the IL family are pro-inflammatory cytokines. They attract leucocytes and amplify proliferation at the inflammation site, synthesize proteolytic enzymes, initiate cytotoxicity, and secrete other pro-inflammatory factors to continue inflammation. These cytokines stimulated IL-6 production to generate anti-inflammatory cytokines and other immune factors to neutralize pro-inflammatory effects []. Imbalance between pro-inflammatory and anti-inflammatory cytokines may possibly cause neuroinflammation and NDs. For example, formation of α-synuclein (SNCA) fibrils were aggregated in PD in neuroinflammation []. Elevated circulating inflammatory cytokines and monocytes with hyper-responsive to immune stimuli were found in HD patients and HD mouse models []. Moreover, several studies discovered excessive pro-inflammatory cytokines in microglia, astrocytes, and neurons, and co-localization with both Aβ plaques and tau, and increasing Aβ and tau phosphorylation in the AD’s brain []. Apart from traditional viewpoints regarding the inflammation, the field of neuroinflammation has broadened to enroll NDs, such as PD, HD, and AD. These diseases lack the signs of classic “inflammation”, such as immune cell infiltration from the blood stream, but they featured cellular and molecular icons of neuroinflammation, including imbalance of pro-inflammatory and anti-inflammatory cytokines expression, microglia activation, etc.
2.3.2. Irregular Tryptophan (TRP) Metabolism
TRP is the precursor of two important metabolic pathways, serotonin synthesis and kynurenine (KYN) synthesis. It is estimated that 95% of mammalian serotonin is found within the gastrointestinal tract [], and only about 1% of dietary TRP is converted to serotonin in the brain []. The serotonin pathway generates melatonin, which may affect neural and endocrine systems that regulate circadian rhythms of behavior, physiology, and sleep patterns [].
The KYN pathway accounts for approximately 90% of TRP catabolism [,]. TRP is firstly oxidized by TRP 2,3-dioxygenase (TDO) or indoleamine 2,3-dioxygenase (IDO) to KYN. There are at least 3 pathways for KYN metabolism: (1) KYN aminotransferase (KAT) pathway: KYN is catabolized by KAT to form KYN acid (KYNA); (2) kynurenine 3-monoxygenase (KMO) pathway: KYN is converted into 3-hydroxykynurenine (3-HK) by KMO. 3-HK is converted into 3-hydroxyanthranilic acid (3-HAA) by KAT; (3) KYNase pathway: KYN is metabolized by kynureninase (KYNase) to form anthranilic acid (AA), which is converted into 3-HAA by anthranilate 3-monooxygenase (AA3MO). 3-HAA is oxidized by 3-hydroxyanthranilic acid oxidase (HAAO) to quinolinic acid (QUIN), which generates NAD+ through quinolinate phosphoribosyltransferase (QPRT) [,]. Additionally, TRP is metabolized to 5-hydroxytryptophan (5-HTP) through TRP hydroxylase (TPH) and tetrahydrobiopterin (BH4). Serotonin is synthesized from 5-HTP via the aromatic acid decarboxylase (AADC) and the vitamin B6. Serotonin is converted into 5-Hydroxyindoleacetic acid (5-HIAA) or 5-hydroxytryptophol (5-HTOL) by monoamine oxidase (MAO), or into N-acetylserotinin by N-acetyl-transferase (NAT). Melatonin is generated via the hydroxyl-indole O methyltransferase (HOMT) (Figure 3).
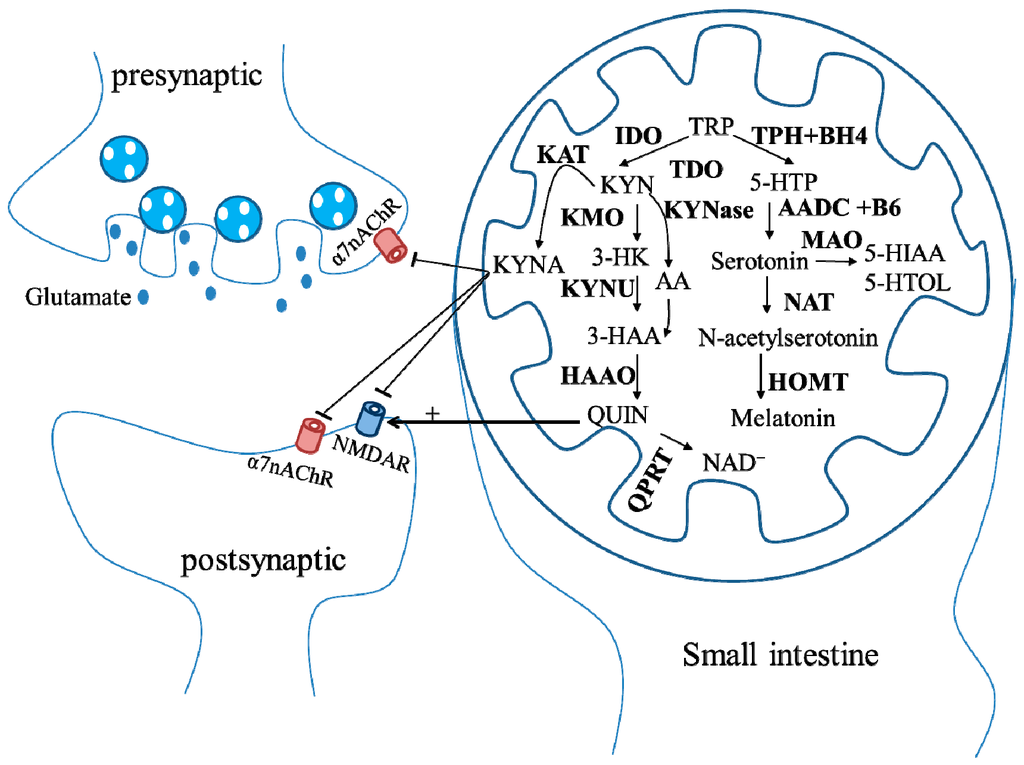
Figure 3.
Schematic representation of the tryptophan (TRP) metabolic pathway. Most TRP is used as the precursor of kynurenine (KYN) pathway, in which TRP is firstly oxidized by TRP 2,3-dioxygenase (TDO) or indoleamine 2,3-dioxygenase (IDO) to the kynurenine (KYN). There are at least three pathways for KYN metabolism. (1) KYN aminotransferase (KAT) pathway: KYN is catabolized by KAT to form KYN acid (KYNA), which antagonizes N-methyl-D-aspartate receptors (NMDAR) and α7 nicotinic receptors (α7nAChR); (2) kynurenine 3-monoxygenase (KMO) pathway: KYN is converted into 3-HK by KMO. 3-HK is converted into 3-HAA by KAT; (3) KYNase pathway: KYN is metabolized by KYNase to form anthranilic acid (AA), which is converted into 3-hydroxyanthranilic acid (3-HAA) by AA3MO. 3-HAA is oxidized by 3-hydroxyanthranilic acid oxidase (HAAO) to quinolinic acid (QUIN), which generates NAD+ through quinolinate phosphoribosyltransferase (QPRT). Additionally, TRP is metabolized to 5-hydroxytryptophan (5-HTP) through TRP hydroxylase (TPH) and tetrahydrobiopterin (BH4). Serotonin is synthesized from 5-HTP via aromatic acid decarboxylase (AADC) and the vitamin B6. Serotonin is converted into 5-Hydroxyindoleacetic acid (5-HIAA) or 5-hydroxytryptophol (5-HTOL) by monoamine oxidase (MAO) or into N-acetylserotinin by N-acetyl-transferase (NAT). Melatonin is generated via the hydroxyl-indole O methyltransferase (HOMT).
KYN pathway plays a crucial role in the neuroinflammation because this pathway is affected by several pro-inflammatory cytokines, which may interfere with enzyme expressions. Consequently, excessive pro-inflammatory cytokines favor the KMO branch of the pathway []. In addition to affecting dopamine, norepinephrine, β-endorphin, serotonin and endocrine, such as cortisol, prolactin and growth hormone, TRP also initiates excitotoxicity through the generation of KYNA to inhibit N-methyl-D-aspartate (NMDA) receptors [] and α7 nicotinic receptors [], and the generation of QUIN to activate N-methyl-D-aspartate (NMDA) receptors [], all linking to the pathogenesis of NDs (Figure 3). Derangements of the TRP metabolism may possibly directly or indirectly lead to accumulation of neurotoxicity, causing PD, HD, or AD.
