
Alternative Splicing in Cardiovascular Disease—A Survey of Recent Findings

 , ,
, ,  and
and
Abstract
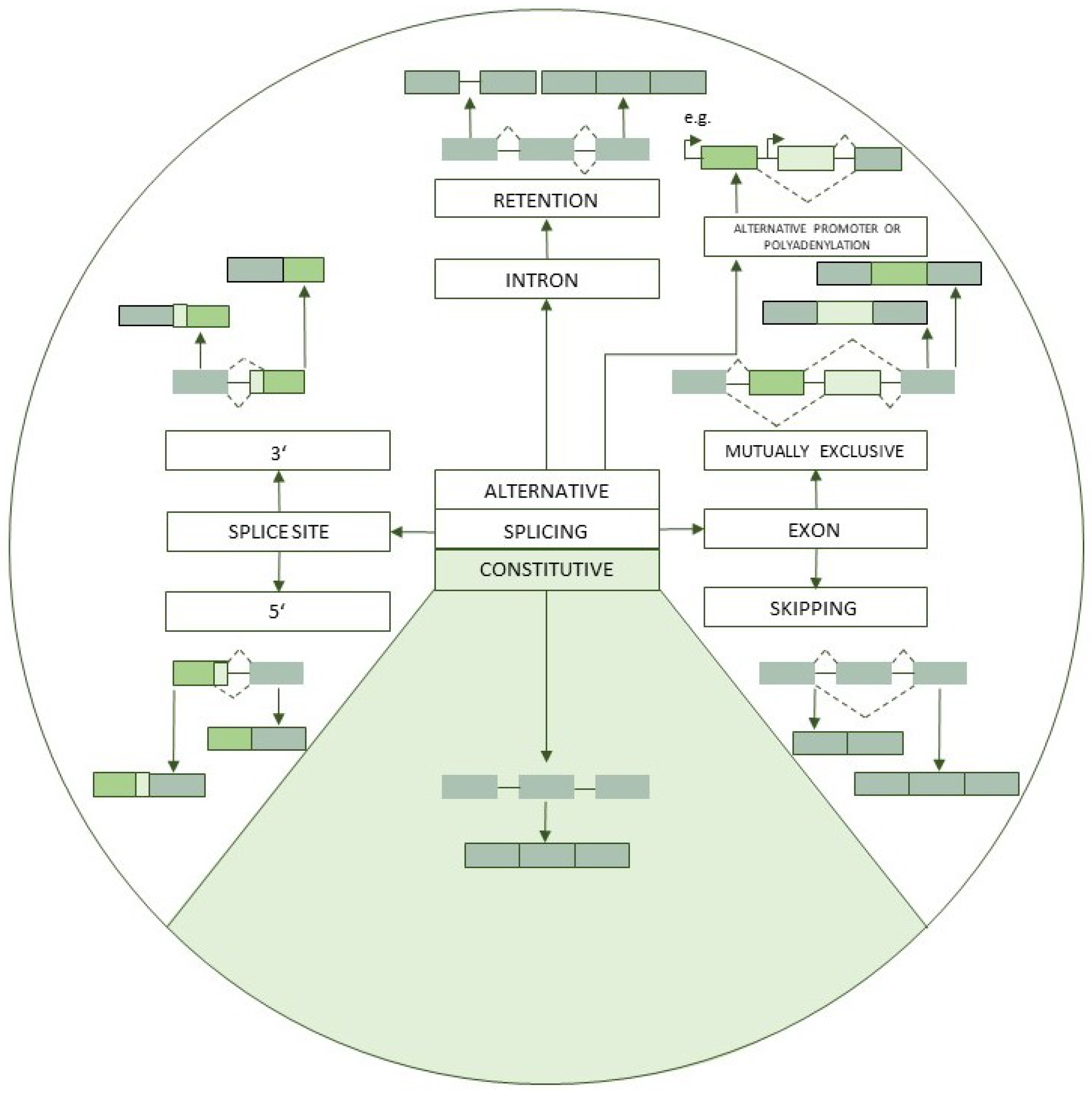
:1. Introduction
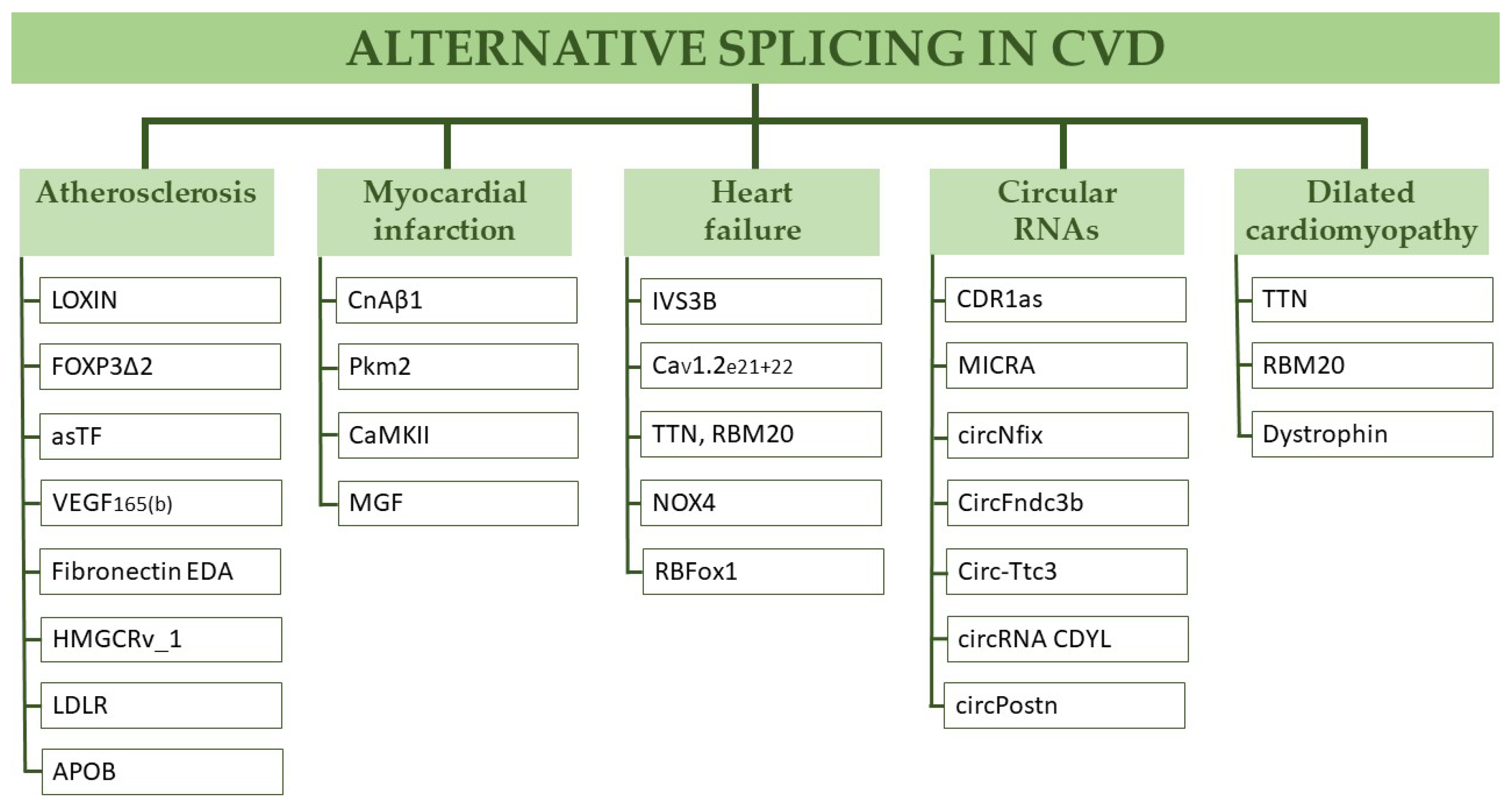
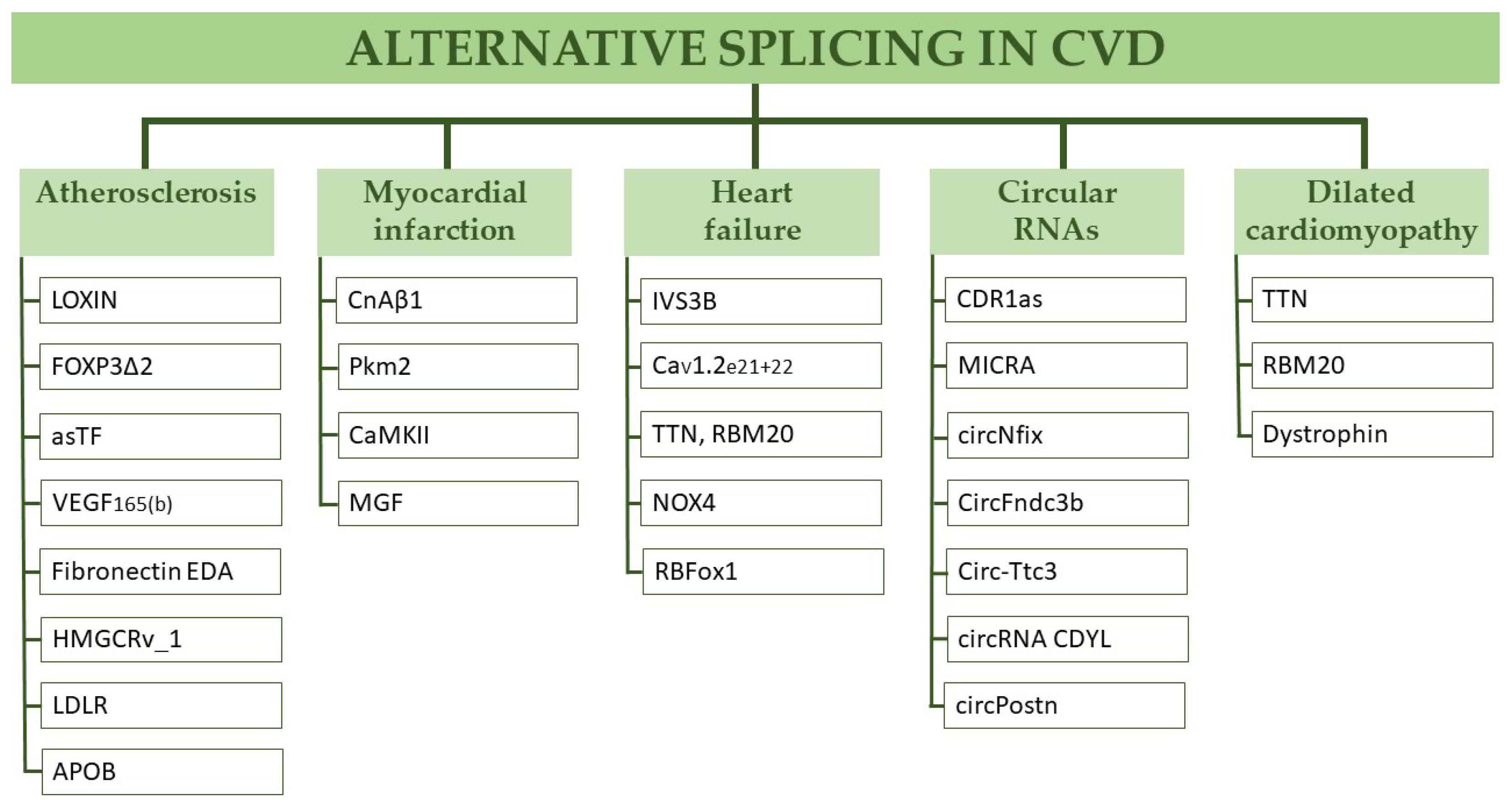
2. Alternative Splicing in Cardiovascular Disease
2.1. Atherosclerosis
2.2. Myocardial Infarction
2.3. Heart Failure
2.4. Dilatative Cardiomyopathy
2.5. Circular RNAs in Myocardial Infarction
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Park, E.; Pan, Z.; Zhang, Z.; Lin, L.; Xing, Y. The Expanding Landscape of Alternative Splicing Variation in Human Populations; Cell Press: Cambridge, MA, USA, 2018; Volume 102, pp. 11–26. [Google Scholar]
- Nilsen, T.W.; Graveley, B.R. Expansion of the eukaryotic proteome by alternative splicing. Nature 2010, 463, 457–463. [Google Scholar] [CrossRef] [Green Version]
- Pajares, M.J.; Ezponda, T.; Catena, R.; Calvo, A.; Pio, R.; Montuenga, L.M. Alternative splicing: An emerging topic in molecular and clinical oncology. Lancet Oncol. 2007, 8, 349–357. [Google Scholar] [CrossRef]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef]
- Xu, Q.; Modrek, B.; Lee, C. Genome-wide detection of tissue-specific alternative splicing in the human transcriptome. Nucleic Acids Res. 2002, 30, 3754–3766. [Google Scholar] [CrossRef] [PubMed]
- Baralle, F.E.; Giudice, J. Alternative splicing as a regulator of development and tissue identity. Nat. Rev. Mol. Cell Biol. 2017, 18, 437–451. [Google Scholar] [CrossRef]
- Wang, H.; Chen, Y.; Li, X.; Chen, G.; Zhong, L.; Chen, G.; Liao, Y.; Liao, W.; Bin, J. Genome-wide analysis of alternative splicing during human heart development. Sci. Rep. 2016, 6, 35520. [Google Scholar] [CrossRef] [Green Version]
- Gamazon, E.R.; Stranger, B.E. Genomics of alternative splicing: Evolution, development and pathophysiology. Hum. Genet. 2014, 133, 679–687. [Google Scholar] [CrossRef] [PubMed]
- Dredge, K.; Polydorides, A.D.; Darnell, R. The splice of life: Alternative splicing and neurological disease. Nat. Rev. Neurosci. 2001, 2, 43–50. [Google Scholar] [CrossRef]
- Bonnal, S.C.; López-Oreja, I.; Valcárcel, J. Roles and mechanisms of alternative splicing in cancer—Implications for care. Nat. Rev. Clin. Oncol. 2020, 17, 457–474. [Google Scholar] [CrossRef] [PubMed]
- Brophy, M.L.; Dong, Y.; Wu, H.; Rahman, H.N.A.; Song, K.; Chen, H. Eating the Dead to Keep Atherosclerosis at Bay. Front. Cardiovasc. Med. 2017, 4, 2. [Google Scholar] [CrossRef] [Green Version]
- Geovanini, G.R.; Libby, P. Atherosclerosis and inflammation: Overview and updates. Clin. Sci. 2018, 132, 1243–1252. [Google Scholar] [CrossRef]
- Rosenson, R.S. Statins in atherosclerosis: Lipid-lowering agents with antioxidant capabilities. Atherosclerosis 2004, 173, 1–12. [Google Scholar] [CrossRef]
