Are Liver Pericytes Just Precursors of Myofibroblasts in Hepatic Diseases? Insights from the Crosstalk between Perivascular and Inflammatory Cells in Liver Injury and Repair

 ,
, {kind=link}
{kind=link}
Abstract
:1. Introduction
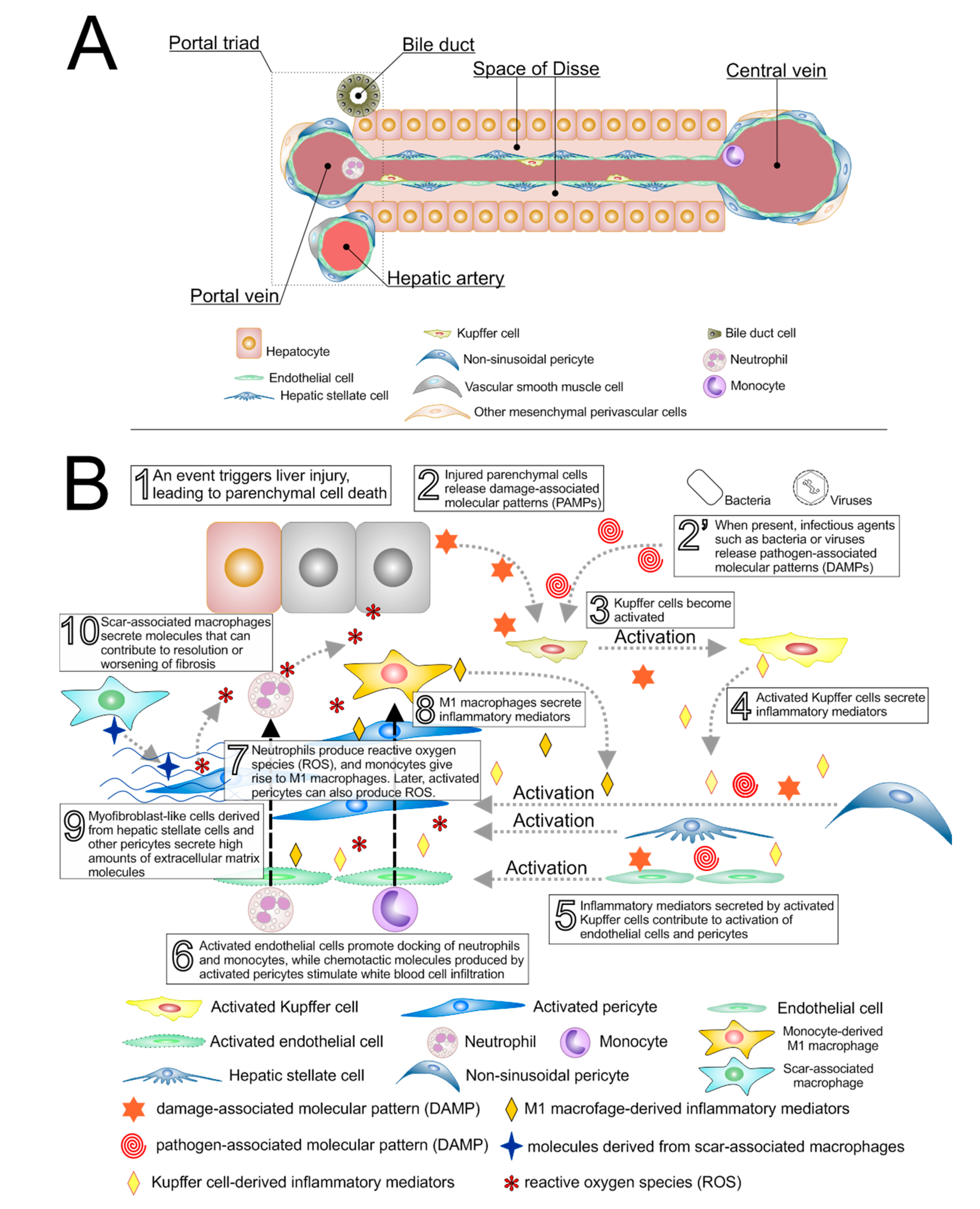
2. Macrophages and Liver Injury
3. Perivascular Cells in Liver Injury
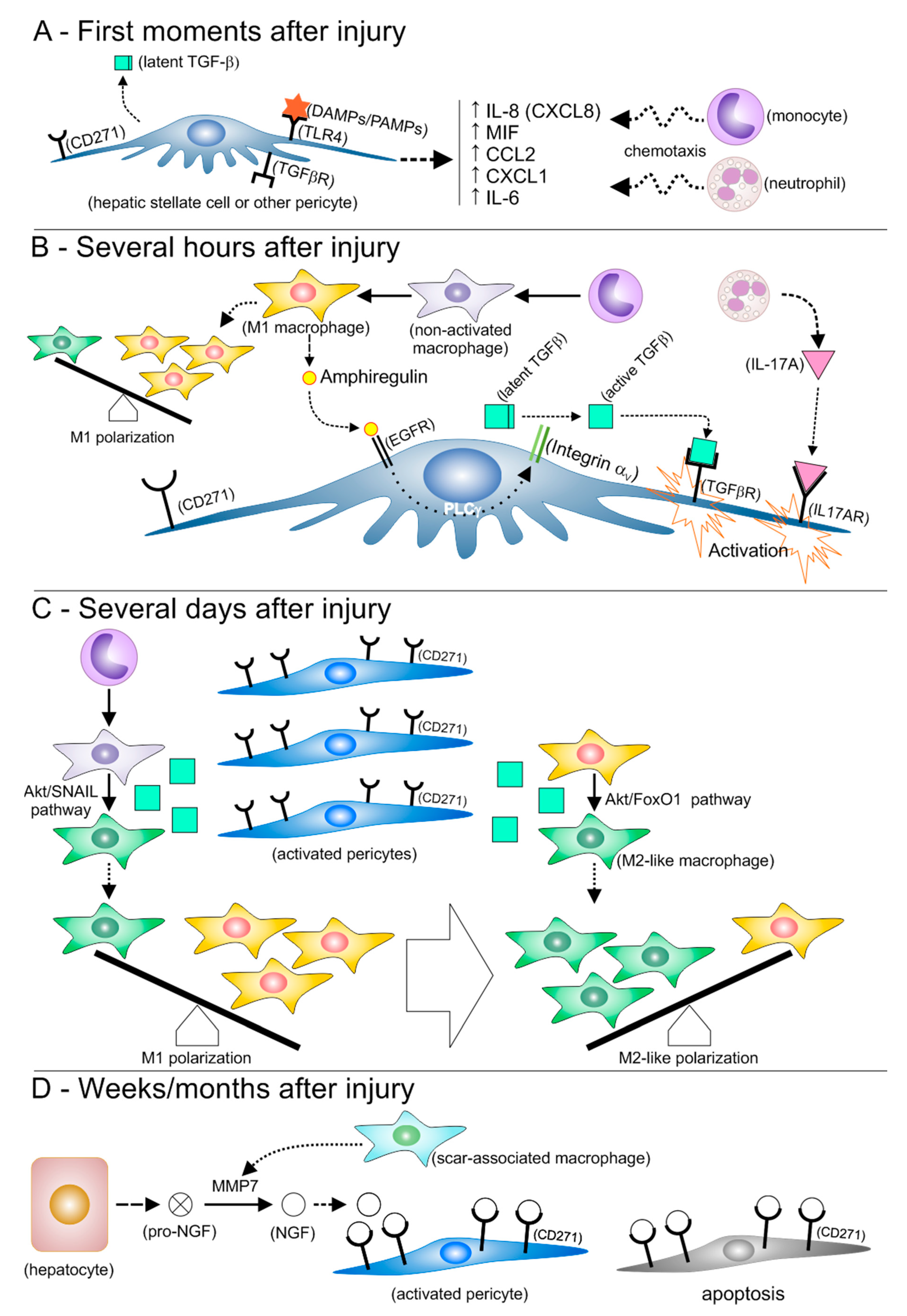
3.1. Pericytes Favor Inflammation at the Early Moments after Injury
3.2. Hypothesis: Activated HSCs Resemble Mesenchymal Stromal Cells, and Contribute to Macrophage Polarization toward an M2-like Phenotype
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mokdad, A.A.; Lopez, A.D.; Shahraz, S.; Lozano, R.; Mokdad, A.H.; Stanaway, J.; Murray, C.J.; Naghavi, M. Liver cirrhosis mortality in 187 countries between 1980 and 2010: A systematic analysis. BMC Med. 2014, 12, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asrani, S.K.; Devarbhavi, H.; Eaton, J.; Kamath, P.S. Burden of liver diseases in the world. J. Hepatol. 2019, 70, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Moon, A.M.; Singal, A.G.; Tapper, E.B. Contemporary Epidemiology of Chronic Liver Disease and Cirrhosis. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2019. [Google Scholar] [CrossRef] [PubMed]
- Stravitz, R.T.; Lee, W.M. Acute liver failure. Lancet 2019, 394, 869–881. [Google Scholar] [CrossRef]
- Tsochatzis, E.A.; Bosch, J.; Burroughs, A.K. Liver cirrhosis. Lancet 2014, 383, 1749–1761. [Google Scholar] [CrossRef]
- McDonald, B.; McAvoy, E.F.; Lam, F.; Gill, V.; de la Motte, C.; Savani, R.C.; Kubes, P. Interaction of CD44 and hyaluronan is the dominant mechanism for neutrophil sequestration in inflamed liver sinusoids. J. Exp. Med. 2008, 205, 915–927. [Google Scholar] [CrossRef] [Green Version]
- Xu, R.; Huang, H.; Zhang, Z.; Wang, F.S. The role of neutrophils in the development of liver diseases. Cell. Mol. Immunol. 2014, 11, 224–231. [Google Scholar] [CrossRef] [Green Version]
- Koyama, Y.; Brenner, D.A. Liver inflammation and fibrosis. J. Clin. Investig. 2017, 127, 55–64. [Google Scholar] [CrossRef]
- Li, H.; You, H.; Fan, X.; Jia, J. Hepatic macrophages in liver fibrosis: Pathogenesis and potential therapeutic targets. BMJ Open Gastroenterol. 2016, 3, e000079. [Google Scholar] [CrossRef]
- Xu, J.; Liu, X.; Koyama, Y.; Wang, P.; Lan, T.; Kim, I.G.; Kim, I.H.; Ma, H.Y.; Kisseleva, T. The types of hepatic myofibroblasts contributing to liver fibrosis of different etiologies. Front. Pharmacol. 2014, 5, 167. [Google Scholar] [CrossRef] [Green Version]
- Ju, C.; Tacke, F. Hepatic macrophages in homeostasis and liver diseases: From pathogenesis to novel therapeutic strategies. Cell. Mol. Immunol. 2016, 13, 316–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchmann, K. Evolution of Innate Immunity: Clues from Invertebrates via Fish to Mammals. Front. Immunol. 2014, 5, 459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antoniades, C.G.; Quaglia, A.; Taams, L.S.; Mitry, R.R.; Hussain, M.; Abeles, R.; Possamai, L.A.; Bruce, M.; McPhail, M.; Starling, C.; et al. Source and characterization of hepatic macrophages in acetaminophen-induced acute liver failure in humans. Hepatology 2012, 56, 735–746. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Dominguez, E.; Samaniego, R.; Flores-Sevilla, J.L.; Campos-Campos, S.F.; Gomez-Campos, G.; Salas, A.; Campos-Pena, V.; Corbi, A.L.; Sanchez-Mateos, P.; Sanchez-Torres, C. CD163L1 and CLEC5A discriminate subsets of human resident and inflammatory macrophages in vivo. J. Leukoc. Biol. 2015, 98, 453–466. [Google Scholar] [CrossRef]
