The Rotenone Models Reproducing Central and Peripheral Features of Parkinson’s Disease


Department of Medical Neurobiology, Okayama University Graduate School of Medicine, Dentistry and Pharmaceutical Sciences, Okayama 700-8558, Japan
*
Author to whom correspondence should be addressed.
NeuroSci 2020, 1(1), 1-14; https://doi.org/10.3390/neurosci1010001
Submission received: 10 July 2020
/
Revised: 31 July 2020
/
Accepted: 1 August 2020
/
Published: 5 August 2020
Abstract
:Parkinson’s disease (PD) is a complex, multi-system, neurodegenerative disorder; PD patients exhibit motor symptoms (such as akinesia/bradykinesia, tremor, rigidity, and postural instability) due to a loss of nigrostriatal dopaminergic neurons, and non-motor symptoms such as hyposmia, autonomic disturbance, depression, and REM sleep behavior disorder (RBD), which precedes motor symptoms. Pathologically, α-synuclein deposition is observed in the central and peripheral nervous system of sporadic PD patients. To clarify the mechanism of neurodegeneration in PD and to develop treatment to slow or stop PD progression, there is a great need for experimental models which reproduce neurological features of PD. Animal models exposed to rotenone, a commonly used pesticide, have received most attention since Greenamyre and his colleagues reported that chronic exposure to rotenone could reproduce the anatomical, neurochemical, behavioral, and neuropathological features of PD. In addition, recent studies demonstrated that rotenone induced neuropathological change not only in the central nervous system but also in the peripheral nervous system in animals. In this article, we review rotenone models especially focused on reproducibility of central and peripheral multiple features of PD. This review also highlights utility of rotenone models for investigation of PD pathogenesis and development of disease-modifying drugs for PD in future.
1. Introduction
Parkinson’s disease (PD) is the second most common neurodegenerative disease. PD has been characterized by loss of nigrostriatal dopaminergic neurons, which causes motor symptoms such as akinesia/bradykinesia, tremor, rigidity, and postural instability. These days, PD is well known as a complex, multi-system, neurodegenerative disorder; PD patients also exhibit non-motor symptoms such as hyposmia, autonomic disturbance, depression, and REM sleep behavior disorder (RBD), which precedes motor symptoms. These prodromal PD symptoms are important for early recognition of incident PD. The pathological hallmark of PD is the presence of Lewy body, which contains α-synuclein, in the cytoplasm of neurons. Braak et al. [1] reported that PD pathology, Lewy bodies and neurites, within the central nervous system (CNS), appeared first in the olfactory bulb and the brainstem (dorsal motor nucleus of the vagus (DMV)), and then spread progressively through the substantia nigra (SN), eventually leading to motor dysfunction, to reach the cerebral cortex. In addition, several reports have demonstrated that PD pathology is also detected within the peripheral nervous system (PNS): the intestinal myenteric plexus, gastric mucosa, cardiac sympathetic nerve, and skin nerve. Constipation is a typical prodromal non-motor symptom in PD, which precedes motor symptoms by 10–20 years [2,3,4]. Therefore, it has been hypothesized that PD pathology propagates from the enteric nervous system (ENS) to the CNS via vagal nerve [5,6,7], although the pathogenesis in sporadic PD remains unknown.
To clarify the mechanism of neurodegeneration in PD and to develop treatment to slow or stop PD progression, there is a great need for experimental models which reproduce neurological features of PD. For the past several decades, researchers reported various animal models using environmental or synthetic neurotoxins, or expressing familial PD-related gene mutations [8,9]. As mentioned above, PD neuropathology is not restricted to the nigrostriatal dopamine (DA) pathway. Accordingly, animal models reproducing central and peripheral features of PD are required. Rotenone is used to develop animal models of PD. Rotenone is a naturally occurring insecticide and pesticide that is derived from the roots of several plants. Rotenone is highly lipophilic and therefore easily crosses the biological membrane, including blood–brain barrier. It is well known that rotenone inhibits selectively mitochondrial complex I, which causes ATP reduction and electron leakage, followed by production of reactive oxygen species [10,11]. Rotenone models have received most attention since Greenamyre and his colleagues reported that chronic exposure to rotenone could reproduce the histological, neurochemical, behavioral, and neuropathological features of PD [12,13]. In addition, various studies demonstrated that rotenone induced neuropathological changes not only in the CNS but also in the PNS in animals. Moreover, the importance of rotenone models is strengthened by epidemiological studies suggesting that pesticide exposure, particularly rotenone and paraquat, increases the risk of PD [14,15]. Here, we review rotenone models especially focused on reproducibility of central and peripheral multiple features of PD.
2. Rotenone-Induced Central and Peripheral Neuropathology
2.1. Neuropathology in the CNS
Remarkable findings were reported by Greenamyre and his colleagues; chronic systemic exposure to rotenone induced highly selective nigrostriatal dopaminergic degeneration accompanied by Lewy body-like cytoplasmic inclusions and exhibited behavioral features of PD such as hypokinesia and rigidity [12,13]. Since then, various studies demonstrated that rotenone treatment by different routes of administration, such as intraperitoneal, subcutaneous, oral, and intranasal injection, induced degeneration of nigrostriatal DA pathway in animals, including results from our laboratory [16,17,18,19,20,21,22,23,24,25,26].
As mentioned above, based on the Braak’s staging hypothesis [1], PD pathology in the CNS is supposed to start from the olfactory bulb and the DMV, then spreads to the SN, and finally reaches the cerebral cortex. To date, there are several reports demonstrating that rotenone administration causes neuropathological change in the CNS that corresponds to Braak’s staging. Sherer et al. [11] reported that systemic rotenone infusion by subcutaneous implantation of osmotic mini pumps modestly elevated protein carbonyl levels as a measure of oxidative damage in the cortex and striatum, but not in the cerebellum and hippocampus. In their rotenone-exposed rats, the largest elevations in soluble protein carbonyls occurred in dopaminergic areas, the midbrain and olfactory bulb. Our previous study also demonstrated that 6 week rotenone (50 mg/kg/day) administration reduced the number of tyrosine hydroxylase (TH)-positive neurons in the glomerular layer of olfactory bulb [21]. However, the dose of rotenone (50 mg/kg/day) in our previous research in 2015 was extremely high; rotenone treatment induced high mortality. Morais et al. examined effects of oral administration of rotenone on olfactory function in mice [27]. They demonstrated that oral administration of rotenone (30 mg/kg) for 7 days disrupted olfactory discrimination. Furthermore, recent studies demonstrated that intranasal administration of rotenone induced hyposmia through lesion of dopaminergic neurons, activation of microglia, and enhanced α-synuclein phosphorylation and its accumulation in the olfactory bulb [28,29]. These results suggest that the inhalation of environmental toxins induces the neurodegeneration of cranial neurons through olfactory transport. Olfactory impairment is a predominant prodromal PD symptom. Considering Braak’s staging theory, the olfactory bulb is focused upon as a gateway to environmental toxin as well as the ENS. Therefore, intranasal administration of rotenone can provide useful PD models.
When the striatal DA levels are decreased by 60–80%, the PD motor symptoms appear [30]. Although the neuropathological mechanisms underlying non-motor symptoms of PD are not well understood, there is evidence suggesting that non-motor symptoms may arise from the disruption of both dopaminergic and nondopaminergic systems, and the involvement of diverse structures outside the nigrostriatal system [31,32]. Multiple neurotransmitter pathways, in addition to the DA, could be involved in the pathogenesis of PD: noradrenergic system in the locus coeruleus, the serotonergic system in the dorsal raphe nuclei, and the cholinergic system of the nucleus basalis of Meynert and the pedunculopontine nucleus [33]. Recently, Almeida et al. [34] reported that exposure to a low dose of rotenone (2 mg/kg/day) by subcutaneous infusion with mini pumps for 1 month increased hyperphosphorylated Tau, α-synuclein, amyloid-beta peptide, and protein carbonylation in the hippocampus, SN, and locus coeruleus of aged Lewis rats. As mentioned above, locus coeruleus is also a degenerative brain region in PD, which precedes the impairment of nigrostriatal DA pathway [35]. In addition, there is another report examining effects of rotenone on nondopaminergic systems; intravenous infusion of rotenone (2.5 mg/kg/day) for 28 days induced loss of striatal dopaminergic fibers, nigral dopaminergic neurons, striatal serotonergic fibers, striatal dopamine- and cAMP-regulated phosphoprotein of 32 kDa (DARPP-32)-positive projection neurons, striatal cholinergic interneurons, cholinergic neurons in the pedunculopontine tegmental nucleus, and noradrenergic neurons in the locus coeruleus in Lewis rats [36]. These results suggest that rotenone can cause neurodegeneration and α-synuclein accumulation in the PD pathology-related regions in the CNS.