4. Conclusions
Since the WHO predicts that NDs will surpass cancer in the rank of top 10 cause of death by 2050, any effective therapy for NDs is needed. Potential drugs or interventions for the treatment of NDs are summarized in the Table 1. A spate of new therapeutics targeting HDAC for treating various types of disorders, such as diabetes, systemic lupus erythematosus (SLE), hepatocellular carcinomas, leukemia and lymphoma, include siRNA HDAC [], SAHA [], PCI-24781 (Abexinostat) [], ITF-2357 (Givinostat) []; MS-275 (Entinostat) [], MGCD 0103 (Mocetinostat) [], LBH-589 (Panobinostat) [], FK228 (Romidepsin) [], AGK2 [], and PXD-101 (Belinostat) []. These novel chemicals have been under investigation as monotherapy or in combinatorial therapy as alternatives or adjuvants to traditional therapies. Nevertheless, epigenetic misregulation, ATM, and neuroinflammation may individually or symphonically involve the pathogenesis of NDs. The use of HDACI may potentially be able to rescue the deteriorated functions of NDs. Genetic ablation and pharmacological inhibitors of ATM can reduce mt-HTT toxicity to protect neurons. Amplifying KYNA and/or mitigating 3-HK and QUIN, and the use of KMO inhibitor reduced the dystonia and dyskinesia and improved striatal dys-functions through the attenuation of neuroinflammation. Synchronically targeting the three mechanisms may not only preserve neuron cells, inhibit cell death, and limit neuroinflammation, leading to slowing or alleviating the symptoms of PD, HD, and AD, but also offer potential beneficial therapy for other neurocognitive disorders in the future.

Table 1.
Potential drugs or interventions for the treatment of neurodegenerative diseases (NDs), including Parkinson’s disease (PD), Huntington’s disease (HD), and Alzheimer’s disease (AD), are classified by mechanisms, including epigenetic misregulation, ATM, and neuroinflammation.
| Mechanisms | Epigenetic Misregulation | Reference | ATM | Reference | Neuroinflammation | Reference |
|---|---|---|---|---|---|---|
| NDs | ||||||
| PD | VPA | [,] | ATM KO | [] | l-KYN | [] |
| TSA | [,] | Ro 61-6048 | [,] | |||
| Butyrate | [,] | |||||
| MS-275 (Entinostat) | [] | |||||
| AGK2 | [] | |||||
| HD | VPA | [] | KU-60019 | [] | SzR-72 | [] |
| TSA | [] | Ro 61-8648 | [] | |||
| Butyrate | [,,,] | |||||
| Pimelic diphenylamide | [] | |||||
| SAHA | [] | |||||
| AGK2 | [] | |||||
| AD | VPA | [] | Coptisine | [] | ||
| TSA | [] | |||||
| MS-275 (Entinostat) | [] |
Acknowledgments
This study was funded by the Ministry of Science and Technology, Taiwan (MOST 103-2320-B-039-021-MY3), Health and welfare surcharge of tobacco products, China Medical University Hospital Cancer Research Center of Excellence (MOHW104-TDUB-212-124-002, Taiwan), Taiwan Ministry of Health and Welfare Clinical Trial and Research Center of Excellence (MOHW104-TDU-B-212-113002) and CMU under the Aim for Top University Plan of the Ministry of Education, Taiwan.
Author Contributions
Hueng-Chuen Fan, Ching-Shiang Chi, Shinn-Zong Lin, and Horng-Jyh Harn proposed the original idea for this work. Shin-Nan Cheng, Hsiu-Fen Lee, and Jeng-Dau Tsai collected information regarding NDs and offered critical viewpoints about the treatments of NDs. Hueng-Chuen Fan and Horng-Jyh Harn wrote and revised this manuscript. Horng-Jyh Harn took responsibility for reviewing the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Fan, H.C.; Hu, C.F.; Juan, C.J.; Chen, S.J. Current proceedings of childhood stroke. Stroke Res. Treat. 2011. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.F.; Fan, H.C.; Chang, C.F.; Chen, S.J. Current approaches to the treatment of head injury in children. Pediatr. Neonatol. 2013, 54, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.C.; Chen, S.J.; Harn, H.J.; Lin, S.Z. Parkinson’s disease: From genetics to treatments. Cell Transplant. 2013, 22, 639–652. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.C.; Ho, L.I.; Chi, C.S.; Chen, S.J.; Peng, G.S.; Chan, T.M.; Lin, S.Z.; Harn, H.J. Polyglutamine (PolyQ) diseases: Genetics to treatments. Cell Transplant. 2014, 23, 441–458. [Google Scholar] [CrossRef] [PubMed]
- Zhongling, F.; Gang, Z.; Lei, Y. Neural stem cells and Alzheimer’s disease: Challenges and hope. Am. J. Alzheimers Dis. Other Dement. 2009, 24, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Menken, M.; Munsat, T.L.; Toole, J.F. The global burden of disease study: Implications for neurology. Arch. Neurol. 2000, 57, 418–420. [Google Scholar] [CrossRef] [PubMed]
- Scheper, W.; Hoozemans, J.J. The unfolded protein response in neurodegenerative diseases: A neuropathological perspective. Acta Neuropathol. 2015, 130, 315–331. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S. Regulation of autophagy by mTOR-dependent and mTOR-independent pathways: Autophagy dysfunction in neurodegenerative diseases and therapeutic application of autophagy enhancers. Biochem. Soc. Trans. 2013, 41, 1103–1130. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, C.; Haynes, C.M.; Yang, Y.; Harding, H.P.; Ron, D. Ubiquitin-like protein 5 positively regulates chaperone gene expression in the mitochondrial unfolded protein response. Genetics 2006, 174, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Saleem, M.; Abbas, K.; Manan, M.; Ijaz, H.; Ahmed, B.; Ali, M.; Hanif, M.; Farooqi, A.A.; Qadir, M.I. Review-Epigenetic therapy for cancer. Pak. J. Pharm. Sci. 2015, 28, 1023–1032. [Google Scholar] [PubMed]
- Schaefer, M.; Lyko, F. Solving the Dnmt2 enigma. Chromosoma 2010, 119, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Hagerty, S.; Cormier, K.A.; Kim, J.; Kung, A.L.; Ferrante, R.J.; Ryu, H. Monoallele deletion of CBP leads to pericentromeric heterochromatin condensation through ESET expression and histone H3 (K9) methylation. Hum. Mol. Genet. 2008, 17, 1774–1782. [Google Scholar] [CrossRef] [PubMed]
- Gryder, B.E.; Sodji, Q.H.; Oyelere, A.K. Targeted cancer therapy: Giving histone deacetylase inhibitors all they need to succeed. Future Med. Chem. 2012, 4, 505–524. [Google Scholar] [CrossRef] [PubMed]
- De Souza, C.; Chatterji, B.P. HDAC inhibitors as novel anti-cancer therapeutics. Recent Pat. Anticancer Drug Discov. 2015, 10, 145–162. [Google Scholar] [CrossRef] [PubMed]
- Minucci, S.; Pelicci, P.G. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer 2006, 6, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Kazantsev, A.G.; Thompson, L.M. Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat. Rev. Drug Discov. 2008, 7, 854–868. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.A.; Denton, E.L.; Arrowsmith, C.H.; Lupien, M.; Schapira, M. A global assessment of cancer genomic alterations in epigenetic mechanisms. Epigenet. Chromatin 2014, 7, 29. [Google Scholar] [CrossRef] [PubMed]
- Chahrour, M.; Jung, S.Y.; Shaw, C.; Zhou, X.; Wong, S.T.; Qin, J.; Zoghbi, H.Y. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 2008, 320, 1224–1229. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.M.; Bird, A. DNA methylation landscapes: Provocative insights from epigenomics. Nat. Rev. Genet. 2008, 9, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Wang, L.; Wang, H.; Xia, L.; Erdjument-Bromage, H.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 2002, 298, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Francis, N.J.; Kingston, R.E.; Woodcock, C.L. Chromatin compaction by a polycomb group protein complex. Science 2004, 306, 1574–1577. [Google Scholar] [CrossRef] [PubMed]
- Vire, E.; Brenner, C.; Deplus, R.; Blanchon, L.; Fraga, M.; Didelot, C.; Morey, L.; van Eynde, A.; Bernard, D.; Vanderwinden, J.M.; et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature 2006, 439, 871–874. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, M.E.; Li, X.; Toy, K.; DuPrie, M.; Ventura, A.C.; Banerjee, M.; Ljungman, M.; Merajver, S.D.; Kleer, C.G. Downregulation of EZH2 decreases growth of estrogen receptor-negative invasive breast carcinoma and requires BRCA1. Oncogene 2009, 28, 843–853. [Google Scholar] [CrossRef] [PubMed]
