Loss of Rb1 Enhances Glycolytic Metabolism in Kras-Driven Lung Tumors In Vivo

 , , and
, , and 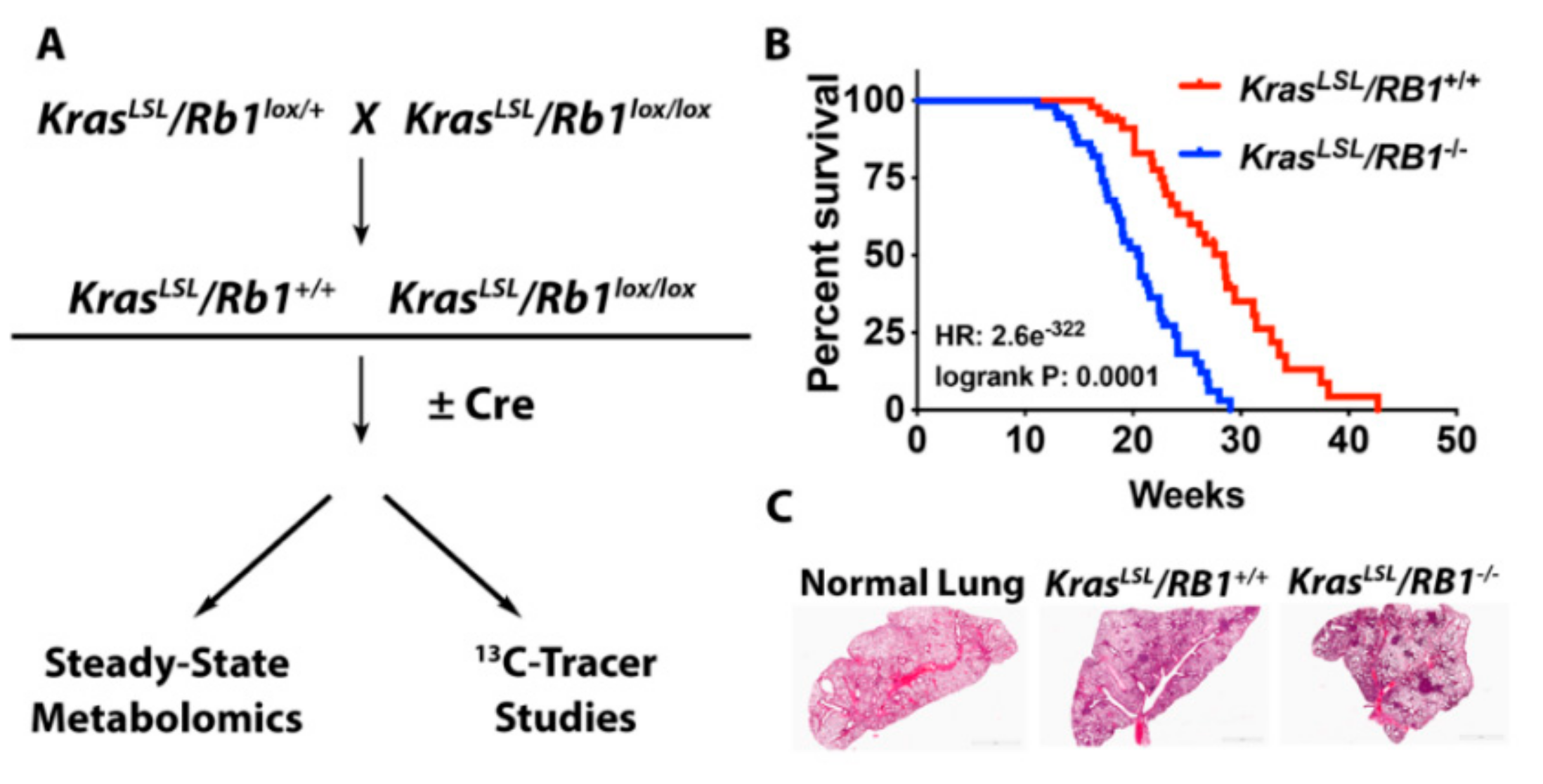{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
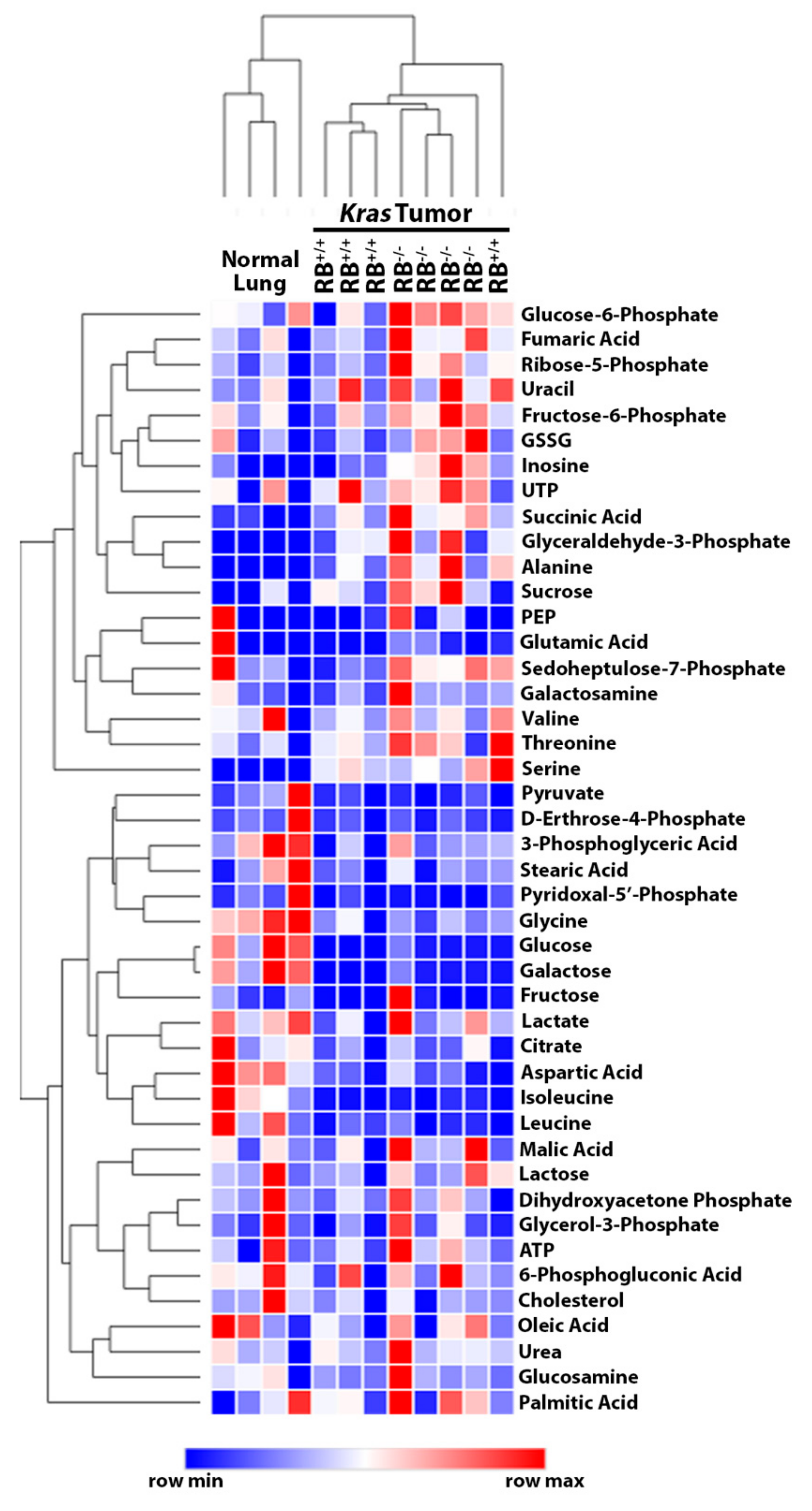
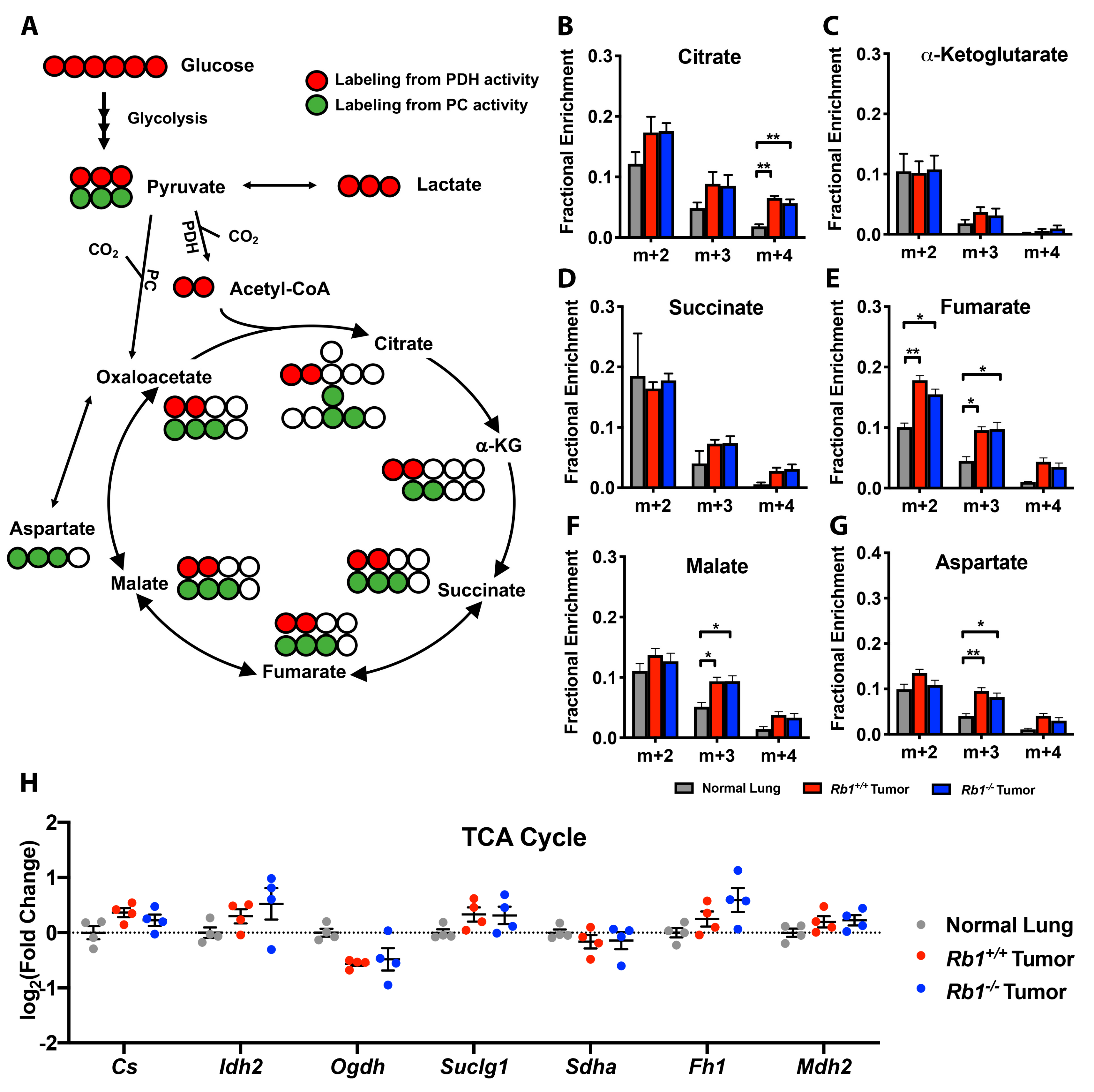
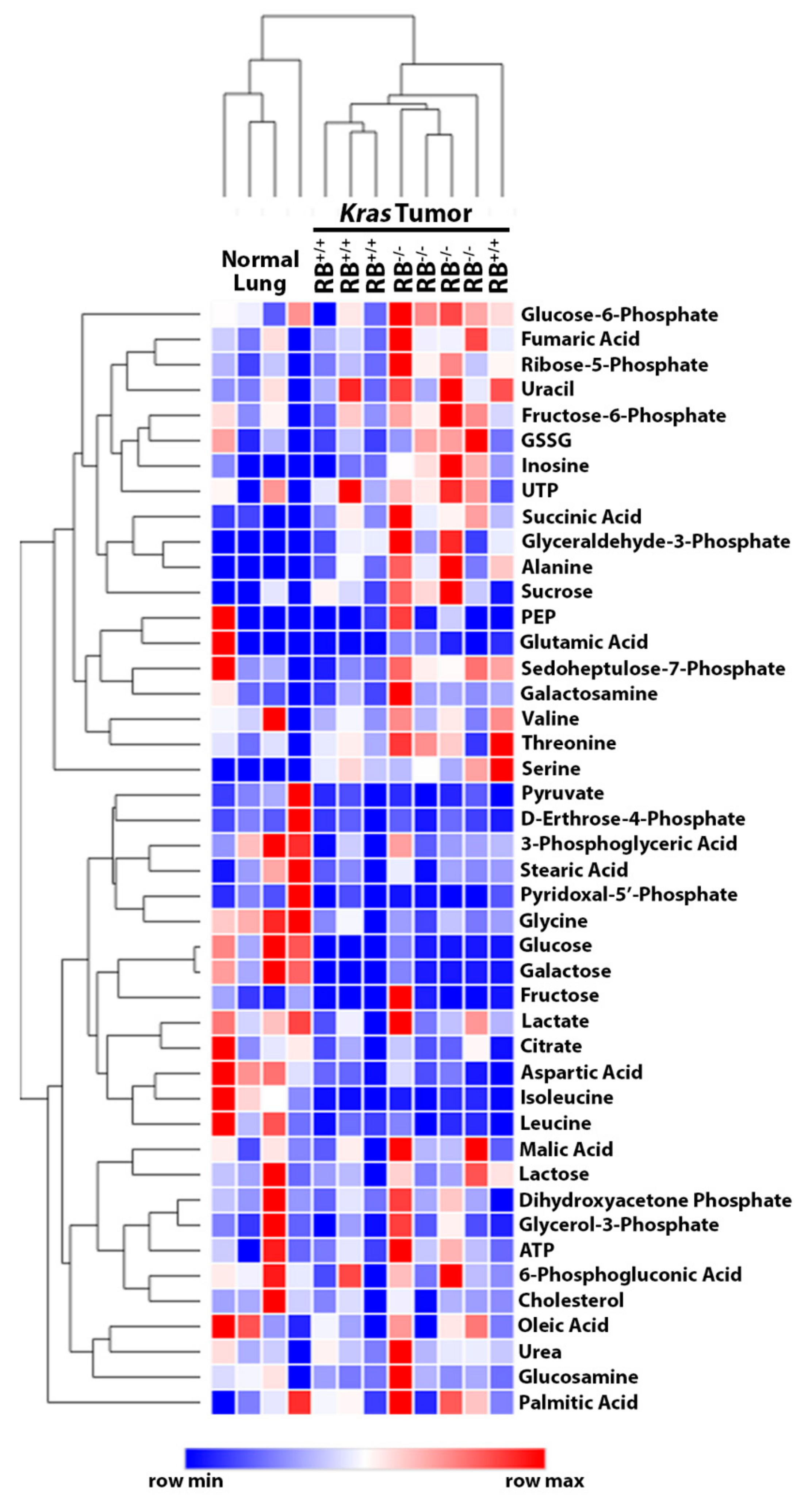
2.1. Steady-State Metabolomics Highlights Metabolic Discrepancies in Rb1-Deficient Lung Tumors In Vivo
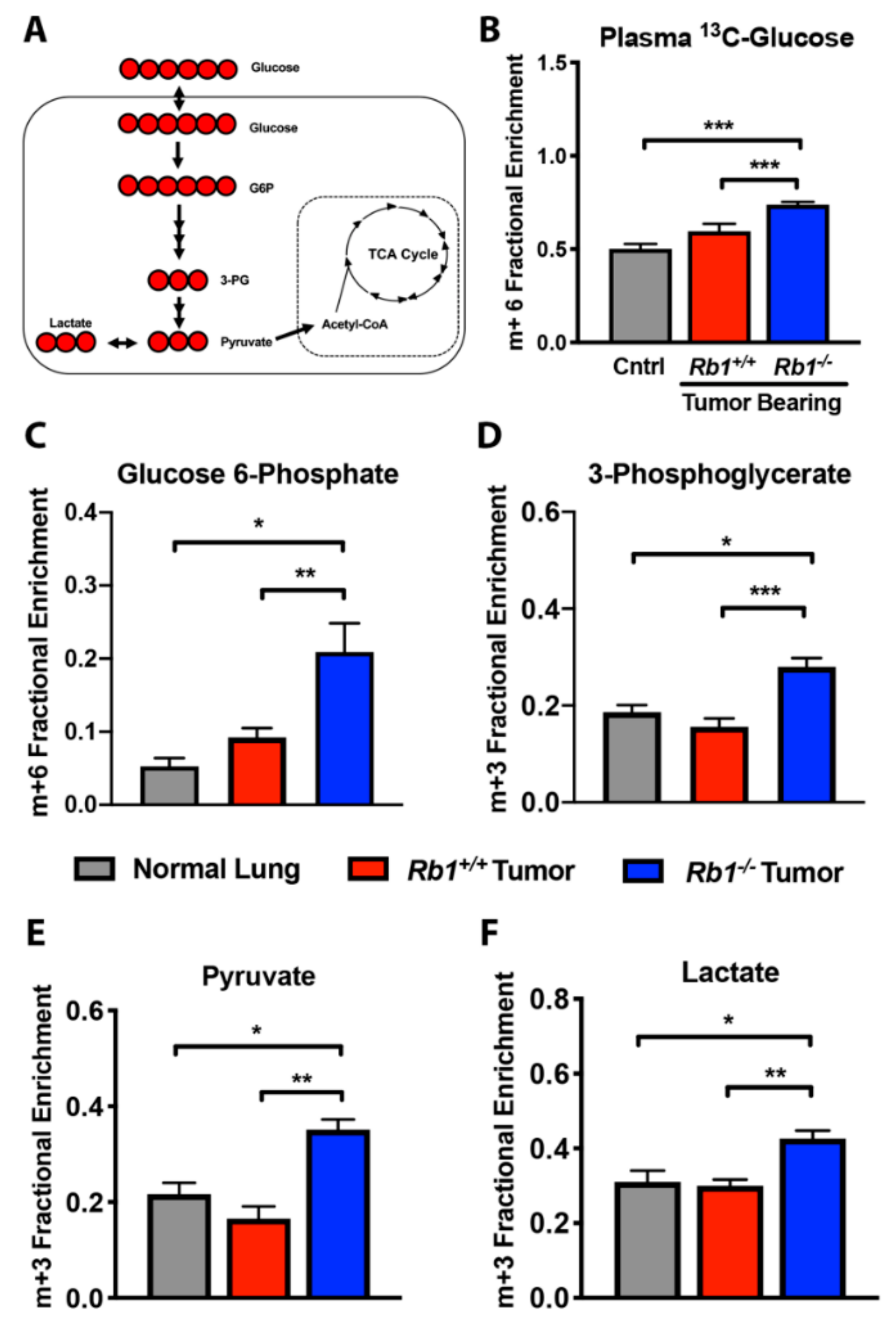
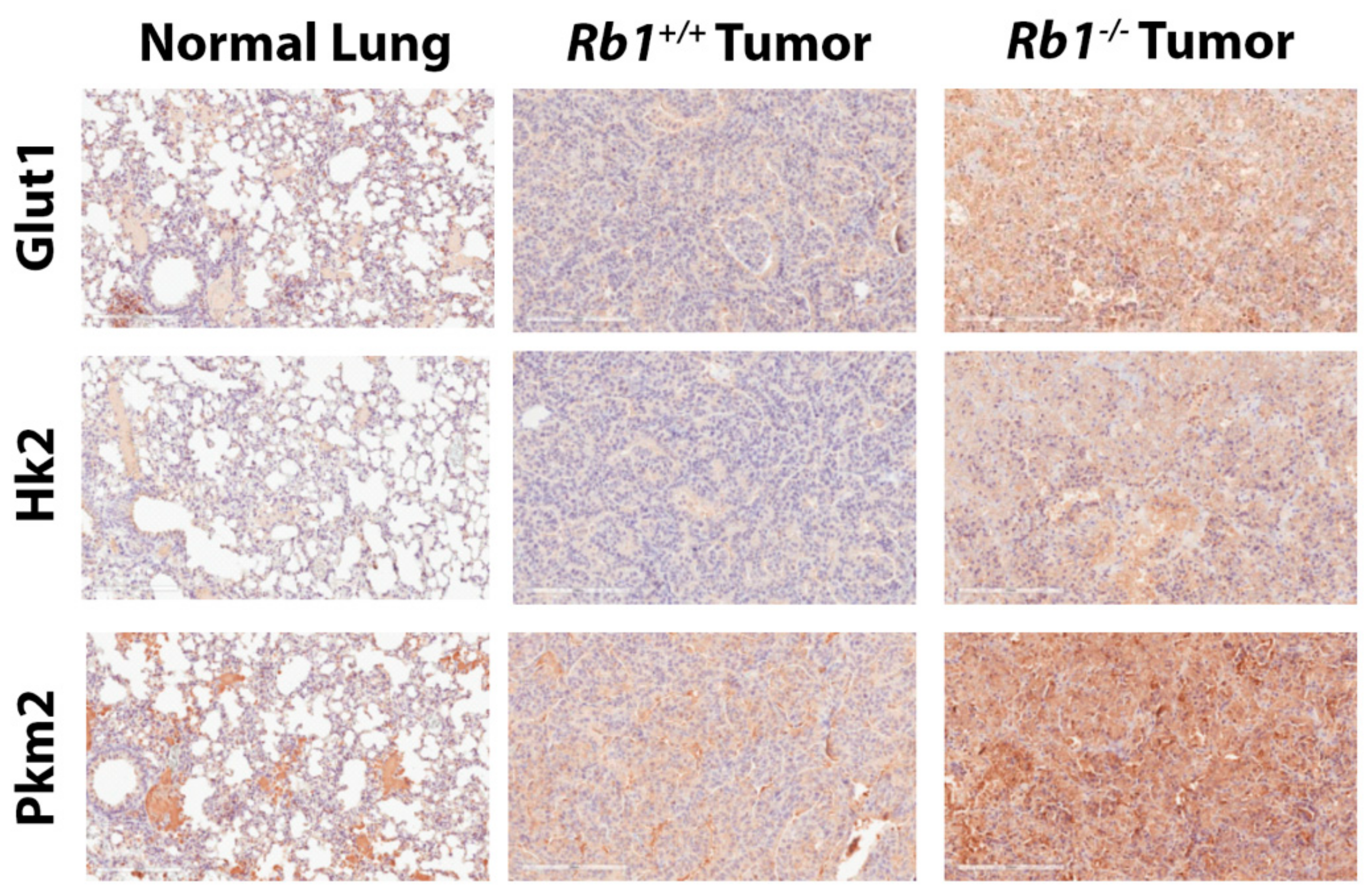
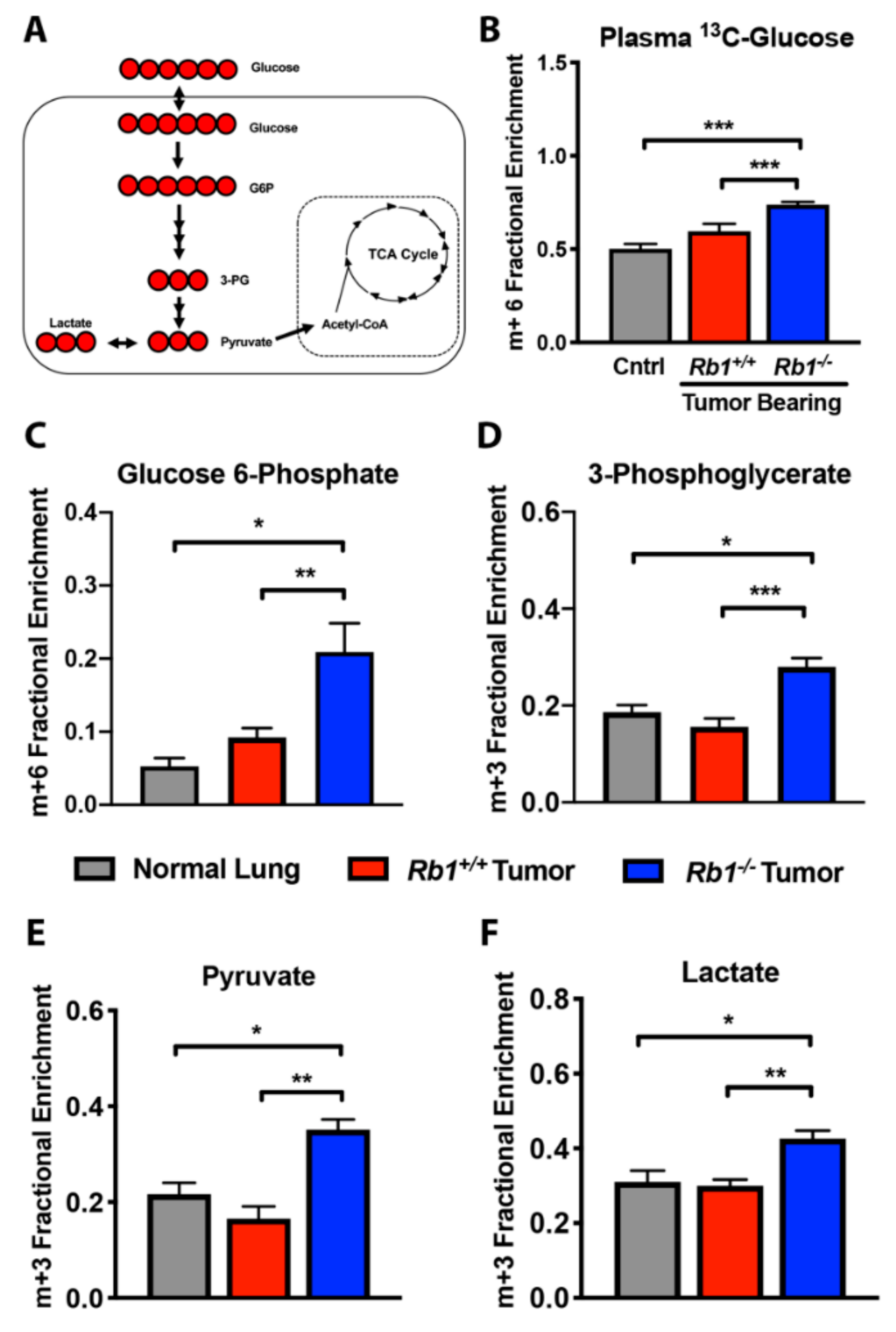
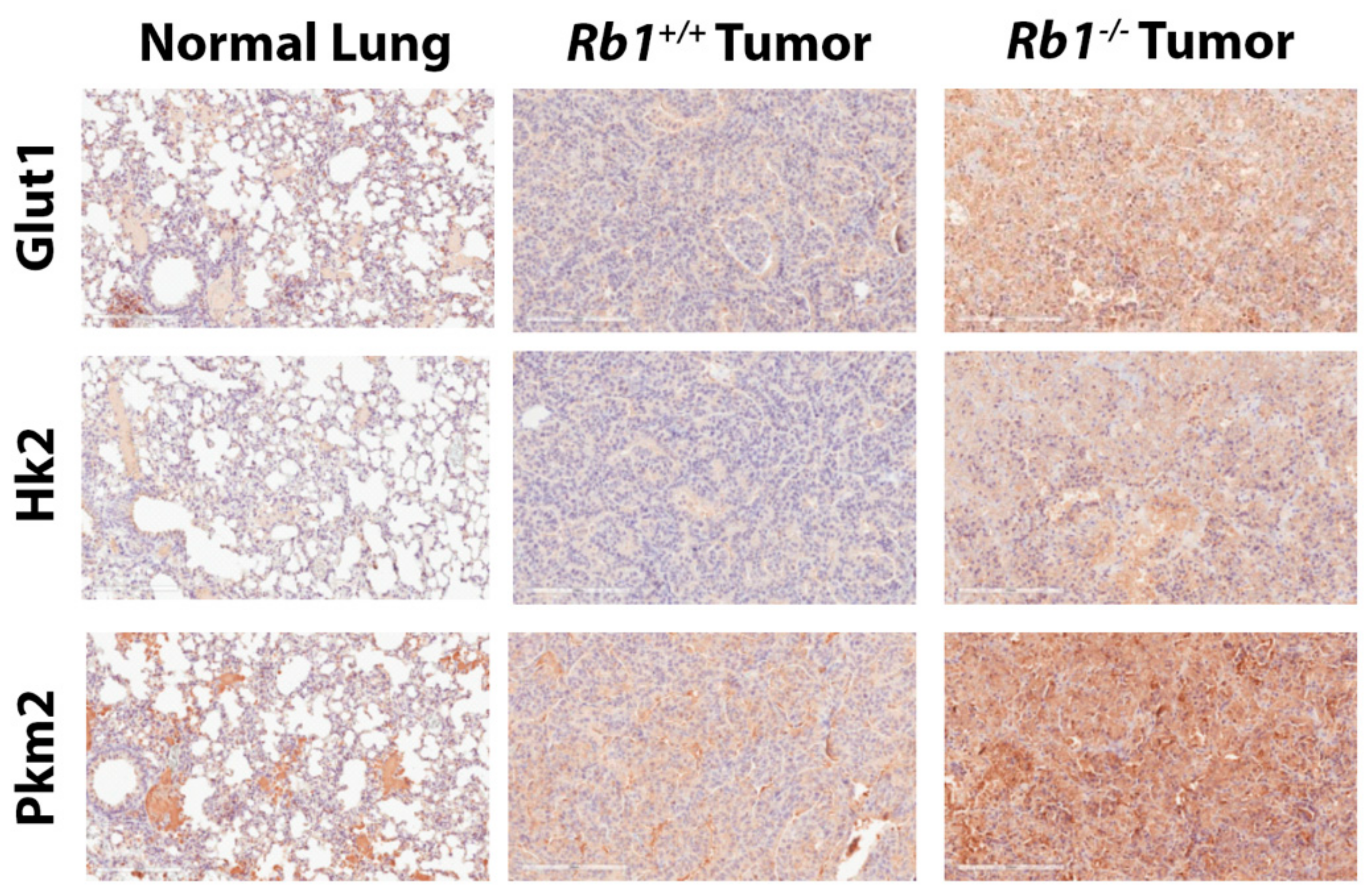
2.2. Loss of Rb1 Enhances Glycolysis in Kras-Driven Lung Tumors
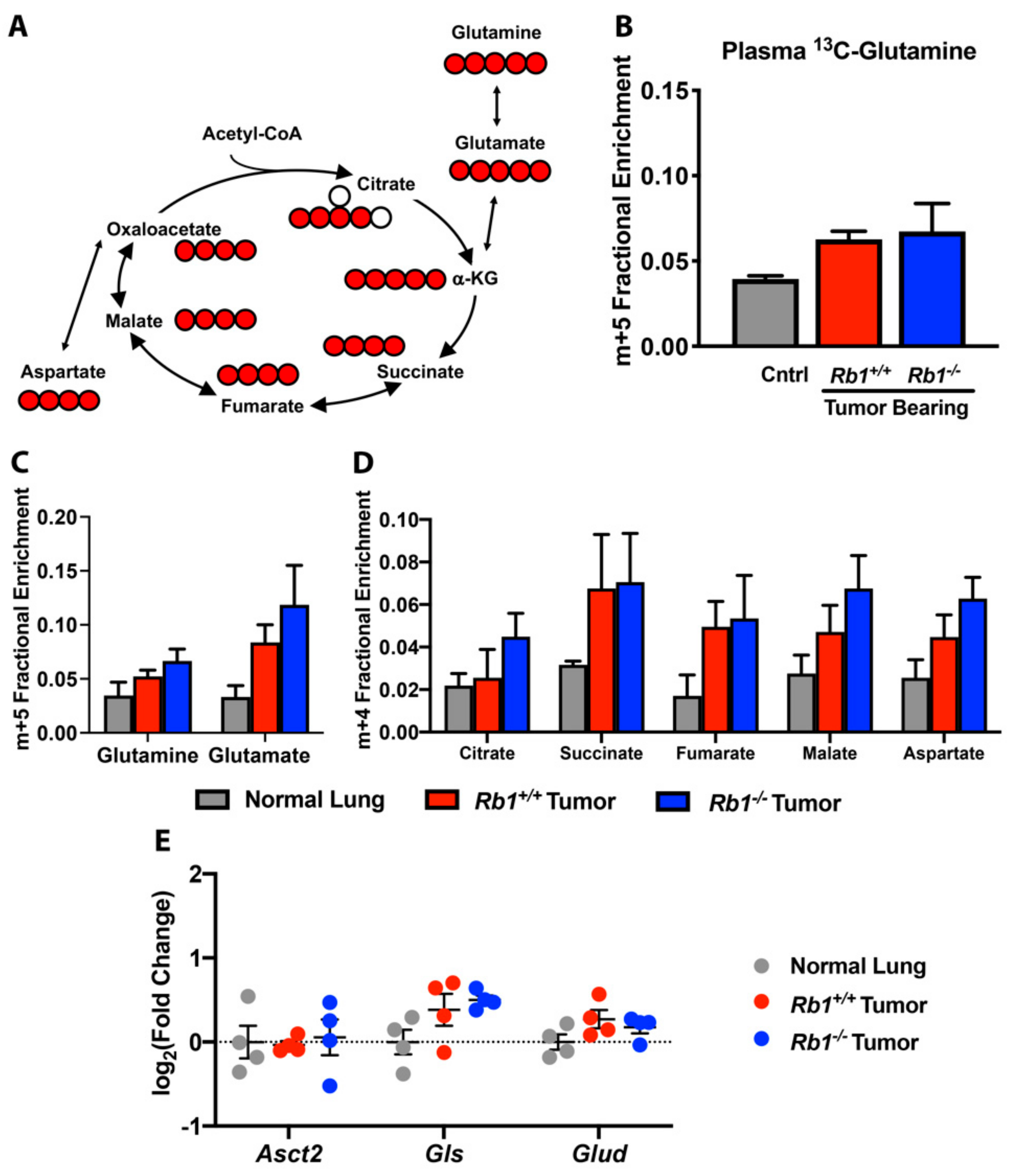
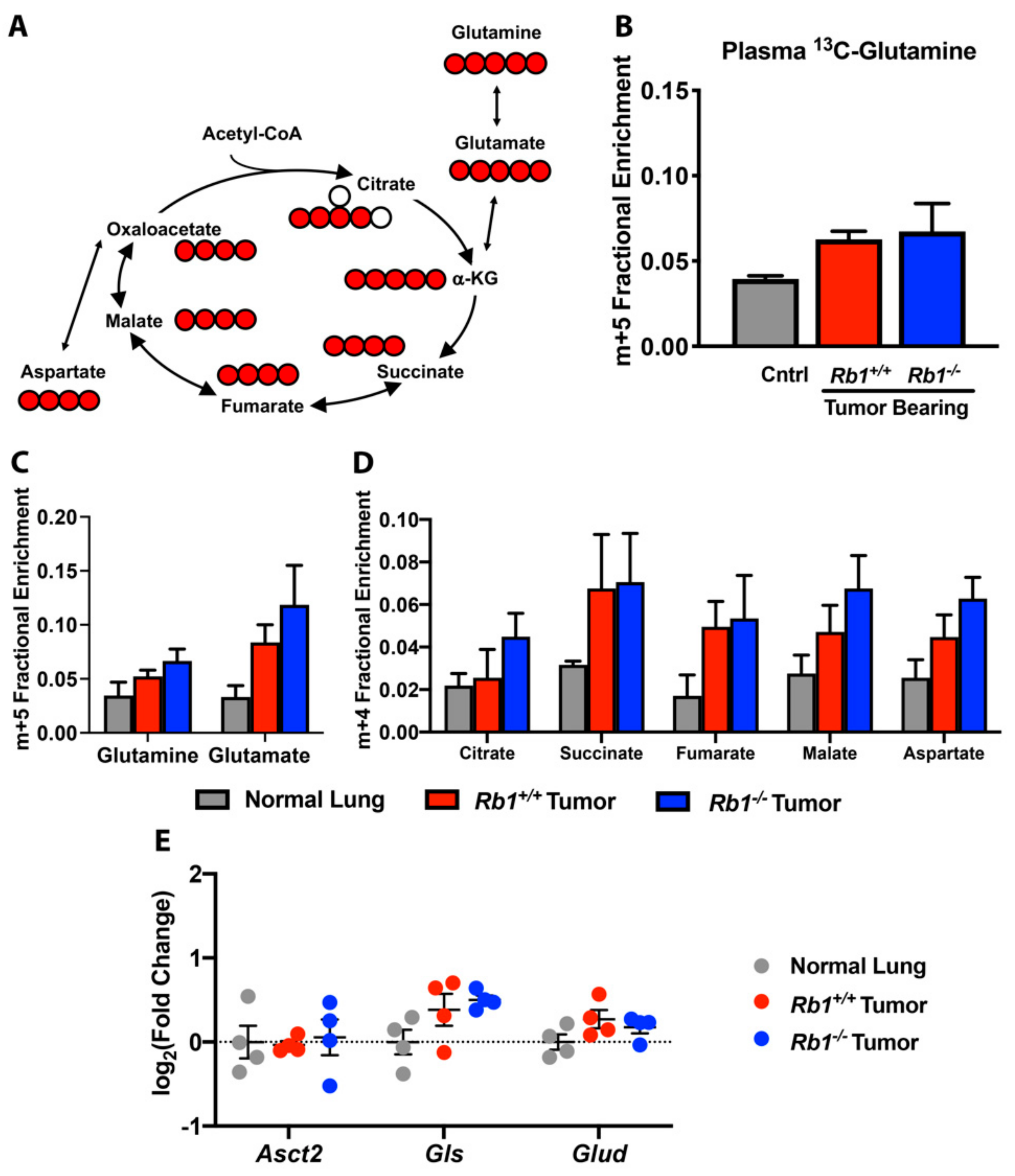
2.3. Rb1 Does Not Regulate Glutamine Utilization In Vivo
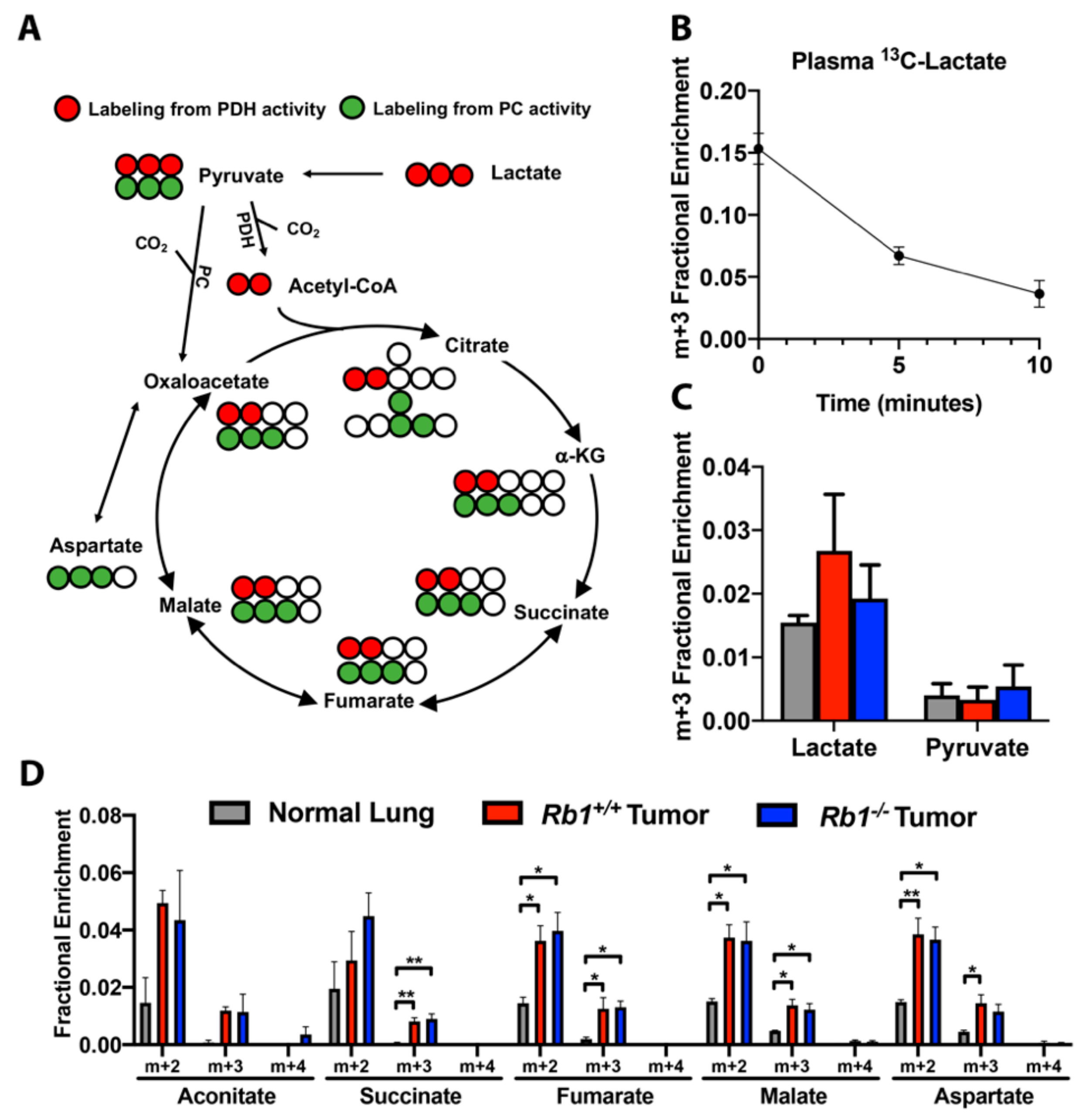
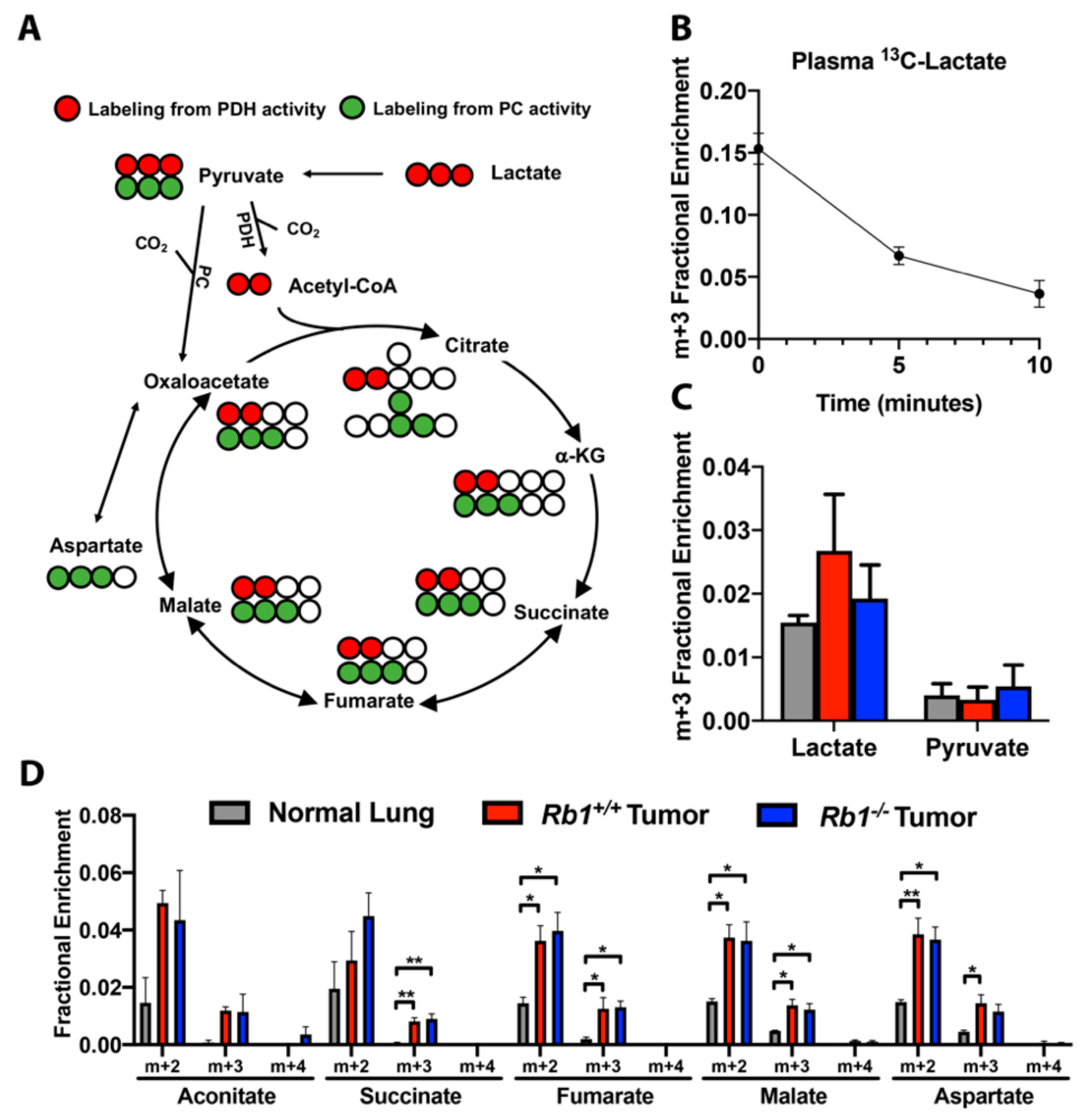
2.4. Loss of Rb1 Does Not Alter Lactate Utilization in Kras-Driven Lung Tumors
3. Discussion
4. Materials and Methods
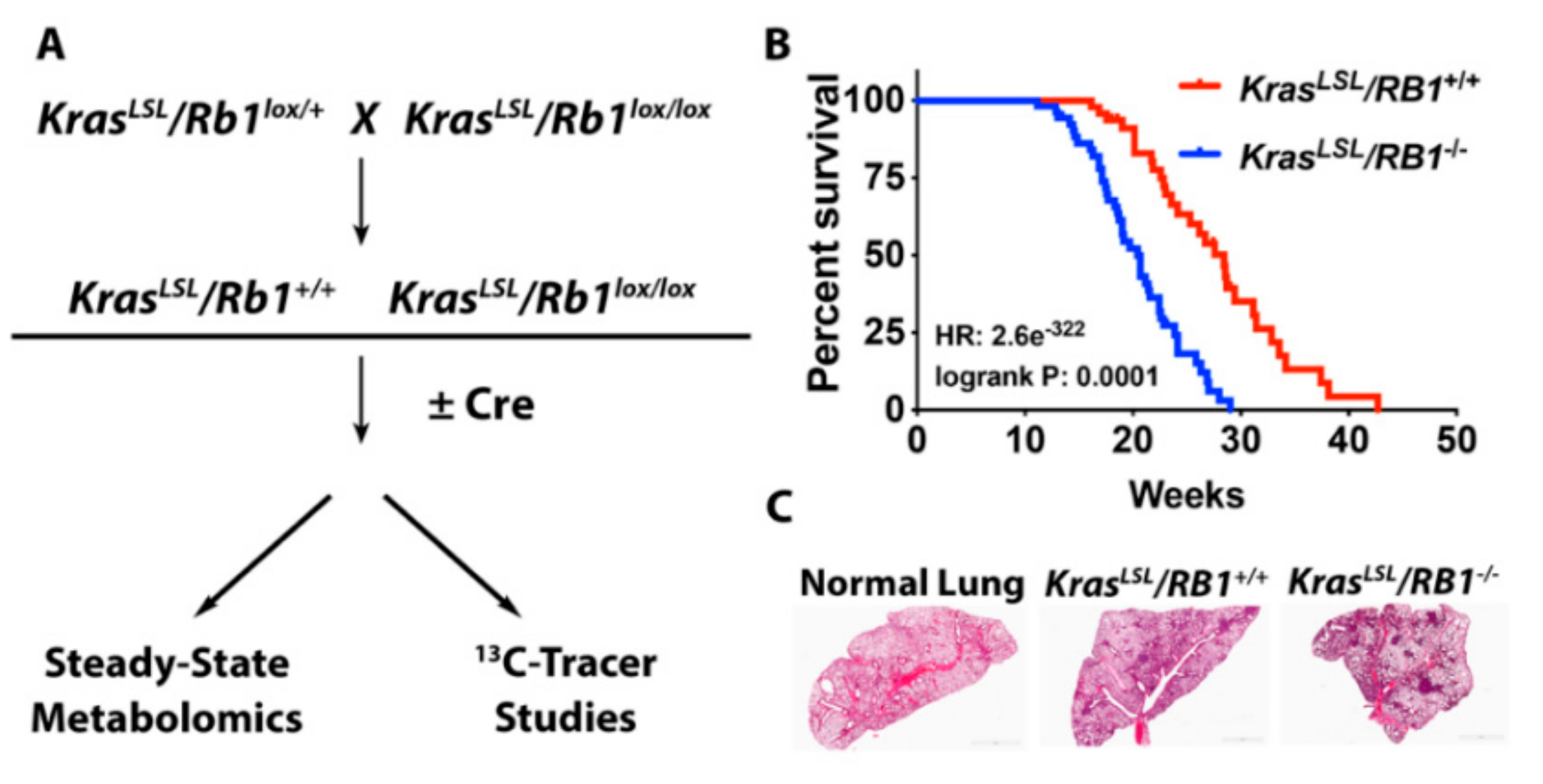
4.1. Mouse Model and Adenoviral Infection
4.2. Kaplan-Meier Analysis
4.3. [U-13C]-Glucose, [U-13C,15N]-Glutamine, and [U-13C]-Lactate Tracer Studies
4.4. Plasma [U-13C]-Glucose, [U-13C,15N]-Glutamine, and [U-13C]-Lactate Analysis
4.5. Metabolite Tissue Sample Preparation for Tracer Studies
4.6. Sample Preparation and Derivatization for Steady-State Metabolomics
4.7. GC-MS Analysis and Data Processing
4.8. 2D-LC-MS/MS Analysis and Data Processing
4.9. Data Analysis for 2D-LC-MS/MS
4.10. 2D-NMR Analysis
4.11. Real Time-PCR
4.12. Immunohistochemistry
4.13. Immunoblotting
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zappa, C.; Mousa, S.A. Non-small cell lung cancer: Current treatment and future advances. Transl. Lung Cancer Res. 2016, 5, 288–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gridelli, C.; Rossi, A.; Carbone, D.P.; Guarize, J.; Karachaliou, N.; Mok, T.; Petrella, F.; Spaggiari, L.; Rosell, R. Non-small-cell lung cancer. Nat. Rev. Dis. Primers 2015, 1, 15009. [Google Scholar] [CrossRef] [PubMed]