2.2. Neuropathology in the PNS
Several studies demonstrated that rotenone induced neuropathological change not only in the CNS but also in the PNS in animals [37,38,39]. The DMV is important in autonomic control of the bowel movement. Constipation is a well-known non-motor symptom in PD, which precedes motor symptoms by 10–20 years [2,3,4]. In addition, several reports have demonstrated that PD pathology is also detected within the ENS [40,41]. Therefore, the DMV can be a key area as a junction of progression of PD pathology from the ENS to the CNS via vagal nerve [5,6,7,42]. Pan-Montojo et al. demonstrated that chronically and intragastric-administered rotenone (5 mg/kg/day) induced α-synuclein accumulation in the DMV and the intermediolateral nucleus of the spinal cord (IML) [38]. However, in their rotenone models, neuronal loss was not observed in the DMV and IML. We recently reported neuropathological change in the DMV of rotenone-treated mice; chronic systemic exposure to a low dose of rotenone (2.5 mg/kg/day) for 4 weeks by subcutaneous implantation of rotenone-filled osmotic mini pump induced degeneration of cholinergic neurons in the DMV, where α-synuclein was accumulated in neuronal soma [19].
Experiments, including our research, showed rotenone exposure induced α-synuclein accumulation in the ENS of mice [19,37,38,39]. Drolet et al. demonstrated that rotenone treatment (2.0 mg/kg, intraperitoneal (i.p.)) for 6 weeks induced loss of small intestine myenteric plexus and α-synuclein aggregation pathology similar to that of enteric Lewy-bodies in PD [37]. Pan-Montojo et al. [38] reported that daily intragastric administration of rotenone (5 mg/kg) induced α-synuclein accumulation in the ENS besides the DMV, IML, and SN with PD-like pathology progression. Furthermore, they demonstrated remarkable findings using their rotenone models; hemivagotomy (i.e., the resection of the truncus vagalis anterior) prevented α-synuclein accumulation in the DMV and nigral dopaminergic neurodegeneration, and then delayed appearance of motor deficits [39]. These results suggest that rotenone can initiate the progression of PD pathology in the ENS and that this progression is based on the transneuronal and retrograde vagal axonal transport of α-synuclein. Our recent study also demonstrated rotenone-induced neuropathology in the ENS; systemic rotenone exposure decreased the intestinal myenteric plexus with α-synuclein accumulation [19].
It is reported that chronic exposure to rotenone (1–2 mg/kg, subcutaneous (s.c.)) for 7–27 days induced loss of nerve fibers and their fragmentation in sciatic nerves of rats [43]. The rotenone-treated rats exhibited peripheral motor nerve dysfunction as reflected by decreased motor nerve conduction velocity, suggesting a continuous motor neuropathy after rotenone exposure. These results suggest that rotenone-induced peripheral neuropathology is also associated with motor dysfunction in addition to degeneration of nigrostriatal pathway.
3. Rotenone Reproduced Motor Features of PD
Various studies demonstrated that rotenone models produced by different routes of administration exhibit PD-like motor deficits originating from degeneration of nigrostriatal DA pathway [12,16,17,19,21,22,23,24,25,26,36,38,39,44,45]. Rats exposed to rotenone by continuous intravenous administration became hypokinetic and had unsteady movement and hunched posture, and some of them developed severe rigidity and exhibited shaking of one or more paws that was similar to rest tremor in PD, which the DA agonist was responsible for [12]. Subcutaneous injection of rotenone also develops motor dysfunction [18]. Richter et al. demonstrated daily subcutaneous injection of rotenone (2.5–5 mg/kg) developed motor dysfunction in mice: decreased locomotor activity, impairment of motor coordination, abnormal gait, reduction of grip strength [46]. Recently, we also demonstrated that chronic subcutaneous infusion of rotenone (2.5 mg/kg/day) for 4 weeks induced motor deficits in mice assessed by open field, rotarod, and cylinder test [19]. Cannon et al. reported that daily intraperitoneal injection of rotenone (3 mg/kg) developed bradykinesia, postural instability, and rigidity, which were attenuated by apomorphine [16]. These results are consistent with those of another study; rats that received daily intraperitoneal injection of rotenone (3 mg/kg) for 11 days exhibited decrease in motor skill, muscle strength, and balance [45]. In addition, Inden et al. reported that daily oral administration of rotenone (30 and 100 mg/kg) induced behavior impairment in mice assessed by the rotarod test [17]. Thus, rotenone can reproduce motor features of PD regardless of the route of administration.
4. Rotenone Reproduced Non-Motor Features of PD
PD is a complex, multi-system, neurodegenerative disorder. To investigate the mechanism of neurodegeneration in PD and to develop novel treatment to stop PD progression, there is a great need for experimental models which exhibit non-motor symptoms corresponding to prodromal PD in addition to motor dysfunction. Recent studies examined reproducibility of non-motor features of PD in rotenone models.
4.1. Olfactory Impairment
Hyposmia is one of the most common, best-characterized non-motor features and a well-known sign of prodromal PD [47,48]. About 80–90% of PD patients have impaired olfaction, which is in line with Braak’s hypothesis. Therefore, hyposmia has received attention as a biomarker for earlier diagnosis of PD. Several studies demonstrated that olfactory impairment was observed in rotenone-treated animals [49,50]. Stereotaxic injection of rotenone into the SN of rats reduced olfactory discrimination ability under the modulation of D2 receptors within the glomerular layer of the olfactory bulb [50]. In addition, Aurich et al. demonstrated neuronal loss of TH-positive neurons in the SN pars compacta could inflict olfactory impairment in rats, which received stereotaxic injection of rotenone [49]. These findings suggest that impairment of DA neurotransmission in the olfactory bulb and the nigrostriatal pathway may have important roles in olfaction disturbance.
4.2. Gastrointestinal Dysfunction
Gastrointestinal dysfunction is a well-known non-motor symptom in PD and includes excessive salivation, dysphagia, impaired gastric emptying, constipation, and impaired defecation, and precedes the presentation of motor symptoms [2,3,4]. Recently, a large-scale prospective study demonstrated that lower bowel movement frequencies predicted the future PD crisis [51]. The cause of the gastrointestinal dysfunction remains unclear. However, Lewy body pathology is detected at almost every level of the gastrointestinal tract of PD patients [40,41,42,52,53]. The bowel movement is controlled by vagal parasympathetic innervation which connects ENS and CNS. Therefore, it has been hypothesized that PD pathology propagates from the ENS to the CNS via vagal nerve [5,6,7,42]. Based on these observations, the presence of PD pathology in the gastrointestinal tract may represent one of the earliest manifestations of the disease. Therefore, developing PD models that exhibit gastrointestinal dysfunction is important for investigation of disease-modifying drugs. Pan-Montojo et al. reported chronic intragastric rotenone administration decreased gut motility, assessed by weighing stool pellets [39]. Tasselli et al. [54] examined effects of oral administration of rotenone on gastrointestinal function in mice; gastrointestinal motility was assessed by measuring gastric emptying, total transit time, fecal pellet output, and bead latency. Although there was no change in gastric emptying and total transit time by rotenone treatment, fecal pellet output was significantly reduced in rotenone-treated mice, which was not associated with changes in wet weight or in the percentage of water content. Morais et al. also reported that oral administration of rotenone (30 mg/kg) for 7 days induced gastrointestinal dysfunction [27]. They evaluated gastrointestinal transit by measuring the distance that Evans blue solution migrated in the intestine from the pylorus to the most distal point of the intestine 6 h after Evans blue administration. Rotenone treatment significantly decreased intestinal transit. These results indicate that rotenone induces gastrointestinal dysfunction, which is understandable because rotenone directly affects enteric neurons by oral/gastrointestinal administration in these experiments.
