
Expanded Reactor Engineering Calculations for the Oxidative Coupling of Methane


Otto H. York Department of Chemical and Materials Engineering, New Jersey Institute of Technology, Newark, NJ 07102, USA
*
Author to whom correspondence should be addressed.
Methane 2022, 1(1), 58-69; https://doi.org/10.3390/methane1010005
Submission received: 12 January 2022
/
Revised: 28 January 2022
/
Accepted: 2 February 2022
/
Published: 11 February 2022
(This article belongs to the Special Issue Methane Conversion Technology)
Abstract
:The catalytic activation of CH4 by limited amounts of O2 produces a mixture of synthesis gas (CO, H2) and light hydrocarbons (C2Hx), the relative amounts of each depending on catalyst type and process conditions. Using an elementary reaction mechanism for the oxidative coupling of methane (OCM) on a La2O3/CeO2 catalyst derived from the literature, this study replaces the activating O2 with moist H2O2 vapor to reduce synthesis gas production while improving C2Hx yields and selectivities. As the H2O2 content of the activating oxidant rises, more of the CH4 conversion occurs in the gas phase instead of with the catalytic surface. In a packed bed reactor (PBR), the use of H2O2 allows the PBR “light-off” to occur using a lower feed temperature. In exchange for a small decline in CH4 conversion, C2Hx selectivity increases while synthesis gas production drops. In a continuous stirred tank reactor (CSTR), H2O2 improves C2Hx over synthesis gas across a wider range of feed temperatures than is possible with the PBR. This suggests the CSTR will likely reduce OCM preheating requirements.
1. Introduction
The expanded use of hydraulic fracturing has resulted in the venting and flaring of large volumes of hydrocarbons, especially natural gas. A lack of local pipeline capacity results in more flaring [1] and fugitive emissions. Public sentiment is prompting government environmental regulators to force the reduction or outright banning of hydrocarbon flaring and fugitive emissions [2] to reduce climate change by global warming. Petroleum and natural gas companies are now actively promoting their efforts to reduce their methane footprints [3]. However, the engineering challenge of CH4 conversion is considerable. Catalytic methods offer several conversion approaches.
Catalytic activation of CH4 is generally classified as indirect or direct. Indirect activation produces synthesis gas (primarily CO and H2) using an oxygen source by reforming (H2O—steam; CO2—dry) or partial oxidation (O2). Synthesis gas can be catalytically converted to useful products such as alcohol (usually CH3OH) or higher hydrocarbons (by Fischer–Tropsch process).
Direct activation of CH4 uses no oxygen source. It directly breaks the very strong CH3–H bond (4.39 × 105 J/mol). For example, methane dehydroaromatization (MDA) uses a Mo/HZSM-5 zeolite [4] catalyst to form C2H4 and aromatics at 950–1030 K. Unfortunately, MDA is thermodynamically limited. In addition, catalyst activity drops quickly due to coke deposition.
An intermediate direct approach is the oxidative coupling of methane (OCM) that uses a very small amount of O2 to activate the CH4 while limiting the coke formation. The OCM catalysts are transition metal oxides on an oxide support, e.g., La2O3/CaO [5] and La2O3/CeO2 [6]. Feed CH4/O2 molar ratios of 7–11 with temperatures ~840–1220 K have been studied. The products include C2+Hx and synthesis gas, with the distribution depending on the catalyst and temperature. More feed O2 favors CH4 conversion, but lowers C2+Hx selectivity in favor of COx.
Gambo et al. [7] reviewed recent advances in OCM, including the use of catalyst nanowires, and identified avenues for further research. Using proprietary nanowire catalysts, Siluria Technologies demonstrated OCM in a continuous flow large demonstration plant [8,9]. The primary goal is the production of C2H4 for subsequent conversion to gasoline and chemicals. Siluria envisions a flexible two-stage OCM reactor in which the first stage is a packed bed reactor (PBR) feeding CH4, O2, and possibly C2H6. The second stage feeds more C2H6 in an endothermic pyrolysis plug flow reactor that uses the first stage exothermicity.
Considering the potential and the constraints of OCM, the reactor type becomes important. Conventional packed beds are not economically viable for OCM [10]. Other configurations such as membrane reactors and fluidized beds should be considered. A recent study [11] compared the packed bed reactor (PBR) and continuous stirred tank reactor (CSTR) for OCM based on calculations with a detailed reaction mechanism [6]. Higher feed temperatures were required to achieve a “light off” of the PBR, while the CSTR required considerably lower feed temperatures to reach nearly comparable conversions. The CSTR—a fluidized bed in likely practice—favored synthesis gas production over C2+Hx as compared to the PBR.
In a difficult experimental study, Liu et al. [12] considered the rapid PBR “light off”. Using careful temperature control and real-time product measurement in a micro-reactor during OCM over La2O3 nanorod catalysts with a feed CH4/O2 = 3, Liu et al. observed that CO2 is the dominant product at the lower temperatures (<853 K) when relative O2 concentrations are high. The system transitions through a window (~873 K) to higher temperatures (>913 K) that favor C2+Hx at the lower O2 levels. In effect, a competition between COx and C2+Hx formation occurs in this window. This suggests that reactor configuration and temperature will be critical for OCM reactor design. These observations also show that, in OCM, the production of byproduct syngas is unavoidable if C2+Hx is the goal.
In typical OCM, some of the limited O2 dissociates on the catalyst surface [13]. An adsorbed •Os then abstracts an H• from CH4 to form the key •CH3 gas phase radical. Oxygen atoms on the metal oxide lattice surface might directly abstract the H• from CH4. In this case, the gas phase O2 replenishes the resulting surface vacancy, leaving behind an adsorbed •Os [7]. In either case, the •CH3 radicals can combine to form C2H6. Further reactions form the desired C2+Hx products, and the undesired COx and coke. An alternate CH3-H bond activator would reduce COx while possibly enhancing C2+Hx formation. Unfortunately, the desired C2+Hx products are more susceptible to oxidation than the reactant CH4. This can occur via gas phase O2 or surface oxygen species [7,13].
An alternate or supplementary oxidant that might be less aggressive toward C2+Hx by reducing surface oxygen species while still activating the CH4 is the •OH gas phase radical. Gas phase hydroxyl radicals can be formed from the gas phase decomposition of co-fed H2O2 vapor: H2O2 + M = 2 •OH + M. Section 3 below summarizes experimental literature on the use of H2O2 for OCM that motivates this paper.
This paper is also a sequel to the Rivera et al. [11] study on OCM. It uses the same elementary reaction mechanism as that developed by Karakaya et al. [6]. The paper considers vapor phase H2O2 as a supplemental or alternative activating oxidizer to reduce syngas production in favor of C2+Hx. Both CSTR and PBR are considered.
2. Kinetic Mechanism and Computational Tool
The detailed reaction mechanism used in this study was developed by Karakaya et al. [6]. It is composed of series and parallel elementary reactions [13] in both the gas and surface phases. The gas phase portion is taken from Chen et al. [14]. The surface portion is inspired by the work of Alexiadis et al. [15].
In the current study, the OCM mechanism was employed in reactor simulations by Detchem® [16]. This program achieves material and energy balances using the mechanism, based on required reactor input data and parameters. In this study, separate adiabatic PBR (modeled as plug flow) and CSTR (modeled as perfectly mixed) runs were conducted with the Detchem® PBED and CSTR applications, respectively. See [11] for a listing of the governing balance equations used in each reactor simulation.
Comparative results were prepared in terms of conversions XCH4, selectivities Sj of useful products (C2Hx, CO, H2) or byproducts (H2O, CO2), and yields Yj:
where Fj = molar flow rates, Fj,in = molar rate at the reactor inlet, and nj is the number of CH4 moles needed to make one mole of product (byproduct). For example, for C2H4, nj = 2; for H2, nj = 0.5.
In the prior study [11] of OCM with O2 (no H2O2) as the activator, twenty cases were considered in separate CSTR and PBR calculations. The process parameters considered are summarized in Table 1. The parametric study included the molar feed CH4/O2 ratio (“low” = 7, “high” = 11; LR and HR, respectively), molar feed rate (“low” = 8.984 × 10−5, “high” = 1.412 × 10−4 mole/s; called LF and HF, respectively), and feed temperature (843–1243). At each of the five feed temperatures, four cases were considered: LR_LF, LR_HF, HR_LF, and HR_HF. These conditions were inspired by published laboratory data [6]. Each reactor simulation assumed the same catalytic site density and total catalytic surface area, resulting in the same 2.42 × 10−6 total moles of sites. A processing rate can be defined as the ratio of total molar feed rate to the total number of catalyst sites. The processing rate range is 37.1–58.3 s−1. These parameters motivate the present study. Larger reactors can be scaled from these conditions.