- Carmena, R.; Duriez, P.; Fruchart, J.-C. Atherogenic Lipoprotein Particles in Atherosclerosis. Circulation 2004, 109, III-2. [Google Scholar] [CrossRef] [Green Version]
- Medina, M.W.; Gao, F.; Ruan, W.; Rotter, J.I.; Krauss, R.M. Alternative Splicing of 3-Hydroxy-3-Methylglutaryl Coenzyme A Reductase Is Associated with Plasma Low-Density Lipoprotein Cholesterol Response to Simvastatin. Circulation 2008, 118, 355–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, C.-Y.; Theusch, E.; Lo, K.; Mangravite, L.M.; Naidoo, D.; Kutilova, M.; Medina, M.W. HNRNPA1 regulates HMGCR alternative splicing and modulates cellular cholesterol metabolism. Hum. Mol. Genet. 2014, 23, 319–332. [Google Scholar] [CrossRef] [Green Version]
- Gao, F.; Ihn, H.E.; Medina, M.W.; Krauss, R.M. A common polymorphism in the LDL receptor gene has multiple effects on LDL receptor function. Hum. Mol. Genet. 2013, 22, 1424–1431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.; Tucker, H.M.; Grear, K.E.; Simpson, J.F.; Manning, A.K.; Cupples, L.A.; Estus, S. A common polymorphism decreases low-density lipoprotein receptor exon 12 splicing efficiency and associates with increased cholesterol. Hum. Mol. Genet. 2007, 16, 1765–1772. [Google Scholar] [CrossRef] [Green Version]
- Medina, M.W.; Gao, F.; Naidoo, D.; Rudel, L.L.; Temel, R.E.; McDaniel, A.L.; Marshall, S.M.; Krauss, R.M. Coordinately Regulated Alternative Splicing of Genes Involved in Cholesterol Biosynthesis and Uptake. PLoS ONE 2011, 6, e19420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khoo, B.; Roca, X.; Chew, S.L.; Krainer, A.R. Antisense oligonucleotide-induced alternative splicing of the APOB mRNA generates a novel isoform of APOB. BMC Mol. Biol. 2007, 8, 3. [Google Scholar] [CrossRef] [Green Version]
- Disterer, P.; Al-Shawi, R.; Ellmerich, S.; Waddington, S.N.; Owen, J.S.; Simons, J.P.; Khoo, B. Exon Skipping of Hepatic APOB Pre-mRNA with Splice-switching Oligonucleotides Reduces LDL Cholesterol In Vivo. Mol. Ther. 2013, 21, 602–609. [Google Scholar] [CrossRef] [Green Version]
- Mango, R. In Vivo and In Vitro Studies Support That a New Splicing Isoform of OLR1 Gene Is Protective Against Acute Myocardial Infarction. Circ. Res. 2005, 97, 152–158. [Google Scholar] [CrossRef] [Green Version]
- Pothineni, N.V.K.; Karathanasis, S.K.; Ding, Z.; Arulandu, A.; Varughese, K.I.; Mehta, J.L. LOX-1 in Atherosclerosis and Myocardial Ischemia: Biology, Genetics, and Modulation. J. Am. Coll. Cardiol. 2017, 69, 2759–2768. [Google Scholar] [CrossRef]
- Biocca, S.; Filesi, I.; Mango, R.; Maggiore, L.; Baldini, F.; Vecchione, L.; Viola, A.; Citro, G.; Federici, G.; Romeo, F.; et al. The splice variant LOXIN inhibits LOX-1 receptor function through hetero-oligomerization. J. Mol. Cell. Cardiol. 2008, 44, 561–570. [Google Scholar] [CrossRef]