Several studies reported that animals treated with rotenone by other routes of injection also exhibit gastrointestinal dysfunction [7,19,37,55]. Drolet et al. [37] reported that rats injected intraperitoneally with rotenone (2.0 mg/kg) for 6 weeks exhibited functional deficit in gastrointestinal motility, which was associated with α-synuclein aggregate pathology and neuron loss in the ENS. In addition, Greene et al. [55] reported that chronic rotenone infusion by osmotic mini pump (3 mg/kg/day) for 22–28 days caused delayed gastric emptying and enteric neuronal dysfunction. In this study, they explored behavioral gastrointestinal motility, electrophysiological function of the ENS, and concurrently, motor behavior and TH expression in the CNS. Rotenone-treated animals had higher residual stomach content after a meal, indicating a significant delay in solid gastric emptying associated with rotenone intoxication, and a transient decrease in colon motility, as assessed by one-hour stool frequency. In addition, rotenone-treated rats exhibited a physiological defect of inhibitory neurons in the ENS, however, the total number of enteric neurons was not changed. These results suggest that rotenone disrupted gastrointestinal motility, which preceded motor dysfunction or CNS neuropathology, corresponding with the PD pathological condition. Our recent research also demonstrated that chronic systemic rotenone infusion using mini pump induced gastrointestinal dysfunction accompanied by enteric neuropathology in mice [19]. Control mice defecated constantly every 5 min, in contrast, all rotenone-treated mice took 15 min until first fecal output. Further, fecal pellets produced by rotenone-treated mice varied in weight. Taken together with these observations, it is indicated that rotenone causes enteric functional disturbance and neuropathology despite systemic rotenone exposure, intraperitoneal and subcutaneous injections, suggesting the ENS is more susceptible and vulnerable to rotenone than the CNS. Recently, an interesting study was performed by Perez-Pardo et al. They reported that both oral and intrastriatal administrations of rotenone induced not only dopaminergic cell loss in the SN and PD-like motor deficits but also delayed intestinal transit, inflammation, and α-synuclein accumulation in the ENS [7,44]. Chronic peripheral inflammation is associated with central neuroinflammation and degeneration. Oral rotenone administration induces chronic inflammation in the periphery, affecting pathological changes in the gut and the brain via a dysfunctional gut–brain axis. On the other hand, environmental factors injected in the brain may start a detrimental process in the brain, subsequently affecting the gastrointestinal tract. Although the mechanism of peripheral neuropathological change by brain injection of rotenone is still unknown, possible bidirectional communication is suggested between the gut and brain for the genesis of PD-like pathology; the brain-to-gut axis could be also involved in PD progression.
4.3. Cardiovascular Dysfunction
Cardiovascular dysfunctions, such as postural hypotension, blood pressure variability, and cardiac dysrhythmias, are also commonly seen in PD patients before the appearance of motor symptoms [32]. In addition, α-synuclein aggregation and distal-dominant degeneration of the cardiac sympathetic nervous system were observed in PD patients [56]. Despite the clinical evidence for altered cardiovascular function in PD, there are relatively few studies examining this change in experimental models of PD [57,58,59]. Zhang et al. examined effects of daily intraperitoneal rotenone injections (1 mg/kg) for 21 days on cardiovascular function and changes in catecholaminergic neurons in the medulla oblongata and nigrostriatal DA system in rats [59]. The number of TH-positive neurons in the SN and DA content in the striatum were decreased 1 week after rotenone treatment. The rotenone-injected rats exhibited motor dysfunction after 13 days of treatments, which is considered to be derived from DA system disruption. Interestingly, rotenone-treated rats showed dysfunction of cardiac autonomic regulation, assessed by electrocardiogram and reduction of mean blood pressure, which preceded nigral dopaminergic neurodegeneration and motor deficits. Moreover, they demonstrated that DA β-hydroxylase (DBH)- and TH-positive neurons in the rostral ventrolateral medulla, but not in the caudal ventrolateral medulla and nucleus tractus solitarii, gradually decreased in the rotenone-treated rats, which began at the 7th day and lasted till the 21st day. These results suggest that rotenone-induced degeneration of catecholaminergic neurons in the medulla oblongata might account for the cardiovascular dysfunction.
4.4. Depression
Depression is common in PD patients and predates the onset of motor symptoms [60]. Several experiments demonstrated depression-like behavior in rotenone models [61,62,63,64,65]. Santiago et al. [63] examined whether nigral injection of rotenone induced depressive-like behaviors and alterations of neurotransmitters similar to those observed in PD. They showed that rotenone produced depressive-like behaviors, anhedonia, and behavioral despair, which were accompanied by hippocampal 5-HT reduction. Morais et al. also reported that daily intraperitoneal administration of rotenone (2.5 mg/kg) for 10 days developed depressive-like behaviors in rats, assessed by forced swim test and sucrose preference test [62]. They also suggested rotenone-induced depression-like behavior was associated with serotonin and noradrenaline turnovers in the striatum and hippocampus. It was also reported that subcutaneous injections of rotenone also induced depression-like behaviors, assessed by forced swim test [61,64].
4.5. Sleep Disorders
Sleep disorders, such as insomnia, RBD, central sleep apnea, restless legs, and nocturnal akinesia, are well-known non-motor symptoms in PD patients [66]. RBD appears at least a decade before the first motor symptoms [5], therefore, it can be a marker of prodromal neurodegenerative disease. There is a report showing sleep disturbance in rotenone models [67]. Chronic subcutaneous infusion of rotenone by implantation of rotenone-filled pump reduced locomotion activity, increased slow-wave sleep during the dark (active) phase and decreased slow-wave sleep during the light (rest) period, and enhanced REM in the dark period. The rotenone-induced sleep disturbance, such as excessive daytime sleepiness and insomnia during the nighttime, was thought to be derived from loss of TH-positive neurons in the SN, because DA has been regarded as a wake promoter.
4.6. Cognitive and Memory Impairment
Cognitive decline and dementia are usually considered to be a non-motor symptom of late-stage PD. Recently, it was reported that poor cognitive functioning is associated with an increased risk of parkinsonism and PD [68], suggesting that cognitive dysfunction could be a sign of prodromal PD. Apolipoprotein E (ApoE) is a protein associated with neurodegenerative disease, including Alzheimer’s disease. A previous meta-analysis of PD patients revealed that ApoE was indeed positively associated with sporadic PD [69]. In addition, alteration in protein levels of ApoE is also associated with the progression of PD [70]. Furthermore, it is known that reduced activity of mitochondrial complex I has also been associated with increased expression of ApoE [71]. Taking these findings into consideration, it is suggested that rotenone treatment can induce cognitive impairment as a consequence of complex I inhibition. Indeed, several studies reported that rotenone-treated animals exhibit cognitive and memory dysfunction [7,72,73]. The first investigation of rotenone-induced cognitive deficits was reported by Kaur et al. [73]. Intraperitoneal injection of rotenone (3 mg/kg) for 30 days disrupted spatial long-term memory, assessed by elevated plus-maze test in rats. In addition, it is reported that intrastriatal injection of rotenone also caused spatial recognition impairment [7]. Rotenone treatment disturbed the ability to react to a spatial novelty in mice. Moreover, Haider et al. recently demonstrated rotenone-induced memory and cognitive impairment in rats [72]. They examined effects of subcutaneous rotenone administration on memory by Morris water maze and cognitive ability by novel object recognition test. Rotenone administration induced memory impairment, testified by significantly increased escape latency, decreased time spent in target quadrant, and crossing over target quadrant as compared with that of control group. Besides, rotenone treatment significantly decreased preference index, assessed by novel object recognition test, indicating cognitive impairment. They also demonstrated that rotenone treatment increased ApoE expression in the striatum of rats. These rotenone models could be useful for research of the relationship between cognitive dysfunction and neuropathological change in the brain.
4.7. Pain and Somatosensory Disturbances
Changes in sensory function and onset of pain are a common feature of PD [74]. About 60–70% of PD patients experience various types of pain, including neuropathic, visceral, musculoskeletal, or dystonic pain [73,75], which often precede motor impairments [76]. Kim et al. reported that intrathecal administration of rotenone produced hyperalgesia in mice [77]. In this study, sensitivity of the hind paw was assessed by measuring foot withdrawal frequencies in response to mechanical stimuli. The first significant hyperalgesia was detected 5–8 h after rotenone treatment and persisted until 48 h post-injection. Thus, the results showed that inhibition of spinal mitochondrial complex I led to a slow and long-lasting hyperalgesia in mice.