- Yang, C.Y.; Chen, J.B.; Tsai, T.F.; Tsai, Y.C.; Tsai, C.Y.; Liang, P.H.; Hsu, T.L.; Wu, C.Y.; Netea, M.G.; Wong, C.H.; et al. CLEC4F is an inducible C-type lectin in F4/80-positive cells and is involved in alpha-galactosylceramide presentation in liver. PLoS ONE 2013, 8, e65070. [Google Scholar] [CrossRef] [Green Version]
- Endo-Umeda, K.; Nakashima, H.; Komine-Aizawa, S.; Umeda, N.; Seki, S.; Makishima, M. Liver X receptors regulate hepatic F4/80 (+) CD11b(+) Kupffer cells/macrophages and innate immune responses in mice. Sci. Rep. 2018, 8, 9281. [Google Scholar] [CrossRef]
- Weston, C.J.; Zimmermann, H.W.; Adams, D.H. The Role of Myeloid-Derived Cells in the Progression of Liver Disease. Front. Immunol. 2019, 10, 893. [Google Scholar] [CrossRef]
- Elchaninov, A.V.; Fatkhudinov, T.K.; Vishnyakova, P.A.; Lokhonina, A.V.; Sukhikh, G.T. Phenotypical and Functional Polymorphism of Liver Resident Macrophages. Cells 2019, 8, 1032. [Google Scholar] [CrossRef] [Green Version]
- Guillot, A.; Tacke, F. Liver Macrophages: Old Dogmas and New Insights. Hepatol. Commun. 2019, 3, 730–743. [Google Scholar] [CrossRef] [Green Version]
- Ramachandran, P.; Dobie, R.; Wilson-Kanamori, J.R.; Dora, E.F.; Henderson, B.E.P.; Luu, N.T.; Portman, J.R.; Matchett, K.P.; Brice, M.; Marwick, J.A.; et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature 2019, 575, 512–518. [Google Scholar] [CrossRef]
- Gomez Perdiguero, E.; Klapproth, K.; Schulz, C.; Busch, K.; Azzoni, E.; Crozet, L.; Garner, H.; Trouillet, C.; de Bruijn, M.F.; Geissmann, F.; et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 2015, 518, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Jenne, C.N.; Kubes, P. Immune surveillance by the liver. Nat. Immunol. 2013, 14, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.; Pluddemann, A. Tissue macrophages: Heterogeneity and functions. BMC Biol. 2017, 15, 53. [Google Scholar] [CrossRef] [PubMed]
- Crispe, I.N. Immune tolerance in liver disease. Hepatology 2014, 60, 2109–2117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Lu, Y.; Zhou, T.; Gu, G.; Xia, Q. Innate Immune Cells in Immune Tolerance After Liver Transplantation. Front. Immunol. 2018, 9, 2401. [Google Scholar] [CrossRef]
- Horst, A.K.; Neumann, K.; Diehl, L.; Tiegs, G. Modulation of liver tolerance by conventional and nonconventional antigen-presenting cells and regulatory immune cells. Cell. Mol. Immunol. 2016, 13, 277–292. [Google Scholar] [CrossRef]
- Karlmark, K.R.; Weiskirchen, R.; Zimmermann, H.W.; Gassler, N.; Ginhoux, F.; Weber, C.; Merad, M.; Luedde, T.; Trautwein, C.; Tacke, F. Hepatic recruitment of the inflammatory Gr1+ monocyte subset upon liver injury promotes hepatic fibrosis. Hepatology 2009, 50, 261–274. [Google Scholar] [CrossRef]
- Roh, Y.S.; Zhang, B.; Loomba, R.; Seki, E. TLR2 and TLR9 contribute to alcohol-mediated liver injury through induction of CXCL1 and neutrophil infiltration. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G30–G41. [Google Scholar] [CrossRef] [Green Version]
- Carlin, L.M.; Stamatiades, E.G.; Auffray, C.; Hanna, R.N.; Glover, L.; Vizcay-Barrena, G.; Hedrick, C.C.; Cook, H.T.; Diebold, S.; Geissmann, F. Nr4a1-dependent Ly6C(low) monocytes monitor endothelial cells and orchestrate their disposal. Cell 2013, 153, 362–375. [Google Scholar] [CrossRef] [Green Version]