- Fiskus, W.; Wang, Y.; Sreekumar, A.; Buckley, K.M.; Shi, H.; Jillella, A.; Ustun, C.; Rao, R.; Fernandez, P.; Chen, J.; et al. Combined epigenetic therapy with the histone methyltransferase EZH2 inhibitor 3-deazaneplanocin A and the histone deacetylase inhibitor panobinostat against human AML cells. Blood 2009, 114, 2733–2743. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Tanaka, K. Regulatory mechanisms involved in the control of ubiquitin homeostasis. J. Biochem. 2010, 147, 793–798. [Google Scholar] [CrossRef] [PubMed]
- Weake, V.M.; Workman, J.L. Histone ubiquitination: Triggering gene activity. Mol. Cell 2008, 29, 653–663. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Wang, X.; Rosenfeld, M.G. Histone H2A ubiquitination in transcriptional regulation and DNA damage repair. Int. J. Biochem. Cell Biol. 2009, 41, 12–15. [Google Scholar] [CrossRef] [PubMed]
- Geng, F.; Tansey, W.P. Polyubiquitylation of histone H2B. Mol. Biol. Cell 2008, 19, 3616–3624. [Google Scholar] [CrossRef] [PubMed]
- Nickel, B.E.; Allis, C.D.; Davie, J.R. Ubiquitinated histone H2B is preferentially located in transcriptionally active chromatin. Biochemistry 1989, 28, 958–963. [Google Scholar] [CrossRef] [PubMed]
- Osley, M.A. Regulation of histone H2A and H2B ubiquitylation. Brief. Funct. Genom. Proteom. 2006, 5, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Nijman, S.M.; Luna-Vargas, M.P.; Velds, A.; Brummelkamp, T.R.; Dirac, A.M.; Sixma, T.K.; Bernards, R. A genomic and functional inventory of deubiquitinating enzymes. Cell 2005, 123, 773–786. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Pao, G.M.; Chen, H.W.; Verma, I.M.; Hunter, T. Enhancement of BRCA1 E3 ubiquitin ligase activity through direct interaction with the BARD1 protein. J. Biol. Chem. 2003, 278, 5255–5263. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, J.; Gill, G. SUMO engages multiple corepressors to regulate chromatin structure and transcription. Epigenetics 2009, 4, 440–444. [Google Scholar] [CrossRef] [PubMed]
- Kalocsay, M.; Hiller, N.J.; Jentsch, S. Chromosome-wide Rad51 spreading and SUMO-H2A.Z-dependent chromosome fixation in response to a persistent DNA double-strand break. Mol. Cell 2009, 33, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Shiio, Y.; Eisenman, R.N. Histone sumoylation is associated with transcriptional repression. Proc. Natl. Acad. Sci. USA 2003, 100, 13225–13230. [Google Scholar] [CrossRef] [PubMed]
- Mossessova, E.; Lima, C.D. Ulp1-SUMO crystal structure and genetic analysis reveal conserved interactions and a regulatory element essential for cell growth in yeast. Mol. Cell 2000, 5, 865–876. [Google Scholar] [CrossRef]
- Geiss-Friedlander, R.; Melchior, F. Concepts in sumoylation: A decade on. Nat. Rev. Mol. Cell Biol. 2007, 8, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Biton, S.; Barzilai, A.; Shiloh, Y. The neurological phenotype of Ataxia-telangiectasia: Solving a persistent puzzle. DNA Repair 2008, 7, 1028–1038. [Google Scholar] [CrossRef] [PubMed]
- Boder, E. Ataxia-telangiectasia: Some historic, clinical and pathologic observations. Birth Defects Orig. Artic. Ser. 1975, 11, 255–270. [Google Scholar] [PubMed]
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Kuzminov, A. Single-strand interruptions in replicating chromosomes cause double-strand breaks. Proc. Natl. Acad. Sci. USA 2001, 98, 8241–8246. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Paull, T.T. Activation and regulation of ATM kinase activity in response to DNA double-strand breaks. Oncogene 2007, 26, 7741–7748. [Google Scholar] [CrossRef] [PubMed]
- Burma, S.; Chen, B.P.; Murphy, M.; Kurimasa, A.; Chen, D.J. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J. Biol. Chem. 2001, 276, 42462–42467. [Google Scholar] [CrossRef] [PubMed]
- Benada, J.; Macurek, L. Targeting the checkpoint to kill cancer cells. Biomolecules 2015, 5, 1912–1937. [Google Scholar] [CrossRef] [PubMed]
- Imbriano, C.; Gurtner, A.; Cocchiarella, F.; Di, A.S.; Basile, V.; Gostissa, M.; Dobbelstein, M.; del Sal, G.; Piaggio, G.; Mantovani, R. Direct p53 transcriptional repression: In vivo analysis of CCAAT-containing G2/M promoters. Mol. Cell. Biol. 2005, 25, 3737–3751. [Google Scholar] [CrossRef] [PubMed]
- Medema, R.H.; Macurek, L. Checkpoint control and cancer. Oncogene 2012, 31, 2601–2613. [Google Scholar] [CrossRef] [PubMed]
- Cortez, D.; Guntuku, S.; Qin, J.; Elledge, S.J. ATR and ATRIP: Partners in checkpoint signaling. Science 2001, 294, 1713–1716. [Google Scholar] [CrossRef] [PubMed]
- Harvey, S.L.; Charlet, A.; Haas, W.; Gygi, S.P.; Kellogg, D.R. Cdk1-dependent regulation of the mitotic inhibitor Wee1. Cell 2005, 122, 407–420. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, P.J. ATM and Ataxia telangiectasia. EMBO Rep. 2004, 5, 772–776. [Google Scholar] [CrossRef] [PubMed]
- Shiloh, Y.; Ziv, Y. The ATM protein kinase: Regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 2013, 14, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Lavin, M.F.; Kozlov, S. ATM activation and DNA damage response. Cell Cycle 2007, 6, 931–942. [Google Scholar] [CrossRef] [PubMed]
- Gatei, M.; Scott, S.P.; Filippovitch, I.; Soronika, N.; Lavin, M.F.; Weber, B.; Khanna, K.K. Role for ATM in DNA damage-induced phosphorylation of BRCA1. Cancer Res. 2000, 60, 3299–3304. [Google Scholar] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Shanbhag, N.M.; Rafalska-Metcalf, I.U.; Balane-Bolivar, C.; Janicki, S.M.; Greenberg, R.A. ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell 2010, 141, 970–981. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Hart, R.P.; Mallimo, E.M.; Swerdel, M.R.; Kusnecov, A.W.; Herrup, K. EZH2-mediated H3K27 trimethylation mediates neurodegeneration in Ataxia-telangiectasia. Nat. Neurosci. 2013, 16, 1745–1753. [Google Scholar] [CrossRef] [PubMed]
- Hensley, K. Neuroinflammation in Alzheimer’s disease: Mechanisms, pathologic consequences, and potential for therapeutic manipulation. J. Alzheimers Dis. 2010, 21, 1–14. [Google Scholar] [PubMed]
- Aguzzi, A.; Barres, B.A.; Bennett, M.L. Microglia: Scapegoat, saboteur, or something else? Science 2013, 339, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Gendelman, H.E. Neural immunity: Friend or foe? J. Neurovirol. 2002, 8, 474–479. [Google Scholar] [CrossRef] [PubMed]
- Zattoni, M.; Mura, M.L.; Deprez, F.; Schwendener, R.A.; Engelhardt, B.; Frei, K.; Fritschy, J.M. Brain infiltration of leukocytes contributes to the pathophysiology of temporal lobe epilepsy. J. Neurosci. 2011, 31, 4037–4050. [Google Scholar] [CrossRef] [PubMed]
- Cacquevel, M.; Lebeurrier, N.; Cheenne, S.; Vivien, D. Cytokines in neuroinflammation and Alzheimer’s disease. Curr. Drug Targets 2004, 5, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Tansey, M.G.; Goldberg, M.S. Neuroinflammation in Parkinson’s disease: Its role in neuronal death and implications for therapeutic intervention. Neurobiol. Dis. 2010, 37, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Trager, U.; Andre, R.; Magnusson-Lind, A.; Miller, J.R.; Connolly, C.; Weiss, A.; Grueninger, S.; Silajdzic, E.; Smith, D.L.; Leavitt, B.R.; et al. Characterisation of immune cell function in fragment and full-length Huntington’s disease mouse models. Neurobiol. Dis. 2014, 73C, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Meraz-Rios, M.A.; Toral-Rios, D.; Franco-Bocanegra, D.; Villeda-Hernandez, J.; Campos-Pena, V. Inflammatory process in Alzheimer’s Disease. Front. Integr. Neurosci. 2013, 7, 59. [Google Scholar] [CrossRef] [PubMed]