3. Alternate Activator H2O2
Garibyan et al. [17] studied OCM over Pb/aerosil, ZnO, and 10% Na2O/ZnO catalysts. Pulses of H2O2 vapor into the CH4/O2 feed increased the C2Hx yield while stabilizing catalytic activity during OCM at 1 atmosphere and 1023 K. With a 1% Au/5% La2O3/CaO catalyst, at 973–1073 K, Eskendirov et al. [18,19] observed that H2O2 increased CH4 conversion, while enhancing C2+ hydrocarbon yields even up to benzene. They speculated that H2O2 decomposition resulted in more •OH radicals for gas phase activation of the CH4. They even observed OCM in the presence of H2O2 vapor without catalyst at temperatures as low as 673 K, with considerable selectivity for C2Hx. These studies motivate this paper.
Considering the importance of making any OCM process as “green” as possible, a potentially sustainable source of H2O2 uses a photo-activated TiO2-Au-Si catalyst while feeding O2 and liquid H2O [20]. Spiegelman and Alvarez [21] developed a simple yet clever technology to produce a continuous vapor stream of H2O2 from a liquid solution of H2O2 in water. Subsequent drying of the vapor stream to raise the H2O2 concentration runs the risk of energetic decomposition, thus posing a safety risk. In a study of the decomposition of H2O2 vapor on various surfaces, Satterfield and Stein [22] generated H2O2 vapor concentrations of up to 0.23 atm in a 1 atm system. Therefore, in the remainder of these calculations, we used a conservative molar H2O/H2O2 = 4 linkage in all cases where H2O2 was used.
The OCM process requires considerable preheating, so the H2O2 decomposition risk also calls into question the feed temperature for the H2O/H2O2 vapor stream. Consider the decomposition: H2O2 + M → 2 •OH + M with a rate constant borrowed from the Chen et al. [14] reaction set for the CH4 gas phase chemistry used in the OCM mechanism [6]. Assume a feed CH4/H2O2 ratio of 11 (the HR case), with the coflowing H2O vapor, and no O2. A simple kinetic calculation shows that, at 1243 K, the H2O2 will be 100% decomposed in 10 microseconds. At 673 K, the time is a more realistic 5 s. This simple calculation suggests that preheating a combined CH4, O2, H2O2, and H2O feed stream would be problematic, especially for a PBR. It also suggests keeping the CH4/O2 and H2O/H2O2 vapor streams separate, with the CH4/O2 stream taking most or all the preheat. Separate feed streams are more easily handled with the CSTR.
Finally, the replacement of O2 by H2O2 maintains the feed CH4-to-O molar ratio, though it somewhat increases the overall H content of the feed. Consider the following overall reactions below. Though simplistic, Equations (1) and (2) show that replacing O2 with H2O2 should increase the production of C2H4 and H2O, while reducing CO and H2. In addition, adiabatic reactor temperatures should be lower.
3.1. Incremental H2O2 Replacing O2 to PBR at Fixed Feed Temperature