5. Concluding Remarks
To clarify the mechanism of neurodegeneration in PD and to develop disease-modifying drugs for PD treatment, animal models which reproduce the pathological hallmarks and clinical features of PD are required. Because PD neuropathology is not restricted to the nigrostriatal DA pathway, reproducibility of central and peripheral features of PD in animal models has been focused upon. Rotenone models of PD have received most attention because the pesticide can reproduce pathological conditions of PD. To date, various studies demonstrated that rotenone treatment by different routes of administration induced central and peripheral neuropathological features and motor and non-motor PD-like symptoms as a consequence of neuropathological changes (Figure 1 and Table 1). Rotenone can induce degeneration of nigrostriatal DA pathway with Lewy pathology and motor dysfunction, which responds to DA agonists. In addition, rotenone also causes time-dependent neuropathological changes in the CNS, such as in the olfactory bulb, hippocampus, locus coeruleus, and pedunculopontine tegmental nucleus, that correspond to Braak’s staging hypothesis. Moreover, rotenone can initiate PD-like pathology and α-synuclein accumulation in the ENS, subsequently inducing propagation of α-synuclein to the DMV, possibly by the transneuronal and retrograde axonal transport. Accordingly, these extensive neurotoxic effects of rotenone can recapitulate non-motor symptoms, such as olfactory, gastrointestinal, and cardiovascular dysfunction, depression, sleep disorders, cognitive impairment, and hyperalgesia, which precede motor deficits, similar to those observed in prodromal PD (Figure 1). The utility of rotenone models in the development of disease-modifying drugs is substantiated in the previous reports. Perez-Pardo et al. demonstrated that uridine and fish oil diet prevented rotenone-induced gastrointestinal pathology and dysfunction in both oral and intrastriatal rotenone exposure in mice [44]. Farombi et al. also reported that kolaviron, a natural anti-inflammatory, anti-oxidative, and anti-apoptotic biflavonoid, ameliorated oxidative stress in the gut and intestinal barrier deficits in stereotaxic rotenone models [78]. Recently, we reported that intake of coffee components, caffeic acid and chlorogenic acid, enhanced the antioxidative properties of enteric glial cells and prevented rotenone-induced neurodegeneration in myenteric plexus [20]. These results indicate that enteric environmental modification exerts protective effects in the ENS. In addition, based on a hypothesis that PD pathology propagates from the ENS to the CNS, these findings suggest a possible food-based therapeutic strategy for early treatment of PD. Moreover, several studies demonstrated that some drugs and flavonoids improved depression-like behavior [61,65,79] and cognitive and memory impairment [72,73] in rotenone models: for example, antidepressive effects of curcumin, melatonin, and ibuprofen, improvement of cognitive and memory impairment by pistachio and carotenoid compound lycopene. Taken together, rotenone models could be useful for the evaluation of candidate drugs for prodromal PD treatment.
Despite these advantages of rotenone models, the adoption of the models, in particular rotenone infusion models, is limiting because of variability in animal sensitivity and the inability of other investigators to consistently reproduce the PD neuropathology. To dissolve this issue, other various routes of administration of rotenone have been developed: intraperitoneal, oral, intragastric, subcutaneous injection, and intranasal inoculation. Especially, oral, intragastric, subcutaneous, and intranasal administrations are unique routes which are based on a background that pesticide exposure, particularly exposure to rotenone and paraquat, increases the risk of PD [14]. However, these administrations still have disadvantages: high mortality of animals, difficulty in method, and health hazard of investigators exposed to the toxin by daily administration. The variability and reproducibility of rotenone PD models would depend on various routes of administrations and applied animal species. In addition, high mortality of animals exposed to rotenone may lead to unstable experimental results. Recently, we established a high-reproducible rotenone model using C57BL/6J mice [19]; the rotenone mouse model was produced by chronic systemic exposure to a low dose of rotenone (2.5 mg/kg/day) for 4 weeks by subcutaneous implantation of rotenone-filled mini pump. Our rotenone models exhibited motor deficits and gastrointestinal dysfunction as a consequence of neurodegeneration with α-synuclein accumulation in the SNpc, DMV, and the intestinal myenteric plexus. The survival rate of rotenone-treated mice was not different from that of control group. The zero mortality is extremely important because that leads to stability of rotenone sensitivity. Furthermore, simple administration of pump implantation enables other investigators to reproduce rotenone toxicity constantly. To date, most of the rotenone models were produced in rats [80]. Although the pathogenesis in sporadic PD remains unknown, there is a consensus that both genetic and environmental factors are thought to contribute to PD pathogenesis. Thus, the rotenone model using C57BL mice, which is a common strain of genetically modified animal, could be expanded to genetic mouse models of PD, and that could provide useful animal models to investigate possible interaction between pesticide exposure and genetic defects. Our low-dose rotenone mouse model would contribute to investigation of PD pathogenesis and development of disease-modifying drugs for PD in the future.
Author Contributions
Writing—original draft, I.M.; writing—review and editing, M.A.; funding acquisition, I.M. and M.A. All authors have read and agree to the published version of the manuscript.
Funding