- Sanger, G.J. 5-Hydroxytryptamine and the gastrointestinal tract: Where next? Trends Pharmacol. Sci. 2008, 29, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Sandyk, R. l-Tryptophan in neuropsychiatric disorders: A review. Int. J. Neurosci. 1992, 67, 127–144. [Google Scholar] [CrossRef] [PubMed]
- Kayumov, L.; Casper, R.F.; Hawa, R.J.; Perelman, B.; Chung, S.A.; Sokalsky, S.; Shapiro, C.M. Blocking low-wavelength light prevents nocturnal melatonin suppression with no adverse effect on performance during simulated shift work. J. Clin. Endocrinol. Metab. 2005, 90, 2755–2761. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, D.M.; Marsh-Richard, D.M.; Mathias, C.W.; Hood, A.J.; Addicott, M.A.; Moeller, F.G.; Morgan, C.J.; Badawy, A.A. Comparison of 50- and 100-g l-tryptophan depletion and loading formulations for altering 5-HT synthesis: Pharmacokinetics, side effects, and mood states. Psychopharmacology 2008, 198, 431–445. [Google Scholar] [CrossRef] [PubMed]
- Sainio, E.L.; Pulkki, K.; Young, S.N. l-Tryptophan: Biochemical, nutritional and pharmacological aspects. Amino Acids 1996, 10, 21–47. [Google Scholar] [CrossRef] [PubMed]
- Reyes, O.J.; Lugo, H.R.; Gonzalez-Esquivel, D.; Ugalde-Muniz, P.; Jimenez-Anguiano, A.; Pineda, B.; Pedraza-Chaverri, J.; Rios, C.; Perez, C.V. Kynurenines with neuroactive and redox properties: Relevance to aging and brain diseases. Oxid. Med. Cell Longev. 2014, 646909. [Google Scholar] [CrossRef]
- Campbell, B.M.; Charych, E.; Lee, A.W.; Moller, T. Kynurenines in CNS disease: Regulation by inflammatory cytokines. Front. Neurosci. 2014, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- Stone, T.W. Neuropharmacology of quinolinic and kynurenic acids. Pharmacol. Rev. 1993, 45, 309–379. [Google Scholar] [PubMed]
- Hilmas, C.; Pereira, E.F.; Alkondon, M.; Rassoulpour, A.; Schwarcz, R.; Albuquerque, E.X. The brain metabolite kynurenic acid inhibits α7 nicotinic receptor activity and increases non-α7 nicotinic receptor expression: Physiopathological implications. J. Neurosci. 2001, 21, 7463–7473. [Google Scholar] [PubMed]
- Stone, T.W.; Mackay, G.M.; Forrest, C.M.; Clark, C.J.; Darlington, L.G. Tryptophan metabolites and brain disorders. Clin. Chem. Lab. Med. 2003, 41, 852–859. [Google Scholar] [CrossRef] [PubMed]
- De Lau, L.M.; Breteler, M.M. Epidemiology of Parkinson’s disease. Lancet Neurol. 2006, 5, 525–535. [Google Scholar] [CrossRef]
- Thomas, B.; Beal, M.F. Parkinson’s disease. Hum. Mol. Genet. 2007, 16, R183–R194. [Google Scholar] [CrossRef] [PubMed]
- Bernheimer, H.; Birkmayer, W.; Hornykiewicz, O.; Jellinger, K.; Seitelberger, F. Brain dopamine and the syndromes of Parkinson and Huntington. Clinical, morphological and neurochemical correlations. J. Neurol. Sci. 1973, 20, 415–455. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. A-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Devine, M.J.; Plun-Favreau, H.; Wood, N.W. Parkinson’s disease and cancer: Two wars, one front. Nat. Rev. Cancer 2011, 11, 812–823. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, L.; Takuma, H.; Tamaoka, A.; Kurisaki, H.; Date, H.; Tsuji, S.; Iwata, A. CpG demethylation enhances α-synuclein expression and affects the pathogenesis of Parkinson’s disease. PLoS ONE 2010, 5, e15522. [Google Scholar] [CrossRef] [PubMed]
- Kontopoulos, E.; Parvin, J.D.; Feany, M.B. A-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum. Mol. Genet. 2006, 15, 3012–3023. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.S.; Wang, C.C.; Bortner, C.D.; Peng, G.S.; Wu, X.; Pang, H.; Lu, R.B.; Gean, P.W.; Chuang, D.M.; Hong, J.S. Valproic acid and other histone deacetylase inhibitors induce microglial apoptosis and attenuate lipopolysaccharide-induced dopaminergic neurotoxicity. Neuroscience 2007, 149, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Marinova, Z.; Ren, M.; Wendland, J.R.; Leng, Y.; Liang, M.H.; Yasuda, S.; Leeds, P.; Chuang, D.M. Valproic acid induces functional heat-shock protein 70 via Class I histone deacetylase inhibition in cortical neurons: A potential role of Sp1 acetylation. J. Neurochem. 2009, 111, 976–987. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Bercury, K.; Cummiskey, J.; Luong, N.; Lebin, J.; Freed, C.R. Phenylbutyrate up-regulates the DJ-1 protein and protects neurons in cell culture and in animal models of Parkinson disease. J. Biol. Chem. 2011, 286, 14941–14951. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Ikemoto, M.; Kawarabayashi, T.; Ikeda, M.; Nishinakagawa, T.; Hosokawa, M.; Shoji, M.; Takahashi, M.; Nakashima, M. A chemical chaperone, sodium 4-phenylbutyric acid, attenuates the pathogenic potency in human α-synuclein A30P + A53T transgenic mice. Parkinsonism Relat. Disord. 2009, 15, 649–654. [Google Scholar] [CrossRef] [PubMed]
- Price, P.A.; Parkes, J.D.; Marsden, C.D. Sodium valproate in the treatment of levodopa-induced dyskinesia. J. Neurol. Neurosurg. Psychiatry 1978, 41, 702–706. [Google Scholar] [CrossRef] [PubMed]
- Nutt, J.; Williams, A.; Plotkin, C.; Eng, N.; Ziegler, M.; Calne, D.B. Treatment of Parkinson’s disease with sodium valproate: Clinical, pharmacological, and biochemical observations. Can. J. Neurol. Sci. 1979, 6, 337–343. [Google Scholar] [PubMed]
- Perlman, S.; Becker-Catania, S.; Gatti, R.A. Ataxia-telangiectasia: Diagnosis and treatment. Semin. Pediatr. Neurol. 2003, 10, 173–182. [Google Scholar] [CrossRef]
- Chun, H.H.; Gatti, R.A. Ataxia-telangiectasia, an evolving phenotype. DNA Repair 2004, 3, 1187–1196. [Google Scholar] [CrossRef] [PubMed]
- Crawford, T.O. Ataxia telangiectasia. Semin. Pediatr. Neurol. 1998, 5, 287–294. [Google Scholar] [CrossRef]
- Woods, C.G.; Taylor, A.M. Ataxia telangiectasia in the British Isles: The clinical and laboratory features of 70 affected individuals. Q. J. Med. 1992, 82, 169–179. [Google Scholar] [PubMed]
- Eilam, R.; Peter, Y.; Groner, Y.; Segal, M. Late degeneration of nigro-striatal neurons in ATM−/− mice. Neuroscience 2003, 121, 83–98. [Google Scholar] [CrossRef]
- Mavrou, A.; Tsangaris, G.T.; Roma, E.; Kolialexi, A. The ATM gene and Ataxia telangiectasia. Anticancer Res. 2008, 28, 401–405. [Google Scholar] [PubMed]
- Veeriah, S.; Taylor, B.S.; Meng, S.; Fang, F.; Yilmaz, E.; Vivanco, I.; Janakiraman, M.; Schultz, N.; Hanrahan, A.J.; Pao, W.; et al. Somatic mutations of the Parkinson’s disease-associated gene PARK2 in glioblastoma and other human malignancies. Nat. Genet. 2010, 42, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Veeriah, S.; Morris, L.; Solit, D.; Chan, T.A. The familial Parkinson disease gene PARK2 is a multisite tumor suppressor on chromosome 6q25.2–27 that regulates cyclin E. Cell Cycle 2010, 9, 1451–1452. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; Itagaki, S.; Boyes, B.E.; McGeer, E.G. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 1988, 38, 1285–1291. [Google Scholar] [CrossRef] [PubMed]
- Imamura, K.; Hishikawa, N.; Sawada, M.; Nagatsu, T.; Yoshida, M.; Hashizume, Y. Distribution of major histocompatibility complex class II-positive microglia and cytokine profile of Parkinson’s disease brains. Acta Neuropathol. 2003, 106, 518–526. [Google Scholar] [CrossRef] [PubMed]
- Nagatsu, T.; Mogi, M.; Ichinose, H.; Togari, A. Changes in cytokines and neurotrophins in Parkinson’s disease. J. Neural Transm. Suppl. 2000, 60, 277–290. [Google Scholar] [PubMed]
- Mogi, M.; Togari, A.; Tanaka, K.; Ogawa, N.; Ichinose, H.; Nagatsu, T. Increase in level of tumor necrosis factor (TNF)-α in 6-hydroxydopamine-lesioned striatum in rats without influence of systemic l-DOPA on the TNF-α induction. Neurosci. Lett. 1999, 268, 101–104. [Google Scholar] [CrossRef]