Rivera et al. [11] showed that, for the parametric range considered (Table 1), the highest CH4 conversions for the PBR were nearly 40% for the LR cases, with little impact of flow rate, for a 1243 K feed temperature. The LR_LF case showed the highest sum_C2Hx (sum of C2H6, C2H4, and C2H2) selectivities and yields. First, for the same 1243 K feed temperature, H2O2 was incrementally substituted for O2. It was assumed that H2O2 vapor will be available at 20 mole percent with the balance of H2O vapor. The starting point was the LR_LF (feed CH4/O2 = 7, feed processing rate 37.1/s) case [11] in the PBR. The PBR bed length was that used in the experiments described elsewhere [6]. This high feed temperature did ignore the H2O2 stability issue. The insights gained, however, will help identify the utility of H2O2 as a potential CH4 activator.
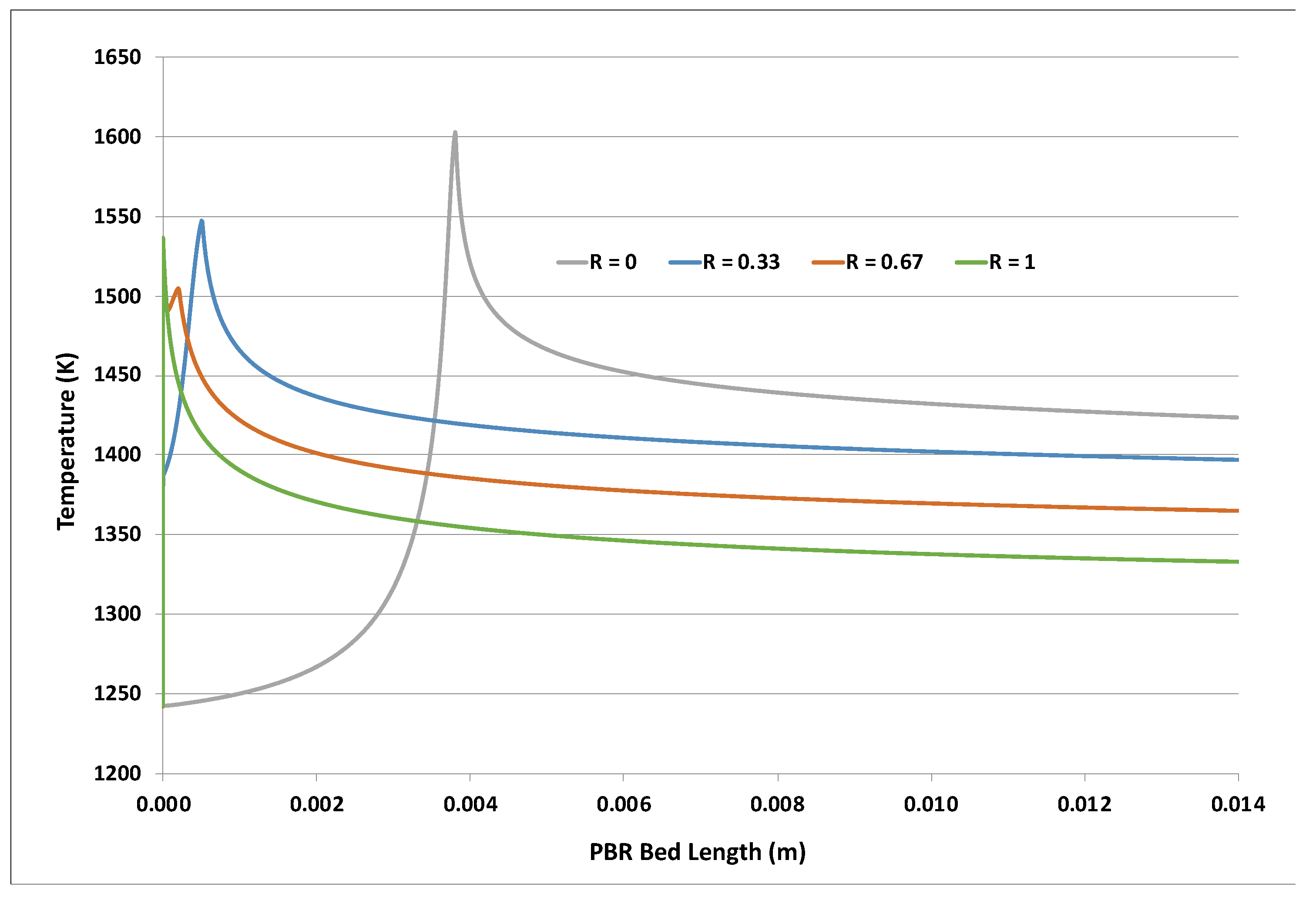
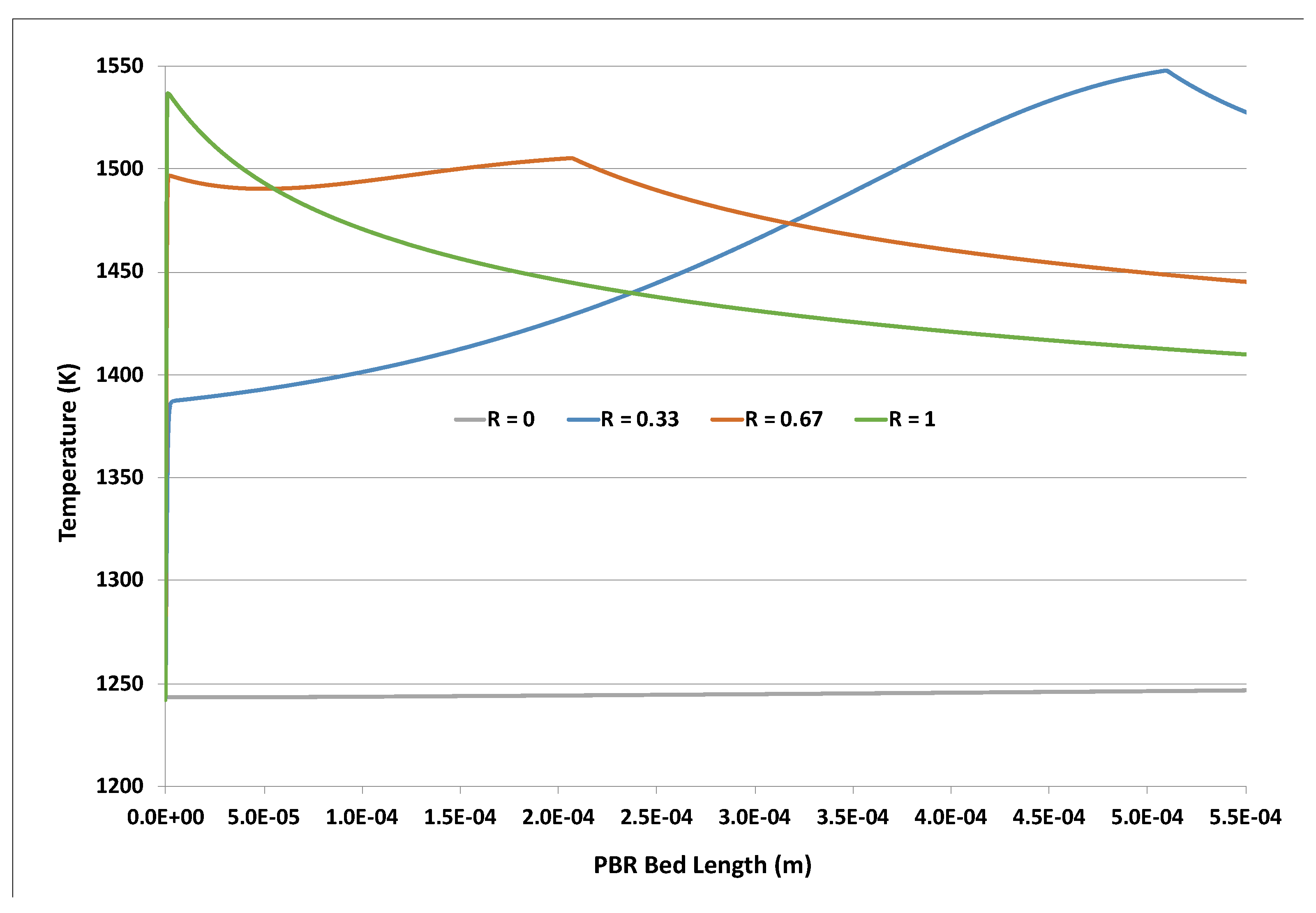
Figure 1 shows that the final PBR bed temperature drops with increasing H2O2 content. This was attributed to the reduced overall reaction exothermicity suggested by Equations (1) and (2). However, the bed temperature peaked much earlier when H2O2 was used in the feed oxidant. A closer examination of the post-entrance region is revealed in Figure 2 below. The H2O2-containing cases showed a near-immediate rise from the 1243 K feed temperature. The very early single peak for the R = 1 (no feed O2) case roughly corresponded to the exhaustion of the H2O2, which was consistent with the extremely rapid H2O2 decomposition described earlier at this temperature. The presence of the feed O2 caused a second local temperature peak further downstream. These peaks (R = 0.33, 0.67 cases) and the much later single peak (R = 0) corresponded approximately to where the O2 ran out, with no further adsorbed •Os.
The R = 1 case (no feed O2) showed almost no adsorbed species, suggesting that all the CH4 conversion effectively occurred in the gas phase (i.e., non-catalytic). On the contrary, for the R = 0.33 and 0.67 cases, while H2O2 was still present, there was a complex parallel/series scheme ongoing with both catalytic and gas-phase reactions occurring. These observations were consistent with the claim that the CH4 conversion is accelerated by •OH gas phase radicals produced from the H2O2 dissociation [18,19].
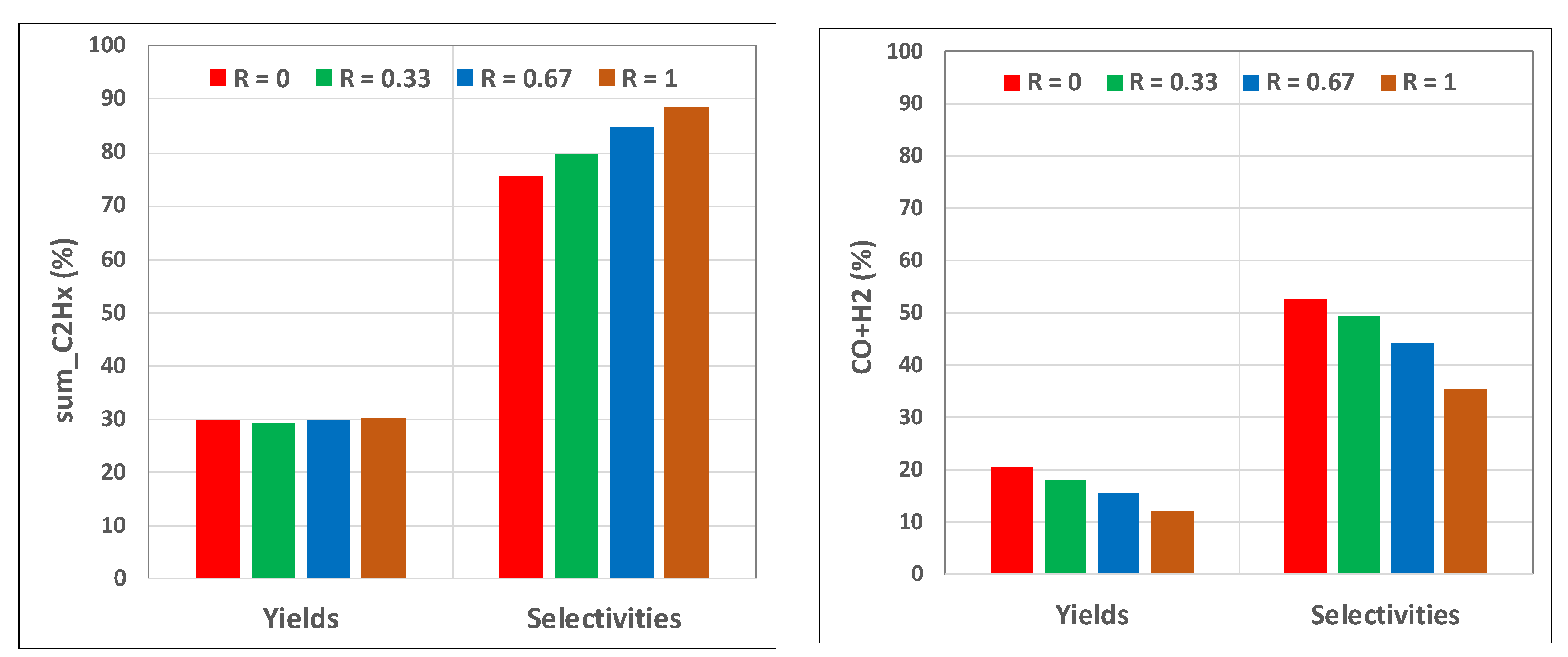
Figure 3 shows that increasing the H2O2 content improved the selectivity of sum_C2Hx, while lowering both the selectivity and yield for syngas (H2 + CO). There was a negligible impact on sum_C2Hx yields. The reduction in syngas was due almost entirely to a reduction in CO. Finally, for these four cases from R = 0 to 1, the CH4 conversions were: 39.3, 36.6, 35.1, and 33.8%, respectively. In all cases, the final O2 and H2O2 conversions were 100%.