- Varol, C.; Mildner, A.; Jung, S. Macrophages: Development and tissue specialization. Annu. Rev. Immunol. 2015, 33, 643–675. [Google Scholar] [CrossRef]
- Collison, J.L.; Carlin, L.M.; Eichmann, M.; Geissmann, F.; Peakman, M. Heterogeneity in the Locomotory Behavior of Human Monocyte Subsets over Human Vascular Endothelium In Vitro. J. Immunol. 2015, 195, 1162–1170. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palma, A.; Jarrah, A.S.; Tieri, P.; Cesareni, G.; Castiglione, F. Gene Regulatory Network Modeling of Macrophage Differentiation Corroborates the Continuum Hypothesis of Polarization States. Front. Physiol. 2018, 9, 1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liaskou, E.; Zimmermann, H.W.; Li, K.K.; Oo, Y.H.; Suresh, S.; Stamataki, Z.; Qureshi, O.; Lalor, P.F.; Shaw, J.; Syn, W.K.; et al. Monocyte subsets in human liver disease show distinct phenotypic and functional characteristics. Hepatology 2013, 57, 385–398. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.; Liu, J.; Xu, Y.; Cao, H. Role of macrophages in experimental liver injury and repair in mice. Exp. Ther. Med. 2019, 17, 3835–3847. [Google Scholar] [CrossRef] [Green Version]
- Montoya, D.; Mehta, M.; Ferguson, B.G.; Teles, R.M.B.; Krutzik, S.R.; Cruz, D.; Pellegrini, M.; Modlin, R.L. Plasticity of antimicrobial and phagocytic programs in human macrophages. Immunology 2019, 156, 164–173. [Google Scholar] [CrossRef] [Green Version]
- Elpek, G.O. Cellular and molecular mechanisms in the pathogenesis of liver fibrosis: An update. World J. Gastroenterol. 2014, 20, 7260–7276. [Google Scholar] [CrossRef]
- Puche, J.E.; Saiman, Y.; Friedman, S.L. Hepatic stellate cells and liver fibrosis. Compr. Physiol. 2013, 3, 1473–1492. [Google Scholar] [CrossRef]
- Higashi, T.; Friedman, S.L.; Hoshida, Y. Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev. 2017, 121, 27–42. [Google Scholar] [CrossRef]
- Pinzani, M. Hepatic stellate (ITO) cells: Expanding roles for a liver-specific pericyte. J. Hepatol. 1995, 22, 700–706. [Google Scholar] [CrossRef]
- Hellerbrand, C. Hepatic stellate cells--the pericytes in the liver. Pflug. Arch. Eur. J. Physiol. 2013, 465, 775–778. [Google Scholar] [CrossRef]
- Poisson, J.; Lemoinne, S.; Boulanger, C.; Durand, F.; Moreau, R.; Valla, D.; Rautou, P.E. Liver sinusoidal endothelial cells: Physiology and role in liver diseases. J. Hepatol. 2017, 66, 212–227. [Google Scholar] [CrossRef] [Green Version]
- Iredale, J.P. Models of liver fibrosis: Exploring the dynamic nature of inflammation and repair in a solid organ. J. Clin. Investig. 2007, 117, 539–548. [Google Scholar] [CrossRef] [Green Version]
- Shenoy, P.S.; Bose, B. Hepatic perivascular mesenchymal stem cells with myogenic properties. J. Tissue Eng. Regen. Med. 2018, 12, e1297–e1310. [Google Scholar] [CrossRef]
- Strauss, O.; Phillips, A.; Ruggiero, K.; Bartlett, A.; Dunbar, P.R. Immunofluorescence identifies distinct subsets of endothelial cells in the human liver. Sci. Rep. 2017, 7, 44356. [Google Scholar] [CrossRef] [Green Version]
- Di Carlo, S.E.; Peduto, L. The perivascular origin of pathological fibroblasts. J. Clin. Investig. 2018, 128, 54–63. [Google Scholar] [CrossRef]
- Wardle, E.N. Kupffer cells and their function. Liver 1987, 7, 63–75. [Google Scholar] [CrossRef]