- Esposito, V.B.A.; Tonini, G.; Vincenzi, B.; Santini, M.; Ambrogi, V.; Mineo, T.C.; Persichetti, P.; Liuzzi, G.; Montesarchio, V.; Wolner, E.; et al. Analysis of Cell Cycle Regulator Proteins in Non-Small Cell Lung Cancer. J. Clin. Pathol. 2004, 57, 58–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eymin, B.; Gazzeri, S. Role of cell cycle regulators in lung carcinogenesis. Cell Adhes. Migr. 2010, 4, 114–123. [Google Scholar] [CrossRef]
- Weinberg, R.A. The Retinoblastoma Protein and Cell Cycle Control. Cell 1995, 81, 323–330. [Google Scholar] [CrossRef] [Green Version]
- Bhateja, P.; Chiu, M.; Wildey, G.; Lipka, M.B.; Fu, P.; Yang, M.C.L.; Ardeshir-Larijani, F.; Sharma, N.; Dowlati, A. Retinoblastoma mutation predicts poor outcomes in advanced non small cell lung cancer. Cancer Med. 2019, 8, 1459–1466. [Google Scholar] [CrossRef] [Green Version]
- Burkhart, D.L.; Sage, J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat. Rev. Cancer 2008, 8, 671–682. [Google Scholar] [CrossRef]
- Clem, B.F.; Chesney, J. Molecular pathways: Regulation of metabolism by RB. Clin. Cancer Res. 2012, 18, 6096–6100. [Google Scholar] [CrossRef] [Green Version]
- Muranaka, H.; Hayashi, A.; Minami, K.; Kitajima, S.; Kohno, S.; Nishimoto, Y.; Nagatani, N.; Suzuki, M.; Kulathunga, L.A.N.; Sasaki, N.; et al. A distinct function of the retinoblastoma protein in the control of lipid composition identified by lipidomic profiling. Oncogenesis 2017, 6, e350. [Google Scholar] [CrossRef]
- Nicolay, B.N.; Dyson, N.J. The multiple connections between pRB and cell metabolism. Curr. Opin. Cell Biol. 2013, 25, 735–740. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, C.; Sasaki, N.; Kitajima, S. Twists in views on RB functions in cellular signaling, metabolism and stem cells. Cancer Sci. 2012, 103, 1182–1188. [Google Scholar] [CrossRef] [PubMed]