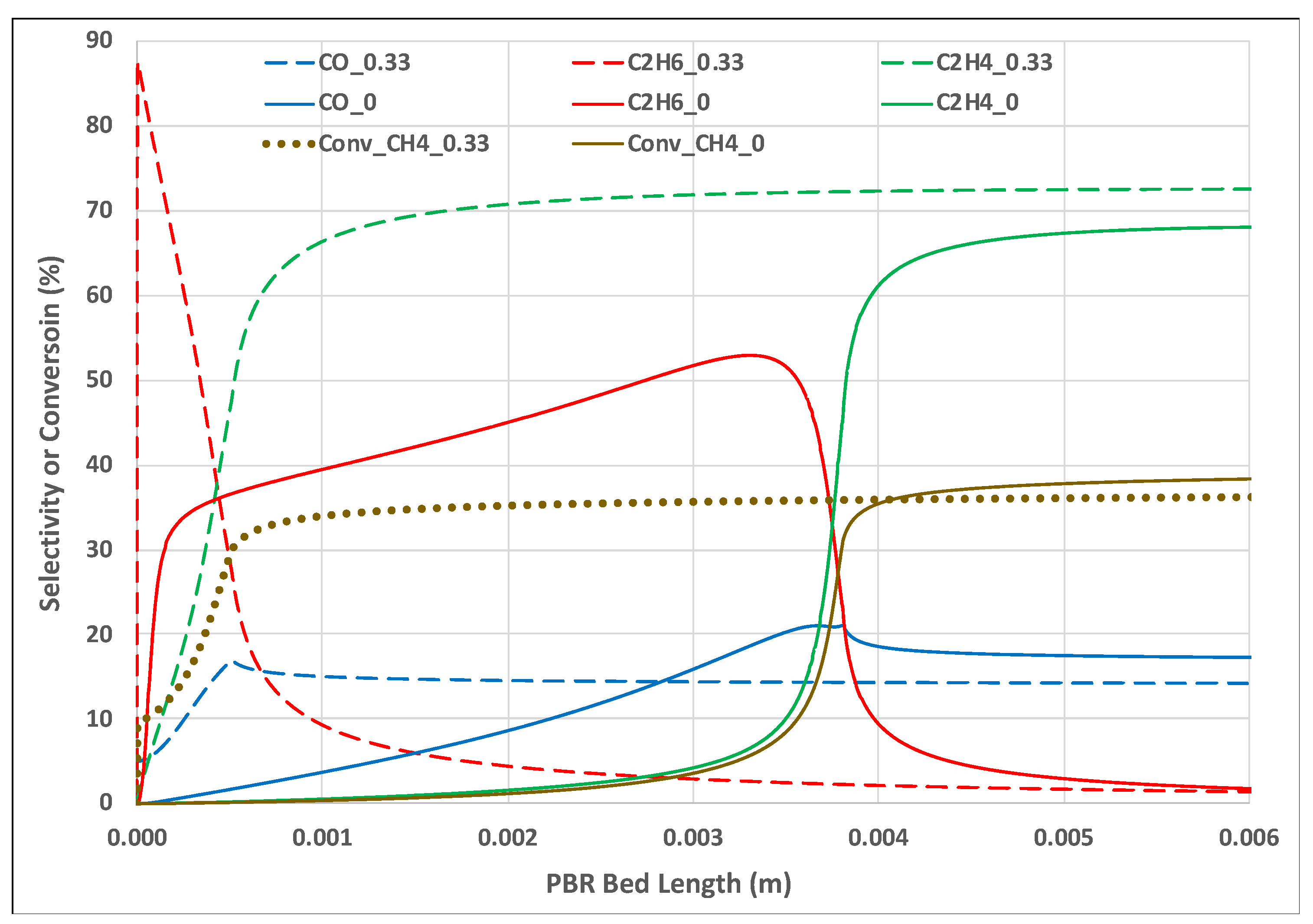
An expanded look at the long-post-entrance region provides more insight into the dramatic impact of substituting some of the feed O2 with H2O2 vapor. Figure 4 shows the selectivities for the CO, C2H6, C2H4, and CH4 conversions for the LR_LF case, at 1243 K feed temperature, for feed ratio cases R = 0 and R = 0.33. The curves were almost unchanged after the 0.006 m bed length was reached.
Some key points can be made here. In the R = 0 case, CO selectivity exceeded C2H4 before the temperature peaked (see Figure 1), but was lower than C2H6. After the peak temperature, consistent with experimental observations by Liu et al. [12], C2H4 exceeded CO. In the R = 0.33 case, the H2O2 (not shown) dissociated immediately upon entry. The resulting •OH radicals abstracted •H atoms from CH4, causing a spike in C2H6 formation, and a quickly rising CH4 conversion. The C2H6 rapidly dehydrogenated to C2H4. The CO peaked at approximately where the temperature peaked. Unlike the R = 0 case, both C2 species exceeded CO prior to the temperature peak. This all occurred much faster than for the R = 0 case. The ultimate CH4 conversion for the R = 0.33 case was only slightly lower than for the R = 0 case, while showing a higher C2H4 selectivity and lower CO. Liu et al. [12], for the R = 0 case, concluded that the selectivities of COx and C2 depended on local O2 concentration and temperature. Using H2O2 added the further complexity of gas phase chemistry to the surface reactions.
3.2. Use of H2O2 to Decrease Feed Temperature to PBR
We now discuss whether the replacement of O2 by H2O2 allows for a lowering of the overall PBR feed temperature, which would be an energy and cost saving. This analysis used the R = 0 and R = 0.33 feeds with the LR_LF case, with the results shown in Table 2.
The partial H2O2 substitution for O2 produced a respectable CH4 conversion at the lower feed temperatures where O2 feed alone showed no OCM activity (R = 0 for 1043 and 943 K feeds). These results at lower feed temperatures were consistent with those observed experimentally [17,18,19]. At the 843 K feed temperature, even the R = 0.33 case was poor.
3.3. Incremental H2O2 Replacing O2 to CSTR
As mentioned above, the CH4/O2 and H2O/H2O2 vapor streams would likely be fed separately into the CSTR due to safety concerns about preheating a vapor stream containing H2O2. For example, for the R = 0.33 and HR_HF case, to achieve an effective (hypothetical) 843 K feed temperature while holding the H2O/H2O2 stream at 373 K, the CH4/O2 stream would be preheated to about 883 K. The HR_HF case was chosen for this CSTR analysis because it showed the best yield and selectivity of sum_C2Hx at the lowest feed temperatures in the earlier study [11].
For the CSTR calculations, the volume was the same as the open (gas) volume of the packed bed, with the same catalyst surface area. The CSTR might be a single-phase ideal fluidized bed, or a perfectly mixed (e.g., jet-stirred) reactor with catalyst on the walls.
