Mpro-SARS-CoV-2 Inhibitors and Various Chemical Reactivity of 1-Bromo- and 1-Chloro-4-vinylbenzene in [3 + 2] Cycloaddition Reactions

 , , , and
, , , and 
Abstract
:1. Introduction
2. Computational Methods
3. Results and Discussion
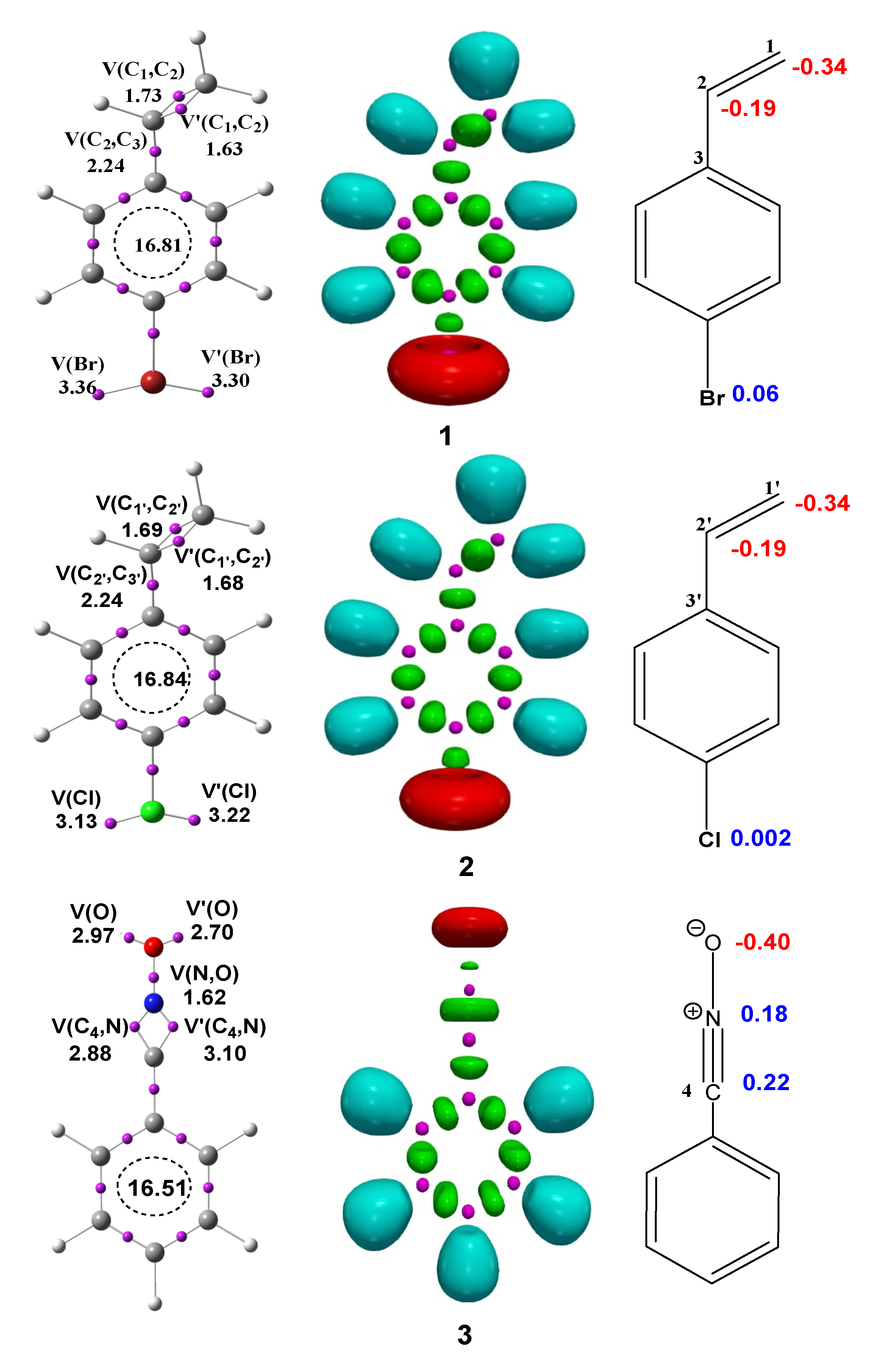
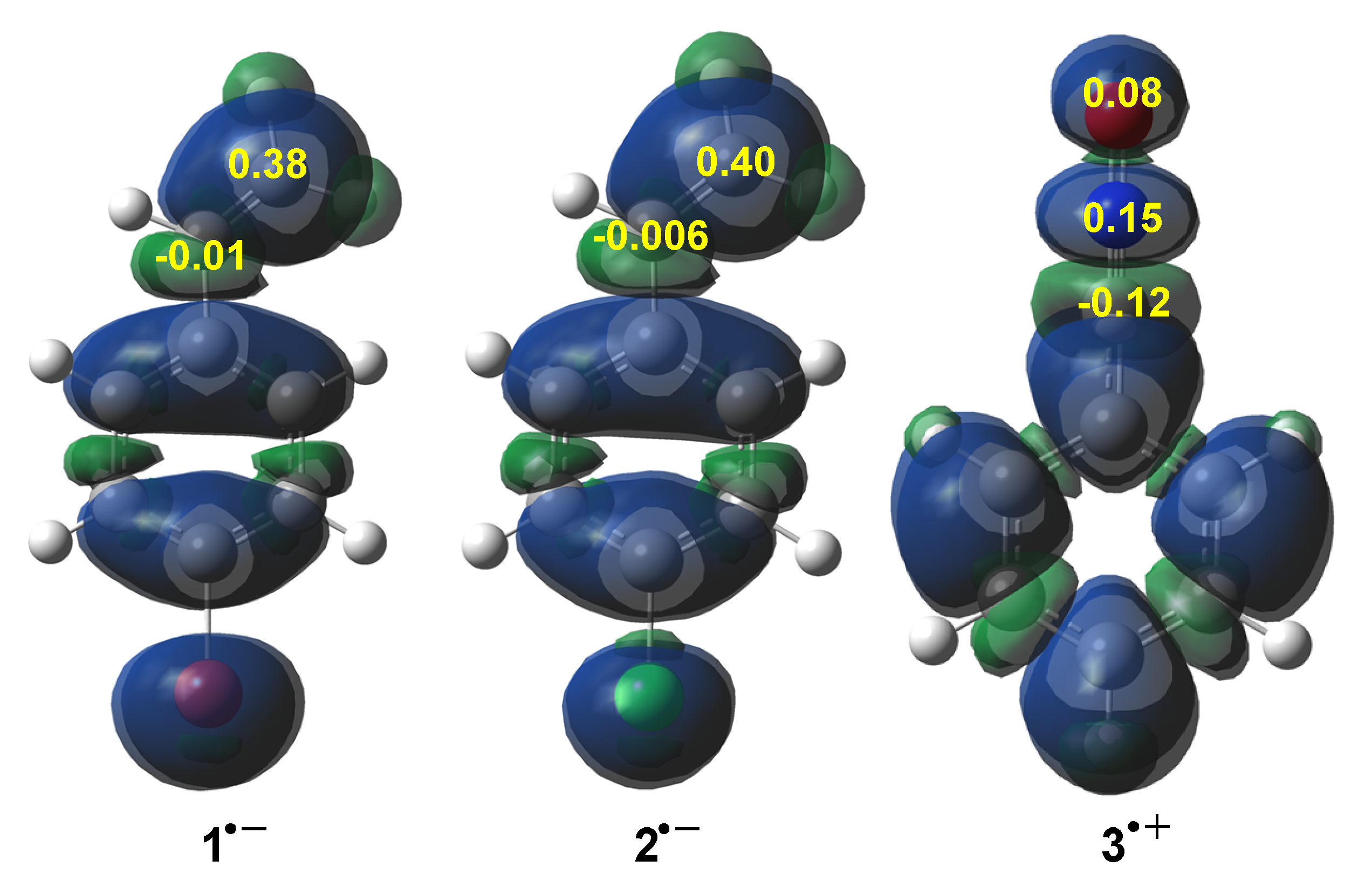
3.1. ELF Analysis of the Electronic Structure