1. Introduction
Since the advent of targeted therapies and precision medicine, companion diagnostics have become an essential tool for patient stratification and selection [
1]. Most of all, cancer treatments driven by biomarkers of clinical relevance have made a big breakthrough for various cancers by providing personalized treatment options and better outcome for patients. For example, molecular diagnoses for mutations in epidermal growth factor receptor gene (
EGFR) and for the anaplastic lymphoma kinase gene (
ALK) rearrangements have opened new therapeutic opportunities for EGFR-positive solid tumors and ALK-positive non-small cell lung cancer, respectively; however, despite the recent attempts to discover novel targets from the uncharacterized genes of human genome, there are still over 43% cancer patients for whom no biomarkers or targeted therapies are available [
2].
ARS-interacting multi-functional protein 2 (AIMP2) is a scaffolding protein that forms a part of the multi-tRNA synthetase complex (MSC) involved in protein translation. When released from MSC, AIMP2 also functions as an anti-proliferative factor via TGF-β, TNF-α, UV, and Wnt signaling [
3,
4,
5,
6,
7,
8]. In contrast to AIMP2, AIMP2-DX2 (hereafter referred to as DX2), an alternative splicing variant of AIMP2 lacking exon 2, shows oncogenic activity, competitively interfering with the tumor suppressive function of AIMP2 upon various signaling [
9,
10]. DX2 is inducible by the carcinogen benzopyrene which causes mutations in and the skipping of exon 2 [
10], and inhibits cancer cell apoptosis by directly interacting with p14/ARF [
11]. High expression of DX2 was significantly associated with the poor prognosis of lung cancer patients and the cancer progression, and knockdown of DX2 using siRNA or a small molecule significantly reduced tumor progression in in vivo [
10,
12], suggesting that DX2 can be a promising cancer target and a diagnostic as well as prognostic marker for anti-cancer therapies. Other than DX2, no genetic alterations of AIMP2 is known to be associated with human cancers, although a homozygotic nonsense mutation of AIMP2 has been reported to be associated with hypomyelinating leukodystrophy-17, an autosomal recessive neurodevelopmental disorder [
13].
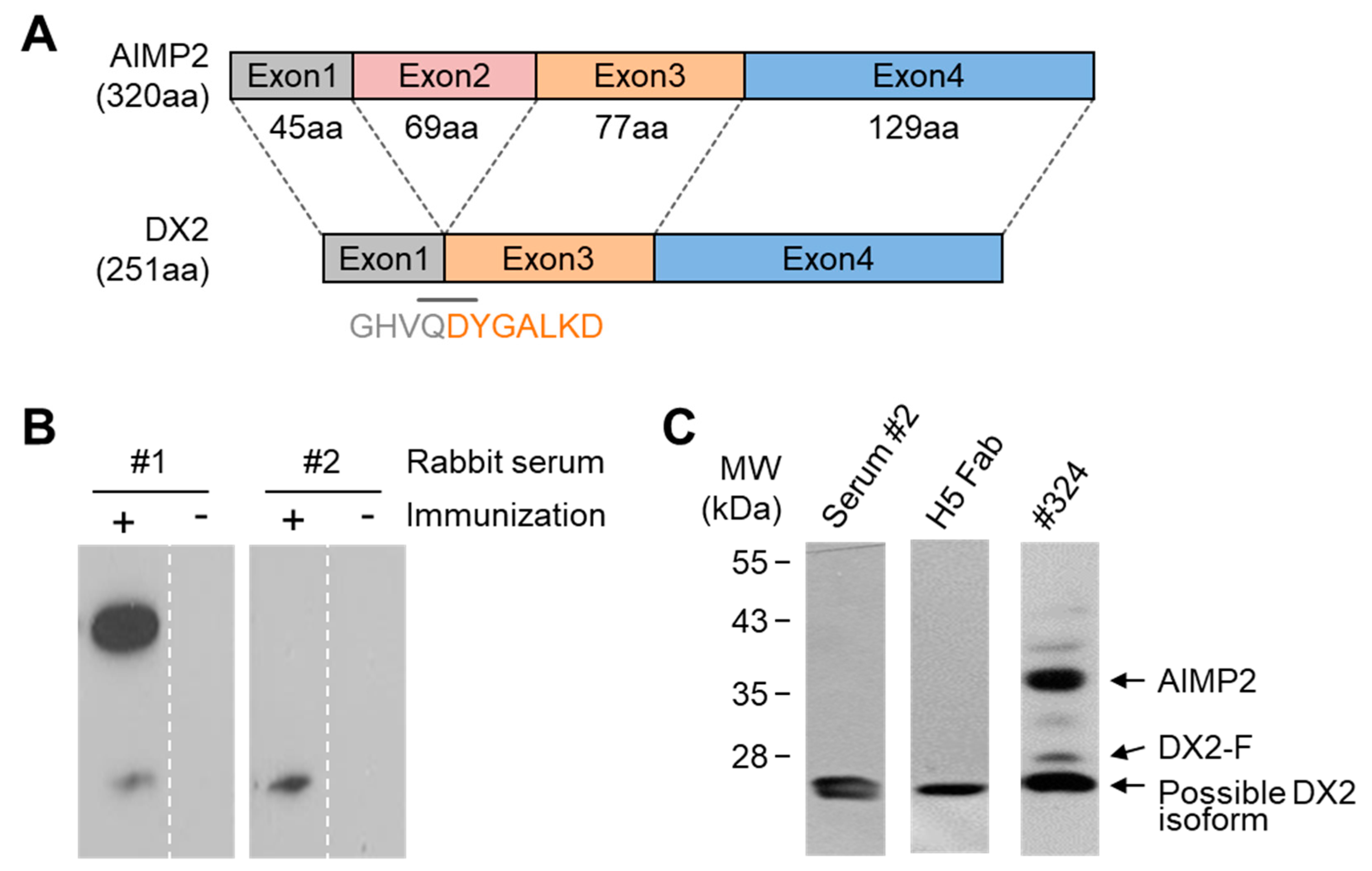
Keeping up with the drug development efforts targeting DX2 [
10,
11], many attempts have been made toward the development of monoclonal antibodies (mAbs) specific to DX2 that can be used as a diagnostic agent. Currently, clone #324 is the only antibody that can be used during immunoblotting to detect DX2. Clone #324 is a mAb obtained by immunization of a mouse with AIMP2 as an immunogen. Although the major protein recognized by clone #324 during immunoblotting is AIMP2, it additionally detected DX2 along with several unidentified bands. So far, all the attempts to develop a DX2-specific antibody by immunization or bio-panning using DX2 peptides or proteins as an antigen have failed. In this study, we report the development of a new anti-DX2 mAb, H5, by rabbit immunization followed by phage display selection. We also suggest that the endogenous DX2 isoform recognized by H5 lacks 33 amino acids (aa) in the N-terminus and is a functional surrogate for DX2. The possible application of the DX2-specific antibody to immunohistochemistry (IHC), immunofluorescence (IF), and enzyme-linked immunosorbent assay (ELISA) indicated the potential utility of this antibody as a tool for research as well as the identification of cancer patients with high levels of DX2.
2. Materials and Methods
2.1. Ethics Statements
All animal studies involving rabbit immunization, euthanasia, and organ harvesting were conducted by AbClon Inc. (Seoul, Korea) in accordance with the guidelines of the National Institute of Health (NIH) “Guide for Care and Use of Animals” and an approved protocol received by the company’s Institution Animal Care and Use Committee. For IHC staining, non-small cell lung cancer (NSCLC) tissues from patients who underwent lung resection between 2000 and 2008 and had submitted written consent providing the residual samples were randomly selected from the institutional tissue archives. The use of tumor tissue was approved by the Institutional Review Board (IRB) of Gangnam Severance Hospital (IRB #3-2014-0299) and was carried out in compliance with the Declaration of Helsinki and Korean Good Clinical Practice guidelines.