- Blanchet, E.; Annicotte, J.S.; Lagarrigue, S.; Aguilar, V.; Clape, C.; Chavey, C.; Fritz, V.; Casas, F.; Apparailly, F.; Auwerx, J.; et al. E2F transcription factor-1 regulates oxidative metabolism. Nat. Cell Biol. 2011, 13, 1146–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, M.C.; Das, D.; Sambandam, N.; Zhang, M.Q.; Nahle, Z. Regulation of the PDK4 isozyme by the Rb-E2F1 complex. J. Biol. Chem. 2008, 283, 27410–27417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolay, B.N.; Danielian, P.S.; Kottakis, F.; Lapek, J.D., Jr.; Sanidas, I.; Miles, W.O.; Dehnad, M.; Tschop, K.; Gierut, J.J.; Manning, A.L.; et al. Proteomic analysis of pRb loss highlights a signature of decreased mitochondrial oxidative phosphorylation. Genes Dev. 2015, 29, 1875–1889. [Google Scholar] [CrossRef] [Green Version]
- Ho, V.M.; Schaffer, B.E.; Karnezis, A.N.; Park, K.S.; Sage, J. The retinoblastoma gene Rb and its family member p130 suppress lung adenocarcinoma induced by oncogenic K-Ras. Oncogene 2009, 28, 1393–1399. [Google Scholar] [CrossRef] [Green Version]
- Davidson, S.M.; Papagiannakopoulos, T.; Olenchock, B.A.; Heyman, J.E.; Keibler, M.A.; Luengo, A.; Bauer, M.R.; Jha, A.K.; O’Brien, J.P.; Pierce, K.A.; et al. Environment Impacts the Metabolic Dependencies of Ras-Driven Non-Small Cell Lung Cancer. Cell Metab. 2016, 23, 517–528. [Google Scholar] [CrossRef]
- Sellers, K.; Fox, M.P.; Bousamra, M., 2nd; Slone, S.P.; Higashi, R.M.; Miller, D.M.; Wang, Y.; Yan, J.; Yuneva, M.O.; Deshpande, R.; et al. Pyruvate carboxylase is critical for non-small-cell lung cancer proliferation. J. Clin. Investig. 2015, 125, 687–698. [Google Scholar] [CrossRef] [Green Version]