- Knyihar-Csillik, E.; Chadaide, Z.; Mihaly, A.; Krisztin-Peva, B.; Fenyo, R.; Vecsei, L. Effect of 6-hydroxydopamine treatment on kynurenine aminotransferase-I (KAT-I) immunoreactivity of neurons and glial cells in the rat substantia nigra. Acta Neuropathol. 2006, 112, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Knyihar-Csillik, E.; Csillik, B.; Pakaski, M.; Krisztin-Peva, B.; Dobo, E.; Okuno, E.; Vecsei, L. Decreased expression of kynurenine aminotransferase-I (KAT-I) in the substantia nigra of mice after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) treatment. Neuroscience 2004, 126, 899–914. [Google Scholar] [CrossRef] [PubMed]
- Luchowski, P.; Luchowska, E.; Turski, W.A.; Urbanska, E.M. 1-Methyl-4-phenylpyridinium and 3-nitropropionic acid diminish cortical synthesis of kynurenic acid via interference with kynurenine aminotransferases in rats. Neurosci. Lett. 2002, 330, 49–52. [Google Scholar] [CrossRef]
- Ogawa, T.; Matson, W.R.; Beal, M.F.; Myers, R.H.; Bird, E.D.; Milbury, P.; Saso, S. Kynurenine pathway abnormalities in Parkinson’s disease. Neurology 1992, 42, 1702–1706. [Google Scholar] [CrossRef] [PubMed]
- Widner, B.; Leblhuber, F.; Fuchs, D. Increased neopterin production and tryptophan degradation in advanced Parkinson’s disease. J. Neural Transm. 2002, 109, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.; Lee, K.S.; Lee, H.J.; Noh, Y.H.; Kim, D.H.; Lee, J.Y.; Cho, S.H.; Yoon, O.J.; Lee, W.B.; Kim, K.Y.; et al. Kynurenic acid attenuates MPP+-induced dopaminergic neuronal cell death via a Bax-mediated mitochondrial pathway. Eur. J. Cell Biol. 2008, 87, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Silva-Adaya, D.; Perez-De La Cruz, V.; Villeda-Hernandez, J.; Carrillo-Mora, P.; Gonzalez-Herrera, I.G.; Garcia, E.; Colin-Barenque, L.; Pedraza-Chaverri, J.; Santamaria, A. Protective effect of l-kynurenine and probenecid on 6-hydroxydopamine-induced striatal toxicity in rats: Implications of modulating kynurenate as a protective strategy. Neurotoxicol. Teratol. 2011, 33, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Connop, B.P.; Boegman, R.J.; Jhamandas, K.; Beninger, R.J. Excitotoxic action of NMDA agonists on nigrostriatal dopaminergic neurons: Modulation by inhibition of nitric oxide synthesis. Brain Res. 1995, 676, 124–132. [Google Scholar] [CrossRef]
- Miranda, A.F.; Boegman, R.J.; Beninger, R.J.; Jhamandas, K. Protection against quinolinic acid-mediated excitotoxicity in nigrostriatal dopaminergic neurons by endogenous kynurenic acid. Neuroscience 1997, 78, 967–975. [Google Scholar] [CrossRef]
- Acuna-Castroviejo, D.; Tapias, V.; Lopez, L.C.; Doerrier, C.; Camacho, E.; Carrion, M.D.; Mora, F.; Espinosa, A.; Escames, G. Protective effects of synthetic kynurenines on 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced parkinsonism in mice. Brain Res. Bull. 2011, 85, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Hamann, M.; Sander, S.E.; Richter, A. Effects of the kynurenine 3-hydroxylase inhibitor Ro 61–8048 after intrastriatal injections on the severity of dystonia in the dtsz mutant. Eur. J. Pharmacol. 2008, 586, 156–159. [Google Scholar] [CrossRef] [PubMed]
- Richter, A.; Hamann, M. The kynurenine 3-hydroxylase inhibitor Ro 61-8048 improves dystonia in a genetic model of paroxysmal dyskinesia. Eur. J. Pharmacol. 2003, 478, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Ouattara, B.; Belkhir, S.; Morissette, M.; Dridi, M.; Samadi, P.; Gregoire, L.; Meltzer, L.T.; Di, P.T. Implication of NMDA receptors in the antidyskinetic activity of cabergoline, CI-1041, and Ro 61–8048 in MPTP monkeys with levodopa-induced dyskinesias. J. Mol. Neurosci. 2009, 38, 128–142. [Google Scholar] [CrossRef] [PubMed]
- Tamim, M.K.; Samadi, P.; Morissette, M.; Gregoire, L.; Ouattara, B.; Levesque, D.; Rouillard, C.; Di, P.T. Effect of non-dopaminergic drug treatment on Levodopa induced dyskinesias in MPTP monkeys: Common implication of striatal neuropeptides. Neuropharmacology 2010, 58, 286–296. [Google Scholar] [CrossRef] [PubMed]
- Myers, R.H.; MacDonald, M.E.; Koroshetz, W.J.; Duyao, M.P.; Ambrose, C.M.; Taylor, S.A.; Barnes, G.; Srinidhi, J.; Lin, C.S.; Whaley, W.L. De novo expansion of a (CAG)n repeat in sporadic Huntington’s disease. Nat. Genet. 1993, 5, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Walker, F.O. Huntington’s disease. Lancet 2007, 369, 218–228. [Google Scholar] [CrossRef]
- Szebenyi, G.; Morfini, G.A.; Babcock, A.; Gould, M.; Selkoe, K.; Stenoien, D.L.; Young, M.; Faber, P.W.; MacDonald, M.E.; McPhaul, M.J.; et al. Neuropathogenic forms of huntingtin and androgen receptor inhibit fast axonal transport. Neuron 2003, 40, 41–52. [Google Scholar] [CrossRef]
- Bennett, E.J.; Shaler, T.A.; Woodman, B.; Ryu, K.Y.; Zaitseva, T.S.; Becker, C.H.; Bates, G.P.; Schulman, H.; Kopito, R.R. Global changes to the ubiquitin system in Huntington’s disease. Nature 2007, 448, 704–708. [Google Scholar] [CrossRef] [PubMed]
- Chafekar, S.M.; Duennwald, M.L. Impaired heat shock response in cells expressing full-length polyglutamine-expanded huntingtin. PLoS ONE 2012, 7, e37929. [Google Scholar] [CrossRef] [PubMed]
- Solans, A.; Zambrano, A.; Rodriguez, M.; Barrientos, A. Cytotoxicity of a mutant huntingtin fragment in yeast involves early alterations in mitochondrial OXPHOS complexes II and III. Hum. Mol. Genet. 2006, 15, 3063–3081. [Google Scholar] [CrossRef] [PubMed]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Ferrante, R.J.; Ryu, H.; Kubilus, J.K.; D’Mello, S.; Sugars, K.L.; Lee, J.; Lu, P.; Smith, K.; Browne, S.; Beal, M.F.; et al. Chemotherapy for the brain: The antitumor antibiotic mithramycin prolongs survival in a mouse model of Huntington’s disease. J. Neurosci. 2004, 24, 10335–10342. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.W.; Yildirim, F.; Yap, Y.S.; Dalin, S.; Matthews, B.J.; Velez, P.J.; Labadorf, A.; Housman, D.E.; Fraenkel, E. Extensive changes in DNA methylation are associated with expression of mutant huntingtin. Proc. Natl. Acad. Sci. USA 2013, 110, 2354–2359. [Google Scholar] [CrossRef] [PubMed]
- Steffan, J.S.; Bodai, L.; Pallos, J.; Poelman, M.; McCampbell, A.; Apostol, B.L.; Kazantsev, A.; Schmidt, E.; Zhu, Y.Z.; Greenwald, M.; et al. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature 2001, 413, 739–743. [Google Scholar] [CrossRef] [PubMed]
- Pallos, J.; Bodai, L.; Lukacsovich, T.; Purcell, J.M.; Steffan, J.S.; Thompson, L.M.; Marsh, J.L. Inhibition of specific HDACs and sirtuins suppresses pathogenesis in a Drosophila model of Huntington’s disease. Hum. Mol. Genet. 2008, 17, 3767–3775. [Google Scholar] [CrossRef] [PubMed]
- Sadri-Vakili, G.; Bouzou, B.; Benn, C.L.; Kim, M.O.; Chawla, P.; Overland, R.P.; Glajch, K.E.; Xia, E.; Qiu, Z.; Hersch, S.M.; et al. Histones associated with downregulated genes are hypo-acetylated in Huntington’s disease models. Hum. Mol. Genet. 2007, 16, 1293–1306. [Google Scholar] [CrossRef] [PubMed]
- Thomas, E.A.; Coppola, G.; Desplats, P.A.; Tang, B.; Soragni, E.; Burnett, R.; Gao, F.; Fitzgerald, K.M.; Borok, J.F.; Herman, D.; et al. The HDAC inhibitor 4b ameliorates the disease phenotype and transcriptional abnormalities in Huntington’s disease transgenic mice. Proc. Natl. Acad. Sci. USA 2008, 105, 15564–15569. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.; Lee, J.; Hagerty, S.W.; Soh, B.Y.; McAlpin, S.E.; Cormier, K.A.; Smith, K.M.; Ferrante, R.J. ESET/SETDB1 gene expression and histone H3 (K9) trimethylation in Huntington’s disease. Proc. Natl. Acad. Sci. USA 2006, 103, 19176–19181. [Google Scholar] [CrossRef] [PubMed]
- West, R.L.; Lee, J.M.; Maroun, L.E. Hypomethylation of the amyloid precursor protein gene in the brain of an Alzheimer’s disease patient. J. Mol. Neurosci. 1995, 6, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Chouliaras, L.; Rutten, B.P.; Kenis, G.; Peerbooms, O.; Visser, P.J.; Verhey, F.; van, O.J.; Steinbusch, H.W.; van den Hove, D.L. Epigenetic regulation in the pathophysiology of Alzheimer’s disease. Prog. Neurobiol. 2010, 90, 498–510. [Google Scholar] [CrossRef] [PubMed]