of BrVB 1, ClVB 2 and BzNO 3
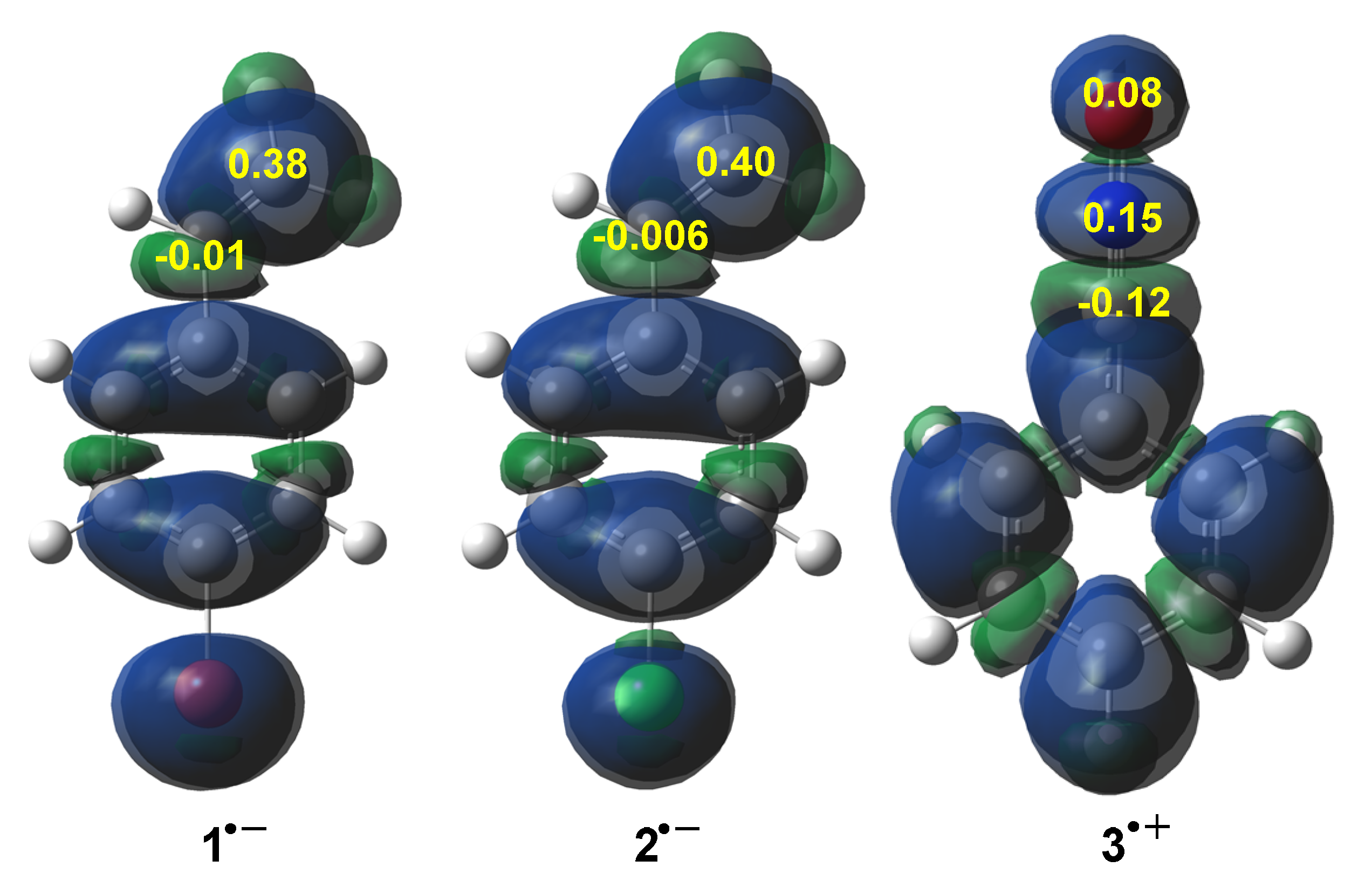
3.2. Analysis of CDFT Reagent Indices