2.2. Rabbit Immunization, mRNA Extraction, and cDNA Synthesis
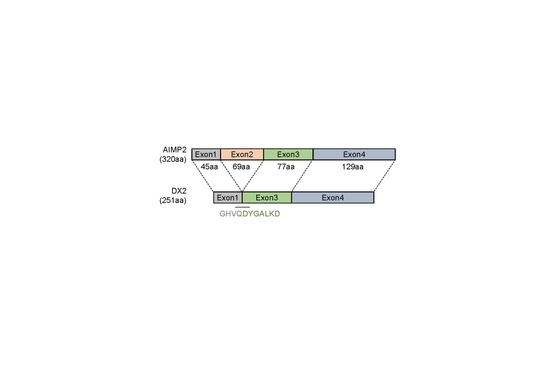
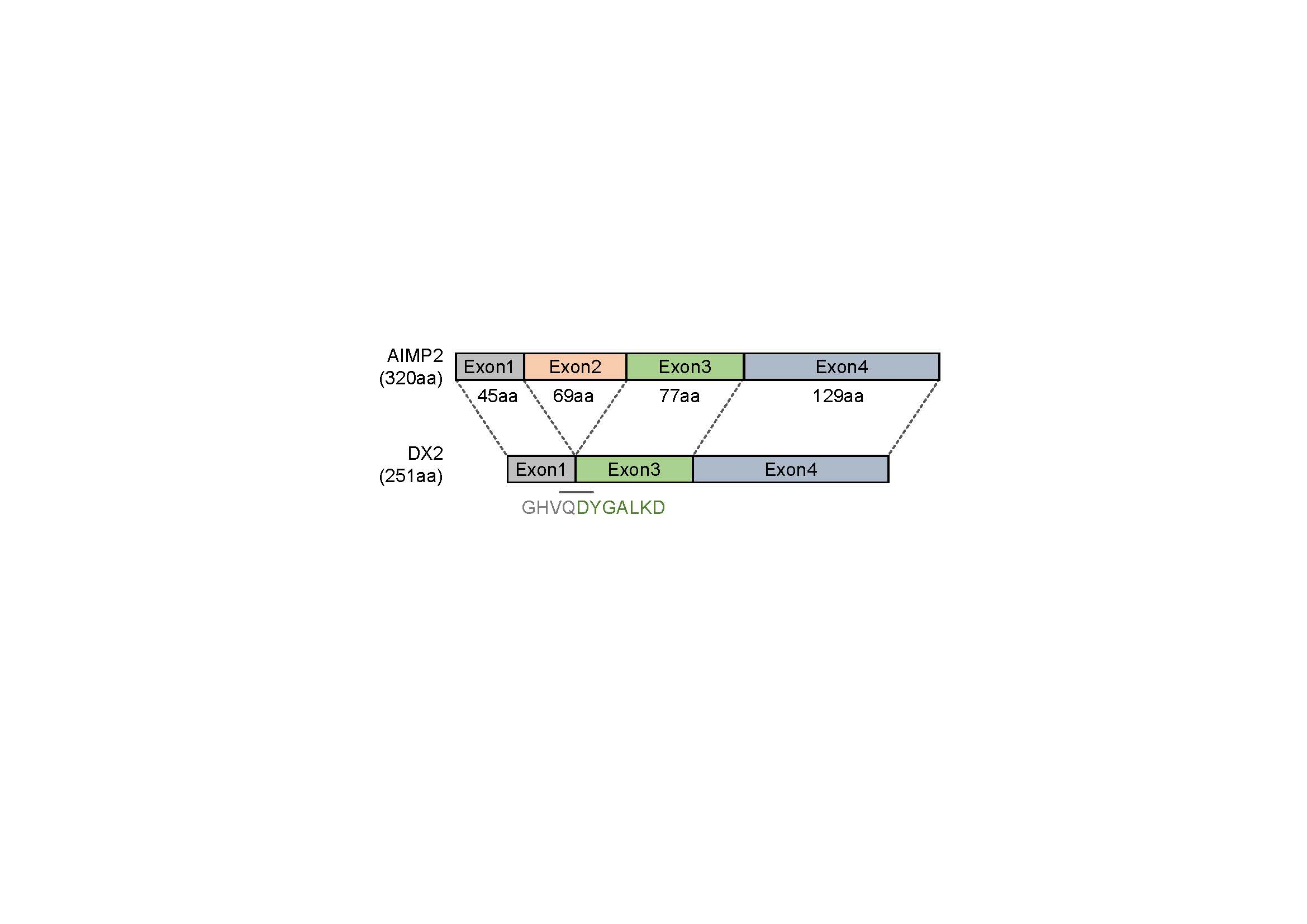
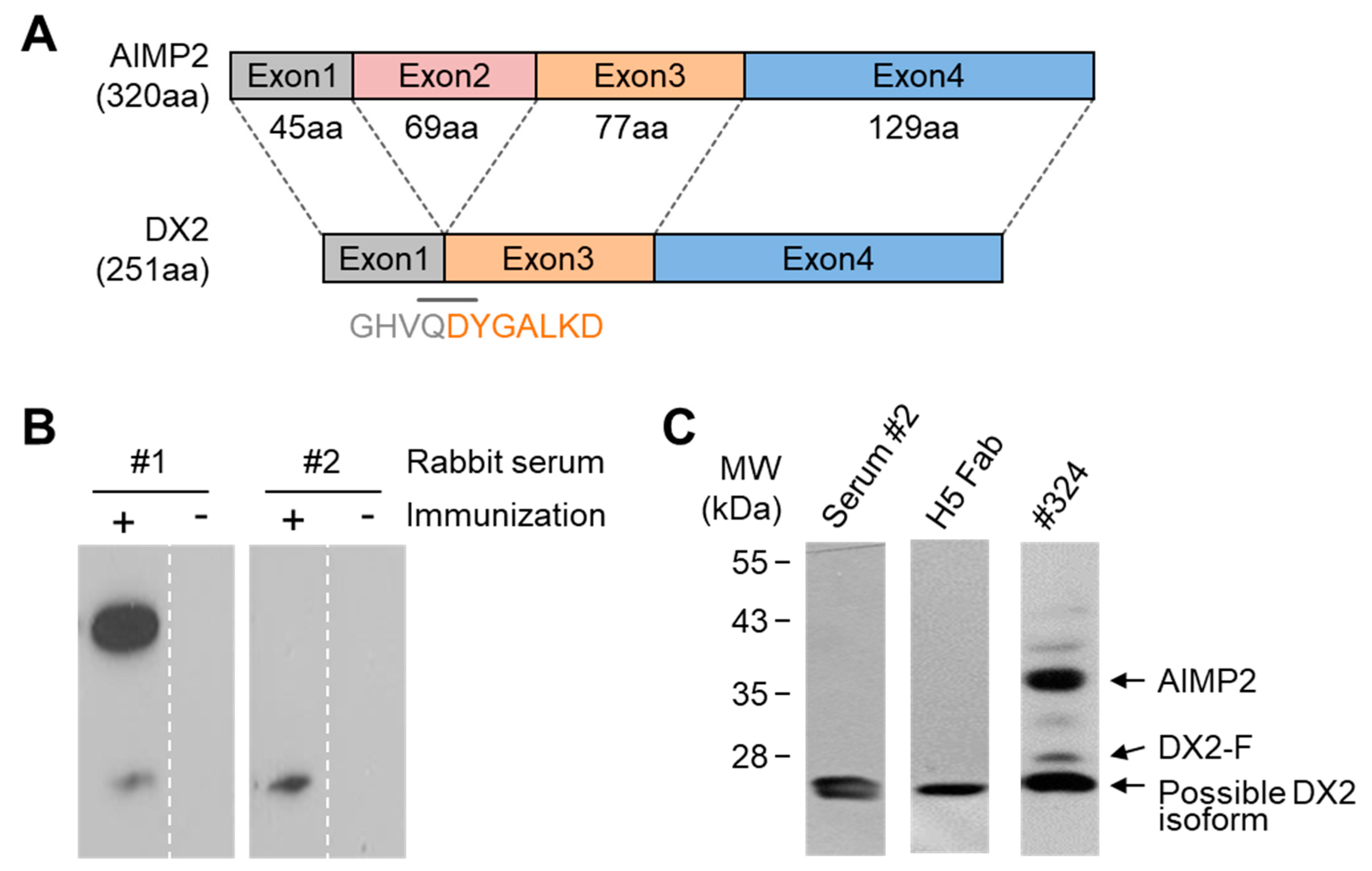
A synthetic peptide corresponding to the sequence spanning the exon 1–3 junction of DX2, GHVQDYGALKDC (C-terminal Cys added for conjugation reaction), was conjugated to keyhole limpet hemocyanin (KLH). A total of 200 µg KLH-conjugated antigen peptide was dissolved in 500 µL phosphate-buffered saline (PBS, pH 7.4) and mixed with 500 µg of adjuvant. Two rabbits were subcutaneously injected with 1 mL of the antigen/adjuvant mixture at 7–10 day intervals. An aliquot of blood was taken from each animal 1 week after each immunization and titrated by ELISA to determine the presence of antigen-specific antibodies. Following the final immunization, the rabbits were exsanguinated, and the spleens and blood sera were harvested. After weighing, splenic tissues were placed in TRI-reagent
® (Sigma-Aldrich) and homogenized. Additional 15 mL of TRI-reagent was added and the homogenate was incubated for 5 min at room temperature (RT). Phase separation was carried out by addition of 1.5 mL of BCP (1-bromo-3-chloropropane). The homogenate was thoroughly mixed for 15 Sec, incubated for 15 min at RT, and centrifuged for 15 min at 12,000 rpm. The supernatant was carefully transferred to a clean tube, avoiding contamination by the fatty layer and pellet separated on the bottom of the tube, and mixed with 7.5 mL of isopropanol for RNA precipitation. The tubes were vortexed and incubated at RT for 10 min, and centrifuged at 12,000 rpm for 10 min at 4 °C to pellet the RNA. The supernatant was removed, and the pellet was washed with 10 mL of 70% ethanol and incubated on ice for 5 min. After centrifugation at 12,000 rpm for 10 min at 4 °C, ethanol was discarded and the pellets were air-dried for 3 min and resuspended in 1 mL of nuclease-free water. Total splenic RNA was further purified by lithium chloride (LiCl) precipitation [
14]. Briefly, RNA was precipitated by adding 53 µL of 7.5 M LiCl to 500 µL of RNA solution, incubating the mixture on ice for 2 h, and centrifuging at 12,000 rpm for 30 min. The pellet was dissolved in 500 µL nuclease-free water, and the RNA was LiCl-precipitated again as above. The pellet was then dissolved in nuclease-free water, and ethanol-precipitated (0.1 volume of 3 M sodium acetate (pH 5.2) and 2.2 volume of ethanol, incubated on ice for 1 h). The precipitated RNA was centrifuged and dissolved in 200 µL nuclease-free water. First-strand cDNA was synthesized using the reverse transcription-polymerase chain reaction (RT-PCR) kit (Thermo Fischer scientific). For complementary DNA (cDNA) synthesis, 1 µL of total RNA was mixed with 1 µL of oligo (dT)
18 primer, 1 µL of 10 mM dNTP mix, 4 µL of 5× RT buffer, 1 µL of Maxima H minus enzyme mix, and water to a final volume of 20 µL. The reaction was performed for 30 min at 50 °C and terminated by heating at 85 °C for 5 min. The first-strand cDNA was immediately used for PCR amplification of antibody variable domain genes.