- White, S.J.; Sala-Newby, G.B.; Newby, A.C. Overexpression of scavenger receptor LOX-1 in endothelial cells promotes atherogenesis in the ApoE−/− mouse model. Cardiovasc. Pathol. 2011, 20, 369–373. [Google Scholar] [CrossRef] [Green Version]
- Veas, C.; Jara, C.; Willis, N.D.; Pérez-Contreras, K.; Gutierrez, N.; Toledo, J.; Fernandez, P.; Radojkovic, C.; Zuñiga, F.A.; Escudero, C.; et al. Overexpression of LOXIN Protects Endothelial Progenitor Cells From Apoptosis Induced by Oxidized Low Density Lipoprotein. J. Cardiovasc. Pharmacol. 2016, 67, 326–335. [Google Scholar] [CrossRef]
- Joly, A.-L.; Seitz, C.; Liu, S.; Kuznetsov, N.V.; Gertow, K.; Westerberg, L.S.; Paulsson-Berne, G.; Hansson, G.K.; Andersson, J. Alternative Splicing of FOXP3 Controls Regulatory T Cell Effector Functions and Is Associated with Human Atherosclerotic Plaque Stability. Circ. Res. 2018, 122, 1385–1394. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Miyara, M.; Costantino, C.; Hafler, D.A. FOXP3+ regulatory T cells in the human immune system. Nat. Rev. Immunol. 2010, 10, 490–500. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Yang, J.; Dong, M.; Zhang, K.; Tu, E.; Gao, Q.; Chen, W.; Zhang, C.; Zhang, Y. Regulatory T cells in cardiovascular diseases. Nat. Rev. Cardiol. 2015, 13, 167–179. [Google Scholar] [CrossRef]
- Lundberg, A.K.; Jonasson, L.; Hansson, G.K.; Mailer, R.K. Activation-induced FOXP3 isoform profile in peripheral CD4+ T cells is associated with coronary artery disease. Atherosclerosis 2017, 267, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Giannarelli, C.; Alique, M.; Rodriguez, D.T.; Yang, N.K.; Jeong, N.; Calcagno, C.; Hutter, R.; Millon, A.; Kovacic, J.C.; Weber, T.; et al. Alternatively spliced tissue factor promotes plaque angiogenesis through the activation of hypoxia-inducible factor-1α and vascular endothelial growth factor signaling. Circulation 2014, 130, 1274–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grover, S.P.; Mackman, N. Tissue factor in atherosclerosis and atherothrombosis. Atherosclerosis 2020, 307, 80–86. [Google Scholar] [CrossRef]
- Srinivasan, R.; Ozhegov, E.; Berg, Y.W.V.D.; Aronow, B.J.; Franco, R.S.; Palascak, M.B.; Fallon, J.; Ruf, W.; Versteeg, H.; Bogdanov, V. Splice variants of tissue factor promote monocyte-endothelial interactions by triggering the expression of cell adhesion molecules via integrin-mediated signaling. J. Thromb. Haemost. 2011, 9, 2087–2096. [Google Scholar] [CrossRef]
- Zhao, N.; Zhang, J. Role of alternative splicing of VEGF-A in the development of atherosclerosis. Aging 2018, 10, 2695–2708. [Google Scholar] [CrossRef] [PubMed]
- Ganta, V.C.; Choi, M.; Farber, C.R.; Annex, B.H. Antiangiogenic VEGF 165 b Regulates Macrophage Polarization via S100A8/S100A9 in Peripheral Artery Disease. Circulation 2019, 139, 226–242. [Google Scholar] [CrossRef]
