
Next-Generation Sequencing (NGS) and Third-Generation Sequencing (TGS) for the Diagnosis of Thalassemia
 ,
,
Abstract
:1. Introduction
2. Conventional DNA Analysis
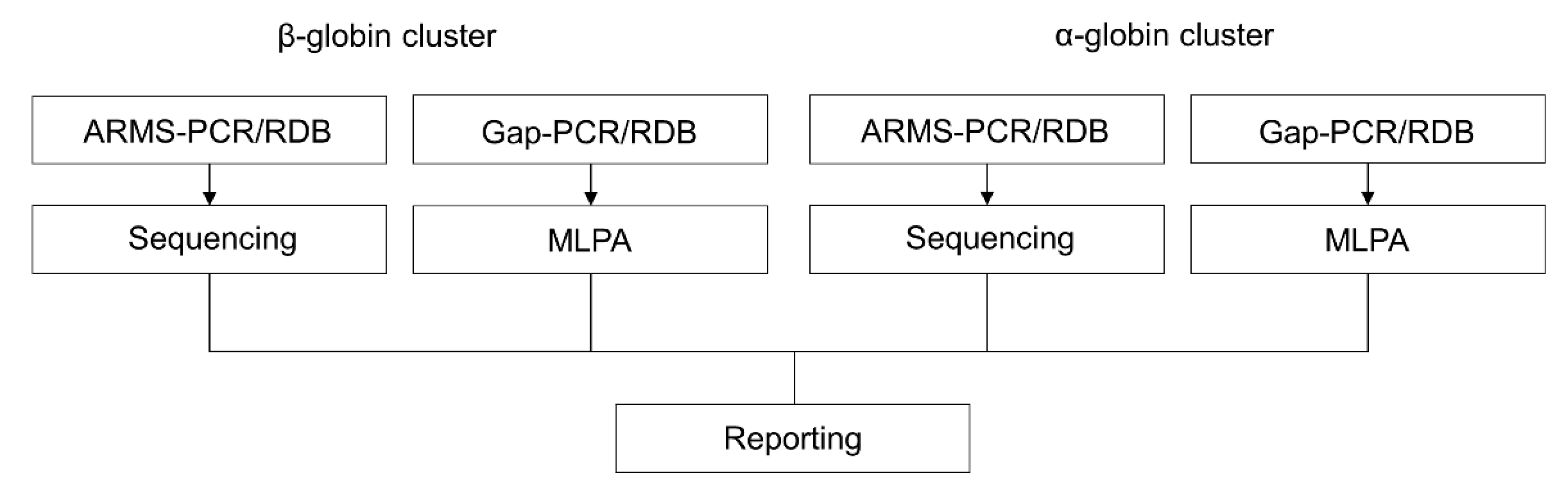
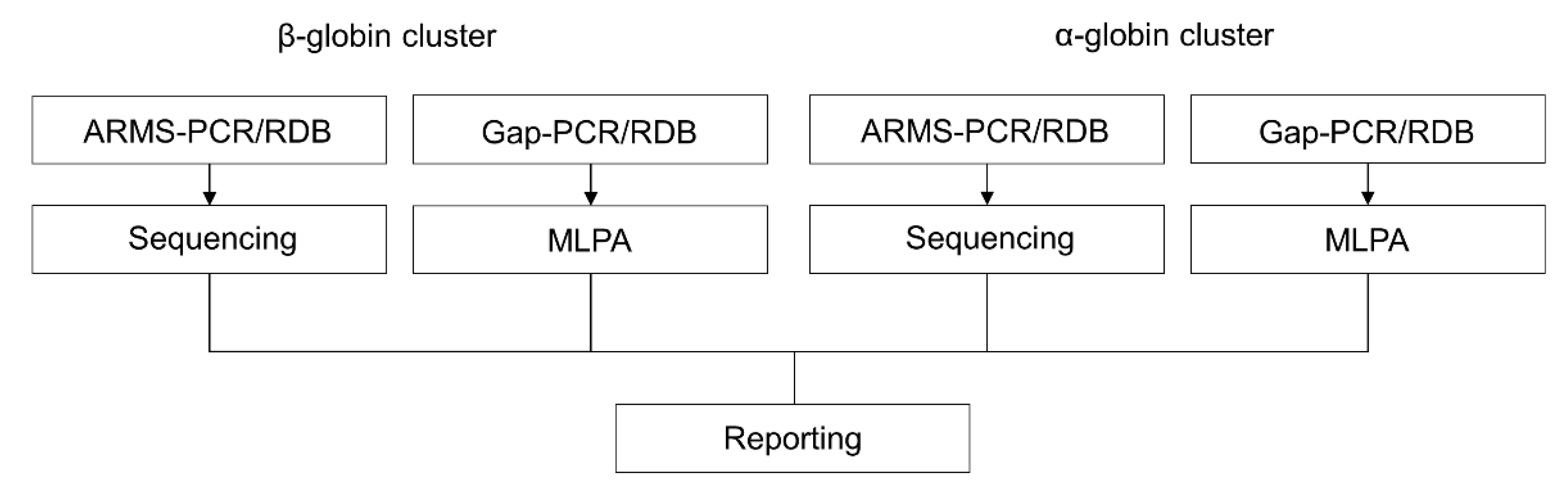
2.1. Reverse Dot-Blot Analysis
2.2. Gap-PCR
2.3. Amplification Refractory Mutation System (ARMS) or Allele-Specific Polymerase Chain Reaction (ASPCR)
2.4. Sanger Sequencing
2.5. Multiplex Ligation Probe-Dependent Analysis
3. Advanced Molecular Techniques towards the Single-Assay DNA Analysis
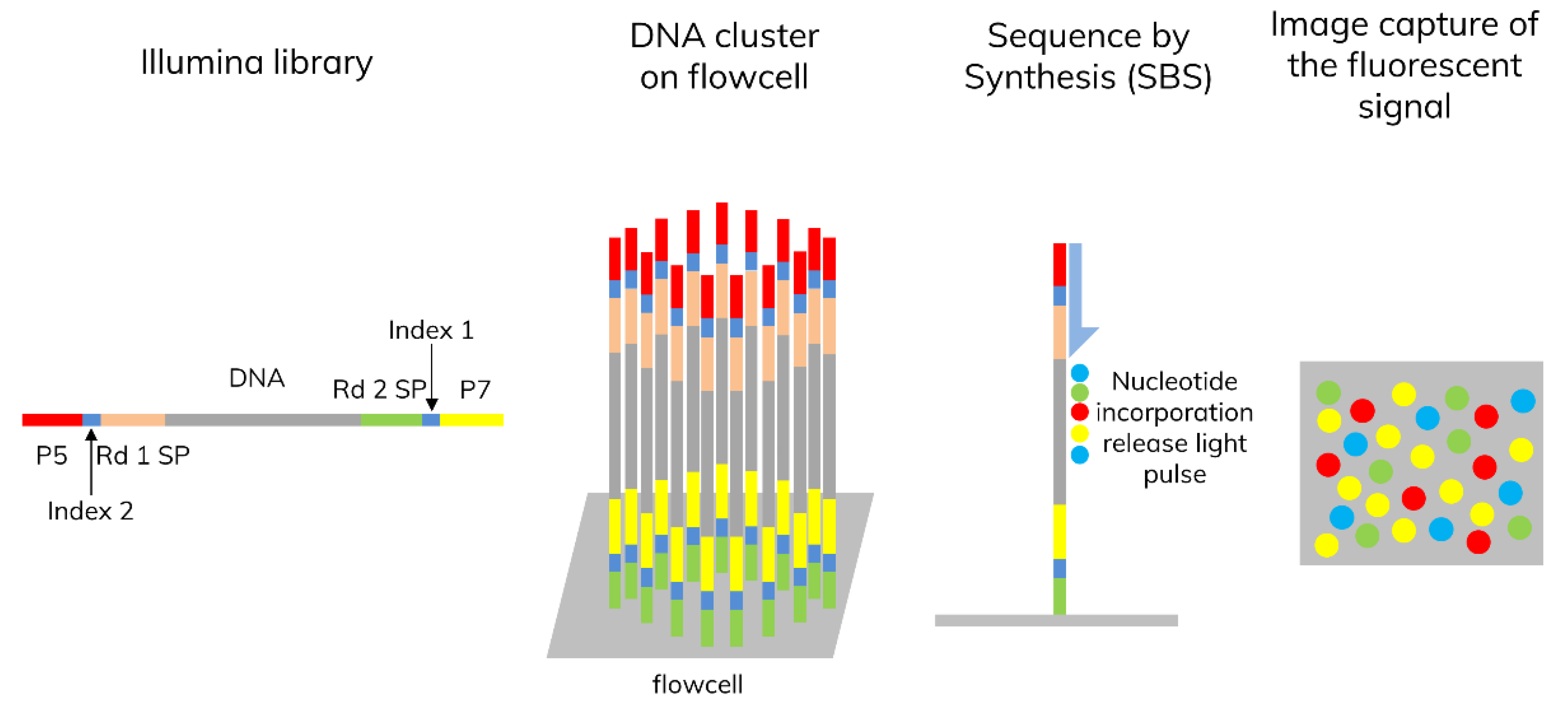
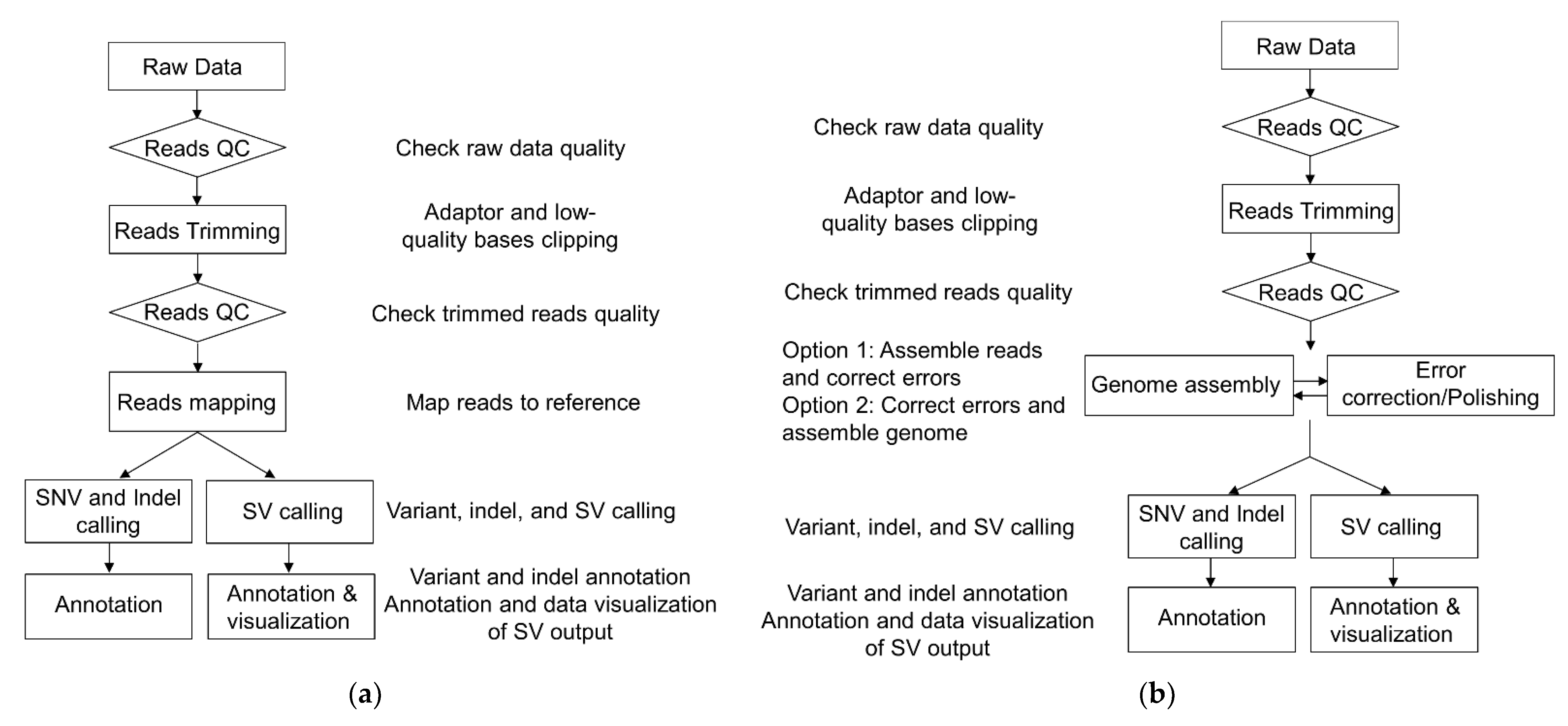
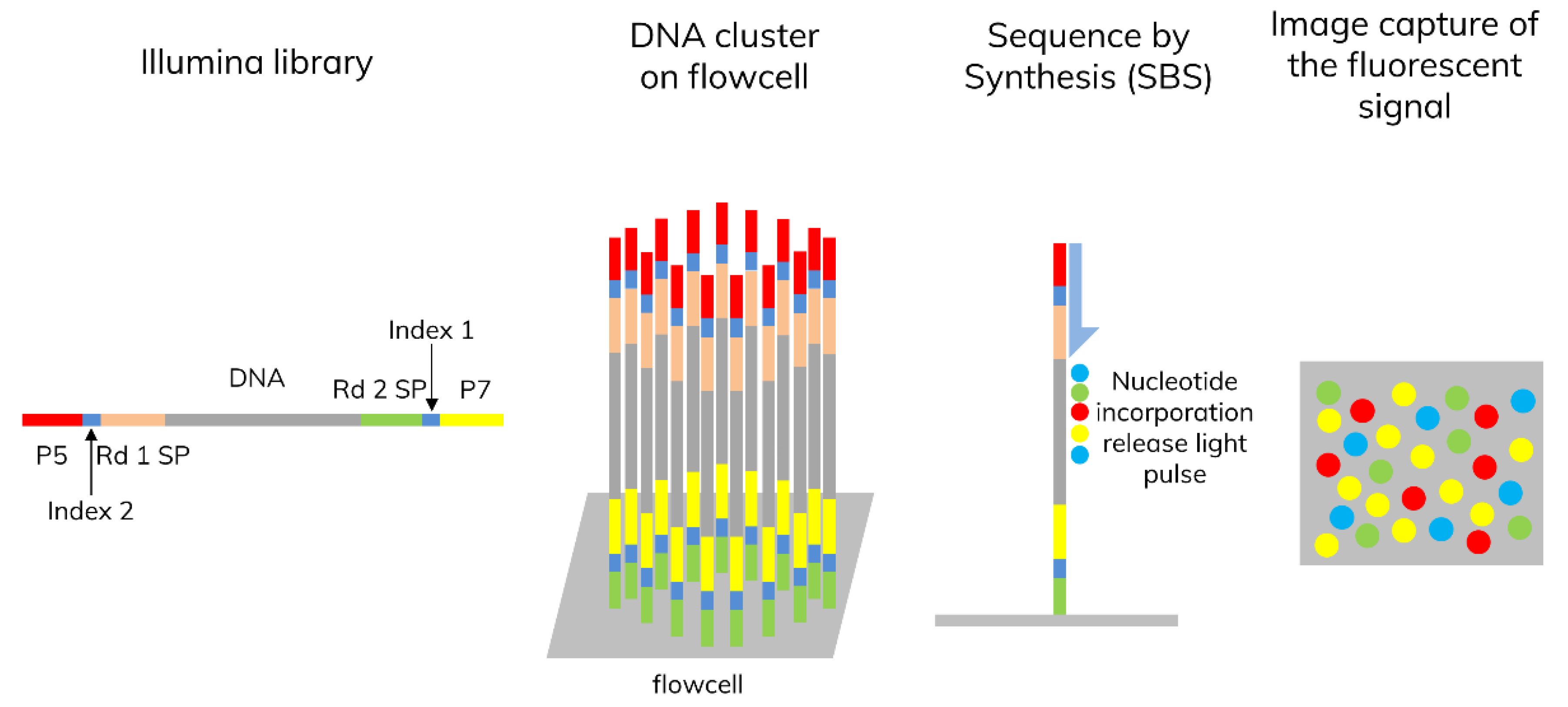
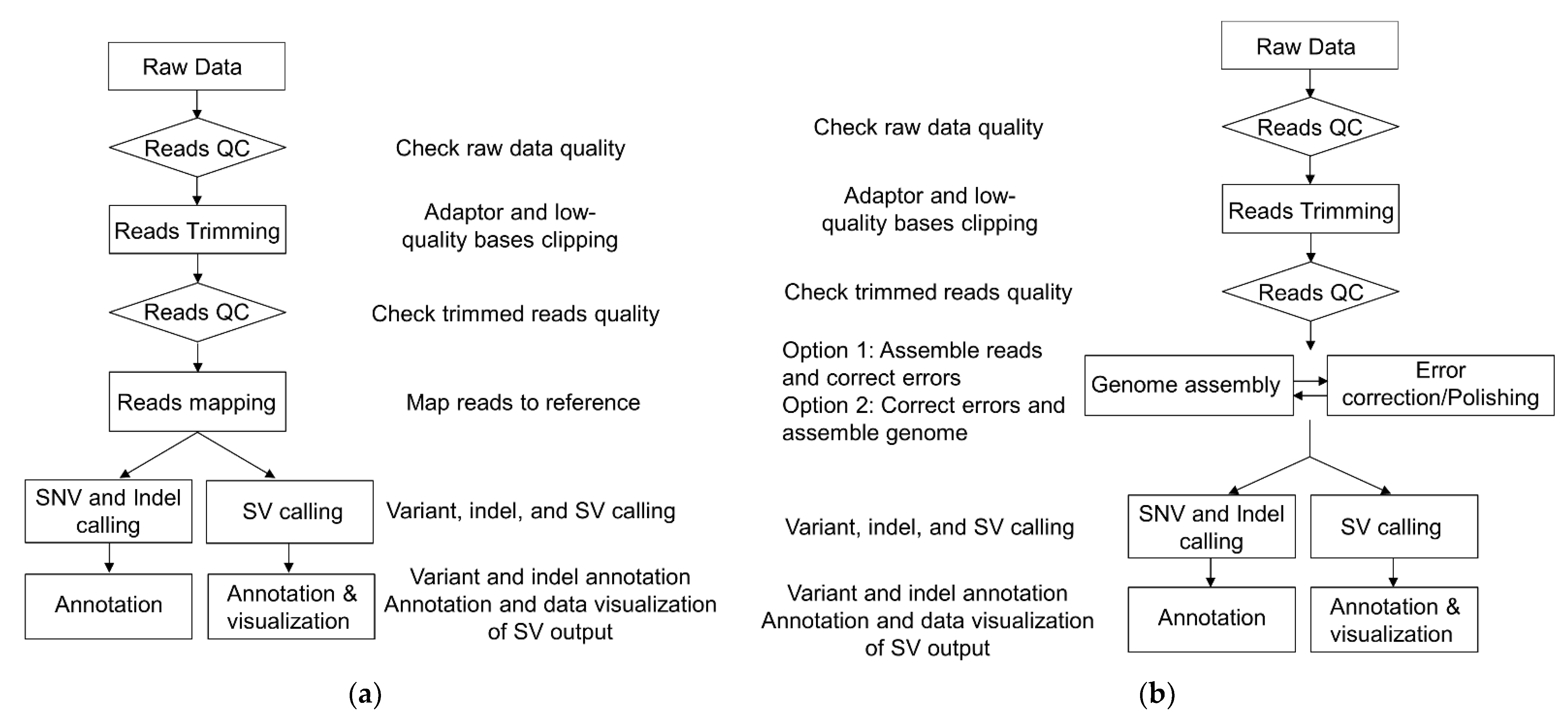
3.1. Next-Generation Sequencing (NGS)
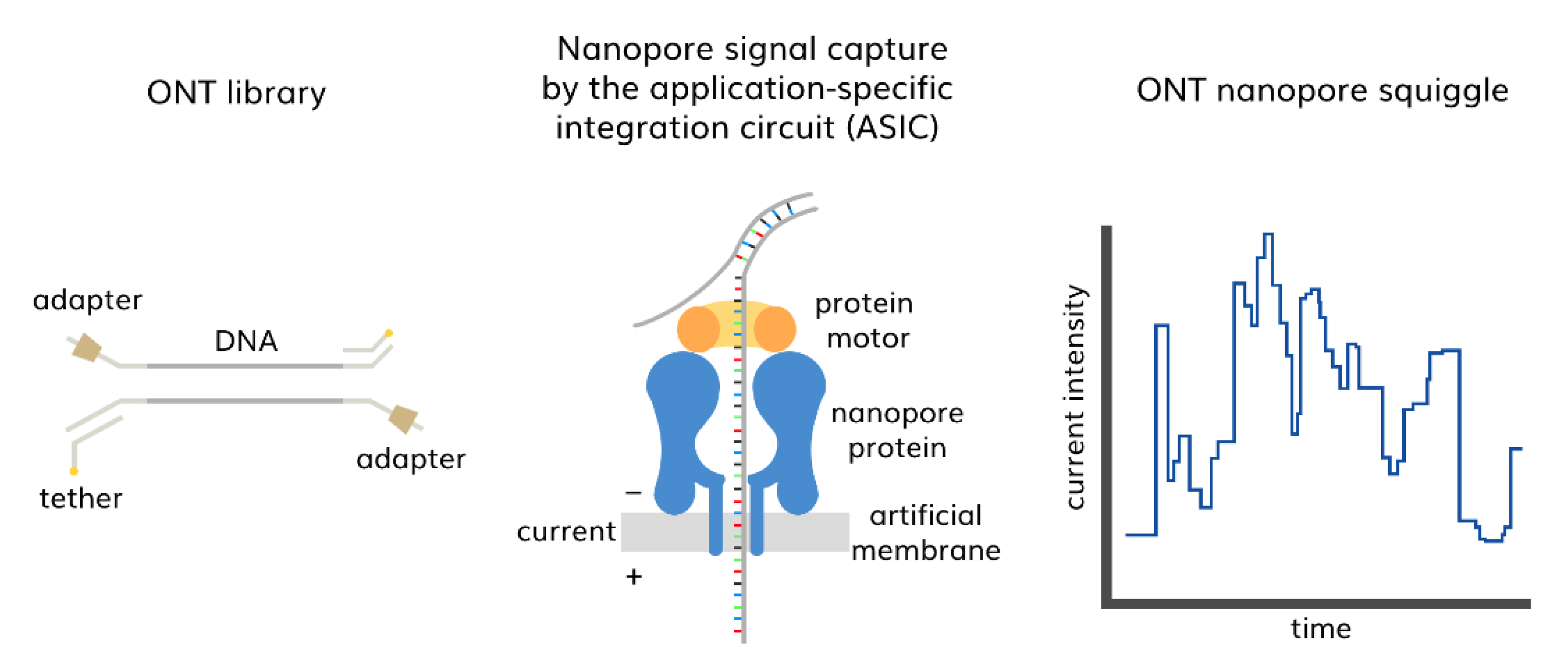
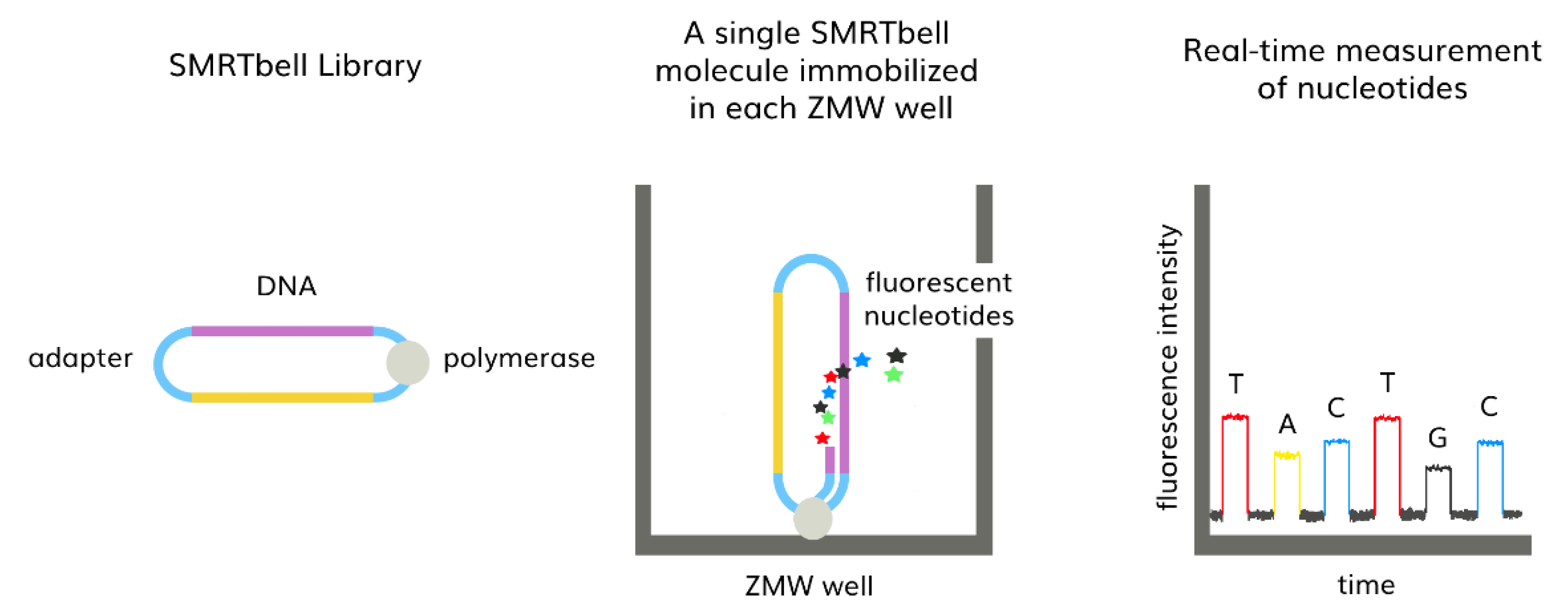
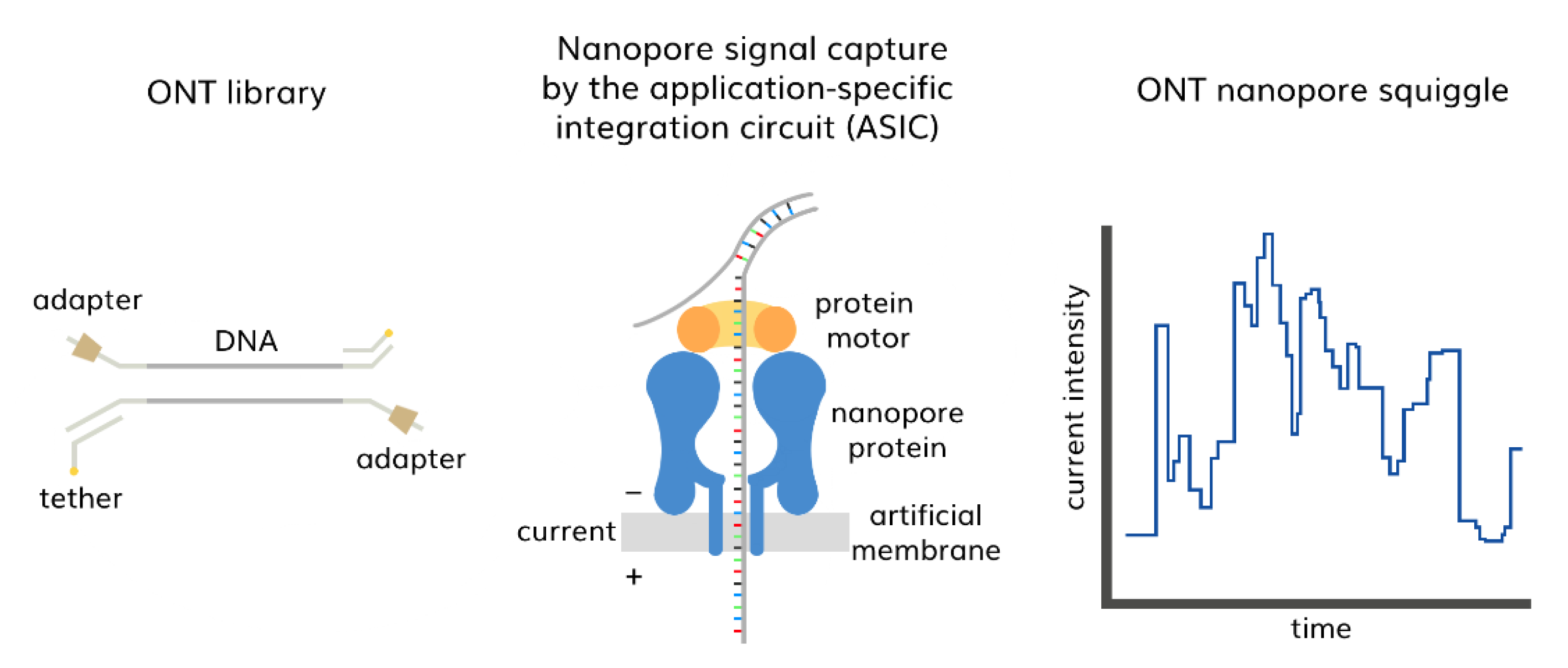
3.2. Third-Generation Sequencing (TGS)
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cooley, T.B.; Witwer, E.R.; Lee, P. Anemia in Children with Splenomegaly and Peculiar Changes in the Bones Report of Cases. Am. J. Dis. Child. 1927, 34, 347–363. [Google Scholar] [CrossRef]
- Whipple, G.H.; Bradford, W.L. Mediterranean Disease-Thalassemia (Erythroblastic Anemia of Cooley): Associated Pigment Abnormalities Simulating Hemochromatosis. J. Pediatr. 1936, 9, 279–311. [Google Scholar] [CrossRef]
- Ribeil, J.-A.; Arlet, J.-B.; Dussiot, M.; Cruz Moura, I.; Courtois, G.; Hermine, O. Ineffective Erythropoiesis in β-Thalassemia. Sci. World J. 2013, 2013, 394295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khandros, E.; Thom, C.S.; D’Souza, J.; Weiss, M.J. Integrated Protein Quality-Control Pathways Regulate Free α-Globin in Murine β-Thalassemia. Blood 2012, 119, 5265–5275. [Google Scholar] [CrossRef] [Green Version]
- Galanello, R.; Origa, R. Beta-Thalassemia. Orphanet J. Rare Dis. 2010, 5, 11. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.; Angelucci, E.; Lucarelli, G.; Aljurf, M.; Snyder, L.M.; Kiefer, C.R.; Ma, L.; Schrier, S.L. Accelerated Programmed Cell Death (Apoptosis) in Erythroid Precursors of Patients with Severe β-Thalassemia (Cooley’s Anemia). Blood 1993, 82, 374–377. [Google Scholar] [CrossRef] [Green Version]
- Nathan, D.G.; Oski, F.A. Hematology of Infancy and Childhood, 4th ed.; W.B. Saunders: Philadelphia, PA, USA, 1993. [Google Scholar]
- Galanello, R.; Cao, A. Relationship between Genotype and Phenotype. Ann. N. Y. Acad. Sci. 1988, 850, 325–333. [Google Scholar] [CrossRef]
- Goossens, M.; Dozy, A.M.; Emburyt, S.H.; Zachariadest, Z.; Hadjiminast, M.G.; Stamatoyannopoulos, G.; Kan, Y.W.A.I. Triplicated A-Globin Loci. Proc. Natl. Acad. Sci. USA 1980, 77, 518–521. [Google Scholar] [CrossRef] [Green Version]
- Henni, T.; Belhani, M.; Morle, F.; Bachir, D.; Tabone, P.; Colonna, P.; Godet, J. Alpha Globin Gene Triplication in Severe Heterozygous Beta Thalassemia. Acta Haematol. 1985, 74, 236–239. [Google Scholar] [CrossRef]
- Weatherall, D.J. The Definition and Epidemiology of Non-Transfusion-Dependent Thalassemia. Blood Rev. 2012, 26, S3–S6. [Google Scholar] [CrossRef]