2.3. Generation of Rabbit Fab (Fragment Antigen-Binding)
First-strand cDNA derived from the rabbit spleen was used as the template to amplify VH (heavy chain variable) and VL (light chain variable) domain genes, and pComb3X-TT phagemid vector [
15] was used for the amplification of human C
κ (constant kappa chain) and CH1 (first constant heavy chain) domains to construct Fab antibody library. Primers used for the amplification of individual domains and their combinations are listed on
Tables S1 and S2, respectively. PCR using
Taq polymerase for the amplification of the variable and the constant domains (~350 bp each) was performed under the following condition: 94 °C for 2 min; 30 cycles of 94 °C for 30 Sec, 56 °C for 30 Sec, and 72 °C for 30 Sec; followed by 72 °C for 7 min. Then, each gene fragment was purified using gel extraction kit (Qiagen) after the PCR reaction products were separated by 1% agarose gel electrophoresis. In the second round of PCR, each of the heavy and the light chain variable fragments was joined to the respective constant domain by overlap extension PCR using
Taq polymerase (New England Biolabs). The PCR program was set under the following condition: 94 °C for 2 min; 25 cycles of 94 °C for 30 Sec, 56 °C for 30 Sec, and 72 °C for 1 min; followed by 72 °C for 10 min. Primers for overlap extension PCR are listed in
Table S3. Approximate sizes of the heavy chain VH-CH1 fragment (Fd) and light chain fragment (LC) were 750 bp and 800 bp, respectively. Finally, the Fd and LC products were joined by PCR again using
Taq polymerase and RSC-SF/dpseq primers. The PCR program was as follows: 94 °C for 2 min; 25 cycles of 94 °C for 30 Sec, 56 °C for 30 Sec, and 72 °C for 1.5 min; followed 72 °C for 10 min. Approximate size of final Fab product was 1500 bp.
2.4. Construction of Fab Library
The rabbit-human chimeric Fab library was cloned into pComb3X phagemid vector. First, the vector and the Fab DNA were digested with SfiI restriction enzyme (New England Biolabs). For digestion of DNA, 15 µg of purified PCR product or vector DNA was mixed with 50 units of SfiI, 20 µL of 10× reaction buffer, and water to a final volume of 200 µL. Digestion mixtures were incubated at 50 °C for 16 h followed by ethanol precipitation overnight at −20 °C. The precipitated DNA was centrifuged and dissolved in 100 µL nuclease-free water, and purified by agarose gel electrophoresis. Digested vector and Fab DNA fragments were ligated overnight (2 μg vector DNA and 3 μg insert DNA (~1:3 molar ratio) in 100 µL total volume) and ethanol precipitated. After centrifugation, the ligated DNA was dissolved in 10 µL of nuclease-free water and transformed to
Escherichia coli (
E. coli) ER2537 cells by electroporation. Transformed bacteria were plated on LB (Luria-Bertani broth)-ampicillin plates with 2% (w/v) glucose and incubated overnight. Next day, the bacterial growth was scraped and resuspended in 5 mL SB (Super Broth) medium, 15% (v/v, final concentration) glycerol was added, and 1 mL aliquots of the bacterial suspension were kept at −80 °C. The immune Fab phage library was rescued from the bacterial stock as previously reported [
16].
2.5. Panning of Phage Displayed Antibody Library
The phage displayed Fab library was panned against DX2-BSA antigen (exon 1/3 junction sequence peptide conjugated to bovine serum albumin [BSA]). For each round of selection, 3 µg of antigen in 1 mL of PBS was coated on an immunotube (NUNC 470319, Thermo Fisher Scientific, Waltham, MA, USA). The tube was coated for 1 h at 37 °C and subsequently blocked by filling the tube with 3% skim milk in PBST (PBS with 0.05% Tween 20, pH 7.4) for 1 h at RT. The blocking solution was removed and the phage library (1012 colony forming units in 1 mL of the blocking solution) was added to the antigen-coated immunotube. The tube was then incubated with shaking at 37 °C for 2 h, and the unbound phages were washed out three times with PBST. The bound phages were eluted with 1 mL of 100 mM TEA (triethanolamine) for 10 min at RT, and the eluted phage solution was neutralized with 500 µL of 1 M Tris-HCl (pH 7.4). The neutralized phages were applied to 8.5 mL mid-log phase ER2537 and incubated for 1 h at 37 °C with shaking at 120 rpm. Infected ER2537 cells were plated on ampicillin-LB agarose plate with 2% (w/v) glucose and grown overnight at 37 °C. The next day, the bacteria were harvested and phage library was rescued. Identical panning steps were repeated three times for the enrichment of antibody specific to the target peptide.
2.6. Screening for Phage Library Clones
Following three rounds of panning selection, output clones were screened by ELISA to identify specific target binders. Individual bacterial colonies were grown in 96-well plates containing 200 µL SB per well supplemented with 100 µg/mL ampicillin at 37 °C with vigorous shaking. After 3–4 h, when most wells became turbid, 1 mM final concentration of IPTG (isopropyl β-D-1-thiogalactopyranoside) was added to each well, and the induced cells were grown at 30 °C overnight with shaking. Prior to induction, replicate plates were prepared using a 96-pin plate replicator (Boekel Scientific, Feasterville-Trevose, PA, USA). Next day, IPTG-induced cells were spun down at 3500× g for 15 min and supernatants were discarded. Periplasmic extracts (PPEs) were obtained by resuspending the cell pellet in 60 μL of cold 1× TES buffer (30 mM Tris-HCl, 1 mM EDTA (ethylenediaminetetraacetic acid), 20% sucrose; pH 8.0), subsequently adding 90 μL of cold 0.2× TES buffer, and incubating on ice for 30 min. PPEs were applied to ELISA plates (25 µL per well) previously coated with either 100 ng/well of the antigen or PBS (blank control) and blocked with 3% skim milk-PBST. Plates were incubated for 1 h at RT, washed three times with PBST, and 25 µL per well of HRP (horseradish peroxidase)-conjugated anti-HA tag antibody (Santa Cruz Biotechnology clone F-7, 1:3000 dilution) was added to each well. After 1 h binding, plates were washed three times with PBST and 25 µL of TMB (3,3′,5,5′-tetramethylbenzidine) solution was added to each well. As blue color developed, 25 µL of 1 M sulfuric acid per well was added to stop the reaction. Absorbance was read at 450 nm in a plate reader.
2.7. Fab and Immunoglobulin G (IgG) Purification
To produce Fab, 20 mL of SB-ampicillin medium was inoculated with 5 µL of E. coli stock of the clone of interest from the replicate plate (see above). When the culture became turbid (absorbance at 600 nm > 0.5), 1 mM IPTG was added and the cells were further cultured overnight at 30 °C. The next day, cells were centrifuged and resuspended in 1 mL of cold 1× TES. Subsequently, 1.5 mL of cold 0.2× TES was added and mixed well, and the cell suspension was incubated on ice for 30 min. After centrifugation, the supernatant was transferred to a 15 mL conical tube, 100 µL of Ni-NTA (Ni2+-coordinated nitrilotriacetic acid) agarose beads was added, and the mixture was incubated at 4 °C with slow rotation for 1 h. The tube was centrifuged, supernatant was discarded, and the beads were washed twice with PBS containing 5 mM imidazole. Fab protein was eluted from the beads in 100 µL fractions using PBS containing 200 mM imidazole (pH 7.4). H5 Fab isolated from the library was also reformatted to human and rabbit IgGs. Heavy and light chain constant regions with an upstream signal sequence for protein secretion had previously been cloned in multiple cloning sites of the dual-promoter plasmid pVITRO1 (Invivogen, San Diego, CA, USA). VH and VL genes of H5 were amplified by PCR and serially cloned to this modified pVITRO1 vector harboring human or rabbit IgG constant region genes. The resulting human H5 IgG (hH5) or rabbit H5 IgG (rH5)-expression vector was transfected to 293F suspension cell line. Briefly, 100 µg of polyethyleneimine (PEI, linear, MW ~25,000; Polysciences Inc., Warrington, PA, USA) in 2.5 mL of sterile PBS was added to 100 µg of the vector DNA in 2.5 mL of sterile PBS. The mixture was incubated for 15 min at RT, and added dropwise to 50 mL of 293F cell culture at 2 × 106 cells/mL. Next day, 50 mL of fresh Freestyle 293 medium (Thermo Fisher Scientific, Waltham, MA, USA) was added to the cells, and the flask was incubated with shaking (~120 rpm) for 5 days at 37 °C under 8% CO2. After 5 days, cells were centrifuged, and H5 IgG was purified from the supernatant using protein A-agarose column chromatography.
2.8. Cell Culture
H460, HCC827, HCC15, H2108, DLD-1, HCC1419, SU8686, MKN74, NCC59, MKN45, and CHO-K1 cells were cultured in RPMI medium. SW620, MDA-MB 231, MIA-Paca3, BxPc3, DU145, and HEK293T cells were cultured in Dulbecco’s modified Eagle medium (DMEM). All of the cell lines were cultured in medium supplemented with 10% FBS and 1% penicillin/streptomycin at 37 °C in 5% CO2 incubator. Freestyle 293 cells for antibody purification were cultured with shaking at 120 rpm in Freestyle 293 Expression Medium at 37 °C under 8% CO2.