- Babaev, V.R.; Porro, F.; Linton, M.F.; Fazio, S.; Baralle, F.E.; Muro, A.F. Absence of regulated splicing of fibronectin EDA exon reduces atherosclerosis in mice. Atherosclerosis 2008, 197, 534–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pankov, R.; Yamada, K.M. Fibronectin at a glance. J. Cell Sci. 2002, 115, 3861–3863. [Google Scholar] [CrossRef] [Green Version]
- Cappellari, G.G.; Barazzoni, R.; Cattin, L.; Muro, A.F.; Zanetti, M. Lack of Fibronectin Extra Domain A Alternative Splicing Exacerbates Endothelial Dysfunction in Diabetes. Sci. Rep. 2016, 6, 37965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reed, G.W.; Rossi, J.E.; Cannon, C.P. Acute Myocardial Infarction. Lancet 2017, 389, 197–210. [Google Scholar] [CrossRef]
- van der Bijl, P.; Abou, R.; Goedemans, L.; Gersh, B.J.; Holmes, D.R.; Marsan, N.A.; Delgado, V.; Bax, J.J. Left Ventricular Post-Infarct Remodeling: Implications for Systolic Function Improvement and Outcomes in the Modern Era. JACC Heart Fail. 2020, 8, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Felkin, L.E.; Narita, T.; Germack, R.; Shintani, Y.; Takahashi, K.; Sarathchandra, P.; López-Olañeta, M.M.; Gómez-Salinero, J.M.; Suzuki, K.; Barton, P.; et al. Calcineurin Splicing Variant Calcineurin Aβ1 Improves Cardiac Function after Myocardial Infarction without Inducing Hypertrophy. Circulation 2011, 123, 2838–2847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lara-Pezzi, E.; Winn, N.; Paul, A.; Mccullagh, K.; Slominsky, E.; Santini, M.P.; Mourkioti, F.; Sarathchandra, P.; Fukushima, S.; Suzuki, K.; et al. A naturally occurring calcineurin variant inhibits FoxO activity and enhances skeletal muscle regeneration. J. Cell Biol. 2007, 179, 1205–1218. [Google Scholar] [CrossRef]
- Padrón-Barthe, L.; Orero, M.V.; Gómez-Salinero, J.M.; Acin-Perez, R.; Cogliati, S.; López-Olañeta, M.; Ortiz-Sánchez, P.; Bonzon-Kulichenko, E.; Vázquez, J.; García-Pavía, P.; et al. Activation of Serine One-Carbon Metabolism by Calcineurin Aβ1 Reduces Myocardial Hypertrophy and Improves Ventricular Function. J. Am. Coll. Cardiol. 2018, 71, 654–667. [Google Scholar] [CrossRef]
- Williams, A.L.; Khadka, V.; Tang, M.; Avelar, A.; Schunke, K.; Menor, M.; Shohet, R.V. HIF1 mediates a switch in pyruvate kinase isoforms after myocardial infarction. Physiol. Genom. 2018, 50, 479–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, A.L.; Walton, C.B.; Pinell, B.; Khadka, V.S.; Dunn, B.; Lee, K.; Anagaran, M.C.T.; Avelar, A.; Shohet, R.V. Ischemic heart injury leads to HIF1-dependent differential splicing of CaMK2γ. Sci. Rep. 2021, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Mavrommatis, E.; Shioura, K.M.; Los, T.; Goldspink, P.H. The E-domain region of mechano-growth factor inhibits cellular apoptosis and preserves cardiac function during myocardial infarction. Mol. Cell. Biochem. 2013, 381, 69–83. [Google Scholar] [CrossRef] [Green Version]