- Rachmilewitz, E.A.; Giardina, P.J. How I Treat Thalassemia. Blood 2011, 118, 3479–3488. [Google Scholar] [CrossRef] [Green Version]
- Musallam, K.M.; Rivella, S.; Vichinsky, E.; Rachmilewitz, E.A. Non-Transfusion-Dependent Thalassemias. Haematologica 2013, 98, 833–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lederer, C.W.; Basak, A.N.; Aydinok, Y.; Christou, S.; El-Beshlawy, A.; Eleftheriou, A.; Fattoum, S.; Felice, A.E.; Fibach, E.; Galanello, R.; et al. An Electronic Infrastructure for Research and Treatment of the Thalassemias and Other Hemoglobinopathies: The Euro-Mediterranean ITHANET Project. Hemoglobin 2009, 33, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Fokkema, I.F.A.C.; Taschner, P.E.M.; Schaafsma, G.C.P.; Celli, J.; Laros, J.F.J.; den Dunnen, J.T. LOVD v.2.0: The next Generation in Gene Variant Databases. Hum. Mutat. 2011, 32, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Giardine, B.M.; Joly, P.; Pissard, S.; Wajcman, H.; Chui, D.H.K.; Hardison, R.C.; Patrinos, G.P. Clinically Relevant Updates of the HbVar Database of Human Hemoglobin Variants and Thalassemia Mutations. Nucleic Acids Res. 2021, 49, D1192–D1196. [Google Scholar] [CrossRef]
- Kan, Y.W.; Lee, K.Y.; Furbetta, M.; Angius, A.; Cao, A. Polymorphism of DNA Sequence in the β-Globin Gene Region. N. Engl. J. Med. 1980, 302, 185–188. [Google Scholar] [CrossRef]
- Kazazian, H.H.; Phillips, J.A.; Boehm, C.D.; Vik, T.A.; Mahoney, M.J.; Ritchey, A.K. Prenatal Diagnosis of β-Thalassemias by Amniocentesis: Linkage Analysis Using Multiple Polymorphic Restriction Endonuclease Sites. Blood 1980, 56, 926–930. [Google Scholar] [CrossRef]
- Geever, R.F.; Wilson, L.B.; Nallaseth, F.S.; Milner, P.F.; Bittner, M. Direct Identification of Sickle Cell Anemia by Blot Hybridization. Proc. Natl. Acad. Sci. USA 1981, 78, 5081–5085. [Google Scholar] [CrossRef] [Green Version]
- Pirastu, M.; Kan, Y.W.; Cao, A.; Conner, B.J.; Teplitz, R.L.; Wallace, R.B. Prenatal Diagnosis of β-Thalassemia. N. Engl. J. Med. 1983, 309, 284–287. [Google Scholar] [CrossRef]
- Kleppe, K.; Ohtsuka, E.; Kleppe, R.; Molineux, I.; Khorana, H.G. Studies on Polynucleotides. XCVI. Repair Replication of Short Synthetic DNA’s as Catalyzed by DNA Polymerases. J. Mol. Biol. 1971, 56, 341–361. [Google Scholar] [CrossRef]
- Saiki, R.K.; Scharf, S.; Faloona, F.; Mullis, K.B.; Horn, G.T.; Erlich, H.A.; Arnheim, N. Enzymatic Amplification of β-Globin Genomic Sequences and Restriction Site Analysis for Diagnosis of Sickle Cell Anemia. Science 1985, 230, 1350–1354. [Google Scholar] [CrossRef] [PubMed]
- Mullis, K.B.; Faloona, F.A.B.T.-M. Specific Synthesis of DNA in Vitro via a Polymerase-Catalyzed Chain Reaction. In Recombinant DNA Part F; Academic Press: Cambridge, MA, USA, 1987; Volume 155, pp. 335–350. ISBN 0076-6879. [Google Scholar]
- Amselem, S.; Nunes, V.; Vidaud, M.; Estivill, X.; Wong, C.; d’Auriol, L.; Vidaud, D.; Galibert, F.; Baiget, M.; Goossens, M. Determination of the Spectrum of β-Thalassemia Genes in Spain by Use of Dot-Blot Analysis of Amplified β-Globin DNA. Am. J. Hum. Genet. 1988, 43, 95–100. [Google Scholar] [PubMed]