2.9. Plasmids, siRNA, and Transfection
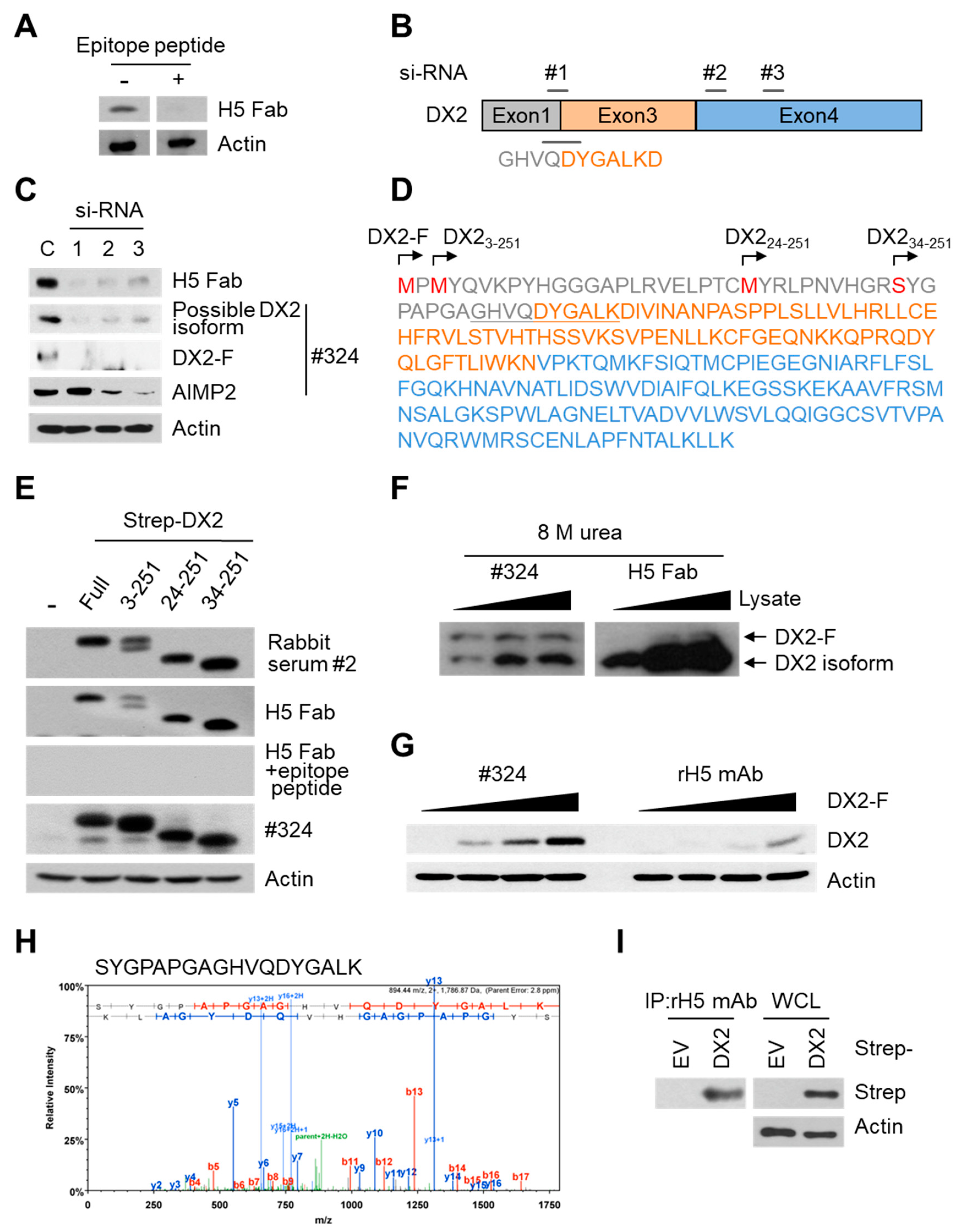
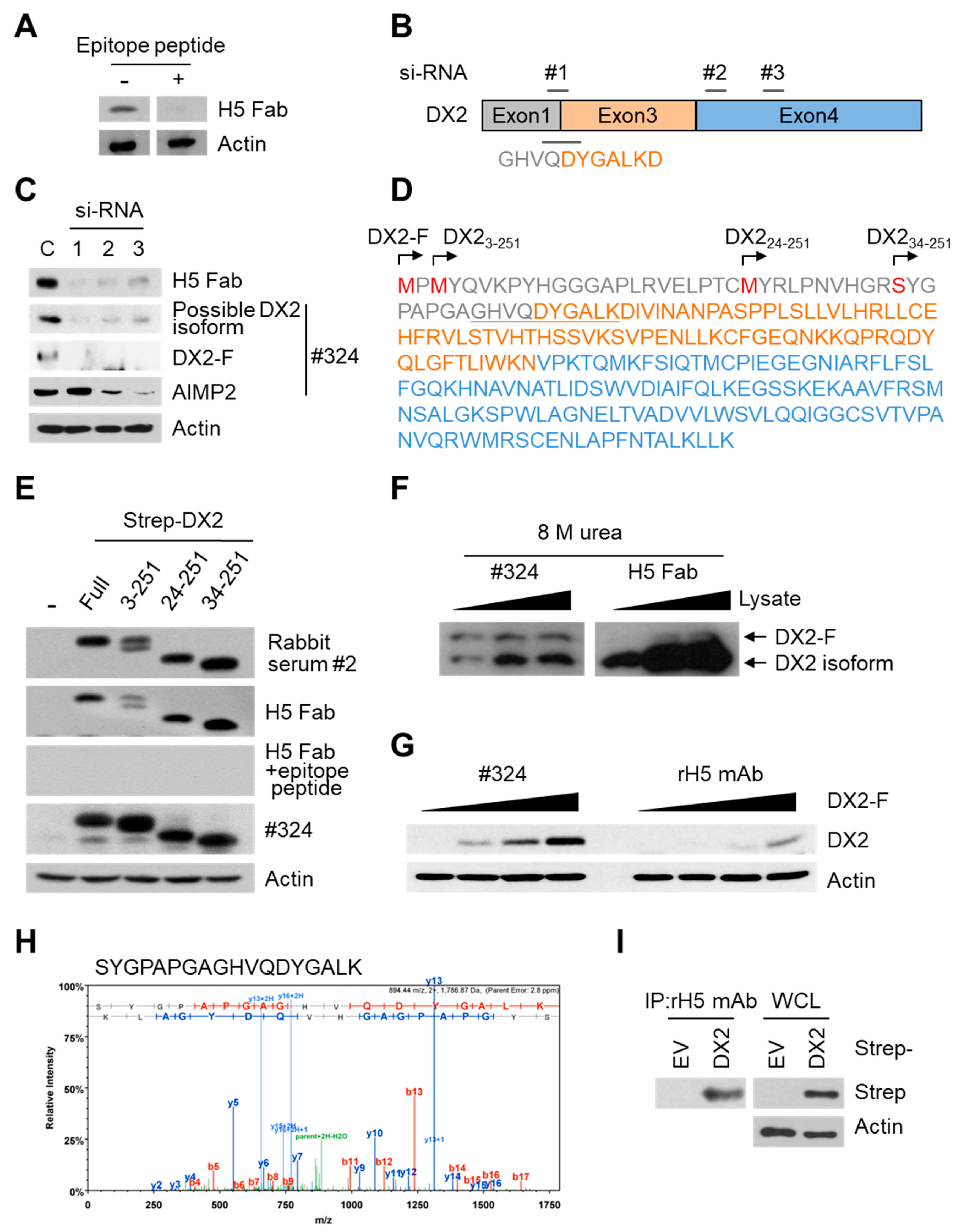
Three different siRNAs were designed and purchased from Oligo Center, ST Pharm Co. Ltd. (Gyeonggi, Korea) The sequences of siRNAs against DX2 were as follows: siRNA-1, CUGGCCACGUGCAGGAUUA; siRNA-2, GGAACAUUGCACGUUUCUU; and siRNA-3, GCUGUCAACGCAACCCUUA. Strep-tagged full-size DX2 (DX2-F), DX22-251, DX223-251, and DX234-251 were cloned in pEXPR IBA105 vector. Tag free DX2 gene was inserted into pcDNA3.1(+) vector using NheI/XhoI sites. To generate N-terminal deleted DX2 isoforms, an initiation codon (ATG) was inserted in front of each DX2 isoform sequence. Each of the siRNAs against DX2 and for negative control (Invitrogen) was transfected into H460 cells using Lipofectamine 2000 (Invitrogen), according to the manufacturer’s protocol. HEK293T cells and CHO-K1 cells were transfected with tag-free DX2 or Strep-tagged DX2 using Turbofect (Thermo Fisher Scientific, Waltham, MA, USA) reagent. The cells were incubated for at least 20 h prior to experiments.
2.10. Protein Purification
Tag-free DX2 protein was purified as previously described [
17]. C-terminal his-tagged DX2
34-251 and AIMP2
34-320 genes were cloned into pET-28a vector and overexpressed in
E. coli BL21(DE3) by induction with 0.5 mM IPTG. After centrifugation, cells were resuspended and sonicated in buffer (35 mM imidazole, 500 mM NaCl, and 20 mM Tris-HCl (pH 7.5)) supplemented with PMSF (phenylmethylsulfonyl fluoride) and protease inhibitor cocktail (Calbiochem, San Diego, CA, USA). After centrifugation, the supernatant was loaded onto a HiTrap chelating HP column (GE Healthcare, Chicago, IL, USA) pre-equilibrated with the same buffer without the protease inhibitors. The column was washed with the equilibration buffer, and the protein was eluted with the elution buffer (1 M imidazole, 500 mM NaCl, and 20 mM Tris-HCl (pH 7.5)). The partially purified proteins were further subjected to size exclusion (HiLoadl 6/600 Superdex75, GE Healthcare, Chicago, IL, USA) and cation exchange (HiTrap Sp HP) chromatographies.
2.11. Limited Proteolysis with Trypsin
Purified DX2 protein and trypsin (Promega, Madison, WI, USA) was mixed in reaction buffer containing 20 mM Tris/HCl (pH 7.5), 150 mM NaCl, 1 mM dithiothreitol (DTT), and 10% glycerol. Reaction ratio of trypsin to DX2 protein was 1:1000 and mixture was incubated for 1 min at 25 °C. Reaction was stopped by adding SDS (sodium dodecyl sulfate) sample buffer and boiled for 10 min at 100 °C to halt enzyme reaction. Samples were subjected to liquid chromatography-mass spectrometry (LC-MS) analysis or Western blotting after SDS-PAGE (sodium dodecyl sulfate-polyacrylamide gel electrophoresis).