- Stavropoulou, A.; Halapas, A.; Sourla, A.; Philippou, A.; Papageorgiou, E.; Papalois, A.; Koutsilieris, M. IGF-1 Expression in Infarcted Myocardium and MGF E Peptide Actions in Rat Cardiomyocytes in Vitro. Mol. Med. 2009, 15, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Ziaeian, B.; Fonarow, G.C. Epidemiology and Aetiology of Heart Failure. Nat. Rev. Cardiol. 2016, 13, 368–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Chen, X.; Margulies, K.; Jeevanandam, V.; Pollack, P.; Bailey, B.A.; Houser, S.R. L-Type Ca2+ Channel α1cSubunit Isoform Switching in Failing Human Ventricular Myocardium. J. Mol. Cell. Cardiol. 2000, 32, 973–984. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Wang, J.-W.; Yu, D.; Soon, J.L.; De Kleijn, D.P.V.; Foo, R.; Liao, P.; Colecraft, H.M.; Soong, T.W. Aberrant Splicing Promotes Proteasomal Degradation of L-type CaV1.2 Calcium Channels by Competitive Binding for CaVβ Subunits in Cardiac Hypertrophy. Sci. Rep. 2016, 6, 35247. [Google Scholar] [CrossRef]
- Li, G.; Wang, J.; Liao, P.; Bartels, P.; Zhang, H.; Yu, D.; Liang, M.C.; Poh, K.K.; Yu, C.Y.; Jiang, F.; et al. Exclusion of alternative exon 33 of CaV1.2 calcium channels in heart is proarrhythmogenic. Proc. Natl. Acad. Sci. USA 2017, 114, E4288–E4295. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Li, G.; Yu, D.; Wong, Y.P.; Yong, T.F.; Liang, M.C.; Liao, P.; Foo, R.; Hoppe, U.C.; Soong, T.W. Characterization of CaV1.2 exon 33 heterozygous knockout mice and negative correlation between Rbfox1 and CaV1.2 exon 33 expressions in human heart failure. Channels 2018, 12, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Zile, M.R.; Baicu, C.F.; Ikonomidis, J.S.; Stroud, R.E.; Nietert, P.J.; Bradshaw, A.D.; Slater, R.; Palmer, B.M.; Van Buren, P.; Meyer, M.; et al. Myocardial Stiffness in Patients with Heart Failure and a Preserved Ejection Fraction: Contributions of Collagen and Titin. Circulation 2015, 131, 1247–1259. [Google Scholar] [CrossRef]
- Bull, M.; Methawasin, M.; Strom, J.; Nair, P.; Hutchinson, K.; Granzier, H.L. Alternative Splicing of Titin Restores Diastolic Function in an HFpEF-Like Genetic Murine Model ( Ttn ΔIAjxn ). Circ. Res. 2016, 119, 764–772. [Google Scholar] [CrossRef] [Green Version]
- Varga, Z.V.; Pipicz, M.; Baán, J.A.; Baranyai, T.; Koncsos, G.; Leszek, P.; Kuśmierczyk, M.; Sanchez-Cabo, F.; García-Pavía, P.; Brenner, G.J.; et al. Alternative Splicing of NOX4 in the Failing Human Heart. Front. Physiol. 2017, 8, 935. [Google Scholar] [CrossRef] [Green Version]
- Gao, C.; Ren, S.; Lee, J.-H.; Qiu, J.; Chapski, D.; Rau, C.D.; Zhou, Y.; Abdellatif, M.; Nakano, A.; Vondriska, T.M.; et al. RBFox1-mediated RNA splicing regulates cardiac hypertrophy and heart failure. J. Clin. Investig. 2016, 126, 195–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultheiss, H.-P.; Fairweather, D.; Caforio, A.L.P.; Escher, F.; Hershberger, R.E.; Lipshultz, S.E.; Liu, P.P.; Matsumori, A.; Mazzanti, A.; McMurray, J.; et al. Dilated cardiomyopathy. Nat. Rev. Dis. Prim. 2019, 5, 32. [Google Scholar] [CrossRef] [PubMed]