- Takebayashi, S.I.; Tanaka, H.; Hino, S.; Nakatsu, Y.; Igata, T.; Sakamoto, A.; Narita, M.; Nakao, M. Retinoblastoma protein promotes oxidative phosphorylation through upregulation of glycolytic genes in oncogene-induced senescent cells. Aging Cell 2015, 14, 689–697. [Google Scholar] [CrossRef]
- Reynolds, M.R.; Lane, A.N.; Robertson, B.; Kemp, S.; Liu, Y.; Hill, B.G.; Dean, D.C.; Clem, B.F. Control of glutamine metabolism by the tumor suppressor Rb. Oncogene 2014, 33, 556–566. [Google Scholar] [CrossRef] [Green Version]
- Faubert, B.; Li, K.Y.; Cai, L.; Hensley, C.T.; Kim, J.; Zacharias, L.G.; Yang, C.; Do, Q.N.; Doucette, S.; Burguete, D.; et al. Lactate Metabolism in Human Lung Tumors. Cell 2017, 171, 358–371. [Google Scholar] [CrossRef] [Green Version]
- Hui, S.; Ghergurovich, J.M.; Morscher, R.J.; Jang, C.; Teng, X.; Lu, W.; Esparza, L.A.; Reya, T.; Le, Z.; Yanxiang Guo, J.; et al. Glucose feeds the TCA cycle via circulating lactate. Nature 2017, 551, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dali-Youcef, N.; Mataki, C.; Coste, A.; Messaddeq, N.; Giroud, S.; Blanc, S.; Koehl, C.; Champy, M.F.; Chambon, P.; Fajas, L.; et al. Adipose tissue-specific inactivation of the retinoblastoma protein protects against diabesity because of increased energy expenditure. Proc. Natl. Acad. Sci. USA 2007, 104, 10703–10708. [Google Scholar] [CrossRef] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Thompson, C.B. Metabolic regulation of cell growth and proliferation. Nat. Rev. Mol. Cell Biol. 2019, 20, 436–450. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Wahab, A.F.; Mahmoud, W.; Al-Harizy, R.M. Targeting glucose metabolism to suppress cancer progression: Prospective of anti-glycolytic cancer therapy. Pharmacol. Res. 2019, 150, 104511. [Google Scholar] [CrossRef] [PubMed]