- Saiki, R.K.; Chang, C.-A.; Levenson, C.H.; Warren, T.C.; Boehm, C.D.; Kazazian, H.H.; Erlich, H.A. Diagnosis of Sickle Cell Anemia and β-Thalassemia with Enzymatically Amplified DNA and Nonradioactive Allele-Specific Oligonucleotide Probes. N. Engl. J. Med. 1988, 319, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Saiki, R.K.; Walsh, P.S.; Levenson, C.H.; Erlich, H.A. Genetic Analysis of Amplified DNA with Immobilized Sequence-Specific Oligonucleotide Probes. Proc. Natl. Acad. Sci. USA 1989, 86, 6230–6234. [Google Scholar] [CrossRef] [Green Version]
- Maggio, A.; Giambona, A.; Cai, S.P.; Wall, J.; Kan, Y.W.; Chehab, F.F. Rapid and Simultaneous Typing of Hemoglobin S, Hemoglobin C, and Seven Mediterranean Beta-Thalassemia Mutations by Covalent Reverse Dot-Blot Analysis: Application to Prenatal Diagnosis in Sicily. Blood 1993, 81, 239–242. [Google Scholar] [CrossRef] [Green Version]
- Cai, S.-P.; Wall, J.; Kan, Y.W.; Chehab, F.F. Reverse Dot Blot Probes for the Screening of β-Thalassernia Mutationsin Asians and American Blacks. Hum. Mutat. 1994, 3, 59–63. [Google Scholar] [CrossRef]
- Giambona, A.; Lo Gioco, P.; Marino, M.; Abate, I.; Di Marzo, R.; Renda, M.; Di Trapani, F.; Messana, F.; Siciliano, S.; Rigano, P. The Great Heterogeneity of Thalassemia Molecular Defects in Sicily. Hum. Genet. 1995, 95, 526–530. [Google Scholar] [CrossRef]
- Sutcharitchan, P.; Saiki, R.; Fucharoen, S.; Winichagoon, P.; Erlich, H.; Embury, S.H. Reverse Dot-Blot Detection of Thai β-Thalassaemia Mutations. Br. J. Haematol. 1995, 90, 809–816. [Google Scholar] [CrossRef]
- Sutcharitchan, P.; Saiki, R.; Huisman, T.H.; Kutlar, A.; McKie, V.; Erlich, H.; Embury, S.H. Reverse Dot-Blot Detection of the African-American Beta-Thalassemia Mutations. Blood 1995, 86, 1580–1585. [Google Scholar] [CrossRef] [Green Version]
- Chan, V.; Yam, I.; Chen, F.E.; Chan, T.K. A Reverse Dot-Blot Method for Rapid Detection of Non-Deletion α Thalassaemia. Br. J. Haematol. 1999, 104, 513–515. [Google Scholar] [CrossRef]
- Bashyam, M.D.; Bashyam, L.; Savithri, G.R.; Gopikrishna, M.; Sangal, V.; Devi, A.R.R. Molecular Genetic Analyses of β-Thalassemia in South India Reveals Rare Mutations in the β-Globin Gene. J. Hum. Genet. 2004, 49, 408–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, M.; Zhu, J.-J.; Wang, Q.; Xie, L.-X.; Lu, M.; Wang, J.-L.; Wang, C.-F.; Zhong, T.-Y.; Zheng, L.; Pan, M.-C.; et al. Development and Evaluation of a Reverse Dot Blot Assay for the Simultaneous Detection of Common Alpha and Beta Thalassemia in Chinese. Blood Cells Mol. Dis. 2012, 48, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Chong, S.S.; Boehm, C.D.; Higgs, D.R.; Cutting, G.R. Single-Tube Multiplex-PCR Screen for Common Deletional Determinants of α-Thalassemia. Blood 2000, 95, 360–362. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Ma, E.S.; Chan, A.Y.; Prior, J.; Erber, W.N.; Chan, L.C.; Chui, D.H.; Chong, S.S. Single-Tube Multiplex-PCR Screen for Anti-3.7 and Anti-4.2 α-Globin Gene Triplications. Clin. Chem. 2003, 49, 1679–1682. [Google Scholar] [CrossRef] [Green Version]
- Craig, J.; Barnetson, R.; Prior, J.; Raven, J.; Thein, S. Rapid Detection of Deletions Causing Delta Beta Thalassemia and Hereditary Persistence of Fetal Hemoglobin by Enzymatic Amplification. Blood 1994, 83, 1673–1682. [Google Scholar] [CrossRef] [Green Version]