- Reichart, D.; Magnussen, C.; Zeller, T.; Blankenberg, S. Dilated Cardiomyopathy: From Epidemiologic to Genetic Phenotypes: A Translational Review of Current Literature. J. Intern. Med. 2019, 286, 362–372. [Google Scholar] [CrossRef] [Green Version]
- Herman, D.S.; Lam, L.; Taylor, M.R.; Wang, L.; Teekakirikul, P.; Christodoulou, D.; Conner, L.; DePalma, S.R.; McDonough, B.; Sparks, E.; et al. Truncations of Titin Causing Dilated Cardiomyopathy. N. Engl. J. Med. 2012, 366, 619–628. [Google Scholar] [CrossRef] [Green Version]
- Granzier, H.L.; Labeit, S. The Giant Protein Titin. Circ. Res. 2004, 94, 284–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, W.; Schäfer, S.; Greaser, M.L.; Radke, M.; Liss, M.; Govindarajan, T.; Maatz, H.; Schulz, H.; Li, S.; Parrish, A.M.; et al. RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat. Med. 2012, 18, 766–773. [Google Scholar] [CrossRef] [PubMed]
- Brauch, K.M.; Karst, M.L.; Herron, K.J.; de Andrade, M.; Pellikka, P.A.; Rodeheffer, R.J.; Michels, V.V.; Olson, T.M. Mutations in Ribonucleic Acid Binding Protein Gene Cause Familial Dilated Cardiomyopathy. J. Am. Coll. Cardiol. 2009, 54, 930–941. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.A.; Reckman, Y.J.; Aufiero, S.; van den Hoogenhof, M.M.; van der Made, I.; Beqqali, A.; Koolbergen, D.R.; Rasmussen, T.B.; van der Velden, J.; Creemers, E.E.; et al. RBM20 Regulates Circular RNA Production From the Titin Gene. Circ. Res. 2016, 119, 996–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Streckfuss-Bömeke, K.; Tiburcy, M.; Fomin, A.; Luo, X.; Li, W.; Fischer, C.; Özcelik, C.; Perrot, A.; Sossalla, S.; Haas, J.; et al. Severe DCM phenotype of patient harboring RBM20 mutation S635A can be modeled by patient-specific induced pluripotent stem cell-derived cardiomyocytes. J. Mol. Cell. Cardiol. 2017, 113, 9–21. [Google Scholar] [CrossRef] [Green Version]
- Briganti, F.; Sun, H.; Wei, W.; Wu, J.; Zhu, C.; Liss, M.; Karakikes, I.; Rego, S.; Cipriano, A.; Snyder, M.; et al. iPSC Modeling of RBM20-Deficient DCM Identifies Upregulation of RBM20 as a Therapeutic Strategy. Cell Rep. 2020, 32, 108117. [Google Scholar] [CrossRef] [PubMed]
- Kamdar, F.; Garry, D.J. Dystrophin-Deficient Cardiomyopathy. J. Am. Coll. Cardiol. 2016, 67, 2533–2546. [Google Scholar] [CrossRef] [PubMed]
- McGreevy, J.W.; Hakim, C.H.; McIntosh, M.A.; Duan, D. Animal models of Duchenne muscular dystrophy: From basic mechanisms to gene therapy. Dis. Model. Mech. 2015, 8, 195–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Q.L.; Rabinowitz, A.; Chen, Y.C.; Yokota, T.; Yin, H.; Alter, J.; Jadoon, A.; Bou-Gharios, G.; Partridge, T. Systemic delivery of antisense oligoribonucleotide restores dystrophin expression in body-wide skeletal muscles. Proc. Natl. Acad. Sci. USA 2005, 102, 198–203. [Google Scholar] [CrossRef] [Green Version]