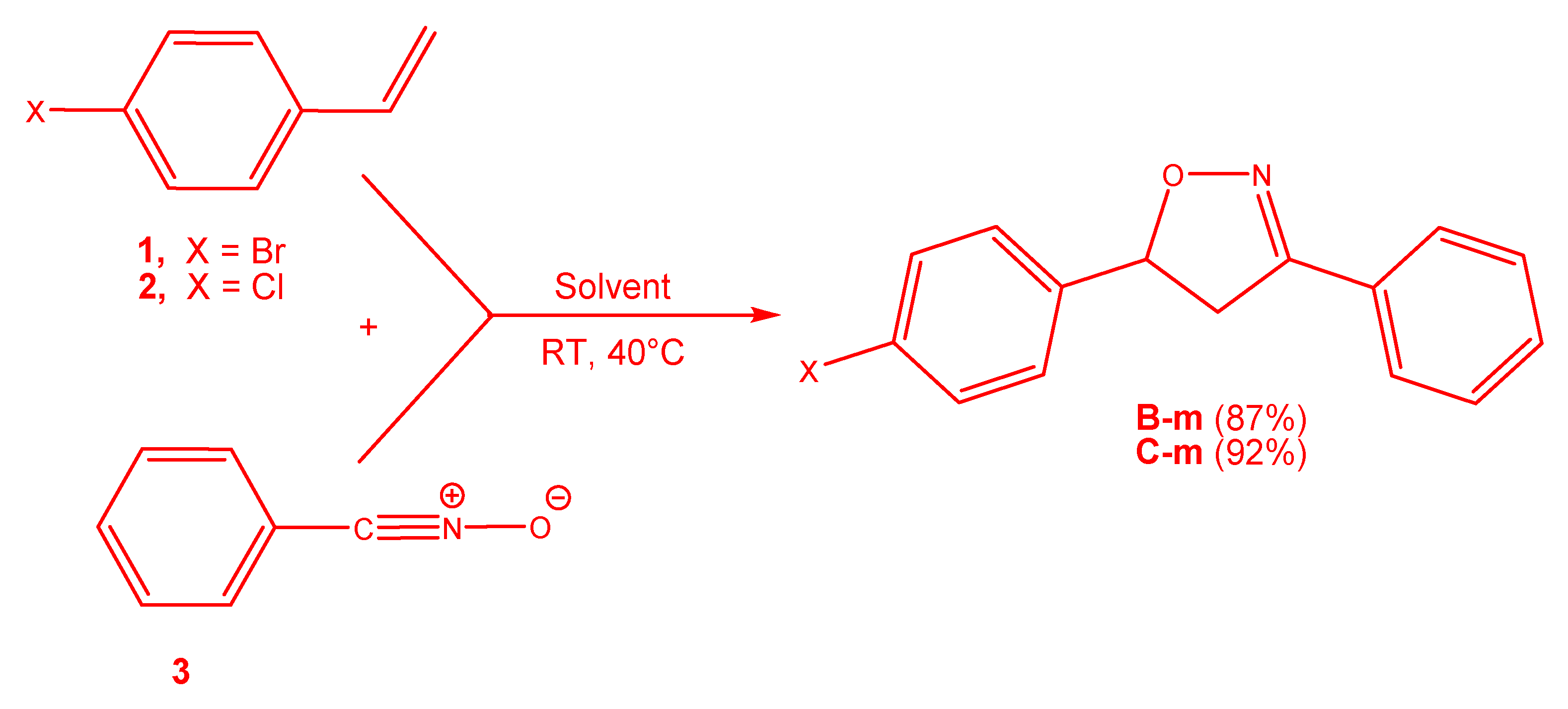
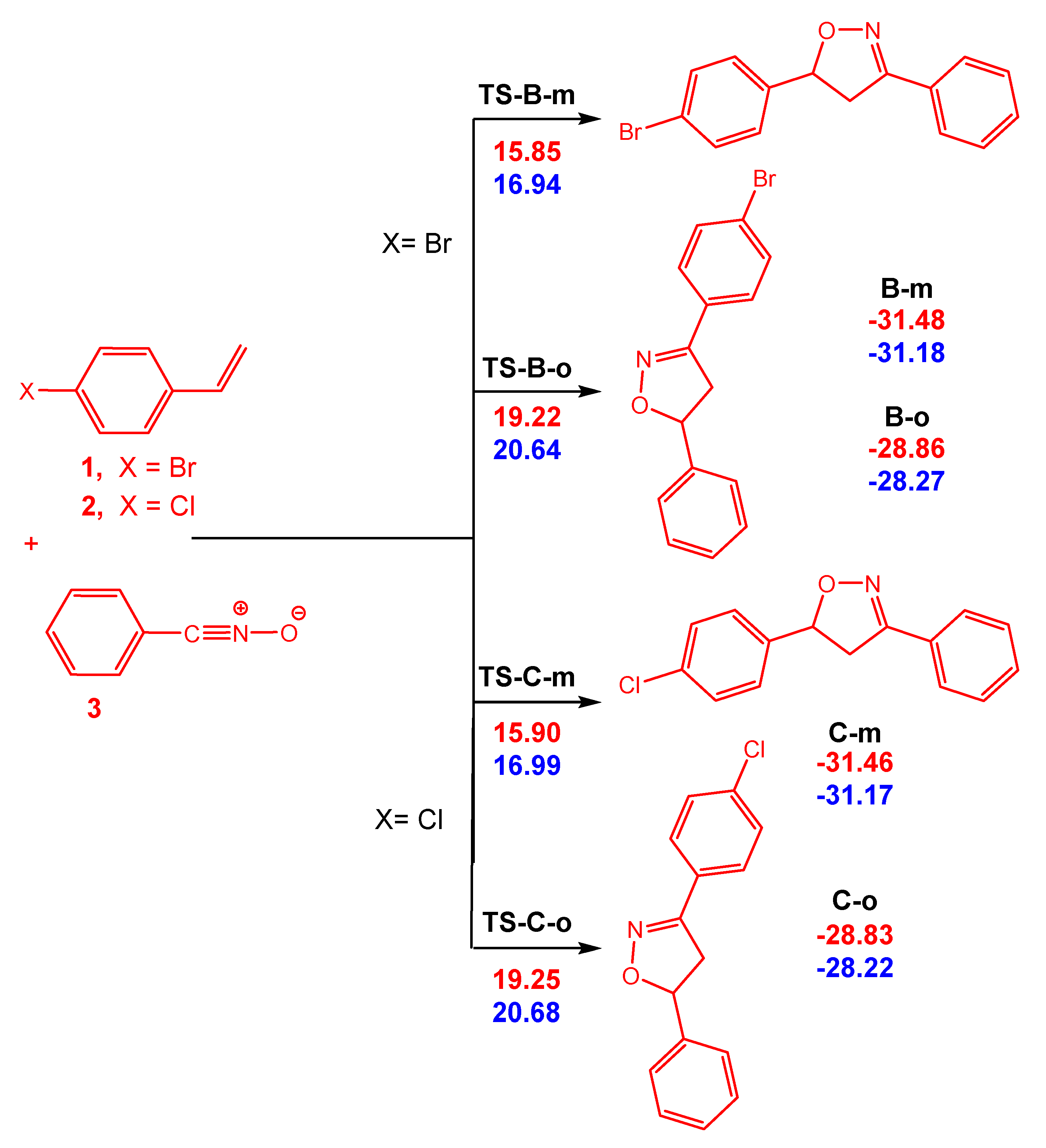
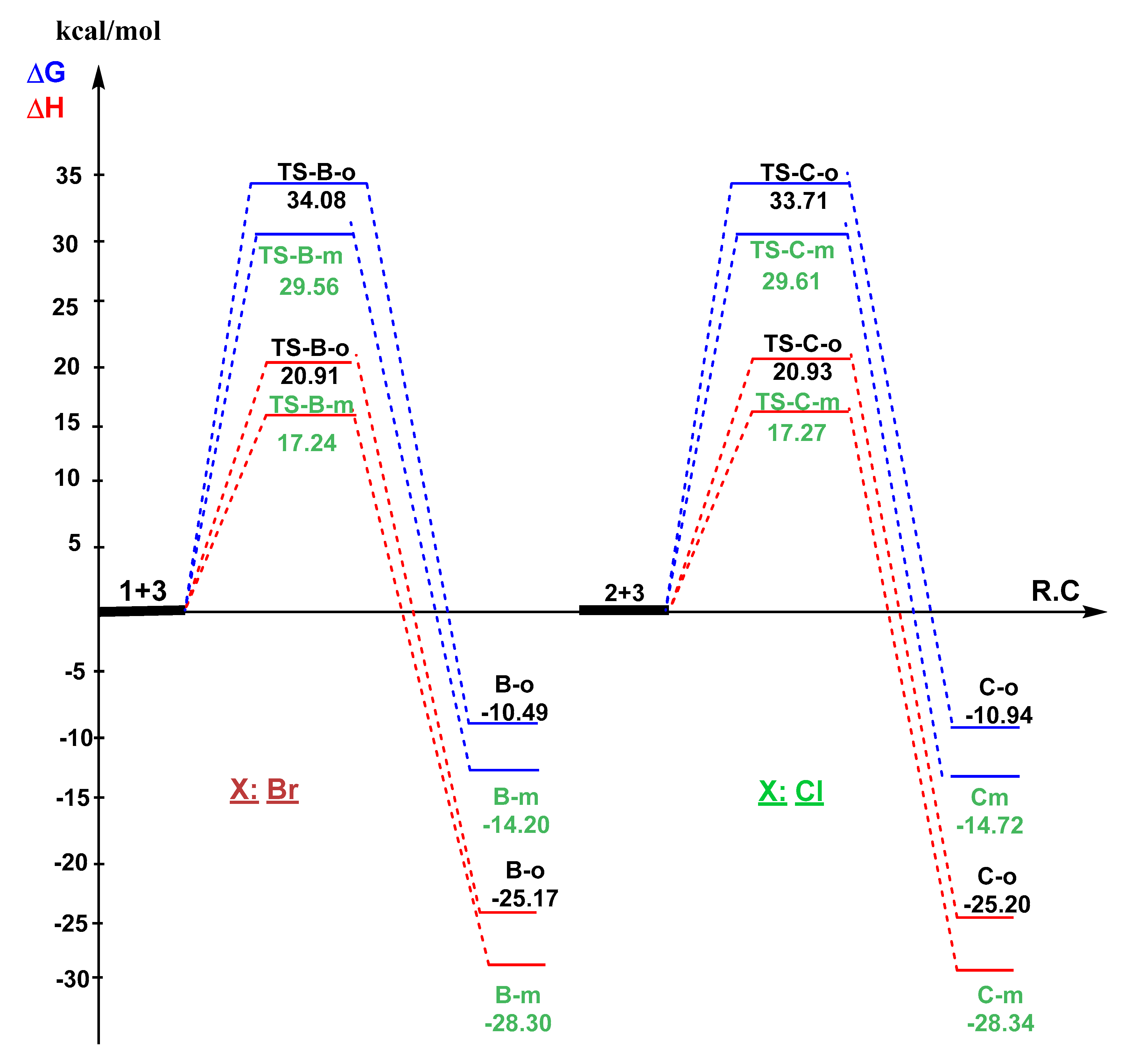