- Garcia Rubino, M.E.; Carrillo, E.; Ruiz Alcala, G.; Dominguez-Martin, A.; Marchal, J.A.; Boulaiz, H. Phenformin as an Anticancer Agent: Challenges and Prospects. Int. J. Mol. Sci. 2019, 20, 3316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Xue, Y.; Xi, Y.R.; Xie, K. Progress in the application and mechanism of metformin in treating non-small cell lung cancer. Oncol. Lett. 2017, 13, 2873–2880. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Mao, W.; Zhai, Y.; Tong, C.; Liu, M.; Ma, L.; Yu, X.; Li, S. Anti-tumor activity of metformin: From metabolic and epigenetic perspectives. Oncotarget 2017, 8, 5619–5628. [Google Scholar] [CrossRef] [Green Version]
- Arrieta, O.; Barron, F.; Padilla, M.S.; Aviles-Salas, A.; Ramirez-Tirado, L.A.; Arguelles Jimenez, M.J.; Vergara, E.; Zatarain-Barron, Z.L.; Hernandez-Pedro, N.; Cardona, A.F.; et al. Effect of Metformin Plus Tyrosine Kinase Inhibitors Compared With Tyrosine Kinase Inhibitors Alone in Patients With Epidermal Growth Factor Receptor-Mutated Lung Adenocarcinoma: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019, 5, e192553. [Google Scholar] [CrossRef]
- Wang, Z.D.; Wei, S.Q.; Wang, Q.Y. Targeting oncogenic KRAS in non-small cell lung cancer cells by phenformin inhibits growth and angiogenesis. Am. J. Cancer Res. 2015, 5, 3339–3349. [Google Scholar]
- Hutcheson, J.; Witkiewicz, A.K.; Knudsen, E.S. The RB tumor suppressor at the intersection of proliferation and immunity: Relevance to disease immune evasion and immunotherapy. Cell Cycle 2015, 14, 3812–3819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitajima, S.; Takahashi, C. Intersection of retinoblastoma tumor suppressor function, stem cells, metabolism, and inflammation. Cancer Sci. 2017, 108, 1726–1731. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Kitajima, S.; Kohno, S.; Yoshida, A.; Tange, S.; Sasaki, S.; Okada, N.; Nishimoto, Y.; Muranaka, H.; Nagatani, N.; et al. Retinoblastoma Inactivation Induces a Protumoral Microenvironment via Enhanced CCL2 Secretion. Cancer Res. 2019, 79, 3903–3915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- San-Millán, I.; Brooks, G.A. Reexamining cancer metabolism: Lactate production for carcinogenesis could be the purpose and explanation of the Warburg Effect. Carcinogenesis 2017, 38, 119–133. [Google Scholar] [CrossRef]
- Morrot, A.; Fonseca, L.M.D.; Salustiano, E.J.; Gentile, L.B.; Conde, L.; Filardy, A.A.; Franklim, T.N.; da Costa, K.M.; Freire-de-Lima, C.G.; Freire-de-Lima, L. Metabolic Symbiosis and Immunomodulation: How Tumor Cell-Derived Lactate May Disturb Innate and Adaptive Immune Responses. Front. Oncol. 2018, 8, 81. [Google Scholar] [CrossRef] [Green Version]
- Romero-Garcia, S.; Moreno-Altamirano, M.M.B.; Prado-Garcia, H.; Sánchez-García, F.J. Lactate Contribution to the Tumor Microenvironment: Mechanisms, Effects on Immune Cells and Therapeutic Relevance. Front. Immunol. 2016, 7, 52. [Google Scholar] [CrossRef] [Green Version]
- Fan, T.W.; Lane, A.N.; Higashi, R.M.; Yan, J. Stable isotope resolved metabolomics of lung cancer in a SCID mouse model. Metab. Off. J. Metab. Soc. 2011, 7, 257–269. [Google Scholar] [CrossRef] [Green Version]
- Lane, A.N.; Yan, J.; Fan, T.W. 13C Tacer studies of Metabolism in Mouse Tumor Xenografts. Bio Protoc. 2015, 5. [Google Scholar] [CrossRef] [Green Version]
- Jung, J.Y.; Oh, M.K. Isotope labeling pattern study of central carbon metabolites using GC/MS. J. Chromatogr. B 2015, 974, 101–108. [Google Scholar] [CrossRef]
- MacRae, J.I.; Sheiner, L.; Nahid, A.; Tonkin, C.; Striepen, B.; McConville, M.J. Mitochondrial metabolism of glucose and glutamine is required for intracellular growth of Toxoplasma gondii. Cell Host Microbe 2012, 12, 682–696. [Google Scholar] [CrossRef] [Green Version]