2.12. LC-MS/MS and Data Analysis
DX2 protein was digested in-gel by trypsin as previously reported [
18]. Peptide sample was analyzed on a LTQ-OrbitrapVelos (Thermo Fisher Scientific, Waltham, MA, USA) connected to an Easy-nano LC II system (Thermo Fisher Scientific, Waltham, MA, USA). The dried peptide sample was resuspended in 70 µL of 0.1% formic acid, and an aliquot (7 μL) was injected to a reverse-phase peptide trap EASY-Column (L 2 cm, ID 100 μm, 5 μm, 120 Å, ReproSil-Pur C18-AQ, Thermo Fisher Scientific, Waltham, MA, USA) and a reversed-phase analytical EASY-Column (L 10 cm, ID 75 μm, 3 μm, 120 Å, ReproSil-Pur C18-AQ, Thermo Fisher Scientific, Waltham, MA, USA), and electrospray ionization was subsequently performed using a 30 μm (internal diameter) nano-bore stainless steel online emitter (Thermo Fisher Scientific, Waltham, MA, USA). The duration of total LC gradient was 60 min. The peptides were eluted in a linear gradient of 10%–40% buffer B (0.1% formic acid in acetonitrile) and 90%–60% buffer A (0.1% formic acid in H
2O) over 40 min and a flow rate of 300 nL/min. The temperature and voltage applied to the capillary was 275 °C and 1.9 V, respectively. All data were acquired with the mass spectrometer operating in automatic data-dependent switching mode. The MS survey was scanned from 350 to 2000 m/z with resolution set to 100,000. All MS/MS samples were analyzed using Sequest (XCorr Only, Thermo Fisher Scientific, Waltham, MA, USA, version v.27, rev. 11), X! Tandem (The GPM, thegpm.org; version CYCLONE (2010.12.01.1)) and the human sequence database (Uniprot 2014). Search parameters were set as follows: Full digestion using trypsin/Lys-C (after KR/-) with up to two missed cleavages, and precursor and fragment mass tolerances of 25 ppm and 1.0 Da, respectively. Each processed data was subsequently transformed to *.sf file with Scaffold 4 Q + S program (Proteome Software Inc., Portland, OR, USA, version 4.6.1). Scaffold program was used to validate MS/MS based peptide and protein identifications and to process the quantitative analysis. Peptide identifications were accepted if they could be established at greater than 80.0% probability by the Peptide Prophet algorithm [
19] with Scaffold delta-mass correction. Protein identifications were accepted if they could be established at greater than 80.0% probability and contained at least two identified peptides. Protein probabilities were assigned by the Protein Prophet algorithm [
20].
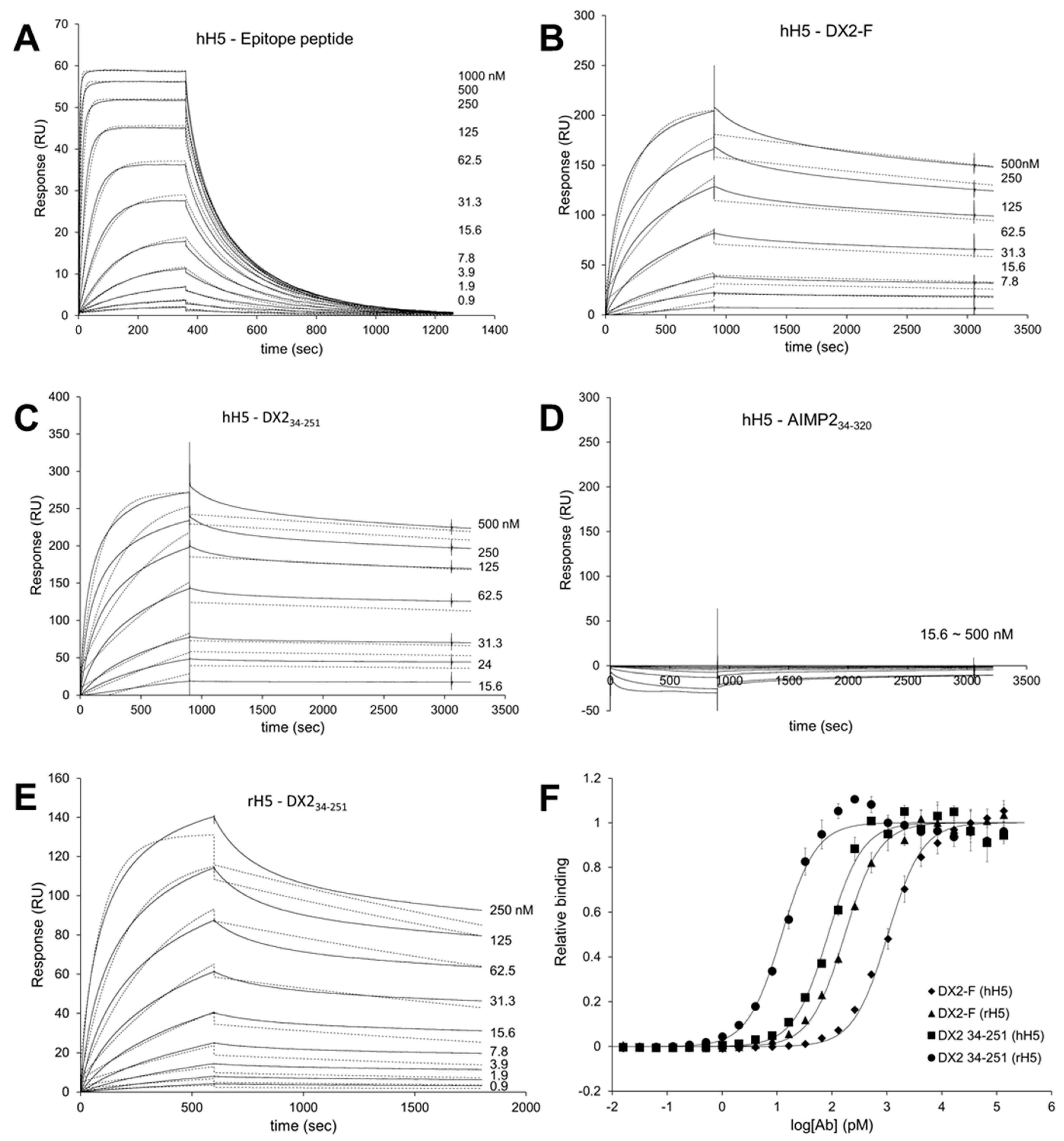
2.13. Surface Plasmon Resonance (SPR)
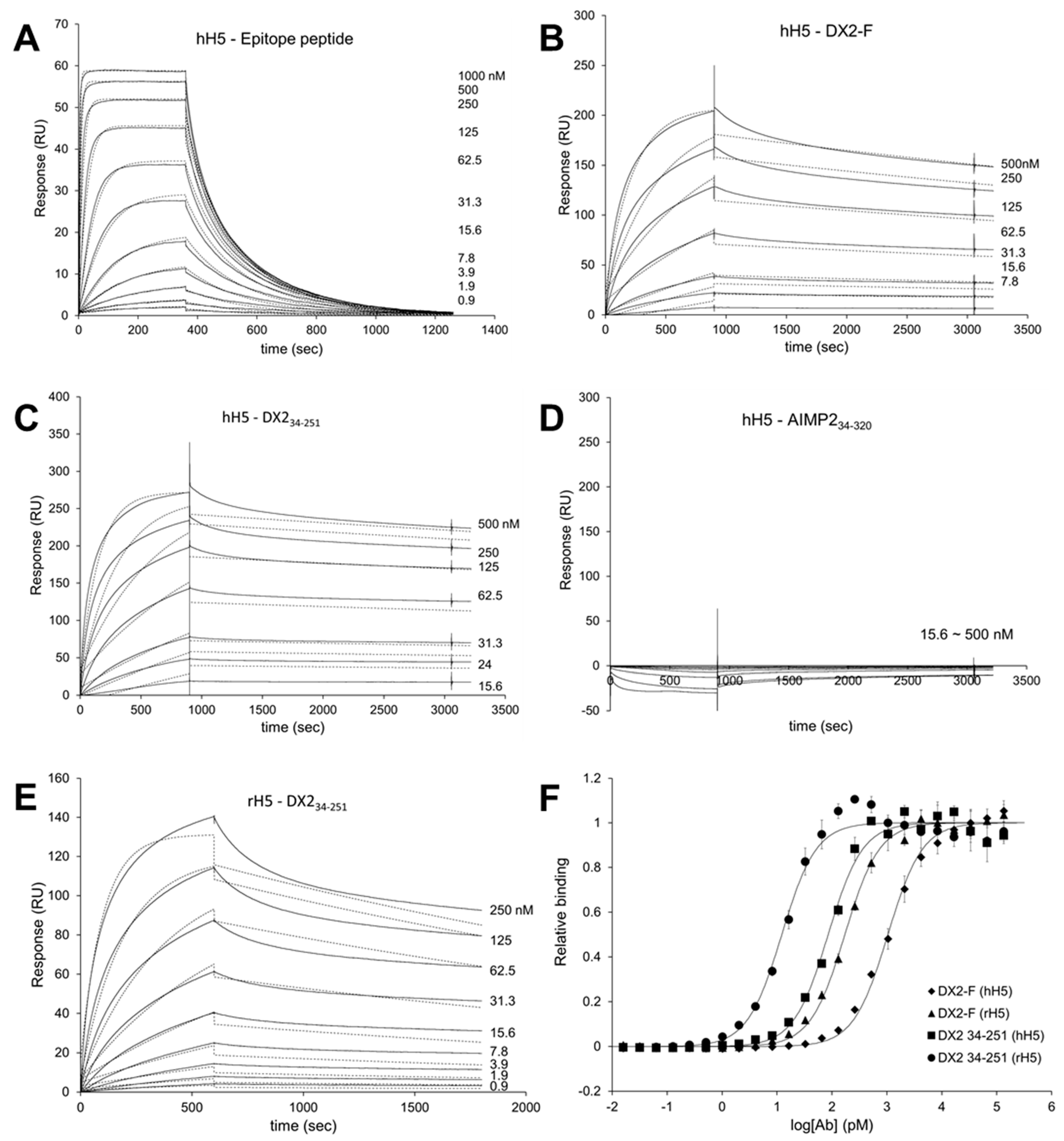
Surface Plasmon Resonance analysis were performed using a Biacore T200 (GE Healthcare, Chicago, IL, USA) equipped with a Series S sensor chip CM5 (GE Healthcare, Chicago, IL, USA). Immobilization was performed using amine coupling kit (GE Healthcare, Chicago, IL, USA) with PBS as a running buffer. Flow cells were activated with a 7 min pulse of a 1:1 mixture of EDC (1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide hydrochloride) and NHS (N-hydroxysuccinimide) according to the manufacturer’s instructions. hH5 mAb was diluted in 10 mM sodium acetate (pH 5.0) and immobilized to the chip, giving a surface density of 3320 response units (RU). Flow cells were then blocked with a 7 min pulse of 1 M ethanolamine-HCl (pH 8.5). For the interaction analysis, epitope peptide was diluted in PBS buffer supplemented with NSB (non-specific binding) reducer (GE Healthcare) and a two-fold serial dilution series was created to generate a concentration range of 0.02 to 1 µM. Proteins (DX2-F, DX234-251, and AIMP234-320) were diluted with 10 mM sodium acetate (pH 5.0) and immobilized to a Series S sensor chip CM5 at an immobilization levels of 590 RU. hH5 or rH5 mAb was diluted in PBS buffer with NSB reducer producing a 2-fold serial dilution series with a concentration range from 0.04 to 1 µM. After each binding cycle, regeneration solution (10 mM glycine-HCl, pH 1.5) was injected to remove any non-covalently bound protein. The binding data was fitted into a 1:1 binding model in Biacore T200 Evaluation software v2.0 (GE Healthcare, Chicago, IL, USA).