- Ferrante, R.J.; Kubilus, J.K.; Lee, J.; Ryu, H.; Beesen, A.; Zucker, B.; Smith, K.; Kowall, N.W.; Ratan, R.R.; Luthi-Carter, R.; et al. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington’s disease mice. J. Neurosci. 2003, 23, 9418–9427. [Google Scholar] [PubMed]
- Mielcarek, M.; Benn, C.L.; Franklin, S.A.; Smith, D.L.; Woodman, B.; Marks, P.A.; Bates, G.P. SAHA decreases HDAC 2 and 4 levels in vivo and improves molecular phenotypes in the R6/2 mouse model of Huntington’s disease. PLoS ONE 2011, 6, e27746. [Google Scholar] [CrossRef] [PubMed]
- McCampbell, A.; Taye, A.A.; Whitty, L.; Penney, E.; Steffan, J.S.; Fischbeck, K.H. Histone deacetylase inhibitors reduce polyglutamine toxicity. Proc. Natl. Acad. Sci. USA 2001, 98, 15179–15184. [Google Scholar] [CrossRef] [PubMed]
- Gardian, G.; Browne, S.E.; Choi, D.K.; Klivenyi, P.; Gregorio, J.; Kubilus, J.K.; Ryu, H.; Langley, B.; Ratan, R.R.; Ferrante, R.J.; et al. Neuroprotective effects of phenylbutyrate in the N171-82Q transgenic mouse model of Huntington’s disease. J. Biol. Chem. 2005, 280, 556–563. [Google Scholar] [CrossRef] [PubMed]
- Ebbel, E.N.; Leymarie, N.; Schiavo, S.; Sharma, S.; Gevorkian, S.; Hersch, S.; Matson, W.R.; Costello, C.E. Identification of phenylbutyrate-generated metabolites in Huntington disease patients using parallel liquid chromatography/electrochemical array/mass spectrometry and off-line tandem mass spectrometry. Anal. Biochem. 2010, 399, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Hogarth, P.; Lovrecic, L.; Krainc, D. Sodium phenylbutyrate in Huntington’s disease: A dose-finding study. Mov Disord. 2007, 22, 1962–1964. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.T.; Liu, G.; Leeds, P.; Chuang, D.M. Combined treatment with the mood stabilizers lithium and valproate produces multiple beneficial effects in transgenic mouse models of Huntington’s disease. Neuropsychopharmacology 2011, 36, 2406–2421. [Google Scholar] [CrossRef] [PubMed]
- Bates, E.A.; Victor, M.; Jones, A.K.; Shi, Y.; Hart, A.C. Differential contributions of Caenorhabditis elegans histone deacetylases to huntingtin polyglutamine toxicity. J. Neurosci. 2006, 26, 2830–2838. [Google Scholar] [CrossRef] [PubMed]
- Hockly, E.; Richon, V.M.; Woodman, B.; Smith, D.L.; Zhou, X.; Rosa, E.; Sathasivam, K.; Ghazi-Noori, S.; Mahal, A.; Lowden, P.A.; et al. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington’s disease. Proc. Natl. Acad. Sci. USA 2003, 100, 2041–2046. [Google Scholar] [CrossRef] [PubMed]
- Luthi-Carter, R.; Taylor, D.M.; Pallos, J.; Lambert, E.; Amore, A.; Parker, A.; Moffitt, H.; Smith, D.L.; Runne, H.; Gokce, O.; et al. SIRT2 inhibition achieves neuroprotection by decreasing sterol biosynthesis. Proc. Natl. Acad. Sci. USA 2010, 107, 7927–7932. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Zaman, K.; Ryu, H.; Conforto, A.; Ratan, R.R. Sequence-selective DNA binding drugs mithramycin A and chromomycin A3 are potent inhibitors of neuronal apoptosis induced by oxidative stress and DNA damage in cortical neurons. Ann. Neurol. 2001, 49, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Stack, E.C.; del Signore, S.J.; Luthi-Carter, R.; Soh, B.Y.; Goldstein, D.R.; Matson, S.; Goodrich, S.; Markey, A.L.; Cormier, K.; Hagerty, S.W.; et al. Modulation of nucleosome dynamics in Huntington’s disease. Hum. Mol. Genet. 2007, 16, 1164–1175. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.H.; Mattis, V.B.; Wang, N.; Al-Ramahi, I.; van den Berg, N.; Fratantoni, S.A.; Waldvogel, H.; Greiner, E.; Osmand, A.; Elzein, K.; et al. Targeting ATM ameliorates mutant Huntingtin toxicity in cell and animal models of Huntington’s disease. Sci. Transl. Med. 2014, 6, 268ra178. [Google Scholar] [CrossRef] [PubMed]
- Illuzzi, J.; Yerkes, S.; Parekh-Olmedo, H.; Kmiec, E.B. DNA breakage and induction of DNA damage response proteins precede the appearance of visible mutant huntingtin aggregates. J. Neurosci. Res. 2009, 87, 733–747. [Google Scholar] [CrossRef] [PubMed]
- Giuliano, P.; de Cristofaro, T.; Affaitati, A.; Pizzulo, G.M.; Feliciello, A.; Criscuolo, C.; de Michele, G.; Filla, A.; Avvedimento, E.V.; Varrone, S. DNA damage induced by polyglutamine-expanded proteins. Hum. Mol. Genet. 2003, 12, 2301–2309. [Google Scholar] [CrossRef] [PubMed]
- Forrest, C.M.; Mackay, G.M.; Stoy, N.; Spiden, S.L.; Taylor, R.; Stone, T.W.; Darlington, L.G. Blood levels of kynurenines, interleukin-23 and soluble human leucocyte antigen-G at different stages of Huntington’s disease. J. Neurochem. 2010, 112, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Schwarcz, R.; Tamminga, C.A.; Kurlan, R.; Shoulson, I. Cerebrospinal fluid levels of quinolinic acid in Huntington’s disease and schizophrenia. Ann. Neurol. 1988, 24, 580–582. [Google Scholar] [CrossRef] [PubMed]
- Guidetti, P.; Luthi-Carter, R.E.; Augood, S.J.; Schwarcz, R. Neostriatal and cortical quinolinate levels are increased in early grade Huntington’s disease. Neurobiol. Dis. 2004, 17, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Guidetti, P.; Schwarcz, R. 3-Hydroxykynurenine and quinolinate: Pathogenic synergism in early grade Huntington’s disease? Adv. Exp. Med. Biol. 2003, 527, 137–145. [Google Scholar] [PubMed]
- Schwarcz, R.; Albin, R.L. Huntington’s disease. In Ionotropic Glutamate Receptors as Therapeutic Targets; Lodge, D., Danysz, C.G., Eds.; FP Graham Publishing Co.: City, TN, USA, 2002; pp. 587–610. [Google Scholar]
- Sathyasaikumar, K.V.; Stachowski, E.K.; Amori, L.; Guidetti, P.; Muchowski, P.J.; Schwarcz, R. Dysfunctional kynurenine pathway metabolism in the R6/2 mouse model of Huntington’s disease. J. Neurochem. 2010, 113, 1416–1425. [Google Scholar] [CrossRef] [PubMed]
- Sapko, M.T.; Guidetti, P.; Yu, P.; Tagle, D.A.; Pellicciari, R.; Schwarcz, R. Endogenous kynurenate controls the vulnerability of striatal neurons to quinolinate: Implications for Huntington’s disease. Exp. Neurol. 2006, 197, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Zhou, D.; Sapp, E.; Aizawa, H.; Ge, P.; Bird, E.D.; Vonsattel, J.P.; DiFiglia, M. Quinolinic acid-induced increases in calbindin D28k immunoreactivity in rat striatal neurons in vivo and in vitro mimic the pattern seen in Huntington’s disease. Neuroscience 1995, 65, 397–407. [Google Scholar] [CrossRef]
- Giorgini, F.; Guidetti, P.; Nguyen, Q.; Bennett, S.C.; Muchowski, P.J. A genomic screen in yeast implicates kynurenine 3-monooxygenase as a therapeutic target for Huntington disease. Nat. Genet. 2005, 37, 526–531. [Google Scholar] [CrossRef] [PubMed]
- Campesan, S.; Green, E.W.; Breda, C.; Sathyasaikumar, K.V.; Muchowski, P.J.; Schwarcz, R.; Kyriacou, C.P.; Giorgini, F. The kynurenine pathway modulates neurodegeneration in a Drosophila model of Huntington’s disease. Curr. Biol. 2011, 21, 961–966. [Google Scholar] [CrossRef] [PubMed]
- Zwilling, D.; Huang, S.Y.; Sathyasaikumar, K.V.; Notarangelo, F.M.; Guidetti, P.; Wu, H.Q.; Lee, J.; Truong, J.; Andrews-Zwilling, Y.; Hsieh, E.W.; et al. Kynurenine 3-monooxygenase inhibition in blood ameliorates neurodegeneration. Cell 2011, 145, 863–874. [Google Scholar] [CrossRef] [PubMed]
- Zadori, D.; Nyiri, G.; Szonyi, A.; Szatmari, I.; Fulop, F.; Toldi, J.; Freund, T.F.; Vecsei, L.; Klivenyi, P. Neuroprotective effects of a novel kynurenic acid analogue in a transgenic mouse model of Huntington’s disease. J. Neural Transm. 2011, 118, 865–875. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Disease International. World Alzheimer Report 2009. Available online: https://www.alz.co.uk/research/files/WorldAlzheimerReport.pdf (accessed on 20 December 2015).