This work was funded by JSPS KAKENHI Grant for Scientific Research (C) (JP25461279, JP16K09673, JP19K07993 to I.M.), the Okayama Medical Foundation (to I.M.) and the grants from All Japan Coffee Association (to M.A.) and Japanese Society of Eucommia (to M.A.).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Braak, H.; Del Tredici, K.; Rub, U.; de Vos, R.A.; Jansen Steur, E.N.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Fasano, A.; Visanji, N.P.; Liu, L.W.; Lang, A.E.; Pfeiffer, R.F. Gastrointestinal dysfunction in Parkinson’s disease. Lancet Neurol. 2015, 14, 625–639. [Google Scholar] [CrossRef]
- Pfeiffer, R.F. Gastrointestinal Dysfunction in Parkinson’s Disease. Curr. Treat. Options Neurol. 2018, 20, 54. [Google Scholar] [CrossRef] [PubMed]
- Ueki, A.; Otsuka, M. Life style risks of Parkinson’s disease: Association between decreased water intake and constipation. J. Neurol. 2004, 251 (Suppl. 7), vII18–vII23. [Google Scholar] [CrossRef]
- Hawkes, C.H.; Del Tredici, K.; Braak, H. A timeline for Parkinson’s disease. Parkinsonism Relat. Disord. 2010, 16, 79–84. [Google Scholar] [CrossRef]
- Klingelhoefer, L.; Reichmann, H. Pathogenesis of Parkinson disease--the gut-brain axis and environmental factors. Nat. Rev. Neurol. 2015, 11, 625–636. [Google Scholar] [CrossRef]
- Perez-Pardo, P.; Kliest, T.; Dodiya, H.B.; Broersen, L.M.; Garssen, J.; Keshavarzian, A.; Kraneveld, A.D. The gut-brain axis in Parkinson’s disease: Possibilities for food-based therapies. Eur. J. Pharmacol. 2017, 817, 86–95. [Google Scholar] [CrossRef]
- Blesa, J.; Phani, S.; Jackson-Lewis, V.; Przedborski, S. Classic and new animal models of Parkinson’s disease. J. Biomed. Biotechnol. 2012, 2012, 845618. [Google Scholar] [CrossRef]
- Tieu, K. A guide to neurotoxic animal models of Parkinson’s disease. Cold Spring Harb. Perspect Med. 2011, 1, a009316. [Google Scholar] [CrossRef]
- Duty, S.; Jenner, P. Animal models of Parkinson’s disease: A source of novel treatments and clues to the cause of the disease. Br. J. Pharmacol. 2011, 164, 1357–1391. [Google Scholar] [CrossRef] [Green Version]
- Sherer, T.B.; Betarbet, R.; Testa, C.M.; Seo, B.B.; Richardson, J.R.; Kim, J.H.; Miller, G.W.; Yagi, T.; Matsuno-Yagi, A.; Greenamyre, J.T. Mechanism of toxicity in rotenone models of Parkinson’s disease. J. Neurosci. 2003, 23, 10756–10764. [Google Scholar] [CrossRef]
- Betarbet, R.; Sherer, T.B.; MacKenzie, G.; Garcia-Osuna, M.; Panov, A.V.; Greenamyre, J.T. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat. Neurosci. 2000, 3, 1301–1306. [Google Scholar] [CrossRef] [PubMed]
- Sherer, T.B.; Kim, J.H.; Betarbet, R.; Greenamyre, J.T. Subcutaneous rotenone exposure causes highly selective dopaminergic degeneration and alpha-synuclein aggregation. Exp. Neurol. 2003, 179, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, A.S.; Tarbutton, G.L.; Levin, J.L.; Plotkin, G.M.; Lowry, L.K.; Nalbone, J.T.; Shepherd, S. Pesticide/environmental exposures and Parkinson’s disease in East Texas. J. Agromed. 2008, 13, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Nandipati, S.; Litvan, I. Environmental Exposures and Parkinson’s Disease. Int. J. Environ. Res. Public Health 2016, 13, 881. [Google Scholar] [CrossRef] [PubMed]
- Cannon, J.R.; Tapias, V.; Na, H.M.; Honick, A.S.; Drolet, R.E.; Greenamyre, J.T. A highly reproducible rotenone model of Parkinson’s disease. Neurobiol. Dis. 2009, 34, 279–290. [Google Scholar] [CrossRef] [Green Version]
- Inden, M.; Kitamura, Y.; Abe, M.; Tamaki, A.; Takata, K.; Taniguchi, T. Parkinsonian rotenone mouse model: Reevaluation of long-term administration of rotenone in C57BL/6 mice. Biol. Pharm. Bull. 2011, 34, 92–96. [Google Scholar] [CrossRef] [Green Version]
- Johnson, M.E.; Bobrovskaya, L. An update on the rotenone models of Parkinson’s disease: Their ability to reproduce the features of clinical disease and model gene-environment interactions. Neurotoxicology 2015, 46, 101–116. [Google Scholar] [CrossRef]
- Miyazaki, I.; Isooka, N.; Imafuku, F.; Sun, J.; Kikuoka, R.; Furukawa, C.; Asanuma, M. Chronic Systemic Exposure to Low-Dose Rotenone Induced Central and Peripheral Neuropathology and Motor Deficits in Mice: Reproducible Animal Model of Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 3254. [Google Scholar] [CrossRef]
- Miyazaki, I.; Isooka, N.; Wada, K.; Kikuoka, R.; Kitamura, Y.; Asanuma, M. Effects of Enteric Environmental Modification by Coffee Components on Neurodegeneration in Rotenone-Treated Mice. Cells 2019, 8, 221. [Google Scholar] [CrossRef] [Green Version]
- Murakami, S.; Miyazaki, I.; Miyoshi, K.; Asanuma, M. Long-Term Systemic Exposure to Rotenone Induces Central and Peripheral Pathology of Parkinson’s Disease in Mice. Neurochem. Res. 2015, 40, 1165–1178. [Google Scholar] [CrossRef] [PubMed]
- Sasajima, H.; Miyazono, S.; Noguchi, T.; Kashiwayanagi, M. Intranasal Administration of Rotenone to Mice Induces Dopaminergic Neurite Degeneration of Dopaminergic Neurons in the Substantia Nigra. Biol. Pharm. Bull. 2017, 40, 108–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, H.; Yanagida, T.; Inden, M.; Takata, K.; Kitamura, Y.; Yamakawa, K.; Sawada, H.; Izumi, Y.; Yamamoto, N.; Kihara, T.; et al. Nicotinic receptor stimulation protects nigral dopaminergic neurons in rotenone-induced Parkinson’s disease models. J. Neurosci. Res. 2009, 87, 576–585. [Google Scholar] [CrossRef] [Green Version]
- Zaitone, S.A.; Ahmed, E.; Elsherbiny, N.M.; Mehanna, E.T.; El-Kherbetawy, M.K.; ElSayed, M.H.; Alshareef, D.M.; Moustafa, Y.M. Caffeic acid improves locomotor activity and lessens inflammatory burden in a mouse model of rotenone-induced nigral neurodegeneration: Relevance to Parkinson’s disease therapy. Pharmacol. Rep. 2019, 71, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Guo, H.; Guo, X.; Ge, D.; Shi, Y.; Lu, X.; Lu, J.; Chen, J.; Ding, F.; Zhang, Q. Involvement of Akt/mTOR in the Neurotoxicity of Rotenone-Induced Parkinson’s Disease Models. Int. J. Environ. Res. Public Health 2019, 16, 3811. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Sun, J.D.; Song, L.K.; Li, J.; Chu, S.F.; Yuan, Y.H.; Chen, N.H. Environment-contact administration of rotenone: A new rodent model of Parkinson’s disease. Behav. Brain Res. 2015, 294, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Morais, L.H.; Hara, D.B.; Bicca, M.A.; Poli, A.; Takahashi, R.N. Early signs of colonic inflammation, intestinal dysfunction, and olfactory impairments in the rotenone-induced mouse model of Parkinson’s disease. Behav. Pharmacol. 2018, 29, 199–210. [Google Scholar] [CrossRef]