- Van Deutekom, J.C.; Janson, A.A.; Ginjaar, I.B.; Frankhuizen, W.S.; Aartsma-Rus, A.; Bremmer-Bout, M.; Dunnen, J.T.D.; Koop, K.; Van Der Kooi, A.J.; Goemans, N.M.; et al. Local Dystrophin Restoration with Antisense Oligonucleotide PRO051. N. Engl. J. Med. 2007, 357, 2677–2686. [Google Scholar] [CrossRef] [Green Version]
- McDonald, C.M.; Wong, B.; Flanigan, K.M.; Wilson, R.; De Kimpe, S.; Lourbakos, A.; Lin, Z.; Campion, G.; The DEMAND V Study Group. Placebo-controlled Phase 2 Trial of Drisapersen for Duchenne Muscular Dystrophy. Ann. Clin. Transl. Neurol. 2018, 5, 913–926. [Google Scholar] [CrossRef] [Green Version]
- Goemans, N.; Mercuri, E.; Belousova, E.; Komaki, H.; Dubrovsky, A.; McDonald, C.; Kraus, J.E.; Lourbakos, A.; Lin, Z.; Campion, G.; et al. A randomized placebo-controlled phase 3 trial of an antisense oligonucleotide, drisapersen, in Duchenne muscular dystrophy. Neuromuscul. Disord. 2018, 28, 4–15. [Google Scholar] [CrossRef] [Green Version]
- Mester-Tonczar, J.; Hašimbegović, E.; Spannbauer, A.; Traxler, D.; Kastner, N.; Zlabinger, K.; Einzinger, P.; Pavo, N.; Goliasch, G.; Gyöngyösi, M. Circular RNAs in Cardiac Regeneration: Cardiac Cell Proliferation, Differentiation, Survival, and Reprogramming. Front. Physiol. 2020, 11, 580465. [Google Scholar] [CrossRef]
- Geng, H.-H.; Li, R.; Su, Y.-M.; Xiao-Ping, J.; Pan, M.; Cai, X.-X.; Ji, X.-P. The Circular RNA Cdr1as Promotes Myocardial Infarction by Mediating the Regulation of miR-7a on Its Target Genes Expression. PLoS ONE 2016, 11, e0151753. [Google Scholar] [CrossRef]
- Li, B.; Li, R.; Zhang, C.; Bian, H.-J.; Wang, F.; Xiao, J.; Liu, S.-W.; Yi, W.; Zhang, M.-X.; Wang, S.-X.; et al. MicroRNA-7a/b Protects against Cardiac Myocyte Injury in Ischemia/Reperfusion by Targeting Poly(ADP-Ribose) Polymerase. PLoS ONE 2014, 9, e90096. [Google Scholar] [CrossRef]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Mester-Tonczar, J.; Winkler, J.; Einzinger, P.; Hasimbegovic, E.; Kastner, N.; Lukovic, D.; Zlabinger, K.; Spannbauer, A.; Traxler, D.; Batkai, S.; et al. Association between Circular RNA CDR1as and Post-Infarction Cardiac Function in Pig Ischemic Heart Failure: Influence of the Anti-Fibrotic Natural Compounds Bufalin and Lycorine. Biomolecules 2020, 10, 1180. [Google Scholar] [CrossRef] [PubMed]