- Zhu, C.W.; Scarmeas, N.; Torgan, R.; Albert, M.; Brandt, J.; Blacker, D.; Sano, M.; Stern, Y. Clinical features associated with costs in early AD: Baseline data from the Predictors Study. Neurology 2006, 66, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Ahn, H.; Han, B.C.; Lee, S.H.; Cho, Y.W.; Kim, C.H.; Hong, E.J.; An, B.S.; Jeung, E.B.; Lee, G.S. Korean red ginseng extracts inhibit NLRP3 and AIM2 inflammasome activation. Immunol. Lett. 2014, 158, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Tanzi, R.E. A brief history of Alzheimer’s disease gene discovery. J. Alzheimers Dis. 2013, 33, S5–S13. [Google Scholar] [PubMed]
- Amtul, Z. Controversies looming over Alzheimer’s research: Do we have consensus over the path to follow? Ageing Res. Rev. 2015, S1568–S1637, 30023–30024. [Google Scholar]
- Katsel, P.; Tan, W.; Fam, P.; Purohit, D.P.; Haroutunian, V. Cell cycle checkpoint abnormalities during dementia: A plausible association with the loss of protection against oxidative stress in Alzheimer’s disease [corrected]. PLoS ONE 2013, 8, e68361. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.T.; Alafuzoff, I.; Bigio, E.H.; Bouras, C.; Braak, H.; Cairns, N.J.; Castellani, R.J.; Crain, B.J.; Davies, P.; del Tredici, K.; et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: A review of the literature. J. Neuropathol. Exp. Neurol. 2012, 71, 362–381. [Google Scholar] [CrossRef] [PubMed]
- Aizenstein, H.J.; Nebes, R.D.; Saxton, J.A.; Price, J.C.; Mathis, C.A.; Tsopelas, N.D.; Ziolko, S.K.; James, J.A.; Snitz, B.E.; Houck, P.R.; et al. Frequent amyloid deposition without significant cognitive impairment among the elderly. Arch. Neurol. 2008, 65, 1509–1517. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.; Paskavitz, J.; Remington, R.; Rasmussen, S.; Shea, T.B. Efficacy of a vitamin/nutriceutical formulation for early-stage Alzheimer’s disease: A 1-year, open-label pilot study with an 16-month caregiver extension. Am. J. Alzheimers Dis. Other Dement. 2008, 23, 571–585. [Google Scholar] [CrossRef] [PubMed]
- Mastroeni, D.; Grover, A.; Delvaux, E.; Whiteside, C.; Coleman, P.D.; Rogers, J. Epigenetic changes in Alzheimer’s disease: Decrements in DNA methylation. Neurobiol. Aging 2010, 31, 2025–2037. [Google Scholar] [CrossRef] [PubMed]
- Mastroeni, D.; Grover, A.; Delvaux, E.; Whiteside, C.; Coleman, P.D.; Rogers, J. Epigenetic mechanisms in Alzheimer’s disease. Neurobiol. Aging 2011, 32, 1161–1180. [Google Scholar] [CrossRef] [PubMed]
- Fuso, A.; Nicolia, V.; Pasqualato, A.; Fiorenza, M.T.; Cavallaro, R.A.; Scarpa, S. Changes in Presenilin 1 gene methylation pattern in diet-induced B vitamin deficiency. Neurobiol. Aging 2011, 32, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Fuso, A.; Seminara, L.; Cavallaro, R.A.; D’Anselmi, F.; Scarpa, S. S-adenosylmethionine/homocysteine cycle alterations modify DNA methylation status with consequent deregulation of PS1 and BACE and β-amyloid production. Mol. Cell. Neurosci. 2005, 28, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Fuso, A.; Nicolia, V.; Ricceri, L.; Cavallaro, R.A.; Isopi, E.; Mangia, F.; Fiorenza, M.T.; Scarpa, S. S-adenosylmethionine reduces the progress of the Alzheimer-like features induced by B-vitamin deficiency in mice. Neurobiol. Aging 2012, 33. [Google Scholar] [CrossRef] [PubMed]
- Serot, J.M.; Christmann, D.; Dubost, T.; Bene, M.C.; Faure, G.C. CSF-folate levels are decreased in late-onset AD patients. J. Neural Transm. 2001, 108, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, B.P.; Bottiglieri, T.; Arning, E.; Ziegler, M.G.; Hansen, L.A.; Masliah, E. Elevated S-adenosylhomocysteine in Alzheimer brain: Influence on methyltransferases and cognitive function. J. Neural Transm. 2004, 111, 547–567. [Google Scholar] [CrossRef] [PubMed]
- Fuso, A.; Nicolia, V.; Cavallaro, R.A.; Ricceri, L.; D’Anselmi, F.; Coluccia, P.; Calamandrei, G.; Scarpa, S. B-vitamin deprivation induces hyperhomocysteinemia and brain S-adenosylhomocysteine, depletes brain S-adenosylmethionine, and enhances PS1 and BACE expression and amyloid-β deposition in mice. Mol. Cell. Neurosci. 2008, 37, 731–746. [Google Scholar] [CrossRef] [PubMed]
- Tohgi, H.; Utsugisawa, K.; Nagane, Y.; Yoshimura, M.; Ukitsu, M.; Genda, Y. The methylation status of cytosines in a tau gene promoter region alters with age to downregulate transcriptional activity in human cerebral cortex. Neurosci. Lett. 1999, 275, 89–92. [Google Scholar] [CrossRef]
- Wang, S.C.; Oelze, B.; Schumacher, A. Age-specific epigenetic drift in late-onset Alzheimer’s disease. PLoS ONE 2008, 3, e2698. [Google Scholar] [CrossRef] [PubMed]
- Moreira, P.R.; Guimaraes, M.M.; Guimaraes, A.L.; Diniz, M.G.; Gomes, C.C.; Brito, J.A.; Gomez, R.S. Methylation of P16, P21, P27, RB1 and P53 genes in odontogenic keratocysts. J. Oral Pathol. Med. 2009, 38, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Muerkoster, S.S.; Werbing, V.; Koch, D.; Sipos, B.; Ammerpohl, O.; Kalthoff, H.; Tsao, M.S.; Folsch, U.R.; Schafer, H. Role of myofibroblasts in innate chemoresistance of pancreatic carcinoma-epigenetic downregulation of caspases. Int. J. Cancer 2008, 123, 1751–1760. [Google Scholar] [CrossRef] [PubMed]
- Tschop, K.; Engeland, K. Cell cycle-dependent transcription of cyclin B2 is influenced by DNA methylation but is independent of methylation in the CDE and CHR elements. FEBS J. 2007, 274, 5235–5249. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.Y.; Schluesener, H.J. Oral administration of histone deacetylase inhibitor MS-275 ameliorates neuroinflammation and cerebral amyloidosis and improves behavior in a mouse model. J. Neuropathol. Exp. Neurol. 2013, 72, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Graff, J.; Kim, D.; Dobbin, M.M.; Tsai, L.H. Epigenetic regulation of gene expression in physiological and pathological brain processes. Physiol. Rev. 2011, 91, 603–649. [Google Scholar] [CrossRef] [PubMed]
- Rao, J.S.; Keleshian, V.L.; Klein, S.; Rapoport, S.I. Epigenetic modifications in frontal cortex from Alzheimer’s disease and bipolar disorder patients. Transl. Psychiatry 2012, 2, e132. [Google Scholar] [CrossRef] [PubMed]
- Lithner, C.U.; Lacor, P.N.; Zhao, W.Q.; Mustafiz, T.; Klein, W.L.; Sweatt, J.D.; Hernandez, C.M. Disruption of neocortical histone H3 homeostasis by soluble Aβ: Implications for Alzheimer’s disease. Neurobiol. Aging 2013, 34, 2081–2090. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Dolan, P.J.; Johnson, G.V. Histone deacetylase 6 interacts with the microtubule-associated protein tau. J. Neurochem. 2008, 106, 2119–2130. [Google Scholar] [CrossRef] [PubMed]
- Simoes-Pires, C.; Zwick, V.; Nurisso, A.; Schenker, E.; Carrupt, P.A.; Cuendet, M. HDAC6 as a target for neurodegenerative diseases: What makes it different from the other HDACs? Mol. Neurodegener. 2013, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Cook, C.; Carlomagno, Y.; Gendron, T.F.; Dunmore, J.; Scheffel, K.; Stetler, C.; Davis, M.; Dickson, D.; Jarpe, M.; DeTure, M.; et al. Acetylation of the KXGS motifs in tau is a critical determinant in modulation of tau aggregation and clearance. Hum. Mol. Genet. 2014, 23, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Govindarajan, N.; Rao, P.; Burkhardt, S.; Sananbenesi, F.; Schluter, O.M.; Bradke, F.; Lu, J.; Fischer, A. Reducing HDAC6 ameliorates cognitive deficits in a mouse model for Alzheimer’s disease. EMBO Mol. Med. 2013, 5, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Morris, B.J. Seven sirtuins for seven deadly diseases of aging. Free Radic. Biol. Med. 2013, 56, 133–171. [Google Scholar] [CrossRef] [PubMed]
- Julien, C.; Tremblay, C.; Emond, V.; Lebbadi, M.; Salem, N., Jr.; Bennett, D.A.; Calon, F. Sirtuin 1 reduction parallels the accumulation of tau in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2009, 68, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Francis, Y.I.; Fa, M.; Ashraf, H.; Zhang, H.; Staniszewski, A.; Latchman, D.S.; Arancio, O. Dysregulation of histone acetylation in the APP/PS1 mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2009, 18, 131–139. [Google Scholar] [PubMed]