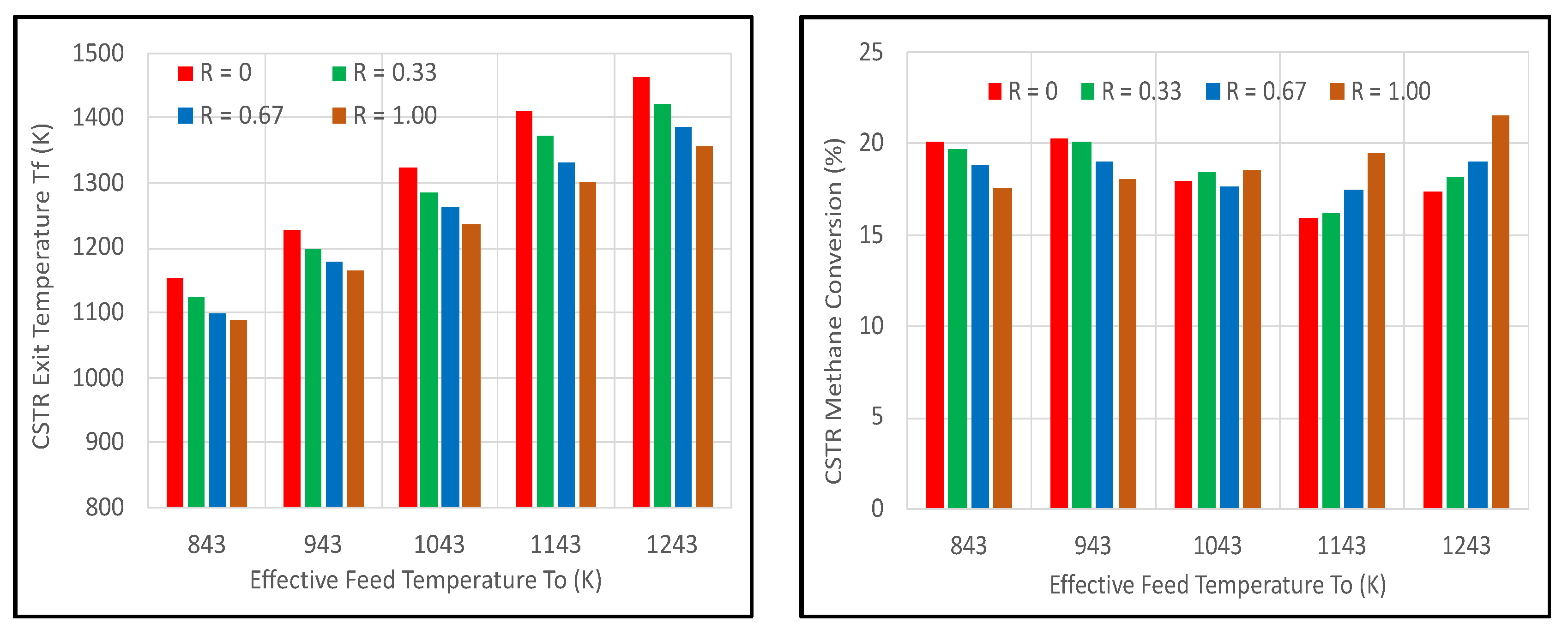
Substitution of all or some of the feed O2 content with H2O2 had a marked impact on the CSTR performance. Figure 5 (left) shows that substituting H2O2 for O2 reduced the exit temperature somewhat (~65–110 K), as might be expected from the lower exothermicity (see Equations (2) and (3)). However, Figure 5 (right) shows a complex story for the impact on CH4 conversion. For effective feed temperatures of 843 and 943 K, switching from O2 to H2O2 reduced CH4 conversion by only ~3 percentage points. With a 1043 K feed temperature, there was little impact on conversion. At 1143 and 1243 K, switching to H2O2 actually increased CH4 conversion. While literature experiments [17,18,19] used a PBR, the results here were still found to be consistent with those observations in terms of the activity of H2O2.
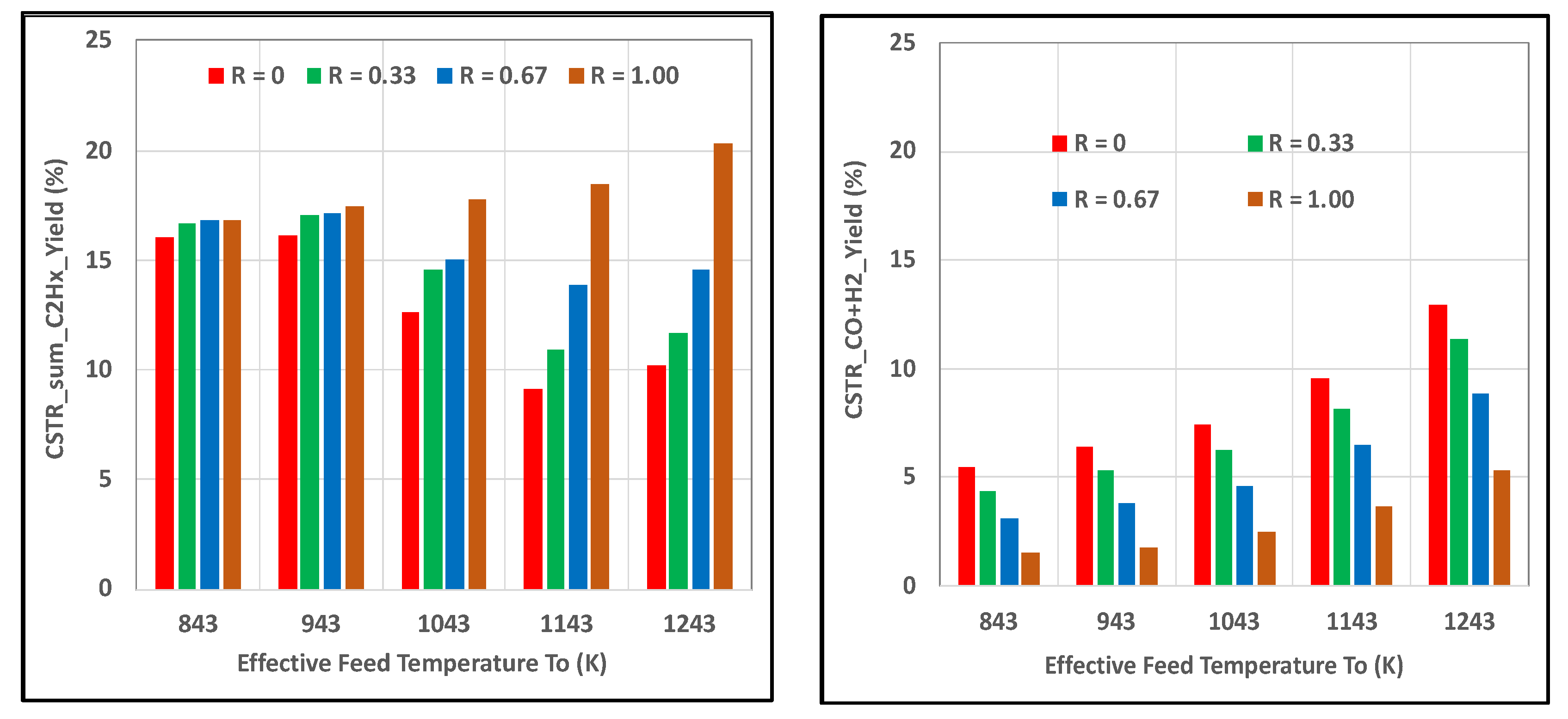
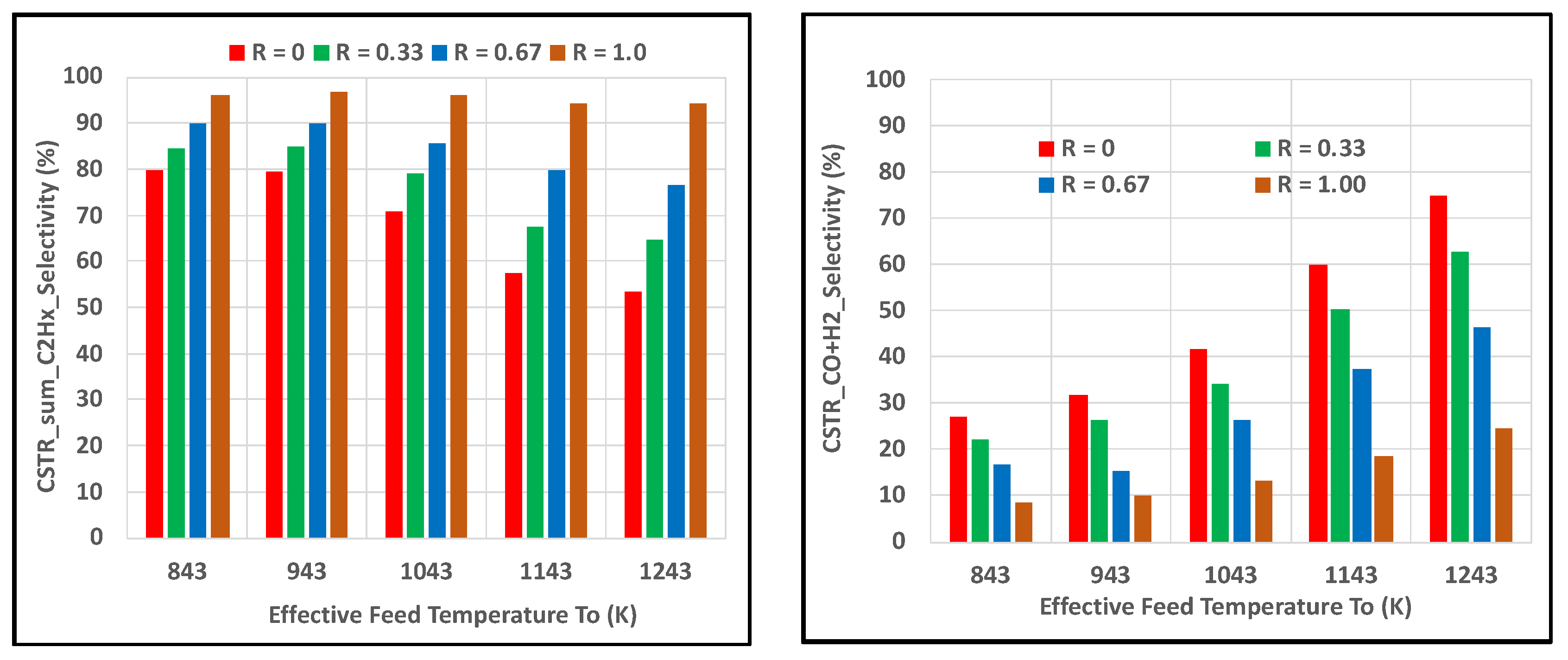
Figure 6 shows that the yield of sum_C2Hx was notably higher than the CO + H2 yield for all R cases, while increasing H2O2 had a greater impact on yields at the higher feed temperatures. These results were consistent with those revealed in Figure 3 for the PBR. Selectivities of sum_C2Hx remained higher than CO + H2, especially at the lower feed temperatures, as seen in Figure 7.