- Li, P.; He, K.; Li, J.; Liu, Z.; Gong, J. The role of Kupffer cells in hepatic diseases. Mol. Immunol. 2017, 85, 222–229. [Google Scholar] [CrossRef]
- Tsutsui, H.; Nishiguchi, S. Importance of Kupffer cells in the development of acute liver injuries in mice. Int. J. Mol. Sci. 2014, 15, 7711–7730. [Google Scholar] [CrossRef] [Green Version]
- Mihm, S. Danger-Associated Molecular Patterns (DAMPs): Molecular Triggers for Sterile Inflammation in the Liver. Int. J. Mol. Sci. 2018, 19, 3104. [Google Scholar] [CrossRef] [Green Version]
- Seki, E.; Tsutsui, H.; Nakano, H.; Tsuji, N.; Hoshino, K.; Adachi, O.; Adachi, K.; Futatsugi, S.; Kuida, K.; Takeuchi, O.; et al. Lipopolysaccharide-induced IL-18 secretion from murine Kupffer cells independently of myeloid differentiation factor 88 that is critically involved in induction of production of IL-12 and IL-1beta. J. Immunol. 2001, 166, 2651–2657. [Google Scholar] [CrossRef] [Green Version]
- Bachmann, M.; Pfeilschifter, J.; Muhl, H. A Prominent Role of Interleukin-18 in Acetaminophen-Induced Liver Injury Advocates Its Blockage for Therapy of Hepatic Necroinflammation. Front. Immunol. 2018, 9, 161. [Google Scholar] [CrossRef] [Green Version]
- Okamura, H.; Tsutsi, H.; Komatsu, T.; Yutsudo, M.; Hakura, A.; Tanimoto, T.; Torigoe, K.; Okura, T.; Nukada, Y.; Hattori, K.; et al. Cloning of a new cytokine that induces IFN-gamma production by T cells. Nature 1995, 378, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Paik, Y.H.; Schwabe, R.F.; Bataller, R.; Russo, M.P.; Jobin, C.; Brenner, D.A. Toll-like receptor 4 mediates inflammatory signaling by bacterial lipopolysaccharide in human hepatic stellate cells. Hepatology 2003, 37, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.C.; Liu, Y.N.; Hu, Y.; Yang, Y.; Chen, Z. Macrophage inflammatory protein-2 as mediator of inflammation in acute liver injury. World J. Gastroenterol. 2017, 23, 3043–3052. [Google Scholar] [CrossRef] [PubMed]
- Mollica Poeta, V.; Massara, M.; Capucetti, A.; Bonecchi, R. Chemokines and Chemokine Receptors: New Targets for Cancer Immunotherapy. Front. Immunol. 2019, 10, 379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rai, V.; Agrawal, D.K. The role of damage- and pathogen-associated molecular patterns in inflammation-mediated vulnerability of atherosclerotic plaques. Can. J. Physiol. Pharmacol. 2017, 95, 1245–1253. [Google Scholar] [CrossRef]
- Hordijk, P.L. Endothelial signalling events during leukocyte transmigration. FEBS J. 2006, 273, 4408–4415. [Google Scholar] [CrossRef]
- Arthur, M.J.; Bentley, I.S.; Tanner, A.R.; Saunders, P.K.; Millward-Sadler, G.H.; Wright, R. Oxygen-derived free radicals promote hepatic injury in the rat. Gastroenterology 1985, 89, 1114–1122. [Google Scholar] [CrossRef]
- Tranah, T.H.; Vijay, G.K.M.; Ryan, J.M.; Abeles, R.D.; Middleton, P.K.; Shawcross, D.L. Dysfunctional neutrophil effector organelle mobilization and microbicidal protein release in alcohol-related cirrhosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 313, G203–G211. [Google Scholar] [CrossRef] [Green Version]