- Nussenzveig, R.H.; Vanhille, D.L.; Hussey, N.; Scott Reading, D.; Agarwal, A.M. Development of a Rapid Multiplex PCR Assay for Identification of the Three Common Hemoglobin-Lepore Variants (Boston-Washington, Baltimore, and Hollandia) and Identification of a New Lepore Variant. Am. J. Hematol. 2012, 87, 74–75. [Google Scholar] [CrossRef]
- Tritipsombut, J.; Phylipsen, M.; Viprakasit, V.; Chalaow, N.; Sanchaisuriya, K.; Giordano, P.C.; Fucharoen, S.; Harteveld, C.L. A Single-Tube Multiplex Gap-Polymerase Chain Reaction for the Detection of Eight β-Globin Gene Cluster Deletions Common in Southeast Asia. Hemoglobin 2012, 36, 571–580. [Google Scholar] [CrossRef]
- Wu, D.Y.; Ugozzoli, L.; Pal, B.K.; Wallace, R.B. Allele-Specific Enzymatic Amplification of Beta-Globin Genomic DNA for Diagnosis of Sickle Cell Anemia. Proc. Natl. Acad. Sci. USA 1989, 86, 2757–2760. [Google Scholar] [CrossRef] [Green Version]
- Newton, C.R.; Graham, A.; Heptinstall, L.E.; Powell, S.J.; Summers, C.; Kalsheker, N.; Smith, J.C.; Markham, A.F. Analysis of Any Point Mutation in DNA. The Amplification Refractory Mutation System (ARMS). Nucleic Acids Res. 1989, 17, 2503–2516. [Google Scholar] [CrossRef]
- Fortina, P.; Dotti, G.; Conant, R.; Monokian, G.; Parrella, T.; Hitchcock, W.; Rappaport, E.; Schwartz, E.; Surrey, S. Detection of the Most Common Mutations Causing Beta-Thalassemia in Mediterraneans Using a Multiplex Amplification Refractory Mutation System (MARMS). PCR Methods Appl. 1992, 2, 163–166. [Google Scholar] [CrossRef]
- Mirasena, S.; Shimbhu, D.; Sanguansermsri, M.; Sanguansermsri, T. Detection of β-Thalassemia Mutations Using a Multiplex Amplification Refractory Mutation System Assay. Hemoglobin 2008, 32, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Hassan, S.; Ahmad, R.; Zakaria, Z.; Zulkafli, Z.; Abdullah, W.Z. Detection of β-Globin Gene Mutations among β-Thalassaemia Carriers and Patients in Malaysia: Application of Multiplex Amplification Refractory Mutation System-Polymerase Chain Reaction. Malaysian J. Med. Sci. 2013, 20, 13–20. [Google Scholar]
- Tan, J.A.M.A.; Tay, J.S.H.; Lin, L.I.; Kham, S.K.Y.; Chia, J.N.; Chin, T.M.; Adb Aziz, N.B.; Wong, H.B. The Amplification Refractory Mutation System (ARMS): A Rapid and Direct Prenatal Diagnostic Technique for β-Thalassaemia in Singapore. Prenat. Diagn. 1994, 14, 1077–1082. [Google Scholar] [CrossRef]
- Chang, J.G.; Liu, H.J.; Huang, J.M.; Yang, T.Y.; Chang, C.P. Multiplex Mutagenically Separated PCR: Diagnosis of Beta-Thalassemia and Hemoglobin Variants. Biotechniques 1997, 22, 520–527. [Google Scholar] [CrossRef]
- Khateeb, B.; Moatter, T.; Shaghil, A.M.; Haroon, S.; Kakepoto, G.N. Genetic Diversity of Beta-Thalassemia Mutations in Pakistani Population. J. Pakistan Med. Assoc. 2000, 50, 293–296. [Google Scholar]
- Eng, B.; Patterson, M.; Walker, L.; Chui, D.H.K.; Waye, J.S. Detection of Severe Nondeletional α-Thalassemia Mutations Using a Single-Tube Multiplex ARMS Assay. Genet. Test. 2001, 5, 327–329. [Google Scholar] [CrossRef]