- Sasajima, H.; Miyazono, S.; Noguchi, T.; Kashiwayanagi, M. Intranasal administration of rotenone in mice attenuated olfactory functions through the lesion of dopaminergic neurons in the olfactory bulb. Neurotoxicology 2015, 51, 106–115. [Google Scholar] [CrossRef]
- Voronkov, D.N.; Kutukova, K.A.; Ivanov, M.V.; Khudoerkov, R.M. Immunomorphological Changes in the Olfactory Bulbs of Rats after Intranasal Administration of Rotenone. Bull. Exp. Biol. Med. 2017, 164, 203–206. [Google Scholar] [CrossRef]
- Dauer, W.; Przedborski, S. Parkinson’s disease: Mechanisms and models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef] [Green Version]
- Jellinger, K.A. Neuropathology of Nonmotor Symptoms of Parkinson’s Disease. Int. Rev. Neurobiol. 2017, 133, 13–62. [Google Scholar] [PubMed]
- Schapira, A.H.V.; Chaudhuri, K.R.; Jenner, P. Non-motor features of Parkinson disease. Nat. Rev. Neurosci. 2017, 18, 435–450. [Google Scholar] [CrossRef] [PubMed]
- Qamar, M.A.; Sauerbier, A.; Politis, M.; Carr, H.; Loehrer, P.; Chaudhuri, K.R. Presynaptic dopaminergic terminal imaging and non-motor symptoms assessment of Parkinson’s disease: Evidence for dopaminergic basis? Parkinsons Dis. 2017, 3, 5. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.F.; Silva, C.M.; D’Unhao, A.M.; Ferrari, M.F. Aged Lewis rats exposed to low and moderate doses of rotenone are a good model for studying the process of protein aggregation and its effects upon central nervous system cell physiology. Arq. Neuropsiquiatr. 2016, 74, 737–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halliday, G.M.; Holton, J.L.; Revesz, T.; Dickson, D.W. Neuropathology underlying clinical variability in patients with synucleinopathies. Acta Neuropathol. 2011, 122, 187–204. [Google Scholar] [CrossRef]
- Hoglinger, G.U.; Feger, J.; Prigent, A.; Michel, P.P.; Parain, K.; Champy, P.; Ruberg, M.; Oertel, W.H.; Hirsch, E.C. Chronic systemic complex I inhibition induces a hypokinetic multisystem degeneration in rats. J. Neurochem. 2003, 84, 491–502. [Google Scholar] [CrossRef] [Green Version]
- Drolet, R.E.; Cannon, J.R.; Montero, L.; Greenamyre, J.T. Chronic rotenone exposure reproduces Parkinson’s disease gastrointestinal neuropathology. Neurobiol. Dis. 2009, 36, 96–102. [Google Scholar] [CrossRef]
- Pan-Montojo, F.; Anichtchik, O.; Dening, Y.; Knels, L.; Pursche, S.; Jung, R.; Jackson, S.; Gille, G.; Spillantini, M.G.; Reichmann, H.; et al. Progression of Parkinson’s disease pathology is reproduced by intragastric administration of rotenone in mice. PLoS ONE 2010, 5, e8762. [Google Scholar] [CrossRef] [Green Version]
- Pan-Montojo, F.; Schwarz, M.; Winkler, C.; Arnhold, M.; O’Sullivan, G.A.; Pal, A.; Said, J.; Marsico, G.; Verbavatz, J.M.; Rodrigo-Angulo, M.; et al. Environmental toxins trigger PD-like progression via increased alpha-synuclein release from enteric neurons in mice. Sci. Rep. 2012, 2, 898. [Google Scholar] [CrossRef] [Green Version]
- Lebouvier, T.; Chaumette, T.; Damier, P.; Coron, E.; Touchefeu, Y.; Vrignaud, S.; Naveilhan, P.; Galmiche, J.P.; Bruley des Varannes, S.; Derkinderen, P.; et al. Pathological lesions in colonic biopsies during Parkinson’s disease. Gut 2008, 57, 1741–1743. [Google Scholar] [CrossRef] [Green Version]
- Shannon, K.M.; Keshavarzian, A.; Mutlu, E.; Dodiya, H.B.; Daian, D.; Jaglin, J.A.; Kordower, J.H. Alpha-synuclein in colonic submucosa in early untreated Parkinson’s disease. Mov. Disord. 2012, 27, 709–715. [Google Scholar] [CrossRef]
- Braak, H.; de Vos, R.A.; Bohl, J.; Del Tredici, K. Gastric alpha-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci. Lett. 2006, 396, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Binienda, Z.K.; Sarkar, S.; Mohammed-Saeed, L.; Gough, B.; Beaudoin, M.A.; Ali, S.F.; Paule, M.G.; Imam, S.Z. Chronic exposure to rotenone, a dopaminergic toxin, results in peripheral neuropathy associated with dopaminergic damage. Neurosci. Lett. 2013, 541, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Perez-Pardo, P.; Dodiya, H.B.; Broersen, L.M.; Douna, H.; van Wijk, N.; Lopes da Silva, S.; Garssen, J.; Keshavarzian, A.; Kraneveld, A.D. Gut-brain and brain-gut axis in Parkinson’s disease models: Effects of a uridine and fish oil diet. Nutr. Neurosci. 2018, 21, 391–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonia Angeline, M.; Chaterjee, P.; Anand, K.; Ambasta, R.K.; Kumar, P. Rotenone-induced parkinsonism elicits behavioral impairments and differential expression of parkin, heat shock proteins and caspases in the rat. Neuroscience 2012, 220, 291–301. [Google Scholar] [CrossRef]
- Richter, F.; Hamann, M.; Richter, A. Chronic rotenone treatment induces behavioral effects but no pathological signs of parkinsonism in mice. J. Neurosci. Res. 2007, 85, 681–691. [Google Scholar] [CrossRef]
- Fullard, M.E.; Morley, J.F.; Duda, J.E. Olfactory Dysfunction as an Early Biomarker in Parkinson’s Disease. Neurosci. Bull. 2017, 33, 515–525. [Google Scholar] [CrossRef]
- Ross, G.W.; Petrovitch, H.; Abbott, R.D.; Tanner, C.M.; Popper, J.; Masaki, K.; Launer, L.; White, L.R. Association of olfactory dysfunction with risk for future Parkinson’s disease. Ann. Neurol. 2008, 63, 167–173. [Google Scholar] [CrossRef]
- Aurich, M.F.; Rodrigues, L.S.; Targa, A.D.S.; Noseda, A.C.D.; Cunha, F.D.W.; Lima, M.M.S. Olfactory impairment is related to REM sleep deprivation in rotenone model of Parkinson’s disease. Sleep Sci. 2017, 10, 47–54. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, L.S.; Targa, A.D.; Noseda, A.C.; Aurich, M.F.; Da Cunha, C.; Lima, M.M. Olfactory impairment in the rotenone model of Parkinson’s disease is associated with bulbar dopaminergic D2 activity after REM sleep deprivation. Front. Cell. Neurosci. 2014, 8, 383. [Google Scholar] [CrossRef] [Green Version]