- Tan, Z.; Qian, X.; Jiang, R.; Liu, Q.; Wang, Y.; Chen, C.; Wang, X.; Ryffel, B.; Sun, B. IL-17A plays a critical role in the pathogenesis of liver fibrosis through hepatic stellate cell activation. J. Immunol. 2013, 191, 1835–1844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kocabayoglu, P.; Lade, A.; Lee, Y.A.; Dragomir, A.C.; Sun, X.; Fiel, M.I.; Thung, S.; Aloman, C.; Soriano, P.; Hoshida, Y.; et al. beta-PDGF receptor expressed by hepatic stellate cells regulates fibrosis in murine liver injury, but not carcinogenesis. J. Hepatol. 2015, 63, 141–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, K.; Huang, Z.; Lin, T.; Liu, S.; Chang, H.; Yan, Z.; Zhang, H.; Liu, C. New Insight into the Anti-liver Fibrosis Effect of Multitargeted Tyrosine Kinase Inhibitors: From Molecular Target to Clinical Trials. Front. Pharmacol. 2015, 6, 300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dewidar, B.; Meyer, C.; Dooley, S.; Meindl-Beinker, A.N. TGF-beta in Hepatic Stellate Cell Activation and Liver Fibrogenesis-Updated 2019. Cells 2019, 8, 1419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paik, Y.H.; Lee, K.S.; Lee, H.J.; Yang, K.M.; Lee, S.J.; Lee, D.K.; Han, K.H.; Chon, C.Y.; Lee, S.I.; Moon, Y.M.; et al. Hepatic stellate cells primed with cytokines upregulate inflammation in response to peptidoglycan or lipoteichoic acid. Lab. Investig. A J. Tech. Methods Pathol. 2006, 86, 676–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edelman, D.A.; Jiang, Y.; Tyburski, J.G.; Wilson, R.F.; Steffes, C.P. Cytokine production in lipopolysaccharide-exposed rat lung pericytes. J. Trauma 2007, 62, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.F.; Mittelsteadt, K.L.; Brauer, R.; McKinney, B.L.; Hallstrand, T.S.; Parks, W.C.; Chen, P.; Schnapp, L.M.; Liles, W.C.; Duffield, J.S.; et al. Lung pericyte-like cells are functional interstitial immune sentinel cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 312, L556–L567. [Google Scholar] [CrossRef] [Green Version]
- Wilson, C.L.; Stephenson, S.E.; Higuero, J.P.; Feghali-Bostwick, C.; Hung, C.F.; Schnapp, L.M. Characterization of human PDGFR-beta-positive pericytes from IPF and non-IPF lungs. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 315, L991–L1002. [Google Scholar] [CrossRef] [Green Version]
- Stark, K.; Eckart, A.; Haidari, S.; Tirniceriu, A.; Lorenz, M.; von Bruhl, M.L.; Gartner, F.; Khandoga, A.G.; Legate, K.R.; Pless, R.; et al. Capillary and arteriolar pericytes attract innate leukocytes exiting through venules and ‘instruct’ them with pattern-recognition and motility programs. Nat. Immunol. 2013, 14, 41–51. [Google Scholar] [CrossRef]
- Kao, Y.H.; Jawan, B.; Goto, S.; Hung, C.T.; Lin, Y.C.; Nakano, T.; Hsu, L.W.; Lai, C.Y.; Tai, M.H.; Chen, C.L. High-mobility group box 1 protein activates hepatic stellate cells in vitro. Transpl. Proc. 2008, 40, 2704–2705. [Google Scholar] [CrossRef]
- Gaceb, A.; Paul, G. Pericyte Secretome. Adv. Exp. Med. Biol 2018, 1109, 139–163. [Google Scholar] [CrossRef] [PubMed]
- Stewart, R.K.; Dangi, A.; Huang, C.; Murase, N.; Kimura, S.; Stolz, D.B.; Wilson, G.C.; Lentsch, A.B.; Gandhi, C.R. A novel mouse model of depletion of stellate cells clarifies their role in ischemia/reperfusion- and endotoxin-induced acute liver injury. J. Hepatol. 