- Bhardwaj, U.; Zhang, Y.-H.; Lorey, F.; McCabe, L.L.; McCabe, E.R.B. Molecular Genetic Confirmatory Testing from Newborn Screening Samples for the Common African-American, Asian Indian, Southeast Asian, and Chinese β-Thalassemia Mutations. Am. J. Hematol. 2005, 78, 249–255. [Google Scholar] [CrossRef]
- Darwish, H.M.; El-Khatib, F.F.; Ayesh, S. Spectrum of β-Globin Gene Mutations Among Thalassemia Patients in the West Bank Region of Palestine. Hemoglobin 2005, 29, 119–132. [Google Scholar] [CrossRef]
- El-Gawhary, S.; El-Shafie, S.; Niazi, M.; Aziz, M.; El-Beshlawy, A. Study of β-Thalassemia Mutations Using the Polymerase Chain Reaction-Amplification Refractory Mutation System and Direct DNA Sequencing Techniques in a Group of Egyptian Thalassemia Patients. Hemoglobin 2007, 31, 63–69. [Google Scholar] [CrossRef]
- Lacerra, G.; Musollino, G.; Di Noce, F.; Prezioso, R. Clementina Carestia Genotyping for Known Mediterranean α-Thalassemia Point Mutations Using a Multiplex Amplification Refractory Mutation System. Haematologica 2007, 92, 254–255. [Google Scholar] [CrossRef] [Green Version]
- Sanger, F.; Coulson, A.R. A Rapid Method for Determining Sequences in DNA by Primed Synthesis with DNA Polymerase. J. Mol. Biol. 1975, 94, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.M.; Fung, S.; Hunkapiller, M.W.; Hunkapiller, T.J.; Hood, L.E. The Synthesis of Oligonucleotides Containing an Aliphatic Amino Group at the 5′ Terminus: Synthesis of Fluorescent DNA Primers for Use in DNA Sequence Analysis. Nucleic Acids Res. 1985, 13, 2399–2412. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.M.; Sanders, J.Z.; Kaiser, R.J.; Hughes, P.; Dodd, C.; Connell, C.R.; Heiner, C.; Kent, S.B.; Hood, L.E. Fluorescence Detection in Automated DNA Sequence Analysis. Nature 1986, 321, 674–679. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.; Dowling, C.E.; Saiki, R.K.; Higuchi, R.G.; Erlich, H.A.; Kazazian, H.H.J. Characterization of Beta-Thalassaemia Mutations Using Direct Genomic Sequencing of Amplified Single Copy DNA. Nature 1987, 330, 384–386. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo, R.; Dowling, C.E.; Wong, C.; Maggio, A.; Kazazian, H.H. The Spectrum of β-Thalassaemia Mutations in Sicily. Br. J. Haematol. 1988, 69, 393–397. [Google Scholar] [CrossRef] [PubMed]
- Drossman, H.; Luckey, J.A.; Kostichka, A.J.; D’Cunha, J.; Smith, L.M. High-Speed Separations of DNA Sequencing Reactions by Capillary Electrophoresis. Anal. Chem. 1990, 62, 900–903. [Google Scholar] [CrossRef] [PubMed]
- Aulehla-Scholz, C.; Basaran, S.; Agaoglu, L.; Arcasoy, A.; Holzgreve, W.; Miny, P.; Ridolfi, F.; Horst, J. Molecular Basis of Beta-Thalassemia in Turkey: Detection of Rare Mutations by Direct Sequencing. Hum. Genet. 1990, 84, 195–197. [Google Scholar] [CrossRef]
- Hsuih, T.C.; Park, Y.N.; Zaretsky, C.; Wu, F.; Tyagi, S.; Kramer, F.R.; Sperling, R.; Zhang, D.Y. Novel, Ligation-Dependent PCR Assay for Detection of Hepatitis C in Serum. J. Clin. Microbiol. 1996, 34, 501–507. [Google Scholar] [CrossRef] [Green Version]
- Carrino, J.J. Multiplex Ligations-Dependent Amplification. 1996. Available online: http://patent.google.com/patent/WO1996015271A1/en (accessed on 25 October 2022).