Equations (2) and (3) suggest that replacing O2 by H2O2 will increase the production of C2H4 and H2O, while reducing CO and H2. Consider the HR_HF case, with the effective feed temperature into the CSTR of 1243 K, with the results shown in Table 3. As the H2O2 fraction in the oxidant increased (i.e., higher R value), the CSTR exit temperature fell, but the CH4 conversion increased. The C2H4 and H2O production increased, while CO and H2 dropped. Finally, the fraction of catalytic sites occupied by adsorbed O atoms (Os) decreased as R increased. Since •H abstraction by •Os is the primary catalytic step for CH4 activation [6] by O2, the drop in •Os fraction was consistent with a shift from heterogeneous catalyzed to homogeneous non-catalyzed conversion pathways at higher R.
3.4. Brief Reactor Comparison Summary
A simple comparison between the PBR and CSTR for OCM with and without H2O2 as an activating oxidant is shown in Table 4. This illustration is based on the LR_LF case (see Table 1). The results will vary somewhat for other cases, but the observations will be similar. This summary considers both the current results and those published earlier with just O2 as an activator [11]. While the claims are based on calculations using the Karakaya et al. [6] mechanism for La2O3/CeO2 catalyst, it is anticipated other OCM catalysts would give rise to similar claims.
Table 4 shows several points. The lowest practical feed temperature is the value below which there is no appreciable CH4 conversion. All remaining values in each column correspond to those temperatures. Replacing a portion of the feed O2 with H2O2 vapor allows the CSTR to achieve good CH4 conversions at the lowest feed temperature. It also allows the PBR to run with a reduced feed temperature. Even at this low feed temperature, the CSTR has a sum_C2Hx selectivity that exceeds the PBR at a much higher temperature. The CSTR also shows reduced syngas (CO + H2) and improved sum_C2Hx selectivity when using the H2O2.
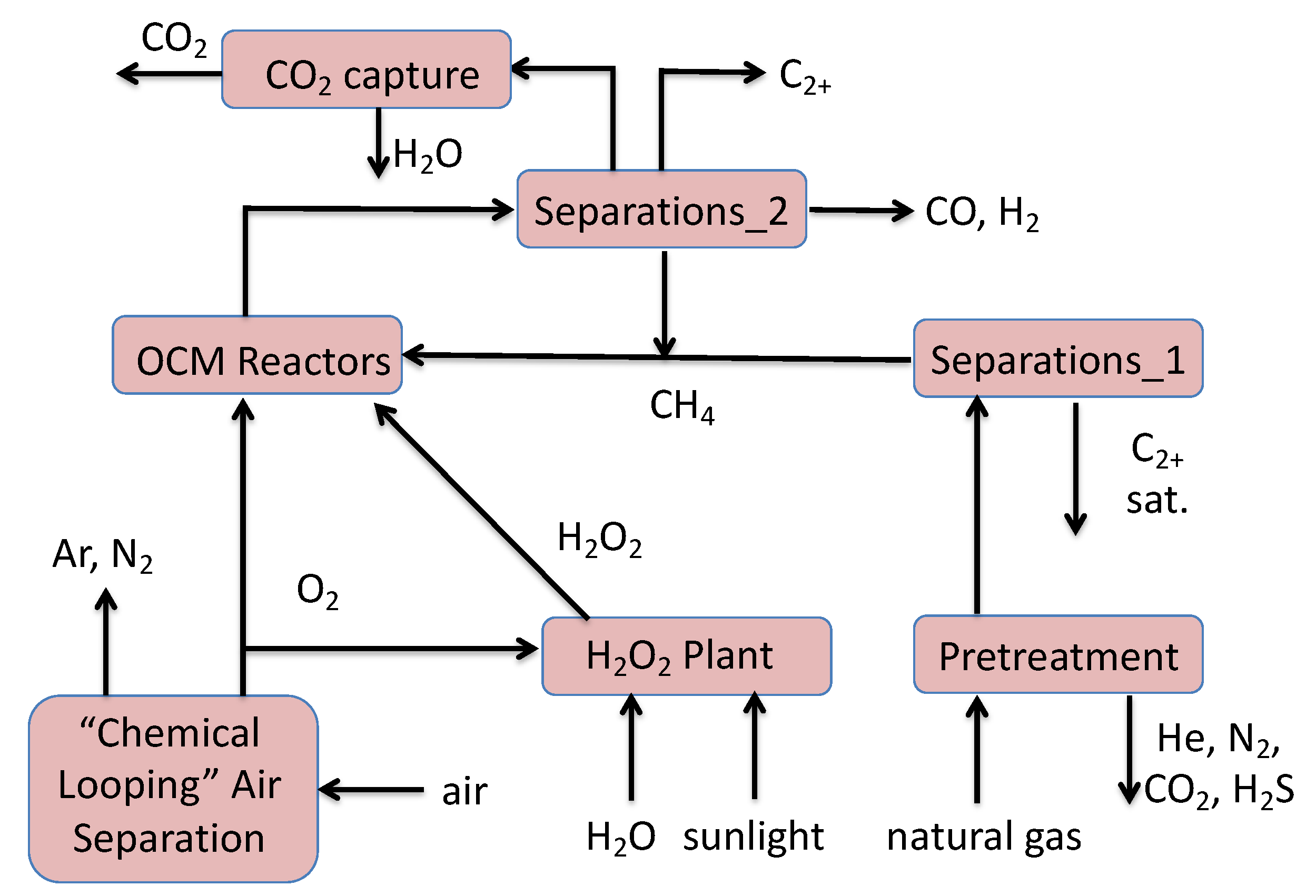
4. Simple Layout of a “Green” OCM Plant
Although Equations (2) and (3) show that OCM via O2 and H2O2 is exothermic, a future sustainable OCM plant must consider O2 and H2O2 production and overall plant heat integration. Figure 8 offers a simple schematic. The OCM reactor feeds CH4 and a combination of O2 and vapor phase moist H2O2. The O2 is produced in a solar powered air separation plant [23] that enjoys the energy and economic savings from chemical looping instead of cryogenic separation [24,25]. Aqueous H2O2 is produced by the solar powered catalyzed reaction of O2 and acidic liquid H2O [20]. Vapor phase H2O2 is stripped out of the liquid by the N2 or He [21] recovered from the natural gas. Heavier-than-CH4 saturated hydrocarbons (C2H6-C5H12) are separated out from the natural gas. Post-OCM reactor processing separates the CO and H2 as synthesis gas and the desirable coupled hydrocarbons (e.g., C2H4 and C2H6). Byproduct CO2 from Separations_2 and Pretreatment can be captured with caustic scrubbing, and subsequently sequestered.