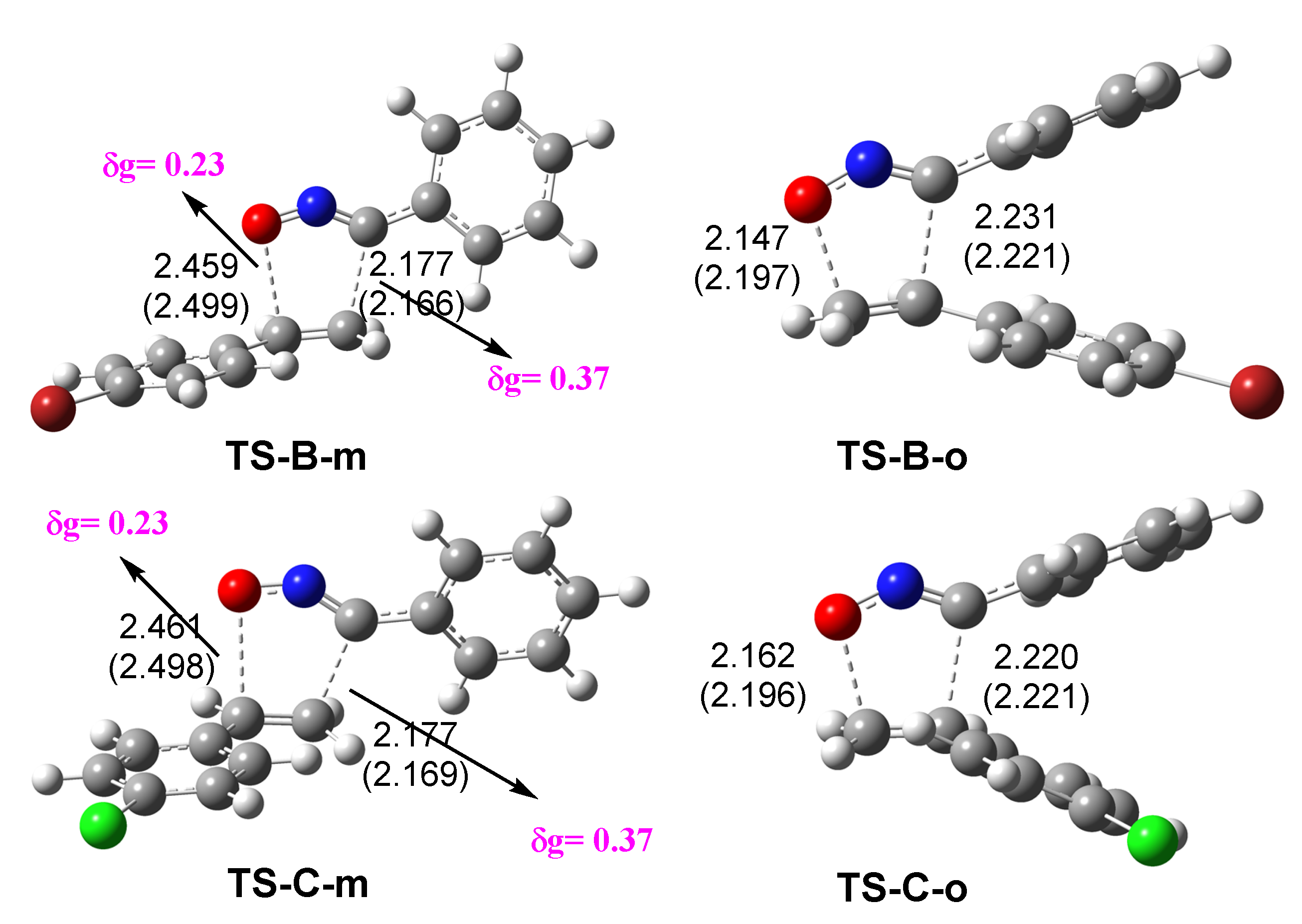
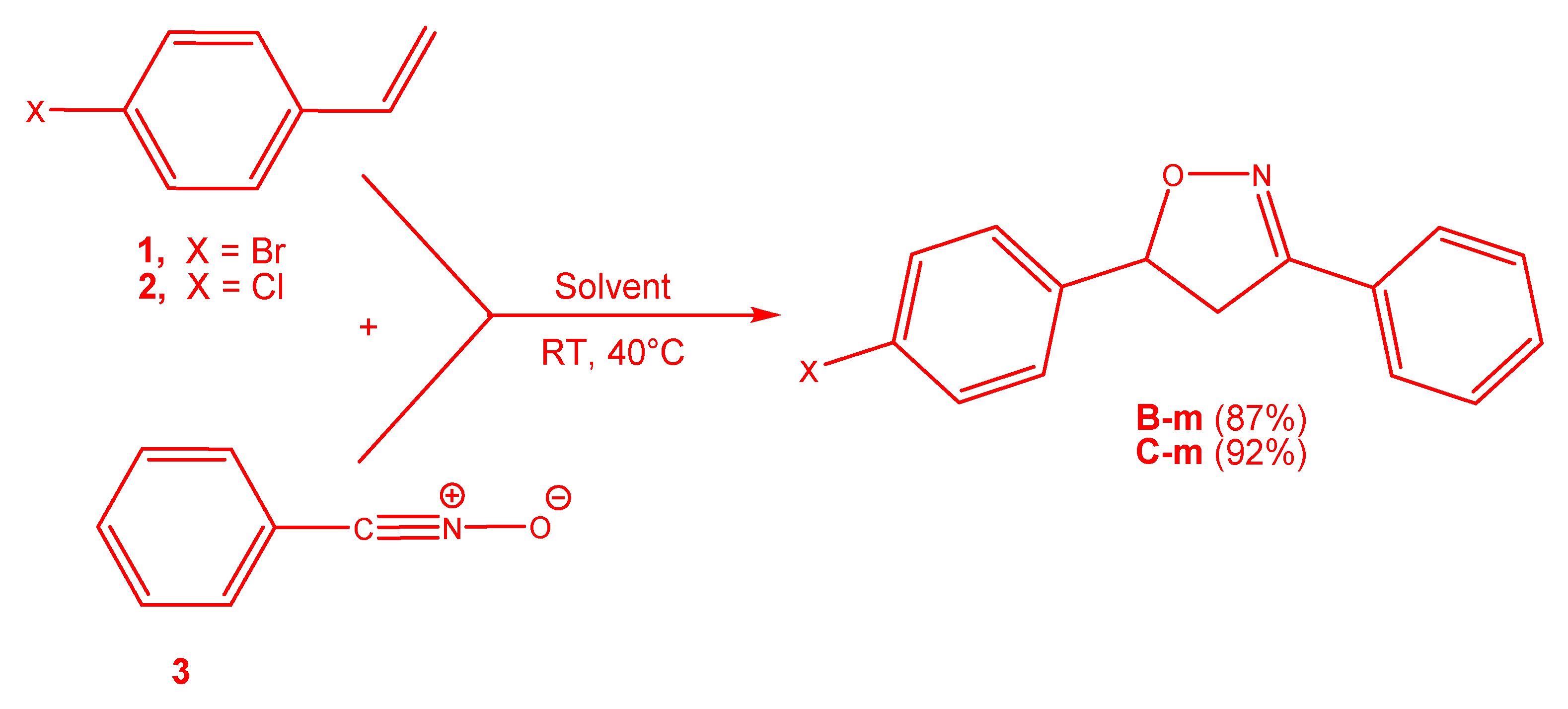
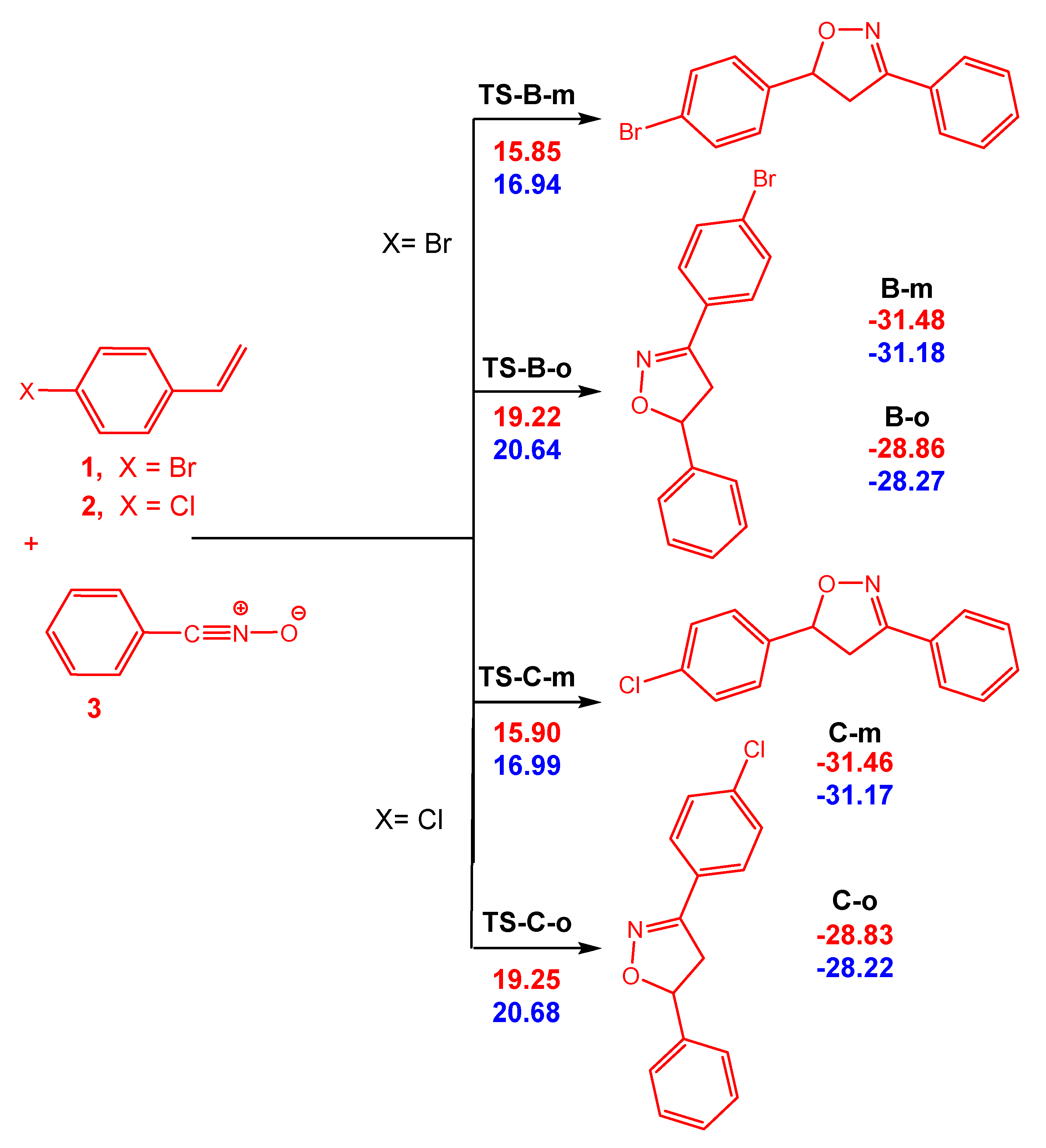