- Schouten, J.P.; McElgunn, C.J.; Waaijer, R.; Zwijnenburg, D.; Diepvens, F.; Pals, G. Relative Quantification of 40 Nucleic Acid Sequences by Multiplex Ligation-Dependent Probe Amplification. Nucleic Acids Res. 2002, 30, e57. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Z.; Zhong, G.; Guan, Z.; Chen, D.; Wu, Z.; Yang, K.; Chen, D.; Liu, Y.; Xu, R.; Chen, J. A Novel 15.8 Kb Deletion α-Thalassemia Confirmed by Long-Read Single-Molecule Real-Time Sequencing: Hematological Phenotypes and Molecular Characterization. Clin. Biochem. 2022, 108, 46–49. [Google Scholar] [CrossRef]
- Luo, S.; Chen, X.; Zeng, D.; Tang, N.; Yuan, D.; Liu, B.; Chen, L.; Zhong, Q.; Li, J.; Liu, Y.; et al. Detection of Four Rare Thalassemia Variants Using Single-Molecule Realtime Sequencing. Front. Genet. 2022, 13, 974999. [Google Scholar] [CrossRef] [PubMed]
- Harteveld, C.L.; Refaldi, C.; Cassinerio, E.; Cappellini, M.D.; Giordano, P.C. Segmental Duplications Involving the α-Globin Gene Cluster Are Causing β-Thalassemia Intermedia Phenotypes in β-Thalassemia Heterozygous Patients. Blood Cells Mol. Dis. 2008, 40, 312–316. [Google Scholar] [CrossRef] [PubMed]
- Sollaino, M.C.; Paglietti, M.E.; Perseu, L.; Giagu, N.; Loi, D.; Galanello, R. Association of a Globin Gene Quadruplication and Heterozygous β Thalassemia in Patients with Thalassemia Intermedia. Haematologica 2009, 94, 1445–1448. [Google Scholar] [CrossRef]
- Jiang, H.; Liu, S.; Zhang, Y.L.; Wan, J.H.; Li, R.; Li, D.Z. Association of an α-Globin Gene Cluster Duplication and Heterozygous β-Thalassemia in a Patient with a Severe Thalassemia Syndrome. Hemoglobin 2015, 39, 102–106. [Google Scholar] [CrossRef]
- Steinberg-Shemer, O.; Ulirsch, J.C.; Noy-Lotan, S.; Krasnov, T.; Attias, D.; Dgany, O.; Laor, R.; Sankaran, V.G.; Tamary, H. Whole-Exome Sequencing Identifies an α-Globin Cluster Triplication Resulting in Increased Clinical Severity of β-Thalassemia. Cold Spring Harb. Mol. Case Stud. 2017, 3, a001941. [Google Scholar] [CrossRef]
- Clark, B.; Shooter, C.; Smith, F.; Brawand, D.; Steedman, L.; Oakley, M.; Rushton, P.; Rooks, H.; Wang, X.; Drousiotou, A.; et al. Beta Thalassaemia Intermedia Due to Co-Inheritance of Three Unique Alpha Globin Cluster Duplications Characterised by next Generation Sequencing Analysis. Br. J. Haematol. 2018, 180, 160–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubiczkova Besse, L.; Sedlarikova, L.; Kryukov, F.; Nekvindova, J.; Radova, L.; Almasi, M.; Pelcova, J.; Minarik, J.; Pika, T.; Pikalova, Z.; et al. Combination of serum microRNA-320a and microRNA-320b as a marker for Waldenström macroglobulinemia. Am. J. Hematol. 2015, 90, E51–E52. [Google Scholar] [CrossRef] [PubMed]
- Shooter, C.; Rooks, H.; Thein, S.L.; Clark, B. Next Generation Sequencing Identifies a Novel Rearrangement in the HBB Cluster Permitting To-the-Base Characterization. Hum. Mutat. 2015, 36, 142–150. [Google Scholar] [CrossRef]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A Framework for Variation Discovery and Genotyping Using Next-Generation DNA Sequencing Data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997v2. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H. Minimap2: Pairwise Alignment for Nucleotide Sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaharia, M.; Bolosky, W.J.; Curtis, K.; Fox, A.; Patterson, D.A.; Shenker, S.; Stoica, I.; Karp, R.M.; Sittler, T. Faster and More Accurate Sequence Alignment with {SNAP}. arXiv 2011, arXiv:1111.5572. [Google Scholar]
- Garrison, E.; Marth, G. Haplotype-Based Variant Detection from Short-Read Sequencing. arXiv 2012, arXiv:1207.3907. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Poplin, R.; Chang, P.-C.; Alexander, D.; Schwartz, S.; Colthurst, T.; Ku, A.; Newburger, D.; Dijamco, J.; Nguyen, N.; Afshar, P.T.; et al. A Universal SNP and Small-Indel Variant Caller Using Deep Neural Networks. Nat. Biotechnol. 2018, 36, 983–987. [Google Scholar] [CrossRef]
- Kim, S.; Scheffler, K.; Halpern, A.L.; Bekritsky, M.A.; Noh, E.; Källberg, M.; Chen, X.; Kim, Y.; Beyter, D.; Krusche, P.; et al. Strelka2: Fast and Accurate Calling of Germline and Somatic Variants. Nat. Methods 2018, 15, 591–594. [Google Scholar] [CrossRef]
- Teo, S.M.; Pawitan, Y.; Ku, C.S.; Chia, K.S.; Salim, A. Statistical Challenges Associated with Detecting Copy Number Variations with Next-Generation Sequencing. Bioinformatics 2012, 28, 2711–2718. [Google Scholar] [CrossRef] [Green Version]
- Boeva, V.; Popova, T.; Bleakley, K.; Chiche, P.; Cappo, J.; Schleiermacher, G.; Janoueix-Lerosey, I.; Delattre, O.; Barillot, E. Control-FREEC: A Tool for Assessing Copy Number and Allelic Content Using next-Generation Sequencing Data. Bioinformatics 2012, 28, 423–425. [Google Scholar] [CrossRef] [Green Version]
- Rausch, T.; Zichner, T.; Schlattl, A.; Stütz, A.M.; Benes, V.; Korbel, J.O. DELLY: Structural Variant Discovery by Integrated Paired-End and Split-Read Analysis. Bioinformatics 2012, 28, i333–i339. [Google Scholar] [CrossRef] [Green Version]
- Talevich, E.; Shain, A.H.; Botton, T.; Bastian, B.C. CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Comput. Biol. 2016, 12, e1004873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plagnol, V.; Curtis, J.; Epstein, M.; Mok, K.Y.; Stebbings, E.; Grigoriadou, S.; Wood, N.W.; Hambleton, S.; Burns, S.O.; Thrasher, A.J.; et al. A Robust Model for Read Count Data in Exome Sequencing Experiments and Implications for Copy Number Variant Calling. Bioinformatics 2012, 28, 2747–2754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krumm, N.; Sudmant, P.H.; Ko, A.; O’Roak, B.J.; Malig, M.; Coe, B.P.; Project, N.E.S.; Quinlan, A.R.; Nickerson, D.A.; Eichler, E.E. Copy Number Variation Detection and Genotyping from Exome Sequence Data. Genome Res. 2012, 22, 1525–1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Majewski, J. FishingCNV: A Graphical Software Package for Detecting Rare Copy Number Variations in Exome-Sequencing Data. Bioinformatics 2013, 29, 1461–1462. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yu, Z.; Ban, R.; Zhang, H.; Iqbal, F.; Zhao, A.; Li, A.; Shi, Q. DeAnnCNV: A Tool for Online Detection and Annotation of Copy Number Variations from Whole-Exome Sequencing Data. Nucleic Acids Res. 2015, 43, W289–W294. [Google Scholar] [CrossRef]
- Derouault, P.; Chauzeix, J.; Rizzo, D.; Miressi, F.; Magdelaine, C.; Bourthoumieu, S.; Durand, K.; Dzugan, H.; Feuillard, J.; Sturtz, F.; et al. CovCopCan: An Efficient Tool to Detect Copy Number Variation from Amplicon Sequencing Data in Inherited Diseases and Cancer. PLoS Comput. Biol. 2020, 16, e1007503. [Google Scholar] [CrossRef] [Green Version]
- Chanwigoon, S.; Piwluang, S.; Wichadakul, D. InCNV: An Integrated Analysis Tool for Copy Number Variation on Whole Exome Sequencing. Evol. Bioinforma. 2020, 16, 1176934320956577. [Google Scholar] [CrossRef]
- Erikson, G.A.; Deshpande, N.; Kesavan, B.G.; Torkamani, A. SG-ADVISER CNV: Copy-Number Variant Annotation and Interpretation. Genet. Med. 2015, 17, 714–718. [Google Scholar] [CrossRef] [Green Version]
- Chandramohan, R.; Kakkar, N.; Roy, A.; Parsons, D.W. ReconCNV: Interactive Visualization of Copy Number Data from High-Throughput Sequencing. Bioinformatics 2021, 37, 1164–1167. [Google Scholar] [CrossRef]
- Requena, F.; Abdallah, H.H.; García, A.; Nitschké, P.; Romana, S.; Malan, V.; Rausell, A. CNVxplorer: A Web Tool to Assist Clinical Interpretation of CNVs in Rare Disease Patients. Nucleic Acids Res. 2021, 49, W93–W103. [Google Scholar] [CrossRef]
- Gel, B.; Serra, E. KaryoploteR: An R/Bioconductor Package to Plot Customizable Genomes Displaying Arbitrary Data. Bioinformatics 2017, 33, 3088–3090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahne, F.; Ivanek, R. Visualizing Genomic Data Using Gviz and Bioconductor BT—Statistical Genomics: Methods and Protocols; Mathé, E., Davis, S., Eds.; Springer: New York, NY, USA, 2016; pp. 335–351. ISBN 978-1-4939-3578-9. [Google Scholar]
- Gel, B.; Magallon, M. CopyNumberPlots: Create Copy-Number Plots Using KaryoploteR Functionality. 2021. Available online: http://github.com/bernatgel/CopyNumberPlots (accessed on 25 October 2022).