5. Conclusions
Using an elementary reaction mechanism for the oxidative coupling of methane (OCM) on a La2O3/CeO2 catalyst borrowed from the literature, this study considered the incremental replacement of the activating O2 with moist H2O2 vapor. Both packed bed reactor (PBR) and continuous stirred tank reactor (CSTR) configurations were used. As the H2O2 content of the oxidant increased, more of the CH4 conversion occurred in the gas phase with less assistance from the catalytic surface. Hydroxyl (•OH) radicals from rapid H2O2 decomposition abstracted •H atoms from CH4 to produce •CH3 radicals. This occurred in parallel to a similar abstraction by oxygen atoms (•Os) adsorbed on the catalyst surface when O2 was fed. In the PBR, H2O2 allowed the “light-off” temperature jump to occur using a lower feed temperature. Even though there was a slight decline in CH4 conversion, the C2Hx selectivity increased while synthesis gas dropped. Since significant preheat was still needed, process safety considerations might dictate that H2O2 vapor is better suited to the continuous stirred tank reactor (CSTR) configuration where the H2O2/H2O vapor stream can be fed at lower temperatures separately from the preheated CH4/O2 stream. In a CSTR, H2O2 significantly improved C2Hx production compared to synthesis gas over all feed temperatures studied, thus showing that OCM is possible with significantly less preheating compared to PBR. A future OCM plant can operate in a more “green” way with the use of solar-activated H2O2 production, and solar-powered O2 production from chemical-looping air separation.
Author Contributions
A.M.—calculations, S.R.—calculations, P.P.—calculations, M.L.—calculations, R.B.—calculations, writing, supervision, total responsibility. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Supporting data are not posted, but are available upon request.
Acknowledgments
The authors appreciate the support of Canan Karakaya in using Detchem®.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Compound samples are not available from the authors.
References
- Yang, S.; Dezember, R. The U.S. Is Overflowing with Natural Gas. Not Everyone Can Get It. Wall Street J. 8 July 2019. Available online: https://www.wsj.com/articles/the-u-s-is-overflowing-with-natural-gas-not-everyone-can-get-it-11562518355 (accessed on 8 July 2019).
- Kohler, J. Colorado Regulators Give Initial OK to Ban on Flaring of Oil, Gas Wells to Curb Methane Pollution. The Denver Post. Available online: https://www.denverpost.com/2020/11/06/colorado-venting-flaring-oil-gas-wells-methane-pollution (accessed on 6 November 2020).
- NMOGA. New Mexico Oil & Gas Association. Available online: https://www.nmoga.org/exxon_mobil_to_use_satellites_to_detect_methane_emissions_in_permian_basin (accessed on 17 December 2021).
- Karakaya, C.; Morejudo, S.H.; Zhu, H.; Kee, R.J. Catalytic chemistry for methane dehydroaromatization (MDA) on a bifunctional Mo/HZM-5 catalyst in a packed bed. Ind. Eng. Chem. Res. 2016, 55, 9895–9906. [Google Scholar] [CrossRef]
- Stansch, Z.; Mleczko, L.; Baerns, M. Comprehensive Kinetics of Oxidative Coupling of Methane over the La2O3/CaO Catalyst. Ind. Eng. Chem. Res. 1997, 36, 2568–2579. [Google Scholar] [CrossRef]
- Karakaya, C.; Zhu, H.; Zohour, B.; Senkan, S.; Kee, R.J. Detailed Reaction Mechanisms for the Oxidative Coupling of Methane over La2O3/CeO2 Nanofiber Fabric Catalysts. Chem. Cat. Chem. 2017, 9, 4538–4551. [Google Scholar]
- Gambo, Y.; Jalil, A.A.; Triwahyono, S.; Abdulrasheed, A.A. Recent Advances and future prospect in catalysts for oxidative coupling of methane to ethylene: A review. J. Ind. Eng. Chem. 2018, 59, 218–229. [Google Scholar] [CrossRef]
- Siluria. 2014. Available online: http://siluria.com/Technology/Demonstration_Plant (accessed on 11 January 2022).
- Department of Energy (DOE). 2017. Available online: https://www.osti.gov/servlets/purl/1414280 (accessed on 11 January 2022).
- Cruellas, A.; Melchiori, T.; Gallucci, F.; van Sint Annaland, M. Advanced reactor concepts for oxidative coupling of methane. Cat. Rev. 2017, 59, 234–294. [Google Scholar] [CrossRef] [Green Version]
- Rivera, S.; Molla, A.; Pera, P.; Landaverde, M.; Barat, R. Reactor engineering calculations with a detailed reaction mechanism for the oxidative coupling of methane. Int. J. Chem. React. Eng. 2020, 18. [Google Scholar] [CrossRef]
- Liu, Z.; Li, J.P.H.; Vovk, E.; Zhu, Y.; Li, S.; Wang, S.; van Bavel, A.P.; Yang, Y. Online Kinetics Study of Oxidative Coupling of Methane over La2O3 for Methane Activation; What is Behind the Distinguished Light-Off Temperatures? ACS Catal. 2018, 8, 11761–11772. [Google Scholar] [CrossRef]
- Beck, B.; Fleischer, V.; Arndt, S.; Hevia, M.; Urakawa, A.; Hugo, P.; Schomacker, R. Oxidative coupling of methane—A complex surface/gas phase mechanism with strong impact on the reaction engineering. Cat. Today 2014, 228, 212–218. [Google Scholar] [CrossRef]
- Chen, Q.; Couwenberg, P.M.; Marin, G.B. Effect of pressure on the oxidative coupling of methane in the absence of catalyst. AIChE J. 1994, 40, 521–535. [Google Scholar] [CrossRef]
- Alexiadis, V.; Thybaut, J.; Kechagiopoulos, P.; Chaar, M.; Van Veen, A.; Muhler, M.; Marin, G. Oxidative coupling of methane: Catalytic behavior assessment via comprehensive microkinetic modeling. Appl. Cat. B Environ. 2014, 150–151, 496–505. [Google Scholar] [CrossRef]
- Deutschmann, O.; Tischer, S.; Correa, C.; Chatterjee, D.; Kleditzsch, S.; Janardhanan, V.M.; Mladenov, N.; Minh, H.D.; Karadeniz, H.; Hettel, M. DETCHEM Software Package, 2.5 ed.; DETCHEM: Karlsruhe, Germany, 2014; Available online: www.detchem.com (accessed on 11 January 2022).
- Garibyan, T.; Muradyan, A.; Grigoryan, R.; Vartikyan, L.; Minasyan, V.; Manukyan, N. New methods of increasing the catalytic activity and selectivity in the oxidative conversion processes of methane and propylene. Cat. Today 1995, 24, 249–250. [Google Scholar] [CrossRef]
- Eskendirov, I.; Coville, N.J.; Sokolovskii, V. Methane oxidative coupling on the Au/La2O3/CaO catalyst in the presence of hydrogen peroxide. Cat. Lett. 1995, 35, 33–37. [Google Scholar] [CrossRef]
- Eskendirov, I.; Coville, N.; Parmaliana, A.; Sokolovskii, V. Direct oxidative conversion of methane into higher hydrocarbons and oxy-products in the presence of hydrogen peroxide. In Natural Gas Conversion IV; de Pontes, M., Espinoza, R., Nicolaides, C., Scholz, J., Scurrell, M., Eds.; Studies in Surface Science and Catalysis; Elsevier: Amsterdam, The Netherlands, 1997; Volume 107, pp. 301–306. [Google Scholar]
- Kaynan, N.; Berke, B.A.; Hazut, O.; Yerushalmi, R. Sustainable photocatalytic production of hydrogen peroxide from water and molecular oxygen. J. Mat. Chem. A. 2014, 2, 13822–13826. [Google Scholar] [CrossRef]
- Spiegelman, J.; Alvarez, D. Cheating Raoult’s Law to Enable Delivery of Hydrogen Peroxide as a Stable Vapor. Available online: https://www.peroxidizer.com/resources/articles/article-Cheating-Raoults-Law-Stabilized-Hydrogen-Peroxide-Vapor.pdf (accessed on 11 January 2022).
- Satterfield, C.; Stein, T. Decomposition of Hydrogen Peroxide Vapor on Relatively Inert Surfaces. Ind. Eng. Chem. 1957, 49, 1173–1180. [Google Scholar] [CrossRef]
- Patzschke, C.F.; Bahzad, H.; Boot-Handford, M.E.; Fennell, P.S. Simulation of a 100-MW solar-powered thermo-chemical air separation system combined with an oxy-fuel power plant for bio-energy with carbon capture and storage (BECCS). Mitig. Adapt. Strateg. Glob. Change 2020, 25, 539–557. [Google Scholar] [CrossRef] [Green Version]
- Qing, M.; Jin, B.; Ma, J.; Zou, X.; Wang, X.; Zheng, C.; Zhao, H. Thermodynamic and economic performance of oxy-combustion power plants integrating chemical looping air separation. Energy 2020, 206, 118136. [Google Scholar] [CrossRef]
- Shah, K.; Moghtaderi, B.; Wall, T. Chemical Looping Air Separation (CLAS) for Oxygen production: Thermodynamic and Economic Aspects. In Proceedings of the Australian Combustion Symposium, Newcastle, Australia, 29 November–1 December 2011; pp. 240–243. [Google Scholar]
Figure 1.