- Fiehn, O. Metabolomics by gas chromatography-mass spectrometry: Combined targeted and untargeted profiling. Curr. Protoc. Mol. Biol. 2016, 114, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Fiehn, O.; Kopka, J.; Dormann, P.; Altmann, T.; Trethewey, R.N.; Willmitzer, L. Metabolite profiling for plant functional genomics. Nat. Biotechnol. 2000, 18, 1157–1161. [Google Scholar] [CrossRef] [PubMed]
- Kind, T.; Wohlgemuth, G.; Lee, D.Y.; Lu, Y.; Palazoglu, M.; Shahbaz, S.; Fiehn, O. FiehnLib: Mass spectral and retention index libraries for metabolomics based on quadrupole and time-of-flight gas chromatography/mass spectrometry. Anal. Chem. 2009, 81, 10038–10048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dagley, M.J.; McConville, M.J. DExSI: A new tool for the rapid quantitation of 13C-labelled metabolites detected by GC-MS. Bioinformatics 2018, 34, 1957–1958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, L.E.; Brizzee, C.O.; Macedo, J.K.; Gentry, M.S.; Sun, R.C. Rapid and sensitive quantitation of glucose and glucose phosphates derived from storage carbohydrates using gas chromatography mass spectrometry. bioRxiv 2019. [Google Scholar] [CrossRef]
- Klavins, K.; Drexler, H.; Hann, S.; Koellensperger, G. Quantitative metabolite profiling utilizing parallel column analysis for simultaneous reversed-phase and hydrophilic interaction liquid chromatography separations combined with tandem mass spectrometry. Anal. Chem. 2014, 86, 4145–4150. [Google Scholar] [CrossRef]
- Adusumilli, R.; Mallick, P. Data Conversion with ProteoWizard msConvert. Methods Mol. Biol. 2017, 1550, 339–368. [Google Scholar]
- Agrawal, S.; Kumar, S.; Sehgal, R.; George, S.; Gupta, R.; Poddar, S.; Jha, A.; Pathak, S. El-MAVEN: A Fast, Robust, and User-Friendly Mass Spectrometry Data Processing Engine for Metabolomics. In High-Throughput Metabolomics: Methods and Protocols; D’Alessandro, A., Ed.; Springer: New York, NY, USA, 2019. [Google Scholar]