2014, 60, 298–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puche, J.E.; Lee, Y.A.; Jiao, J.; Aloman, C.; Fiel, M.I.; Munoz, U.; Kraus, T.; Lee, T.; Yee, H.F., Jr.; Friedman, S.L. A novel murine model to deplete hepatic stellate cells uncovers their role in amplifying liver damage in mice. Hepatology 2013, 57, 339–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassiman, D.; Denef, C.; Desmet, V.J.; Roskams, T. Human and rat hepatic stellate cells express neurotrophins and neurotrophin receptors. Hepatology 2001, 33, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Siao, C.J.; Lorentz, C.U.; Kermani, P.; Marinic, T.; Carter, J.; McGrath, K.; Padow, V.A.; Mark, W.; Falcone, D.J.; Cohen-Gould, L.; et al. ProNGF, a cytokine induced after myocardial infarction in humans, targets pericytes to promote microvascular damage and activation. J. Exp. Med. 2012, 209, 2291–2305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passino, M.A.; Adams, R.A.; Sikorski, S.L.; Akassoglou, K. Regulation of hepatic stellate cell differentiation by the neurotrophin receptor p75NTR. Science 2007, 315, 1853–1856. [Google Scholar] [CrossRef] [Green Version]
- Kendall, T.J.; Hennedige, S.; Aucott, R.L.; Hartland, S.N.; Vernon, M.A.; Benyon, R.C.; Iredale, J.P. p75 Neurotrophin receptor signaling regulates hepatic myofibroblast proliferation and apoptosis in recovery from rodent liver fibrosis. Hepatology 2009, 49, 901–910. [Google Scholar] [CrossRef]
- Oakley, F.; Trim, N.; Constandinou, C.M.; Ye, W.; Gray, A.M.; Frantz, G.; Hillan, K.; Kendall, T.; Benyon, R.C.; Mann, D.A.; et al. Hepatocytes express nerve growth factor during liver injury: Evidence for paracrine regulation of hepatic stellate cell apoptosis. Am. J. Pathol. 2003, 163, 1849–1858. [Google Scholar] [CrossRef]
- Addo, L.; Tanaka, H.; Yamamoto, M.; Toki, Y.; Ito, S.; Ikuta, K.; Sasaki, K.; Ohtake, T.; Torimoto, Y.; Fujiya, M.; et al. Hepatic nerve growth factor induced by iron overload triggers defenestration in liver sinusoidal endothelial cells. Biochim. Biophys. Acta 2015, 1852, 175–183. [Google Scholar] [CrossRef] [Green Version]
- Tsai, M.S.; Lee, P.H.; Sun, C.K.; Chiu, T.C.; Lin, Y.C.; Chang, I.W.; Chen, P.H.; Kao, Y.H. Nerve growth factor upregulates sirtuin 1 expression in cholestasis: A potential therapeutic target. Exp. Mol. Med. 2018, 50, e426. [Google Scholar] [CrossRef] [Green Version]
- Trim, N.; Morgan, S.; Evans, M.; Issa, R.; Fine, D.; Afford, S.; Wilkins, B.; Iredale, J. Hepatic stellate cells express the low affinity nerve growth factor receptor p75 and undergo apoptosis in response to nerve growth factor stimulation. Am. J. Pathol. 2000, 156, 1235–1243. [Google Scholar] [CrossRef] [Green Version]
- Duffield, J.S.; Forbes, S.J.; Constandinou, C.M.; Clay, S.; Partolina, M.; Vuthoori, S.; Wu, S.; Lang, R.; Iredale, J.P. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J. Clin. Investig. 2005, 115, 56–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- da Silva Meirelles, L.; Bellagamba, B.C.; Camassola, M.; Nardi, N.B. Mesenchymal stem cells and their relationship to pericytes. Front. Biosci. 2016, 21, 130–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kordes, C.; Sawitza, I.; Gotze, S.; Haussinger, D. Hepatic stellate cells support hematopoiesis and are liver-resident mesenchymal stem cells. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2013, 31, 290–304. [Google Scholar] [CrossRef] [PubMed]
- Kordes, C.; Sawitza, I.; Gotze, S.; Herebian, D.; Haussinger, D. Hepatic stellate cells contribute to progenitor cells and liver regeneration. J. Clin. Investig. 2014, 124, 5503–5515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crisan, M.; Yap, S.; Casteilla, L.; Chen, C.W.; Corselli, M.; Park, T.S.; Andriolo, G.; Sun, B.; Zheng, B.; Zhang, L.; et al. A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell 2008, 3, 301–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- da Silva Meirelles, L.; Malta, T.M.; de Deus Wagatsuma, V.M.; Palma, P.V.; Araujo, A.G.; Ribeiro Malmegrim, K.C.; Morato de Oliveira, F.; Panepucci, R.A.; Silva, W.A., Jr.; Kashima Haddad, S.; et al. Cultured Human Adipose Tissue Pericytes and Mesenchymal Stromal Cells Display a Very Similar Gene Expression Profile. Stem Cells Dev. 2015, 24, 2822–2840. [Google Scholar] [CrossRef] [Green Version]