- Long, Z.M.; Zhao, L.; Jiang, R.; Wang, K.J.; Luo, S.F.; Zheng, M.; Li, X.F.; He, G.Q. Valproic acid modifies synaptic structure and accelerates neurite outgrowth via the glycogen synthase kinase-3β signaling pathway in an Alzheimer’s disease model. CNS Neurosci. Ther. 2015, 21, 887–897. [Google Scholar] [CrossRef] [PubMed]
- De Jager, P.L.; Srivastava, G.; Lunnon, K.; Burgess, J.; Schalkwyk, L.C.; Yu, L.; Eaton, M.L.; Keenan, B.T.; Ernst, J.; McCabe, C.; et al. Alzheimer’s disease: Early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat. Neurosci. 2014, 17, 1156–1163. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Cohen, M.L.; Lerner, A.J.; Yang, Y.; Herrup, K. DNA damage and cell cycle events implicate cerebellar dentate nucleus neurons as targets of Alzheimer’s disease. Mol. Neurodegener. 2010, 5, 60. [Google Scholar] [CrossRef] [PubMed]
- Suberbielle, E.; Sanchez, P.E.; Kravitz, A.V.; Wang, X.; Ho, K.; Eilertson, K.; Devidze, N.; Kreitzer, A.C.; Mucke, L. Physiologic brain activity causes DNA double-strand breaks in neurons, with exacerbation by amyloid-β. Nat. Neurosci. 2013, 16, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Rimkus, S.A.; Katzenberger, R.J.; Trinh, A.T.; Dodson, G.E.; Tibbetts, R.S.; Wassarman, D.A. Mutations in String/CDC25 inhibit cell cycle re-entry and neurodegeneration in a Drosophila model of Ataxia telangiectasia. Genes Dev. 2008, 22, 1205–1220. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Herrup, K. Loss of neuronal cell cycle control in Ataxia-telangiectasia: A unified disease mechanism. J. Neurosci. 2005, 25, 2522–2529. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, A.; Minami, A.; Kitagishi, Y.; Ogura, Y.; Matsuda, S. BRCA1 and p53 tumor suppressor molecules in Alzheimer’s disease. Int. J. Mol. Sci. 2015, 16, 2879–2892. [Google Scholar] [CrossRef] [PubMed]
- Pavard, S.; Metcalf, C.J. Negative selection on BRCA1 susceptibility alleles sheds light on the population genetics of late-onset diseases and aging theory. PLoS ONE 2007, 2, e1206. [Google Scholar] [CrossRef] [PubMed]
- Petersen, A.J.; Rimkus, S.A.; Wassarman, D.A. ATM kinase inhibition in glial cells activates the innate immune response and causes neurodegeneration in Drosophila. Proc. Natl. Acad. Sci. USA 2012, 109, E656–E664. [Google Scholar] [CrossRef] [PubMed]
- Kuljis, R.O.; Xu, Y.; Aguila, M.C.; Baltimore, D. Degeneration of neurons, synapses, and neuropil and glial activation in a murine ATM knockout model of Ataxia-telangiectasia. Proc. Natl. Acad. Sci. USA 1997, 94, 12688–12693. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Stoica, G.; Yan, M.; Scofield, V.L.; Qiang, W.; Lynn, W.S.; Wong, P.K. ATM deficiency induces oxidative stress and endoplasmic reticulum stress in astrocytes. Lab. Investig. 2005, 85, 1471–1480. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.; Cooper, N.R.; Webster, S.; Schultz, J.; McGeer, P.L.; Styren, S.D.; Civin, W.H.; Brachova, L.; Bradt, B.; Ward, P. Complement activation by β-amyloid in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1992, 89, 10016–10020. [Google Scholar] [CrossRef] [PubMed]
- Griffin, W.S.; Stanley, L.C.; Ling, C.; White, L.; MacLeod, V.; Perrot, L.J.; White, C.L., III; Araoz, C. Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc. Natl. Acad. Sci. USA 1989, 86, 7611–7615. [Google Scholar] [CrossRef] [PubMed]
- Niranjan, R. Molecular basis of etiological implications in Alzheimer’s disease: Focus on neuroinflammation. Mol. Neurobiol. 2013, 48, 412–428. [Google Scholar] [CrossRef] [PubMed]
- Alsadany, M.A.; Shehata, H.H.; Mohamad, M.I.; Mahfouz, R.G. Histone deacetylases enzyme, copper, and IL-8 levels in patients with Alzheimer’s disease. Am. J. Alzheimers Dis. Other Dement. 2013, 28, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Widner, B.; Leblhuber, F.; Walli, J.; Tilz, G.P.; Demel, U.; Fuchs, D. Tryptophan degradation and immune activation in Alzheimer’s disease. J. Neural Transm. 2000, 107, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.S.; Paris, D.; Mathura, V.; Quadros, A.N.; Crawford, F.C.; Mullan, M.J. Inflammatory cytokine levels correlate with amyloid load in transgenic mouse models of Alzheimer’s disease. J. Neuroinflamm. 2005, 2, 9. [Google Scholar] [CrossRef] [PubMed]
- Lue, L.F.; Walker, D.G.; Rogers, J. Modeling microglial activation in Alzheimer’s disease with human postmortem microglial cultures. Neurobiol. Aging 2001, 22, 945–956. [Google Scholar] [CrossRef]
- Brosseron, F.; Krauthausen, M.; Kummer, M.; Heneka, M.T. Body fluid cytokine levels in mild cognitive impairment and Alzheimer’s disease: A comparative overview. Mol. Neurobiol. 2014, 50, 534–544. [Google Scholar] [CrossRef] [PubMed]
- Guillemin, G.J.; Smythe, G.A.; Veas, L.A.; Takikawa, O.; Brew, B.J. A β 1-42 induces production of quinolinic acid by human macrophages and microglia. Neuroreport 2003, 14, 2311–2315. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Tao, B.B.; Yang, Y.Y.; Du, L.S.; Yang, S.S.; He, X.J.; Zhu, Y.W.; Yan, J.K.; Yang, Q. The IDO inhibitor coptisine ameliorates cognitive impairment in a mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2015, 43, 291–302. [Google Scholar] [PubMed]
- McGeer, P.L.; McGeer, E.G. The amyloid cascade-inflammatory hypothesis of Alzheimer disease: Implications for therapy. Acta Neuropathol. 2013, 126, 479–497. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Seo, D.; Choi, K.J.; Andersen, J.B.; Won, M.A.; Kitade, M.; Gomez-Quiroz, L.E.; Judge, A.D.; Marquardt, J.U.; Raggi, C.; et al. Antitumor effects in hepatocarcinoma of isoform-selective inhibition of HDAC2. Cancer Res. 2014, 74, 4752–4761. [Google Scholar] [CrossRef] [PubMed]
- Morschhauser, F.; Terriou, L.; Coiffier, B.; Bachy, E.; Varga, A.; Kloos, I.; Lelievre, H.; Sarry, A.L.; Depil, S.; Ribrag, V. Phase 1 study of the oral histone deacetylase inhibitor abexinostat in patients with Hodgkin lymphoma, non-Hodgkin lymphoma, or chronic lymphocytic leukaemia. Invest. New Drugs 2015, 33, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Regna, N.L.; Chafin, C.B.; Hammond, S.E.; Puthiyaveetil, A.G.; Caudell, D.L.; Reilly, C.M. Class I and II histone deacetylase inhibition by ITF2357 reduces SLE pathogenesis in vivo. Clin. Immunol. 2014, 151, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Grayson, D.R.; Kundakovic, M.; Sharma, R.P. Is there a future for histone deacetylase inhibitors in the pharmacotherapy of psychiatric disorders? Mol. Pharmacol. 2010, 77, 126–135. [Google Scholar] [CrossRef] [PubMed]
- El-Khoury, V.; Pierson, S.; Szwarcbart, E.; Brons, N.H.; Roland, O.; Cherrier-De, W.S.; Plawny, L.; van Dyck, E.; Berchem, G. Disruption of autophagy by the histone deacetylase inhibitor MGCD0103 and its therapeutic implication in B-cell chronic lymphocytic leukemia. Leukemia 2014, 28, 1636–1646. [Google Scholar] [CrossRef]
- Ganai, S.A. Panobinostat: The small molecule metalloenzyme inhibitor with marvelous anticancer activity. Curr. Top. Med. Chem. 2016, 16, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Bates, S.E.; Zhan, Z.; Steadman, K.; Obrzut, T.; Luchenko, V.; Frye, R.; Robey, R.W.; Turner, M.; Gardner, E.R.; Figg, W.D.; et al. Laboratory correlates for a phase II trial of romidepsin in cutaneous and peripheral T-cell lymphoma. Br. J. Haematol. 2010, 148, 256–267. [Google Scholar] [CrossRef]
- Outeiro, T.F.; Kontopoulos, E.; Altmann, S.M.; Kufareva, I.; Strathearn, K.E.; Amore, A.M.; Volk, C.B.; Maxwell, M.M.; Rochet, J.C.; McLean, P.J.; et al. Sirtuin 2 inhibitors rescue α-synuclein-mediated toxicity in models of Parkinson’s disease. Science 2007, 317, 516–519. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).