2.14. Enzyme-Linked Immunosorbent Assay (ELISA)
Each protein was diluted in 0.05 M sodium carbonate buffer (pH 9.6) to a final concentration of 1.5 ng/µL, and 100 µL/well was added to 96-well ELISA plates (Thermo Fisher Scientific, Waltham, MA, USA) and incubated overnight at 4 °C. After washing three times with 1× PBST, wells were blocked with 1% BSA in PBST for 1 h at RT. The plates were incubated with serially diluted hH5 or rH5 mAb for 1 h at RT, washed three times with PBST, and incubated with anti-human or anti-rabbit IgG secondary antibody (1:10,000 dilution). After 1 h binding, plates were washed three times with PBST and 100 µL of TMB substrate solution was added to each well. As blue color developed 10 min later, 1 M sulfuric acid was added (50 µL/well) to stop the reaction, and absorbance was read at 450 nm in a plate reader. The data were fitted to the 4-parameter logistic curve model using GraphPad Prism (GraphPad Software, San Diego, CA, USA).
2.15. Immunoprecipitation
H460 or HEK293T cells were rinsed with cold PBS, lysed in lysis buffer (50 mM Tris-HCl, 250 mM NaCl, 0.5% NP-40, and 5 mM EDTA) for 20 min at 4 °C, and centrifuged at 13,000 rpm for 30 min at 4 °C. The supernatant was incubated with rH5 mAb overnight. Protein A agarose beads (Invitrogen, Carlsbad, CA, USA) were added, and the mixture was incubated with rotation at 4 °C for 2 h. The beads were washed with cold lysis buffer five times, and the precipitates were subjected to SDS-PAGE.
2.16. Immunoblotting
Cells were lysed in cold RIPA (radioimmunoprecipitation assay) lysis buffer (25 mM Tris-HCl [pH 7.4], 125 M NaCl, 0.5% NP-40, 0.25% sodium deoxycholate, and 0.5% SDS, supplemented with protease inhibitors) for 20 min at 4 °C. After centrifugation at 13,000 rpm for 30 min at 4 °C, the supernatant proteins were quantified by Bradford assay (BioRad, Hercules, CA, USA). Proteins were separated by 10%–15% SDS-PAGE and transferred to 0.45 µm nitrocellulose or PVDF (polyvinylidene difluoride) membrane. After blocking, the membrane was incubated with primary antibody solution overnight at 4 °C, followed by washing three times with PBST and binding by HRP-conjugated secondary antibody for 1 h at RT. The membrane was washed three times with PBST, and subjected to luminescence detection using ECL (enhanced chemiluminescence) solution (GE Healthcare, Chicago, IL, USA). For the competition binding study, epitope peptide (100 µg/mL) was added during primary antibody (H5 Fab) binding. ImageJ software was used for the quantification of the detected band.
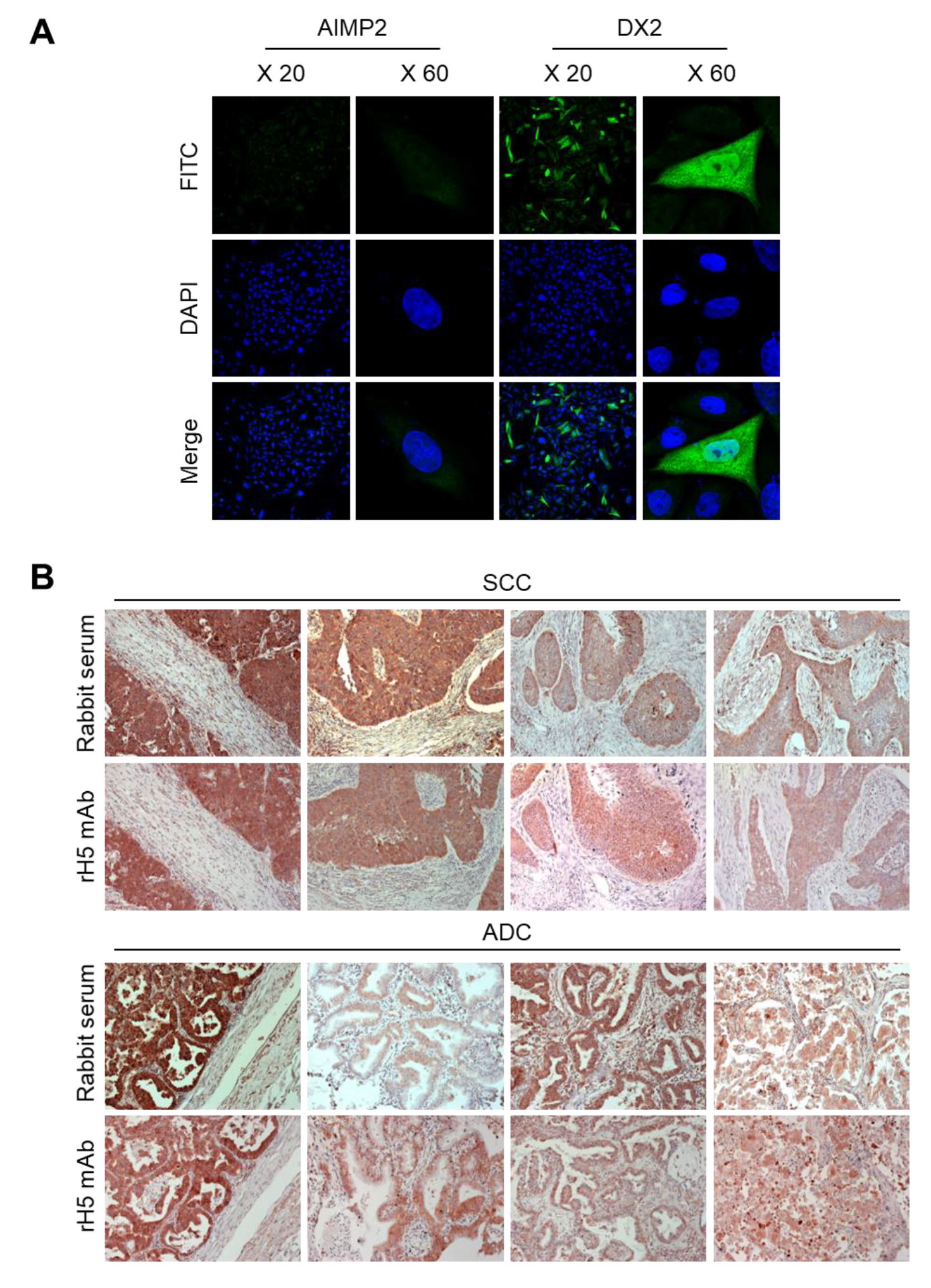
2.17. Immunofluorescence Staining
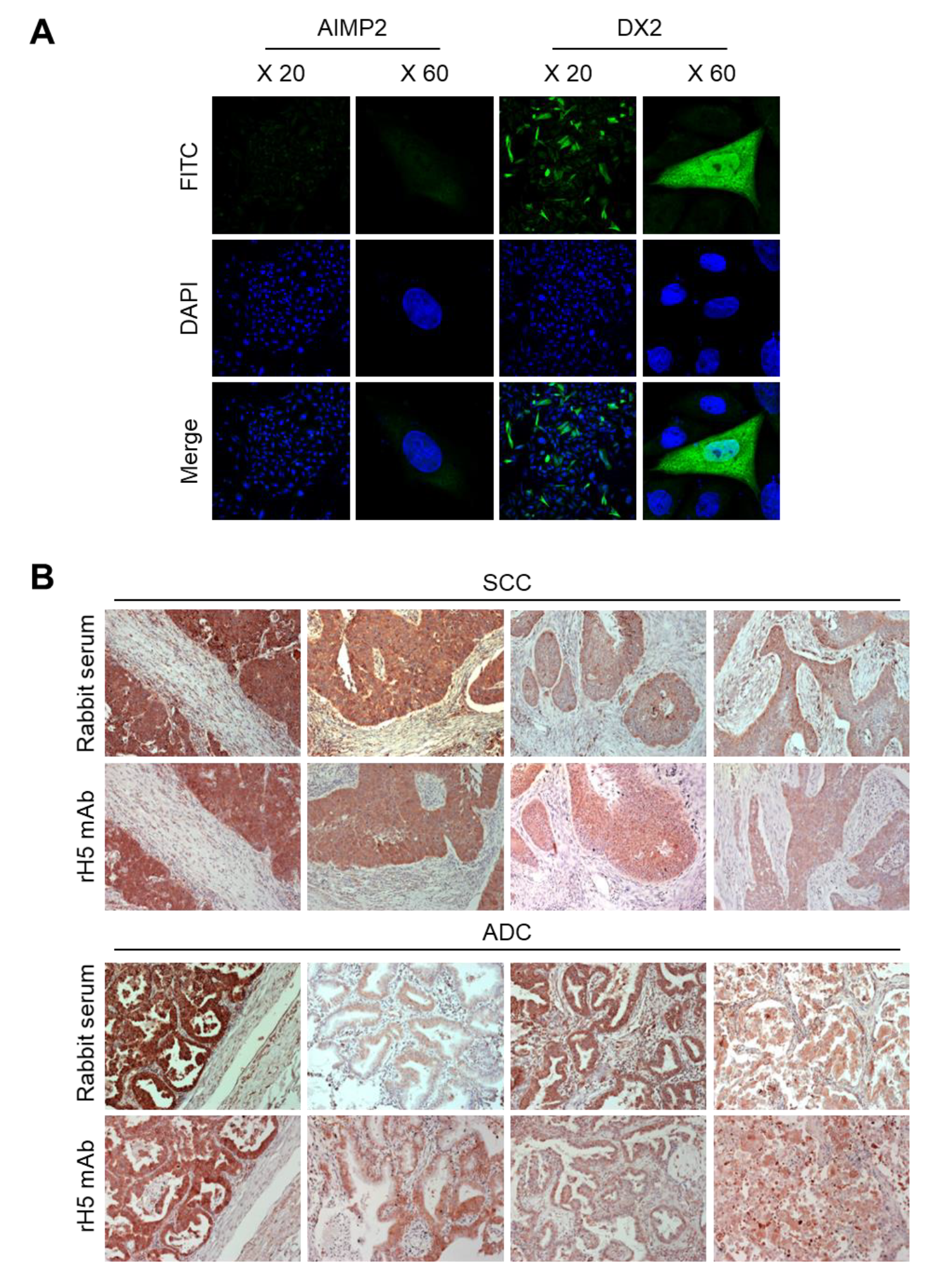
CHO-K1 cells were transfected with the DX2 or AIMP2 expression plasmid, and cells (1.2 × 105) were seeded in 12-well plates. After cell attachment, cultured media was completely removed, cells were fixed and permeabilized with 100% methanol for 7 min, and blocked with 3% CAS-Blocking solution (Life Technologies, Carlsbad, CA, USA, Cat. 008120) for 15 min at RT. Cells were subsequently treated with rH5 mAb (1:100 dilution) for 1 h at RT and washed with PBS for three times. Alexa 488-conjugated anti-rabbit IgG (Invitrogen, Carlsbad, CA, USA, 1:500 dilution) was added and incubated for 1 h at RT. After washing, 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI, 2 μg/mL) was used for nucleus staining. Stained cells were mounted on slides, covered with cover glass, and the fluorescence was detected by confocal microscope (Nikon, Tokyo, Japan) at 20× and 60× resolution.
2.18. Immunohistochemistry (IHC) Staining
IHC staining was performed using slides containing human lung tissues prepared at Severance Hospital-Gangnam, Seoul, Korea. Slides were deparaffinized as follows: Tissues were treated in xylene three times for 5 min each, 100% ethanol two times for 2 min each, 95% ethanol for 2 min, 90% ethanol for 2 min, 70% ethanol for 2 min, distilled water for 2 min, and 1× PBS for 5 min. The slides were then treated with 0.3% hydrogen peroxide for 10 min and rinsed with 1× PBS for 5 min. Next, they were microwaved in 0.1 M citrate buffer (pH 6.0) for 3.5 min, cooled down for 10 Sec, and boiled again for 10 Sec for antigen retrieval. These steps were repeated for 10 times and the slides were kept at RT for 20 min until the temperature dropped. After rinsing the slides with fleshly prepared 1× PBS three times for 5 min each, liquid barrier around the tissues were drawn