- Abbott, R.D.; Petrovitch, H.; White, L.R.; Masaki, K.H.; Tanner, C.M.; Curb, J.D.; Grandinetti, A.; Blanchette, P.L.; Popper, J.S.; Ross, G.W. Frequency of bowel movements and the future risk of Parkinson’s disease. Neurology 2001, 57, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Gelpi, E.; Navarro-Otano, J.; Tolosa, E.; Gaig, C.; Compta, Y.; Rey, M.J.; Marti, M.J.; Hernandez, I.; Valldeoriola, F.; Rene, R.; et al. Multiple organ involvement by alpha-synuclein pathology in Lewy body disorders. Mov. Disord. 2014, 29, 1010–1018. [Google Scholar] [CrossRef] [PubMed]
- Lebouvier, T.; Neunlist, M.; Bruley des Varannes, S.; Coron, E.; Drouard, A.; N’Guyen, J.M.; Chaumette, T.; Tasselli, M.; Paillusson, S.; Flamand, M.; et al. Colonic biopsies to assess the neuropathology of Parkinson’s disease and its relationship with symptoms. PLoS ONE 2010, 5, e12728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tasselli, M.; Chaumette, T.; Paillusson, S.; Monnet, Y.; Lafoux, A.; Huchet-Cadiou, C.; Aubert, P.; Hunot, S.; Derkinderen, P.; Neunlist, M. Effects of oral administration of rotenone on gastrointestinal functions in mice. Neurogastroenterol. Motil. 2013, 25, e183–e193. [Google Scholar] [CrossRef] [PubMed]
- Greene, J.G.; Noorian, A.R.; Srinivasan, S. Delayed gastric emptying and enteric nervous system dysfunction in the rotenone model of Parkinson’s disease. Exp. Neurol. 2009, 218, 154–161. [Google Scholar] [CrossRef] [Green Version]
- Orimo, S.; Uchihara, T.; Nakamura, A.; Mori, F.; Kakita, A.; Wakabayashi, K.; Takahashi, H. Axonal alpha-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson’s disease. Brain 2008, 131, 642–650. [Google Scholar] [CrossRef] [Green Version]
- Fukumitsu, N.; Suzuki, M.; Fukuda, T.; Kiyono, Y.; Kajiyama, S.; Saji, H. Reduced 125I-meta-iodobenzylguanidine uptake and norepinephrine transporter density in the hearts of mice with MPTP-induced parkinsonism. Nucl. Med. Biol. 2006, 33, 37–42. [Google Scholar] [CrossRef]
- Takatsu, H.; Nishida, H.; Matsuo, H.; Watanabe, S.; Nagashima, K.; Wada, H.; Noda, T.; Nishigaki, K.; Fujiwara, H. Cardiac sympathetic denervation from the early stage of Parkinson’s disease: Clinical and experimental studies with radiolabeled MIBG. J. Nucl. Med. 2000, 41, 71–77. [Google Scholar]
- Zhang, Z.; Du, X.; Xu, H.; Xie, J.; Jiang, H. Lesion of medullary catecholaminergic neurons is associated with cardiovascular dysfunction in rotenone-induced Parkinson’s disease rats. Eur. J. Neurosci. 2015, 42, 2346–2355. [Google Scholar] [CrossRef]
- Shiba, M.; Bower, J.H.; Maraganore, D.M.; McDonnell, S.K.; Peterson, B.J.; Ahlskog, J.E.; Schaid, D.J.; Rocca, W.A. Anxiety disorders and depressive disorders preceding Parkinson’s disease: A case-control study. Mov. Disord. 2000, 15, 669–677. [Google Scholar] [CrossRef]
- Madiha, S.; Haider, S. Curcumin restores rotenone induced depressive-like symptoms in animal model of neurotoxicity: Assessment by social interaction test and sucrose preference test. Metab. Brain Dis. 2019, 34, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Morais, L.H.; Lima, M.M.; Martynhak, B.J.; Santiago, R.; Takahashi, T.T.; Ariza, D.; Barbiero, J.K.; Andreatini, R.; Vital, M.A. Characterization of motor, depressive-like and neurochemical alterations induced by a short-term rotenone administration. Pharmacol. Rep. 2012, 64, 1081–1090. [Google Scholar] [CrossRef]
- Santiago, R.M.; Barbieiro, J.; Lima, M.M.; Dombrowski, P.A.; Andreatini, R.; Vital, M.A. Depressive-like behaviors alterations induced by intranigral MPTP, 6-OHDA, LPS and rotenone models of Parkinson’s disease are predominantly associated with serotonin and dopamine. Prog. Neuropsychopharmacol. Biol. Psychiatry 2010, 34, 1104–1114. [Google Scholar] [CrossRef] [PubMed]
- Shin, M.S.; Kim, T.W.; Lee, J.M.; Sung, Y.H.; Lim, B.V. Treadmill exercise alleviates depressive symptoms in rotenone-induced Parkinson disease rats. J. Exerc. Rehabil. 2017, 13, 124–129. [Google Scholar] [CrossRef] [Green Version]
- Zaminelli, T.; Gradowski, R.W.; Bassani, T.B.; Barbiero, J.K.; Santiago, R.M.; Maria-Ferreira, D.; Baggio, C.H.; Vital, M.A. Antidepressant and antioxidative effect of Ibuprofen in the rotenone model of Parkinson’s disease. Neurotox Res. 2014, 26, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Bohnen, N.I.; Hu, M.T.M. Sleep Disturbance as Potential Risk and Progression Factor for Parkinson’s Disease. J. Parkinsons Dis. 2019, 9, 603–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, P.L.; Tsai, C.H.; Lu, M.K.; Liu, H.J.; Chen, Y.C.; Chang, F.C. Interleukin-1beta mediates sleep alteration in rats with rotenone-induced parkinsonism. Sleep 2007, 30, 413–425. [Google Scholar] [CrossRef] [Green Version]
- Darweesh, S.K.L.; Wolters, F.J.; Postuma, R.B.; Stricker, B.H.; Hofman, A.; Koudstaal, P.J.; Ikram, M.K.; Ikram, M.A. Association Between Poor Cognitive Functioning and Risk of Incident Parkinsonism: The Rotterdam Study. JAMA Neurol. 2017, 74, 1431–1438. [Google Scholar] [CrossRef]
- Huang, X.; Chen, P.C.; Poole, C. APOE-[epsilon]2 allele associated with higher prevalence of sporadic Parkinson disease. Neurology 2004, 62, 2198–2202. [Google Scholar] [CrossRef] [Green Version]
- Ghebremedhin, E.; Del Tredici, K.; Vuksic, M.; Rub, U.; Thal, D.R.; Burbach, G.J.; Rosenberger, A.; Bickeboller, H.; Deller, T.; de Vos, R.A.; et al. Relationship of apolipoprotein E and age at onset to Parkinson disease neuropathology. J. Neuropathol. Exp. Neurol. 2006, 65, 116–123. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.K.; Ji, Z.S.; Dodson, S.E.; Miranda, R.D.; Rosenblum, C.I.; Reynolds, I.J.; Freedman, S.B.; Weisgraber, K.H.; Huang, Y.; Mahley, R.W. Apolipoprotein E4 domain interaction mediates detrimental effects on mitochondria and is a potential therapeutic target for Alzheimer disease. J. Biol. Chem. 2011, 286, 5215–5221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haider, S.; Madiha, S.; Batool, Z. Amelioration of motor and non-motor deficits and increased striatal APoE levels highlight the beneficial role of pistachio supplementation in rotenone-induced rat model of PD. Metab. Brain Dis. 2020. [Google Scholar] [CrossRef] [PubMed]