Impact of H2O2 content in feed oxidant; PBR LR_LF case at 1243 K feed; curves are different feed molar values R ≡ H2O2/(O2 + H2O2).
Figure 1.
Impact of H2O2 content in feed oxidant; PBR LR_LF case at 1243 K feed; curves are different feed molar values R ≡ H2O2/(O2 + H2O2).

Figure 2.
Post-entrance region of PBR for LR_LF case at 1243 K feed; curves are different feed molar values R ≡ H2O2/(O2 + H2O2).
Figure 2.
Post-entrance region of PBR for LR_LF case at 1243 K feed; curves are different feed molar values R ≡ H2O2/(O2 + H2O2).

Figure 3.
Impact of H2O2 content in feed oxidant; PBR LR_LF case at 1243 K feed; R ≡ H2O2/(O2 + H2O2). Values are based on PBR outlet; (left) sumC2Hx = C2H6 + C2H4 + C2H2; (right) CO + H2.
Figure 3.
Impact of H2O2 content in feed oxidant; PBR LR_LF case at 1243 K feed; R ≡ H2O2/(O2 + H2O2). Values are based on PBR outlet; (left) sumC2Hx = C2H6 + C2H4 + C2H2; (right) CO + H2.

Figure 4.
PBR long-post-entrance region for LR_LF case, 1243 K feed; key sensitivities and conversions for R = 0, 0.33 oxidant ratios.
Figure 4.
PBR long-post-entrance region for LR_LF case, 1243 K feed; key sensitivities and conversions for R = 0, 0.33 oxidant ratios.

Figure 5.
Impact of H2O2 content in feed oxidant on CSTR exit temperature (left) and CH4 conversion (right) for HR_HF case where molar R = H2O2/(O2 + H2O2).
Figure 5.
Impact of H2O2 content in feed oxidant on CSTR exit temperature (left) and CH4 conversion (right) for HR_HF case where molar R = H2O2/(O2 + H2O2).

Figure 6.
Impact of H2O2 content in feed oxidant on sum_C2Hx and CO + H2 yields in CSTR for HR_HF case where molar R = H2O2/(O2 + H2O2); (left) sumC2Hx = C2H6 + C2H4 + C2H2; (right) CO + H2.
Figure 6.
Impact of H2O2 content in feed oxidant on sum_C2Hx and CO + H2 yields in CSTR for HR_HF case where molar R = H2O2/(O2 + H2O2); (left) sumC2Hx = C2H6 + C2H4 + C2H2; (right) CO + H2.

Figure 7.
Impact of H2O2 content in feed oxidant on sum_C2Hx and CO + H2 selectivities in CSTR for HR_HF case where molar R = H2O2/(O2 + H2O2); (left) sumC2Hx = C2H6 + C2H4 + C2H2; (right) CO + H2.
Figure 7.
Impact of H2O2 content in feed oxidant on sum_C2Hx and CO + H2 selectivities in CSTR for HR_HF case where molar R = H2O2/(O2 + H2O2); (left) sumC2Hx = C2H6 + C2H4 + C2H2; (right) CO + H2.

Figure 8.
Hypothetical “green” OCM plant utilizing solar-powered H2O2 production and chemical looping for air separation.
Figure 8.
Hypothetical “green” OCM plant utilizing solar-powered H2O2 production and chemical looping for air separation.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Process parameters of this study, inspired by Karakaya et al. [6].
Table 1.
Process parameters of this study, inspired by Karakaya et al. [6].
| Low Feed Ratio “LR” CH4/O2 or CH4/H2O2 = 7 | High Feed Ratio “HR” CH4/O2 or CH4/H2O2 = 11 | |||
| Low Feed Processing Rate “LF” 37.1/s | High Feed Processing Rate “HF” 58.3/s | |||
| Feed Temperatures Tin (K) | ||||
| 843 | 943 | 1043 | 1143 | 1243 |
Table 2.
Impact of partial replacement of feed O2 by H2O2 at various feed temperatures for PBR running LR_LF case.
Table 2.
Impact of partial replacement of feed O2 by H2O2 at various feed temperatures for PBR running LR_LF case.
| Feed Temperature (K) | 1243 | 1143 | 1043 | 1043 | 943 | 943 |
| R value | 0 | 0 | 0 | 0.33 | 0 | 0.33 |
| Max Temp (K) | 1603 | 1547 | 1065 | 1414 | 944 | 1345 |
| Exit Temp (K) | 1424 | 1423 | 1065 | 1343 | 944 | 1321 |
| CH4 Conv (%) | 39.3 | 33.8 | 0.891 | 26.4 | 0.044 | 21.8 |
| sum_C2Hx Selec (%) | 75.6 | 72.3 | 58.7 | 70.0 | 36.7 | 62.6 |
| sum_C2Hx Yield (%) | 29.7 | 24.4 | 0.522 | 18.5 | 0.016 | 13.6 |
| CO + H2 Sel. (%) | 52.4 | 51.6 | 24.3 | 54.5 | 10.2 | 57.8 |
| CO + H2 Yield (%) | 20.6 | 17.5 | 0.217 | 14.4 | 0.004 | 12.6 |
Table 3.
Impact of progressive replacement of feed O2 by H2O2 at 1243 K effective feed temperatures for CSTR running HR_HF case. * H2O values are corrected for H2O in feed for R ≠ 0 cases.
Table 3.
Impact of progressive replacement of feed O2 by H2O2 at 1243 K effective feed temperatures for CSTR running HR_HF case. * H2O values are corrected for H2O in feed for R ≠ 0 cases.
| R Value | 0 | 0.33 | 0.67 | 1 |
| Exit Temp (K) | 1463 | 1421 | 1386 | 1356 |
| CH4 Conv (%) | 17.3 | 18.1 | 19.0 | 21.5 |
| C2H4 Selec (%) | 46.1 | 52.6 | 58.8 | 70.0 |
| C2H4 Yield (%) | 7.98 | 9.50 | 11.2 | 15.1 |
| H2O Selec (%) * | 29.1 | 34.3 | 38.5 | 42.1 |
| H2O Yield (%) * | 5.03 | 6.19 | 7.32 | 9.07 |
| CO Selec (%) | 35.1 | 26.7 | 14.9 | 0.10 |
| CO Yield (%) | 6.05 | 4.82 | 2.84 | 0.02 |
| H2 Selec (%) | 39.8 | 36.1 | 31.5 | 24.5 |
| H2 Yield (%) | 6.89 | 6.51 | 5.99 | 5.27 |
| 24.5 | 15.8 | 9.61 | 0.48 |
Table 4.
Brief comparison of PBR vs. CSTR for OCM using the LR_LF case and R = 0.33 for the O2/H2O2 runs.
Table 4.
Brief comparison of PBR vs. CSTR for OCM using the LR_LF case and R = 0.33 for the O2/H2O2 runs.
| PFR | PFR | CSTR | CSTR | |
|---|---|---|---|---|
| O2 | O2/H2O2 | O2 | O2/H2O2 | |
| Lowest practical feed temperature (K) | 1143 | 943 | 843 | 843 |
| CH4 conversion | 34 | 22 | 23 | 28 |
| Sum_C2Hx selectivity | 72 | 63 | 55 | 80 |
| CO + H2 selectivity | 52 | 58 | 57 | 33 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Molla, A.; Rivera, S.; Pera, P.; Landaverde, M.; Barat, R. Expanded Reactor Engineering Calculations for the Oxidative Coupling of Methane. Methane 2022, 1, 58-69. https://doi.org/10.3390/methane1010005
AMA Style
Molla A, Rivera S, Pera P, Landaverde M, Barat R. Expanded Reactor Engineering Calculations for the Oxidative Coupling of Methane. Methane. 2022; 1(1):58-69. https://doi.org/10.3390/methane1010005
Chicago/Turabian StyleMolla, Andrin, Sonya Rivera, Phillip Pera, Michael Landaverde, and Robert Barat. 2022. "Expanded Reactor Engineering Calculations for the Oxidative Coupling of Methane" Methane 1, no. 1: 58-69. https://doi.org/10.3390/methane1010005