using DAKO pen (DAKO). PBS with 2% normal goat serum (Jaximmuno) and 2% BSA (Sigma, Cat. A9647-100G) was added to each slide for 30 min at 4 °C for blocking. After removing all blocking solution, slides were incubated with primary antibody (rH5 mAb) within the boundary of liquid barrier and incubated overnight at 4 °C. Next day, the slides were rinsed with 1× PBS three times for 5 min each and the tissues were incubated with labeled polymer-HRP anti-mouse/rabbit antibody (DAKO) for 1 h at 4 °C. Tissues were washed with 1× PBS again, and treated with a mixture of 1 mL DAB+ substrate buffer and 20 µL DAB (3,3’-diaminobenzidine) chromogen for color fixation for 1 min under dark condition. Subsequently, counterstaining was performed by the treatment with Meyer’s hematoxylin (Sigma-Aldrich, St. Louis, MO, USA) for 1 min and rinse with running tap water. Tissues were dehydrated as follows: 70% ethanol (v/v) for 2 min, 90% ethanol for 2 min, 95% ethanol for 2 min, 100% ethanol twice for 2 min each, and xylene for three times for 5 min each. Drops of SUB-X mounting medium (Electron Microscopy Sciences, Hatfield, PA, USA) was added on tissues to place cover glass over them. Slides were scanned with Pannoramic MIDI (3D HISTECH).
2.19. Cell Viability Assay
HEK293T cells (4 × 104) transfected with empty vector (EV) or DX2 mutant expression vector were seeded in 96-well plates and cultured for 24 h. MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide, 5 mg/mL, Amresco) was added and incubated for 1 h at 37 °C. After discarding the media, 100 μL dimethylsulfoxide (DMSO, Duchefa) was added to each well for the solubilization of precipitated formazan. Absorbance at 560 nm was measured using a microplate reader. The experiments were repeated three times independently.
4. Discussion
Previous studies have shown that DX2 compromises the tumor suppressive activity of AIMP2 and promotes tumor progression [
10]. It is already known that high expression of DX2 in patients with cancer is significantly correlated with poor prognosis and chemoresistance in lung cancer and ovarian cancer, respectively [
7,
25], and knockdown or suppression of DX2 induced tumor regression [
10,
12], suggesting that DX2 could be a potential therapeutic target and diagnostic marker. Over the past years, multiple efforts to create an antibody against DX2 have proven unsuccessful despite its potential applicability as a diagnostic tool, due to the poor stability and immunogenicity of DX2 protein. Various forms of purified DX2 proteins with different tags and modifications to improve its solubility and stability, as well as DX2 exon 1/3 junction sequence peptides of different lengths were used as immunogens for rabbits and mice as well as for antibody library panning; however, no antibody with specificity toward DX2 could be identified.
In this study, we immunized rabbits with a short peptide, GHVQDYGALKD, specific to exon 1/3 junction of DX2. Remarkably, one of the two immunized rabbits yielded a polyserum that showed a strong and specific immunoblotting band roughly corresponding to the molecular weight of DX2. We generated a phage Fab library from the splenic cDNA of the rabbit, from which H5 mAb was isolated. The size of the Fab library prepared for this study was estimated to be 7.2 × 107 clones as deduced from the number of E. coli transformants. While this number is somewhat low for an antibody library, the immunized antibody repertoire was already enriched with target binders and prominent binding clones could be isolated, even with VH-VL shuffling that might considerably dilute native VH-VL pairings in the original immune repertoire. The library was panned against the exon 1/3 junction sequence peptide conjugated to BSA, and multiple peptide binders were identified by ELISA screening of the panning output clones. Of these, only one clone could detect the endogenous DX2 isoform protein in H460 cell lysate in immunoblot assay, possibly due to the difference between the conformations of the BSA-conjugated peptide and the corresponding sequence in SDS-denatured protein, or because some of the clones recognized a part of the linker structure that conjugated the peptide to BSA. The conformational dependence of the antibody binding to the exon 1/3 junction sequence may also be the reason for the dependence of H5 binding to DX2 on the N-terminal sequence of the antigen, possibly via flexibility-derived conformational differences.