- Kaur, H.; Chauhan, S.; Sandhir, R. Protective effect of lycopene on oxidative stress and cognitive decline in rotenone induced model of Parkinson’s disease. Neurochem. Res. 2011, 36, 1435–1443. [Google Scholar] [CrossRef] [PubMed]
- Djaldetti, R.; Shifrin, A.; Rogowski, Z.; Sprecher, E.; Melamed, E.; Yarnitsky, D. Quantitative measurement of pain sensation in patients with Parkinson disease. Neurology 2004, 62, 2171–2175. [Google Scholar] [CrossRef] [PubMed]
- Valek, L.; Auburger, G.; Tegeder, I. Sensory neuropathy and nociception in rodent models of Parkinson’s disease. Dis. Model. Mech. 2019, 12, dmm039396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strobel, A.V.; Tankisi, H.; Finnerup, N.B.; Fuglsang-Frederiksen, A.; Jennum, P.; Svendsen, K.B.; Kirov, F.I.; Otto, M. Somatosensory function is impaired in patients with idiopathic REM sleep behaviour disorder. Sleep Med. 2018, 42, 83–89. [Google Scholar] [CrossRef]
- Kim, H.Y.; Chung, J.M.; Chung, K. Increased production of mitochondrial superoxide in the spinal cord induces pain behaviors in mice: The effect of mitochondrial electron transport complex inhibitors. Neurosci. Lett. 2008, 447, 87–91. [Google Scholar] [CrossRef] [Green Version]
- Farombi, E.O.; Awogbindin, I.O.; Olorunkalu, P.D.; Ogbuewu, E.; Oyetunde, B.F.; Agedah, A.E.; Adeniyi, P.A. Kolaviron protects against nigrostriatal degeneration and gut oxidative damage in a stereotaxic rotenone model of Parkinson’s disease. Psychopharmacol. (Berl) 2020. [Google Scholar] [CrossRef]
- Bassani, T.B.; Gradowski, R.W.; Zaminelli, T.; Barbiero, J.K.; Santiago, R.M.; Boschen, S.L.; da Cunha, C.; Lima, M.M.; Andreatini, R.; Vital, M.A. Neuroprotective and antidepressant-like effects of melatonin in a rotenone-induced Parkinson’s disease model in rats. Brain Res. 2014, 1593, 95–105. [Google Scholar] [CrossRef]
- Cicchetti, F.; Drouin-Ouellet, J.; Gross, R.E. Environmental toxins and Parkinson’s disease: What have we learned from pesticide-induced animal models? Trends Pharmacol. Sci. 2009, 30, 475–483. [Google Scholar] [CrossRef]
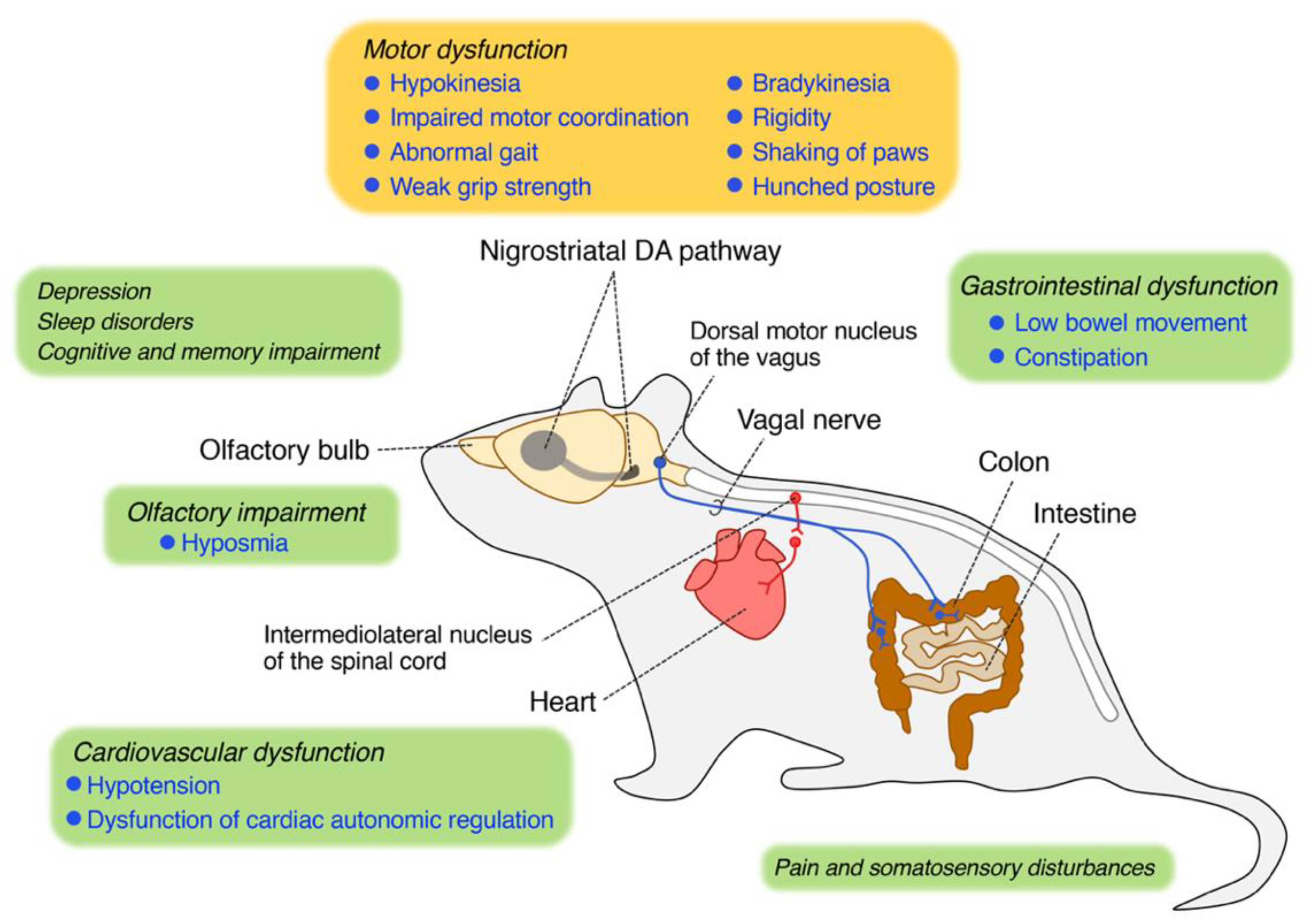
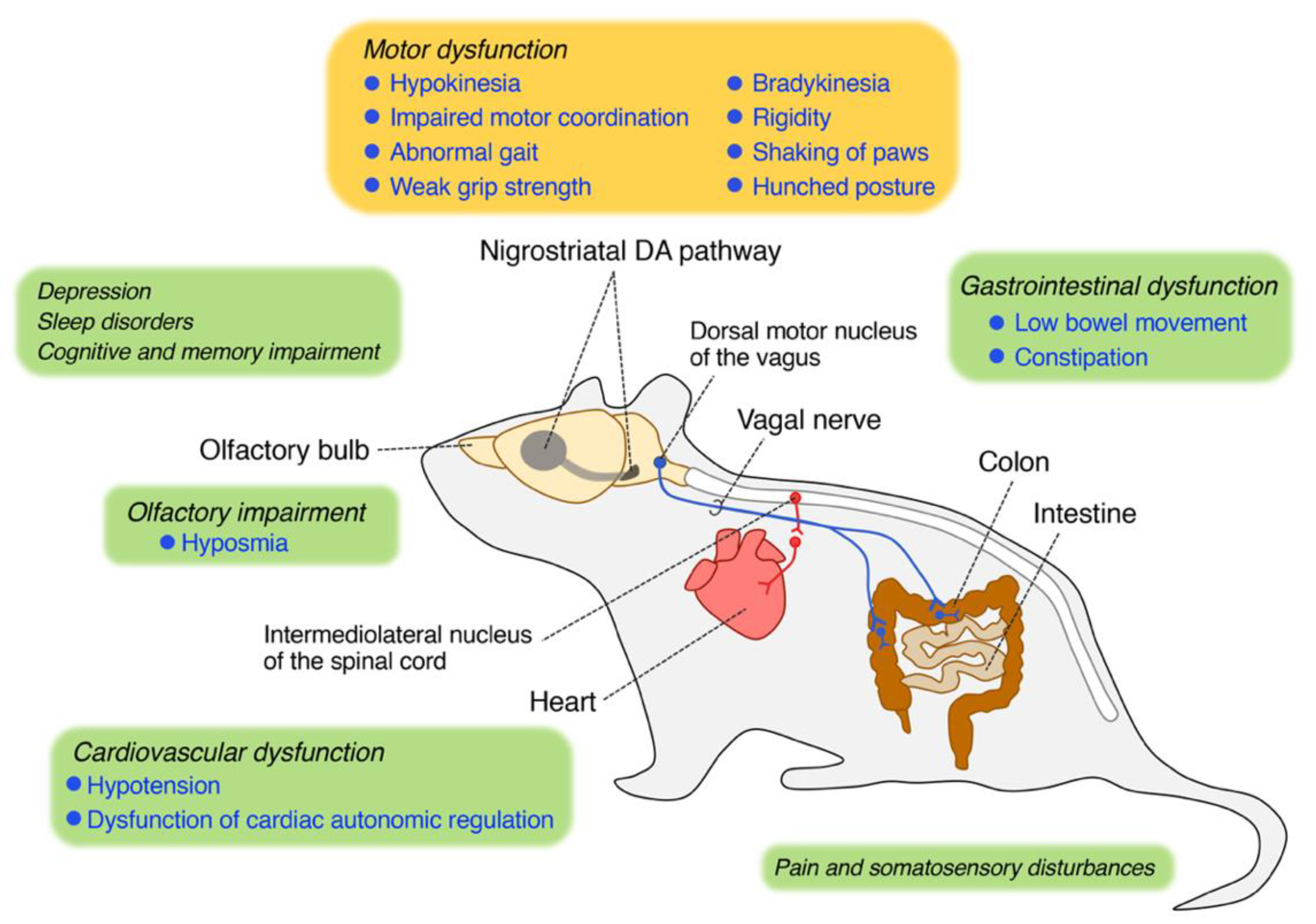
Figure 1.
Neuropathological regions and associated behavioral manifestation in rotenone animal models. Rotenone treatment induces central and peripheral neuropathological features and motor and non-motor Parkinson’s disease (PD)-like symptoms.
Figure 1.
Neuropathological regions and associated behavioral manifestation in rotenone animal models. Rotenone treatment induces central and peripheral neuropathological features and motor and non-motor Parkinson’s disease (PD)-like symptoms.

{kind=link}
Table 1.
Summary of reproducibility of central and peripheral features of PD in rotenone models.
| Animal | Dose Route | Central Neuropathology | Peripheral Neuropathology | Motor Symptom | Non-motor Symptom | Ref | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DA | OB | Other | DMV | ENS | Other | OI | GI | Other | ||||
| SD, Lewis rat | 2–3 mg/kg i.v. | + | + | [12] | ||||||||
| Lewis rat | 1–3 mg/kg s.c. | + | Hip, LC | [11,13,34] | ||||||||
| Lewis rat | 2.75–3 mg/kg i.p. | + | + | [16] | ||||||||
| Wistar rat | 2.5 mg/kg i.n. | + | [29] | |||||||||
| Wistar rat | 12 ug/site intranigral | + | + | Hip | + | + | Dep | [49,50,63] | ||||
| Lewis rat | 2.5 mg/kg i.v. | + | PP, LC Hip Cb, Cx | + | [36] | |||||||
| Lewis rat | 1, 2 mg/kg i.p. | + | + | [37] | ||||||||
| Lewis rat | 3 mg/kg s.c. | - | + | [55] | ||||||||
| SD rat | 2 mg/kg s.c. | + | ScN | + | [43] | |||||||
| Wistar rat | 1, 2.5 mg/kg i.p. | + | RVLM | + | CV Dep | [59,62,65] | ||||||
| Wistar rat | 1.5 mg/kg s.c. | + | Hip | Dep | [61] | |||||||
| SD rat | 3 mg/kg s.c. | + | DR | + | Dep Sleep | [64,67] | ||||||
| Wistar rat | 1.5, 3 mg/kg s.c. | + | + | CI | [72,73] | |||||||
| C57BL mouse | 10, 30 mg/kg p.o. | + | + | + | + | [17,23,44,54] | ||||||
| Swiss mouse | 1 mg/kg s.c. | + | + | [24,45] | ||||||||
| ICR mouse | 1, 3 mg/kg i.p. | + | + | [25] | ||||||||
| C57BL mouse | 5 mg/kg s.c. | - | + | [46] | ||||||||
| BALB/c mouse | 0.35 mg/kg i.n. | + | + | + | + | [22,28] | ||||||
| C57BL mouse | 50 mg/kg s.c. | + | + | + | + | [21] | ||||||
| C57BL mouse | 5 mg/kg contact | + | + | + | + | + | + | [26] | ||||
| Swiss mouse | 30 mg/kg p.o. | + | + | + | + | + | [27] | |||||
| C57BL mouse | 5 mg/kg p.o. | + | + | + | IML | + | + | [38,39] | ||||
| C57BL mouse | 2.5 mg/kg s.c. | + | + | + | + | + | [19,20] | |||||
| C57BL mouse | 5.4 µg/site intrastriatal | + | + | + | + | CI | [7,44] | |||||
| C57BL mouse | 50–2500 pmol intrathecal | Pain | [77] | |||||||||
DA: nigrostriatal DA pathway; OB: olfactory bulb; DMV: dorsal motor nucleus of the vagus; ENS: enteric nervous system; Hip: hippocampus; LC: locus coeruleus; PP: pedunculopontine nucleus; Cb: cerebellum; Cx: cortex; RVLM: rostral ventrolateral medulla; DR: dorsal raphe; IML: intermediolateral nucleus of the spinal cord; ScN: sciatic nerves; OI: olfactory impairment; GI: gastrointestinal impairment; CV: cardiovascular dysfunction; Dep: depression; Sleep: sleep disorders; CI: cognitive impairment; i.v.: intravenous; s.c.: subcutaneous; i.n.: intranasal; i.p.: intraperitoneal; p.o.: per oral. +: positive; -: negative; blank: not examined.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Miyazaki, I.; Asanuma, M. The Rotenone Models Reproducing Central and Peripheral Features of Parkinson’s Disease. NeuroSci 2020, 1, 1-14. https://doi.org/10.3390/neurosci1010001
AMA Style
Miyazaki I, Asanuma M. The Rotenone Models Reproducing Central and Peripheral Features of Parkinson’s Disease. NeuroSci. 2020; 1(1):1-14. https://doi.org/10.3390/neurosci1010001
Chicago/Turabian StyleMiyazaki, Ikuko, and Masato Asanuma. 2020. "The Rotenone Models Reproducing Central and Peripheral Features of Parkinson’s Disease" NeuroSci 1, no. 1: 1-14. https://doi.org/10.3390/neurosci1010001