- Shang, X.; Peng, Z.; Ye, Y.; Asan; Zhang, X.; Chen, Y.; Zhu, B.; Cai, W.; Chen, S.; Cai, R.; et al. Rapid Targeted Next-Generation Sequencing Platform for Molecular Screening and Clinical Genotyping in Subjects with Hemoglobinopathies. EBioMedicine 2017, 23, 150–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, P.; Yu, X.; Huang, H.; Zeng, W.; He, X.; Liu, M.; Huang, B. Evaluation of Ion Torrent Next-Generation Sequencing for Thalassemia Diagnosis. J. Int. Med. Res. 2020, 48, 0300060520967778. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Li, J.; Lai, Q.; Yu, Y. Combined Use of Gap-PCR and next-Generation Sequencing Improves Thalassaemia Carrier Screening among Premarital Adults in China. J. Clin. Pathol. 2020, 73, 488–492. [Google Scholar] [CrossRef]
- He, J.; Song, W.; Yang, J.; Lu, S.; Yuan, Y.; Guo, J.; Zhang, J.; Ye, K.; Yang, F.; Long, F.; et al. Next-Generation Sequencing Improves Thalassemia Carrier Screening among Premarital Adults in a High Prevalence Population: The Dai Nationality, China. Genet. Med. 2017, 19, 1022–1031. [Google Scholar] [CrossRef]
- Fan, D.M.; Yang, X.; Huang, L.M.; Ouyang, G.J.; Yang, X.X.; Li, M. Simultaneous Detection of Target CNVs and SNVs of Thalassemia by Multiplex PCR and Next-generation Sequencing. Mol. Med. Rep. 2019, 19, 2837–2848. [Google Scholar] [CrossRef] [Green Version]
- Zebisch, A.; Schulz, E.; Grosso, M.; Lombardo, B.; Acierno, G.; Sill, H.; Iolascon, A. Identification of a Novel Variant of Epsilon-Gamma-Delta-Beta Thalassemia Highlights Limitations of next Generation Sequencing. Am. J. Hematol. 2015, 90, E52–E54. [Google Scholar] [CrossRef]
- Fromer, M.; Moran, J.L.; Chambert, K.; Banks, E.; Bergen, S.E.; Ruderfer, D.M.; Handsaker, R.E.; McCarroll, S.A.; O’Donovan, M.C.; Owen, M.J.; et al. Discovery and Statistical Genotyping of Copy-Number Variation from Whole-Exome Sequencing Depth. Am. J. Hum. Genet. 2012, 91, 597–607. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Chan, G.C.-F. NGS4THAL, a One-Stop Molecular Diagnosis and Carrier Screening Tool for Thalassemia and Other Hemoglobinopathies by next-Generation Sequencing. Res. Sq. 2022, 24, 1089–1099. [Google Scholar] [CrossRef]
- Jiang, F.; Lyu, G.Z.; Zhang, V.W.; Li, D.Z. Identification of Thalassemia Gene Cluster Deletion by Long-Read Whole-Genome Sequencing (LR-WGS). Int. J. Lab. Hematol. 2021, 43, 859–865. [Google Scholar] [CrossRef]
- Li, Q.; Mao, Y.; Li, S.; Du, H.; He, W.; He, J.; Kong, L.; Zhang, J.; Liang, B.; Liu, J. Haplotyping by Linked-Read Sequencing (HLRS) of the Genetic Disease Carriers for Preimplantation Genetic Testing without a Proband or Relatives. BMC Med. Genomics 2020, 13, 117. [Google Scholar] [CrossRef] [PubMed]
- Korlach, J.; Turner, S.W. Single-Molecule Sequencing BT—Encyclopedia of Biophysics; Roberts, G.C.K., Ed.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 2344–2347. ISBN 978-3-642-16712-6. [Google Scholar]
- Eid, J.; Fehr, A.; Gray, J.; Luong, K.; Lyle, J.; Otto, G.; Peluso, P.; Rank, D.; Baybayan, P.; Bettman, B.; et al. Real-Time DNA Sequencing from Single Polymerase Molecules. Science 2009, 323, 133–138. [Google Scholar] [CrossRef] [PubMed]
- PacBio SMRT Sequencing—How It Works. Available online: https://www.pacb.com/smrt-science/attachment/infographic_smrt-sequencing-how-it-works/%0Awww.pacb.com (accessed on 25 October 2022).
- Wenger, A.M.; Peluso, P.; Rowell, W.J.; Chang, P.-C.; Hall, R.J.; Concepcion, G.T.; Ebler, J.; Fungtammasan, A.; Kolesnikov, A.; Olson, N.D.; et al. Accurate Circular Consensus Long-Read Sequencing Improves Variant Detection and Assembly of a Human Genome. Nat. Biotechnol. 2019, 37, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, Y.; Bollas, A.; Wang, Y.; Au, K.F. Nanopore Sequencing Technology, Bioinformatics and Applications. Nat. Biotechnol. 2021, 39, 1348–1365. [Google Scholar] [CrossRef]
- Tomasz Dobrzycki Selecting the Right Library Prep Method for Your Experiment. Available online: https://nanoporetech.com/resource-centre/video/lc22/selecting-the-right-library-prep-method-for-your-experiment (accessed on 31 October 2022).
- Oxford Nanopore Technologies How It Works. Available online: https://nanoporetech.com/how-it-works (accessed on 12 October 2022).
- Carneiro, M.O.; Russ, C.; Ross, M.G.; Gabriel, S.B.; Nusbaum, C.; DePristo, M.A. Pacific Biosciences Sequencing Technology for Genotyping and Variation Discovery in Human Data. BMC Genomics 2012, 13, 375. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.H.; Jiang, P.; Sun, K.; Cheng, Y.K.Y.; Chan, K.C.A.; Leung, T.Y.; Chiu, R.W.K.; Lo, Y.M.D. Noninvasive Prenatal Testing by Nanopore Sequencing of Maternal Plasma DNA: Feasibility Assessment. Clin. Chem. 2015, 61, 1305–1306. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Jain, C.; Aluru, S. A Comprehensive Evaluation of Long Read Error Correction Methods. BMC Genomics 2020, 21, 889. [Google Scholar] [CrossRef]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of Long, Error-Prone Reads Using Repeat Graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef]
- Ruan, J.; Li, H. Fast and Accurate Long-Read Assembly with Wtdbg2. Nat. Methods 2020, 17, 155–158. [Google Scholar] [CrossRef]
- Shafin, K.; Pesout, T.; Lorig-Roach, R.; Haukness, M.; Olsen, H.E.; Bosworth, C.; Armstrong, J.; Tigyi, K.; Maurer, N.; Koren, S.; et al. Nanopore Sequencing and the Shasta Toolkit Enable Efficient de Novo Assembly of Eleven Human Genomes. Nat. Biotechnol. 2020, 38, 1044–1053. [Google Scholar] [CrossRef]
- Morisse, P.; Marchet, C.; Limasset, A.; Lecroq, T.; Lefebvre, A. Scalable Long Read Self-Correction and Assembly Polishing with Multiple Sequence Alignment. Sci. Rep. 2021, 11, 761. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.-L.; Chen, Y.; Xie, S.-Q.; Chen, K.-N.; Wang, Y.; Han, Y.; Luo, F.; Xie, Z. MECAT: Fast Mapping, Error Correction, and de Novo Assembly for Single-Molecule Sequencing Reads. Nat. Methods 2017, 14, 1072–1074. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and Accurate Long-Read Assembly via Adaptive k-Mer Weighting and Repeat Separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berlin, K.; Koren, S.; Chin, C.-S.; Drake, J.P.; Landolin, J.M.; Phillippy, A.M. Assembling Large Genomes with Single-Molecule Sequencing and Locality-Sensitive Hashing. Nat. Biotechnol. 2015, 33, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.-S.; Peluso, P.; Sedlazeck, F.J.; Nattestad, M.; Concepcion, G.T.; Clum, A.; Dunn, C.; O’Malley, R.; Figueroa-Balderas, R.; Morales-Cruz, A.; et al. Phased Diploid Genome Assembly with Single-Molecule Real-Time Sequencing. Nat. Methods 2016, 13, 1050–1054. [Google Scholar] [CrossRef] [Green Version]
- Chaisson, M.J.; Tesler, G. Mapping Single Molecule Sequencing Reads Using Basic Local Alignment with Successive Refinement (BLASR): Application and Theory. BMC Bioinformatics 2012, 13, 238. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Nie, F.; Xie, S.-Q.; Zheng, Y.-F.; Dai, Q.; Bray, T.; Wang, Y.-X.; Xing, J.-F.; Huang, Z.-J.; Wang, D.-P.; et al. Efficient Assembly of Nanopore Reads via Highly Accurate and Intact Error Correction. Nat. Commun. 2021, 12, 60. [Google Scholar] [CrossRef]
- Shafin, K.; Pesout, T.; Chang, P.-C.; Nattestad, M.; Kolesnikov, A.; Goel, S.; Baid, G.; Kolmogorov, M.; Eizenga, J.M.; Miga, K.H.; et al. Haplotype-Aware Variant Calling with PEPPER-Margin-DeepVariant Enables High Accuracy in Nanopore Long-Reads. Nat. Methods 2021, 18, 1322–1332. [Google Scholar] [CrossRef]
- Ahsan, M.U.; Liu, Q.; Fang, L.; Wang, K. NanoCaller for Accurate Detection of SNPs and Indels in Difficult-to-Map Regions from Long-Read Sequencing by Haplotype-Aware Deep Neural Networks. Genome Biol. 2021, 22, 261. [Google Scholar] [CrossRef]
- Su, J.; Zheng, Z.; Ahmed, S.S.; Lam, T.-W.; Luo, R. Clair3-Trio: High-Performance Nanopore Long-Read Variant Calling in Family Trios with Trio-to-Trio Deep Neural Networks. Brief. Bioinform. 2022, bbac301. [Google Scholar] [CrossRef]