- Meirelles, L.d.S.; Fontes, A.M.; Covas, D.T.; Caplan, A.I. Mechanisms involved in the therapeutic properties of mesenchymal stem cells. Cytokine Growth Factor Rev. 2009, 20, 419–427. [Google Scholar] [CrossRef]
- da Silva Meirelles, L.; Caplan, A.I.; Nardi, N.B. In search of the in vivo identity of mesenchymal stem cells. Stem Cells 2008, 26, 2287–2299. [Google Scholar] [CrossRef] [Green Version]
- Caplan, A.I. New MSC: MSCs as pericytes are Sentinels and gatekeepers. J. Orthop. Res. 2017, 35, 1151–1159. [Google Scholar] [CrossRef] [Green Version]
- Chinnadurai, R.; Sands, J.; Rajan, D.; Liu, X.; Arafat, D.; Das, R.; Anania, F.A.; Gibson, G.; Kisseleva, T.; Galipeau, J. Molecular Genetic and Immune Functional Responses Distinguish Bone Marrow Mesenchymal Stromal Cells from Hepatic Stellate Cells. Stem Cells 2019, 37, 1075–1082. [Google Scholar] [CrossRef] [PubMed]
- Tu, Z.; Li, Y.; Smith, D.S.; Sheibani, N.; Huang, S.; Kern, T.; Lin, F. Retinal pericytes inhibit activated T cell proliferation. Investig. Ophthalmol. Vis. Sci. 2011, 52, 9005–9010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, J.; Hisamatsu, T.; Shimamura, K.; Yoneno, K.; Adachi, M.; Naruse, H.; Igarashi, T.; Higuchi, H.; Matsuoka, K.; Kitazume, M.T.; et al. Activated hepatic stellate cells mediate the differentiation of macrophages. Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 2013, 43, 658–669. [Google Scholar] [CrossRef] [PubMed]
- Chiossone, L.; Conte, R.; Spaggiari, G.M.; Serra, M.; Romei, C.; Bellora, F.; Becchetti, F.; Andaloro, A.; Moretta, L.; Bottino, C. Mesenchymal Stromal Cells Induce Peculiar Alternatively Activated Macrophages Capable of Dampening Both Innate and Adaptive Immune Responses. Stem Cells 2016, 34, 1909–1921. [Google Scholar] [CrossRef] [Green Version]
- Perugorria, M.J.; Latasa, M.U.; Nicou, A.; Cartagena-Lirola, H.; Castillo, J.; Goni, S.; Vespasiani-Gentilucci, U.; Zagami, M.G.; Lotersztajn, S.; Prieto, J.; et al. The epidermal growth factor receptor ligand amphiregulin participates in the development of mouse liver fibrosis. Hepatology 2008, 48, 1251–1261. [Google Scholar] [CrossRef]
- Minutti, C.M.; Modak, R.V.; Macdonald, F.; Li, F.; Smyth, D.J.; Dorward, D.A.; Blair, N.; Husovsky, C.; Muir, A.; Giampazolias, E.; et al. A Macrophage-Pericyte Axis Directs Tissue Restoration via Amphiregulin-Induced Transforming Growth Factor Beta Activation. Immunity 2019, 50, 645–654. [Google Scholar] [CrossRef] [Green Version]
- Cao, Z.; Ye, T.; Sun, Y.; Ji, G.; Shido, K.; Chen, Y.; Luo, L.; Na, F.; Li, X.; Huang, Z.; et al. Targeting the vascular and perivascular niches as a regenerative therapy for lung and liver fibrosis. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.F.; Qiao, M.; Schroder, K.; Zhao, Q.; Asmis, R. Nox4 is a novel inducible source of reactive oxygen species in monocytes and macrophages and mediates oxidized low density lipoprotein-induced macrophage death. Circ. Res. 2010, 106, 1489–1497. [Google Scholar] [CrossRef] [Green Version]