- Vausort, M.; Salgado-Somoza, A.; Zhang, L.; Leszek, P.; Scholz, M.; Teren, A.; Burkhardt, R.; Thiery, J.; Wagner, D.R.; Devaux, Y. Myocardial Infarction-Associated Circular RNA Predicting Left Ventricular Dysfunction. J. Am. Coll. Cardiol. 2016, 68, 1247–1248. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Li, X.; Zheng, H.; Si, X.; Li, B.; Wei, G.; Li, C.; Chen, Y.; Chen, Y.; Liao, W.; et al. Loss of Super-Enhancer-Regulated circRNA Nfix Induces Cardiac Regeneration After Myocardial Infarction in Adult Mice. Circulation 2019, 139, 2857–2876. [Google Scholar] [CrossRef]
- Garikipati, V.N.S.; Verma, S.K.; Cheng, Z.; Liang, D.; Truongcao, M.M.; Cimini, M.; Yue, Y.; Huang, G.; Wang, C.; Benedict, C.; et al. Circular RNA CircFndc3b modulates cardiac repair after myocardial infarction via FUS/VEGF-A axis. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef]
- Cai, L.-D.; Qi, B.; Wu, X.; Peng, S.; Zhou, G.; Wei, Y.; Xu, J.; Chen, S.; Liu, S. Circular RNA Ttc3 regulates cardiac function after myocardial infarction by sponging miR-15b. J. Mol. Cell. Cardiol. 2019, 130, 10–22. [Google Scholar] [CrossRef]
- Hullinger, T.G.; Montgomery, R.L.; Seto, A.G.; Dickinson, B.A.; Semus, H.M.; Lynch, J.M.; Dalby, C.M.; Robinson, K.; Stack, C.; Latimer, P.A.; et al. Inhibition of miR-15 Protects Against Cardiac Ischemic Injury. Circ. Res. 2012, 110, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wang, Z.; Cheng, Q.; Wang, Z.; Lv, X.; Wang, Z.; Li, N. Circular RNA (circRNA) CDYL Induces Myocardial Regeneration by ceRNA After Myocardial Infarction. Med. Sci. Monit. 2020, 26, e923188-1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, N.; Wang, M.-Y.; Wu, Y.-B.; Cui, H.-M.; Wei, S.-X.; Liu, B.; Wang, R. Circular RNA POSTN Promotes Myocardial Infarction-Induced Myocardial Injury and Cardiac Remodeling by Regulating miR-96-5p/BNIP3 Axis. Front. Cell Dev. Biol. 2021, 8, 1907. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| circRNA | Model | Target |
|---|---|---|
| CDR1as | Mouse, pig | miR-7a |
| MICRA | Human | miR-150, not definitively proven |
| circNfix | Mouse | miR-214 |
| CircFndc3b | Mouse | VEGF signaling |
| Circ-Ttc3 | Mouse | miR-15b |
| circRNA CDYL | Mouse | miR-4793-5p |
| circPostn | Mouse | miR-96-5p |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hasimbegovic, E.; Schweiger, V.; Kastner, N.; Spannbauer, A.; Traxler, D.; Lukovic, D.; Gyöngyösi, M.; Mester-Tonczar, J. Alternative Splicing in Cardiovascular Disease—A Survey of Recent Findings. Genes 2021, 12, 1457. https://doi.org/10.3390/genes12091457
Hasimbegovic E, Schweiger V, Kastner N, Spannbauer A, Traxler D, Lukovic D, Gyöngyösi M, Mester-Tonczar J. Alternative Splicing in Cardiovascular Disease—A Survey of Recent Findings. Genes. 2021; 12(9):1457. https://doi.org/10.3390/genes12091457
Chicago/Turabian StyleHasimbegovic, Ena, Victor Schweiger, Nina Kastner, Andreas Spannbauer, Denise Traxler, Dominika Lukovic, Mariann Gyöngyösi, and Julia Mester-Tonczar. 2021. "Alternative Splicing in Cardiovascular Disease—A Survey of Recent Findings" Genes 12, no. 9: 1457. https://doi.org/10.3390/genes12091457