- Salabei, J.K.; Lorkiewicz, P.K.; Mehra, P.; Gibb, A.A.; Haberzettl, P.; Hong, K.U.; Wei, X.; Zhang, X.; Li, Q.; Wysoczynski, M.; et al. Type 2 Diabetes Dysregulates Glucose Metabolism in Cardiac Progenitor Cells. J. Biol. Chem. 2016, 291, 13634–13648. [Google Scholar] [CrossRef] [Green Version]
- Wei, X.; Lorkiewicz, P.K.; Shi, B.; Salabei, J.K.; Hill, B.G.; Kim, S.; McClain, C.J.; Zhang, X. Analysis of stable isotope assisted metabolomics data acquired by high resolution mass spectrometry. Anal. Methods 2017, 9, 2275–2283. [Google Scholar] [CrossRef] [Green Version]
- Lorkiewicz, P.K.; Gibb, A.A.; Rood, B.R.; He, L.; Zheng, Y.; Clem, B.F.; Zhang, X.; Hill, B.G. Integration of flux measurements and pharmacological controls to optimize stable isotope-resolved metabolomics workflows and interpretation. Sci. Rep. 2019, 9, 1–17. [Google Scholar] [CrossRef]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Conroy, L.R.; Dougherty, S.; Kruer, T.; Metcalf, S.; Lorkiewicz, P.; He, L.; Yin, X.; Zhang, X.; Arumugam, S.; Young, L.E.A.; et al. Loss of Rb1 Enhances Glycolytic Metabolism in Kras-Driven Lung Tumors In Vivo. Cancers 2020, 12, 237. https://doi.org/10.3390/cancers12010237
Conroy LR, Dougherty S, Kruer T, Metcalf S, Lorkiewicz P, He L, Yin X, Zhang X, Arumugam S, Young LEA, et al. Loss of Rb1 Enhances Glycolytic Metabolism in Kras-Driven Lung Tumors In Vivo. Cancers. 2020; 12(1):237. https://doi.org/10.3390/cancers12010237
Chicago/Turabian StyleConroy, Lindsey R., Susan Dougherty, Traci Kruer, Stephanie Metcalf, Pawel Lorkiewicz, Liqing He, Xinmin Yin, Xiang Zhang, Sengodagounder Arumugam, Lyndsay E.A. Young, and et al. 2020. "Loss of Rb1 Enhances Glycolytic Metabolism in Kras-Driven Lung Tumors In Vivo" Cancers 12, no. 1: 237. https://doi.org/10.3390/cancers12010237