- Eming, S.A.; Wynn, T.A.; Martin, P. Inflammation and metabolism in tissue repair and regeneration. Science 2017, 356, 1026–1030. [Google Scholar] [CrossRef] [Green Version]
- Hesketh, M.; Sahin, K.B.; West, Z.E.; Murray, R.Z. Macrophage Phenotypes Regulate Scar Formation and Chronic Wound Healing. Int. J. Mol. Sci. 2017, 18, 1545. [Google Scholar] [CrossRef] [Green Version]
- Meng, C.; Liu, G.; Mu, H.; Zhou, M.; Zhang, S.; Xu, Y. Amphiregulin may be a new biomarker of classically activated macrophages. Biochem. Biophys. Res. Commun. 2015, 466, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Gong, D.; Shi, W.; Yi, S.J.; Chen, H.; Groffen, J.; Heisterkamp, N. TGFbeta signaling plays a critical role in promoting alternative macrophage activation. BMC Immunol. 2012, 13, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Wang, H.; Wang, X.; Jiang, G.; Liu, H.; Zhang, G.; Wang, H.; Fang, R.; Bu, X.; Cai, S.; et al. TGF-beta induces M2-like macrophage polarization via SNAIL-mediated suppression of a pro-inflammatory phenotype. Oncotarget 2016, 7, 52294–52306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, L.; Liu, X.; Zheng, Q.; Kong, M.; Zhang, X.; Hu, R.; Lou, J.; Ren, F.; Chen, Y.; Zheng, S.; et al. M2-like macrophages in the fibrotic liver protect mice against lethal insults through conferring apoptosis resistance to hepatocytes. Sci. Rep. 2017, 7, 10518. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Eggert, T.; Budhu, A.; Forgues, M.; Takai, A.; Dang, H.; Ye, Q.; Lee, J.S.; Kim, J.H.; Greten, T.F.; et al. Hepatic stellate cell and monocyte interaction contributes to poor prognosis in hepatocellular carcinoma. Hepatology 2015, 62, 481–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Tao, Y.; Wu, Y.; Zhao, X.; Ye, W.; Zhao, D.; Fu, L.; Tian, C.; Yang, J.; He, F.; et al. Neutrophils promote the development of reparative macrophages mediated by ROS to orchestrate liver repair. Nat. Commun. 2019, 10, 1076. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Qiu, H.; Xue, M.; Zhang, S.; Zhang, X.; Xu, J.; Chen, J.; Yang, Y.; Xie, J. MSC-secreted TGF-beta regulates lipopolysaccharide-stimulated macrophage M2-like polarization via the Akt/FoxO1 pathway. Stem Cell Res. Ther. 2019, 10, 345. [Google Scholar] [CrossRef]
- Haussinger, D.; Kordes, C. Space of Disse: A stem cell niche in the liver. Biol. Chem. 2019. [Google Scholar] [CrossRef]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
da Silva Meirelles, L.; Marson, R.F.; Solari, M.I.G.; Nardi, N.B. Are Liver Pericytes Just Precursors of Myofibroblasts in Hepatic Diseases? Insights from the Crosstalk between Perivascular and Inflammatory Cells in Liver Injury and Repair. Cells 2020, 9, 188. https://doi.org/10.3390/cells9010188
da Silva Meirelles L, Marson RF, Solari MIG, Nardi NB. Are Liver Pericytes Just Precursors of Myofibroblasts in Hepatic Diseases? Insights from the Crosstalk between Perivascular and Inflammatory Cells in Liver Injury and Repair. Cells. 2020; 9(1):188. https://doi.org/10.3390/cells9010188
Chicago/Turabian Styleda Silva Meirelles, Lindolfo, Renan Fava Marson, Maria Inês Gonzalez Solari, and Nance Beyer Nardi. 2020. "Are Liver Pericytes Just Precursors of Myofibroblasts in Hepatic Diseases? Insights from the Crosstalk between Perivascular and Inflammatory Cells in Liver Injury and Repair" Cells 9, no. 1: 188. https://doi.org/10.3390/cells9010188