The clone H5 could be utilized in immunoblotting, immunoprecipitation, ELISA, IF, and IHC experiments, showing its applicability to research purposes as well as to clinical diagnosis. While clone #324 recognizes DX2 as well as AIMP2, H5 monoclonal antibody specifically detects DX2. Although the epitope of clone #324 had not been identified yet, it is in the exon 3-exon 4 region since clone #324 was produced by immunizing a mouse with AIMP2
84-225 which includes parts of exons 2 and 4 and the entire exon 3 [
6], and can detect AIMP2, DX2-F, and DX2 isoforms altogether. Compared with clone #324, H5 showed much weaker recognition of DX2-F, which suggested that N-terminal region of DX2-F interacts with the exon 1/3 junction and thereby interferes with H5′s access to the region. The exon 1-exon 2 region of AIMP2 includes a leucine-zipper motif [
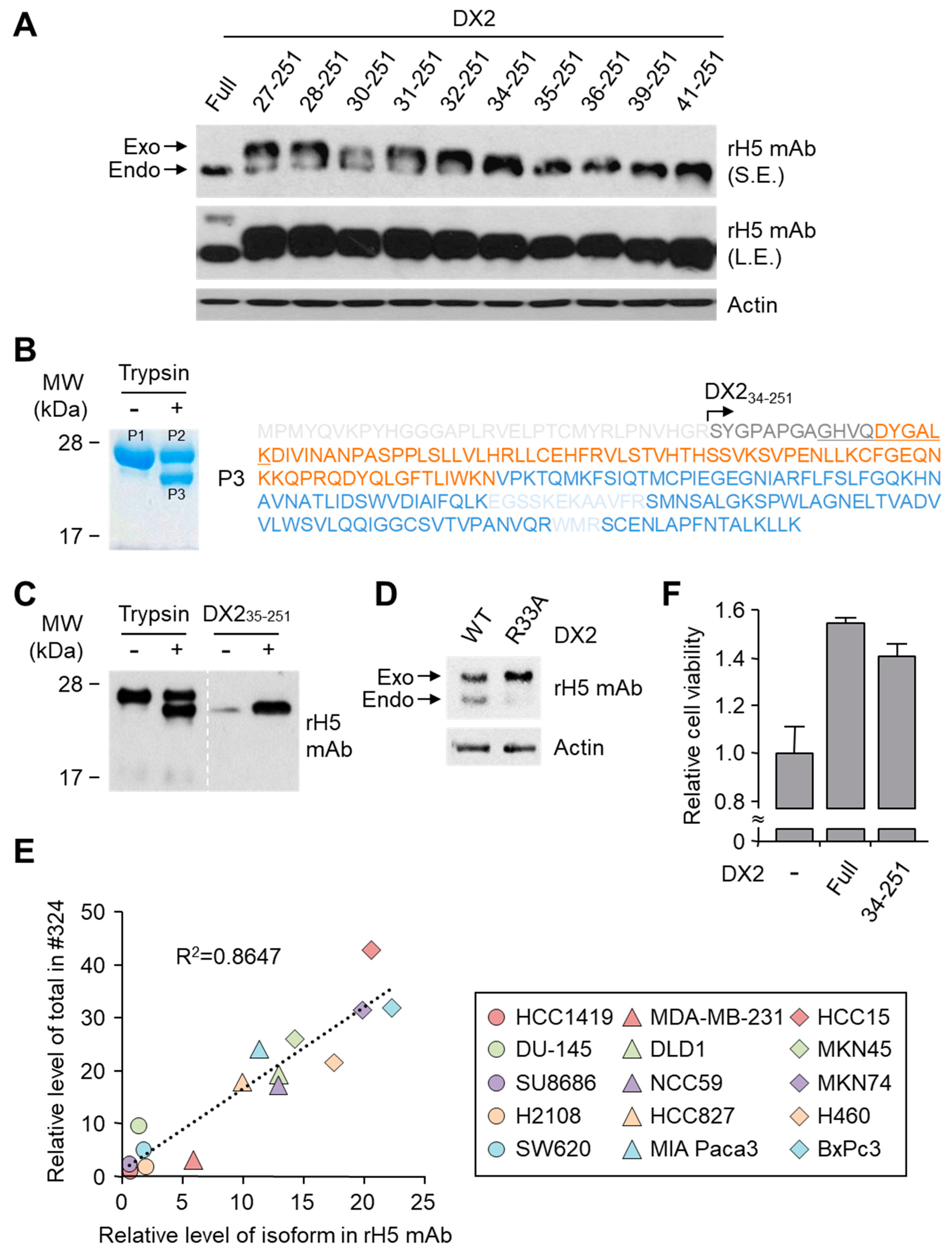
26], but following splicing of the exon 2, it is plausible that the remaining exon 1 loses structural stability. Actually DX2-F was difficult to be crystalized (unpublished data) and an insertion of N-terminal tag significantly enhanced the detectability of DX2-F by H5, corroborating the importance of structural stability of DX2 N-terminus for its recognition by H5. We searched the possible identity of the endogenous DX2 isoform by analyzing the N-terminal digestion by proteases, but not C-terminal digestion, since C-terminal exon 3-exon 4 consists of a stable GST (glutathione S-transferase) domain [
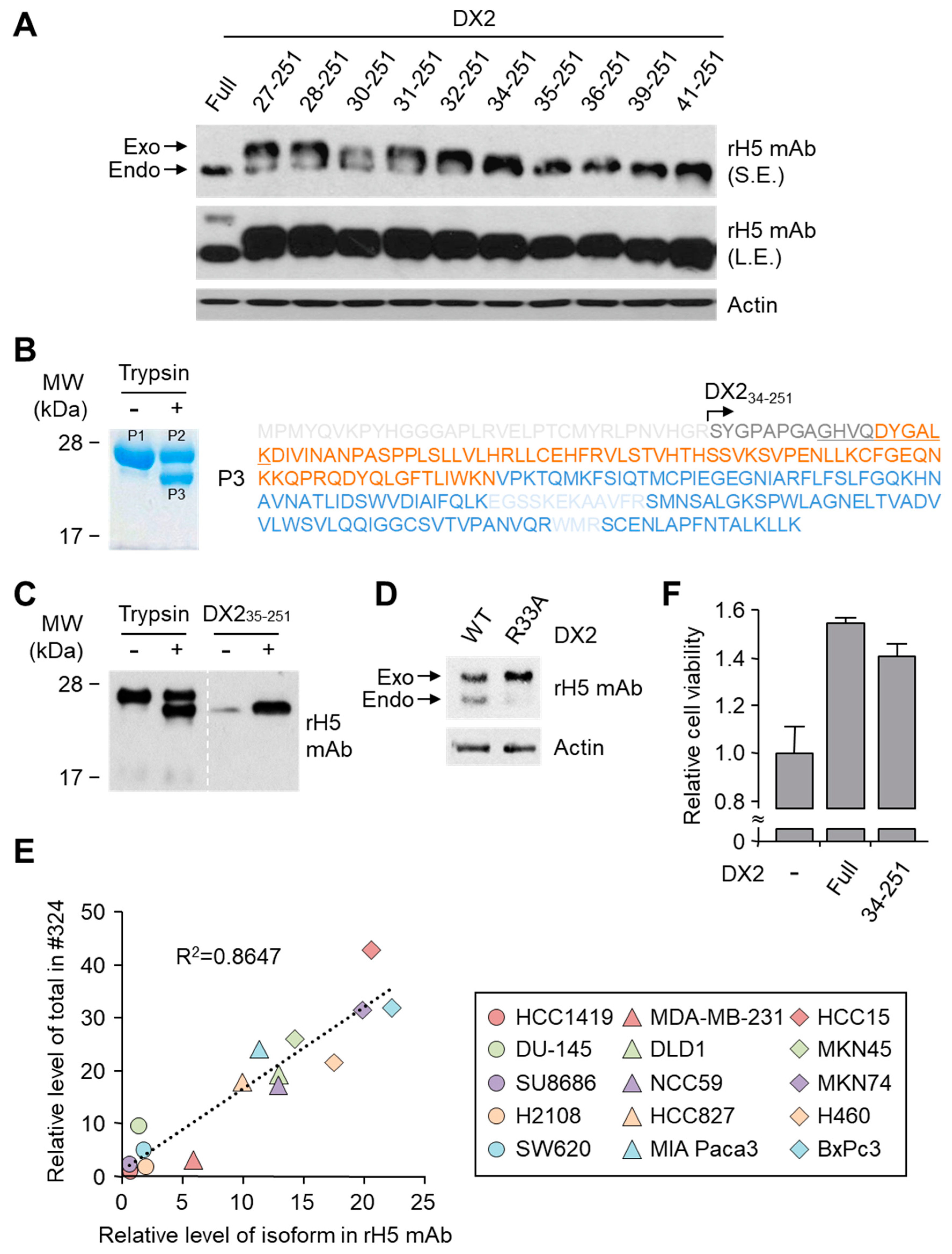
26]. It is likely that the N-terminus of DX2-F is unstable in intracellular environment and proteolytically cleaved to the shorter isoform, which may explain the low level of detection by #324 and the marginal recognition by H5 of DX2-F in H460 cell lysate by immunoblotting. We focused on identifying the N-terminal sequence of the DX2 isoform. Based on the results from mutation analysis, limited digestion, and mass spectrometry, we propose the isoform detected by H5 mAb to be DX2
34-251, which could be generated by cleavage between Arg33 and Ser34 by proteases, such as trypsin (
Figure 3). It is unclear how the DX2-F is actually processed by proteolytic enzymes to generate DX2
34-251 within cancer cells. Several reports have suggested that cancer cells express proteolytic enzymes, including trypsin [
27,
28]; however, more studies are required to elucidate the underlying mechanism of DX2 isoform generation and its relationship to protease enzymes such as trypsin.
While DX2-F is detected much more weakly by H5 than by clone #324, DX2
34-251 is effectively recognized by H5 and functions like DX2-F by enhancing cell proliferation (
Figure 3F). The level of DX2
34-251 is strongly correlated with the total level of DX2 (DX2-F and its isoform combined). Taken together, these results suggested that DX2
34-251 could be used as a surrogate marker for DX2-F and sensitively detected by H5. The development of H5 mAb expanded the limitation of clone #324, which is only applicable to immunoblotting because the antibody detects both AIMP2 and DX2. In the era of precision medicine for cancer therapy, it is expected that the H5 antibody has the potential to be a useful research tool as well as a diagnostic agent for several solid tumors associated with DX2 overexpression, in IHC, cytology staining, or point-of-care testing kits. H5 mAb is currently being tested in multiple studies for the diagnosis of cancers including hematological and colorectal cancers, with increased numbers of patients to ensure proper clinical validation of DX2 and H5 (manuscripts submitted or in preparation).

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}