- Ramachandran, A.; Lumetta, S.S.; Klee, E.W.; Chen, D. HELLO: Improved Neural Network Architectures and Methodologies for Small Variant Calling. BMC Bioinformatics 2021, 22, 404. [Google Scholar] [CrossRef] [PubMed]
- Smolka, M.; Paulin, L.F.; Grochowski, C.M.; Mahmoud, M.; Behera, S.; Gandhi, M.; Hong, K.; Pehlivan, D.; Scholz, S.W.; Carvalho, C.M.B.; et al. Comprehensive Structural Variant Detection: From Mosaic to Population-Level (Sniffles2). bioRxiv 2022. [Google Scholar] [CrossRef]
- Cao, S.; Jiang, T.; Liu, Y.; Liu, S.; Wang, Y. Re-Genotyping Structural Variants through an Accurate Force-Calling Method. bioRxiv 2022. [Google Scholar] [CrossRef]
- Heller, D.; Vingron, M. SVIM: Structural Variant Identification Using Mapped Long Reads. Bioinformatics 2019, 35, 2907–2915. [Google Scholar] [CrossRef]
- Tham, C.Y.; Tirado-Magallanes, R.; Goh, Y.; Fullwood, M.J.; Koh, B.T.H.; Wang, W.; Ng, C.H.; Chng, W.J.; Thiery, A.; Tenen, D.G.; et al. NanoVar: Accurate Characterization of Patients’ Genomic Structural Variants Using Low-Depth Nanopore Sequencing. Genome Biol. 2020, 21, 56. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Mao, A.; Liu, H.; Gui, B.; Choy, K.W.; Huang, H.; Yu, Q.; Zhang, X.; Chen, M.; Lin, N.; et al. Long-Molecule Sequencing: A New Approach for Identification of Clinically Significant DNA Variants in α-Thalassemia and β-Thalassemia Carriers. J. Mol. Diagnostics 2020, 22, 1087–1095. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Gu, W.; Chen, P.; Li, Y.; Liu, Y.; Tian, M.; Zhou, Q.; Qi, H.; Zhang, Y.; He, J.; et al. A More Universal Approach to Comprehensive Analysis of Thalassemia Alleles (CATSA). J. Mol. Diagnostics 2021, 23, 1195–1204. [Google Scholar] [CrossRef]
- Peng, C.; Zhang, H.; Ren, J.; Chen, H.; Du, Z.; Zhao, T.; Mao, A.; Xu, R.; Lu, Y.; Wang, H.; et al. Analysis of Rare Thalassemia Genetic Variants Based on Third-Generation Sequencing. Sci. Rep. 2022, 12, 9907. [Google Scholar] [CrossRef]
- Li, Y.; Liang, L.; Qin, T.; Tian, M. Detection of Hemoglobin H Disease by Long Molecule Sequencing. J. Clin. Lab. Anal. 2022, 36, e24687. [Google Scholar] [CrossRef]
- Jiang, F.; Liu, W.; Zhang, L.; Guo, Y.; Chen, M.; Zeng, X.; Wang, Y.; Li, Y.; Xian, J.; Du, B.; et al. Noninvasive Prenatal Testing for β-Thalassemia by Targeted Nanopore Sequencing Combined with Relative Haplotype Dosage (RHDO): A Feasibility Study. Sci. Rep. 2021, 11, 5714. [Google Scholar] [CrossRef]
- Christopher, H.; Burns, A.; Josephat, E.; Makani, J.; Schuh, A.; Nkya, S. Using DNA Testing for the Precise, Definite, and Low-Cost Diagnosis of Sickle Cell Disease and Other Haemoglobinopathies: Findings from Tanzania. BMC Genomics 2021, 22, 902. [Google Scholar] [CrossRef]
- Liu, S.; Wang, H.; Leigh, D.; Cram, D.S.; Wang, L.; Yao, Y. Third-Generation Sequencing: Any Future Opportunities for PGT? J. Assist. Reprod. Genet. 2021, 38, 357–364. [Google Scholar] [CrossRef]
- Sirén, J.; Monlong, J.; Chang, X.; Novak, A.M.; Eizenga, J.M.; Markello, C.; Sibbesen, J.A.; Hickey, G.; Chang, P.-C.; Carroll, A.; et al. Pangenomics Enables Genotyping of Known Structural Variants in 5202 Diverse Genomes. Science 2021, 374, abg8871. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Feng, X.; Chu, C. The Design and Construction of Reference Pangenome Graphs with Minigraph. Genome Biol. 2020, 21, 265. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Antonacci-Fulton, L.; Howe, K.; Lawson, H.A.; Lucas, J.K.; Phillippy, A.M.; Popejoy, A.B.; Asri, M.; Carson, C.; Chaisson, M.J.P.; et al. The Human Pangenome Project: A Global Resource to Map Genomic Diversity. Nature 2022, 604, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Coldren, C.; Karunamurthy, A.; Kip, N.S.; Klee, E.W.; Lincoln, S.E.; Leon, A.; Pullambhatla, M.; Temple-Smolkin, R.L.; Voelkerding, K.V.; et al. Standards and Guidelines for Validating Next-Generation Sequencing Bioinformatics Pipelines: A Joint Recommendation of the Association for Molecular Pathology and the College of American Pathologists. J. Mol. Diagnostics 2018, 20, 4–27. [Google Scholar] [CrossRef] [Green Version]
- Santani, A.; Simen, B.B.; Briggs, M.; Lebo, M.; Merker, J.D.; Nikiforova, M.; Vasalos, P.; Voelkerding, K.; Pfeifer, J.; Funke, B. Designing and Implementing NGS Tests for Inherited Disorders: A Practical Framework with Step-by-Step Guidance for Clinical Laboratories. J. Mol. Diagnostics 2019, 21, 369–374. [Google Scholar] [CrossRef] [Green Version]
- Roy, N.B.A.; Da Costa, L.; Russo, R.; Bianchi, P.; del Mar Mañú-Pereira, M.; Fermo, E.; Andolfo, I.; Clark, B.; Proven, M.; Sanchez, M.; et al. The Use of Next-Generation Sequencing in the Diagnosis of Rare Inherited Anaemias: A Joint BSH/EHA Good Practice Paper. HemaSphere 2022, 6, e739. [Google Scholar] [CrossRef]
- International Organization for Standardization [ISO] ISO 20397-1:2022 Biotechnology—Massively Parallel Sequencing. Available online: https://www.iso.org/standard/74054.html (accessed on 14 November 2022).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tool | Algorithm | Highlight |
|---|---|---|
| Control-FREEC | LASSO-based, Gaussian mixture models (GMM) | Output BAF from SAM pileup or ratio and copy number calls of each segment |
| Use GC content and mappability profiles to normalize read count if control sample is unavailable | ||
| DELLY2 | Graph-based paired-end clustering and k-mer filtering for split-read analysis | Call SV from distinct insert sizes PE libraries |
| Output VCF containing SV quality prediction | ||
| Support short and long reads | ||
| CNVkit | Circular Binary Segmentation (CBS), HaarSeg, HMM | Primarily for hybrid capture sequencing |
| Use on- and off-target reads to call CNV | ||
| Support amplicon sequencing-based TS | ||
| Multiple segmentation algorithms to choose from | ||
| ExomeDepth | Beta-binomial model, HMM, maximum likelihood Viterbi algorithm | An R package works on Windows and UNIX systems |
| Source read count data from multiple samples to build optimized reference sets | ||
| CoNIFER | Singular value decomposition and z-scores reads per thousand bases per million read sequenced (SVD-ZRPKM) | Use Matplotlib and Pyplot to generate arbitrary segment of the SVD-ZRPKM data |
| Calculate batch effect biases by concurrently analyzing multiple samples suitable for large sample sets | ||
| FishingCNV | PCA | Support CLI and GUI for Windows and UNIX systems |
| Compare coverage depth in test samples and use PCA to remove batch effect |
| Feature | Conventional | NGS | TGS |
|---|---|---|---|
| DNA usage | High | Low | Low |
| Mutation detection | Method-dependent | Simultaneous | Simultaneous |
| Haplotype-phasing | Not relevant | Yes 1 | Yes |
| TAT | Long | Short | Short |
| Per sample cost | Variable | Uniform | Uniform |
| Technical difficulty | Low | High | High |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hassan, S.; Bahar, R.; Johan, M.F.; Mohamed Hashim, E.K.; Abdullah, W.Z.; Esa, E.; Abdul Hamid, F.S.; Zulkafli, Z. Next-Generation Sequencing (NGS) and Third-Generation Sequencing (TGS) for the Diagnosis of Thalassemia. Diagnostics 2023, 13, 373. https://doi.org/10.3390/diagnostics13030373
Hassan S, Bahar R, Johan MF, Mohamed Hashim EK, Abdullah WZ, Esa E, Abdul Hamid FS, Zulkafli Z. Next-Generation Sequencing (NGS) and Third-Generation Sequencing (TGS) for the Diagnosis of Thalassemia. Diagnostics. 2023; 13(3):373. https://doi.org/10.3390/diagnostics13030373
Chicago/Turabian StyleHassan, Syahzuwan, Rosnah Bahar, Muhammad Farid Johan, Ezzeddin Kamil Mohamed Hashim, Wan Zaidah Abdullah, Ezalia Esa, Faidatul Syazlin Abdul Hamid, and Zefarina Zulkafli. 2023. "Next-Generation Sequencing (NGS) and Third-Generation Sequencing (TGS) for the Diagnosis of Thalassemia" Diagnostics 13, no. 3: 373. https://doi.org/10.3390/diagnostics13030373