3.3. Exploration of the Reaction Paths Associated with 32CA Reaction of BrVB 1 and ClVB 2 with BzNO 3
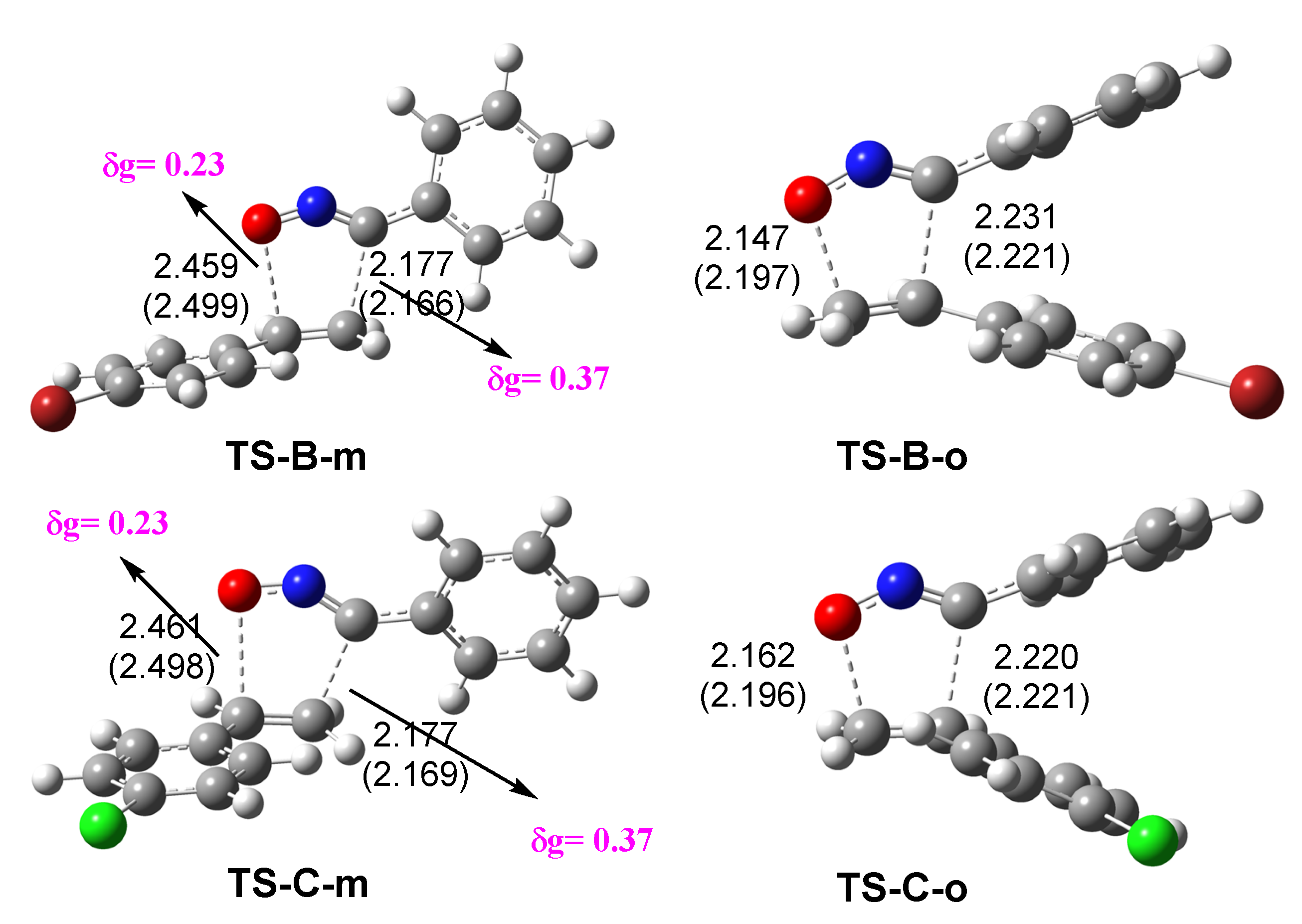
3.4. Bonding Evolution Theory (BET) Study of the 32CA Reactions of BrVB 1 and ClVB 2 with BzNO 3
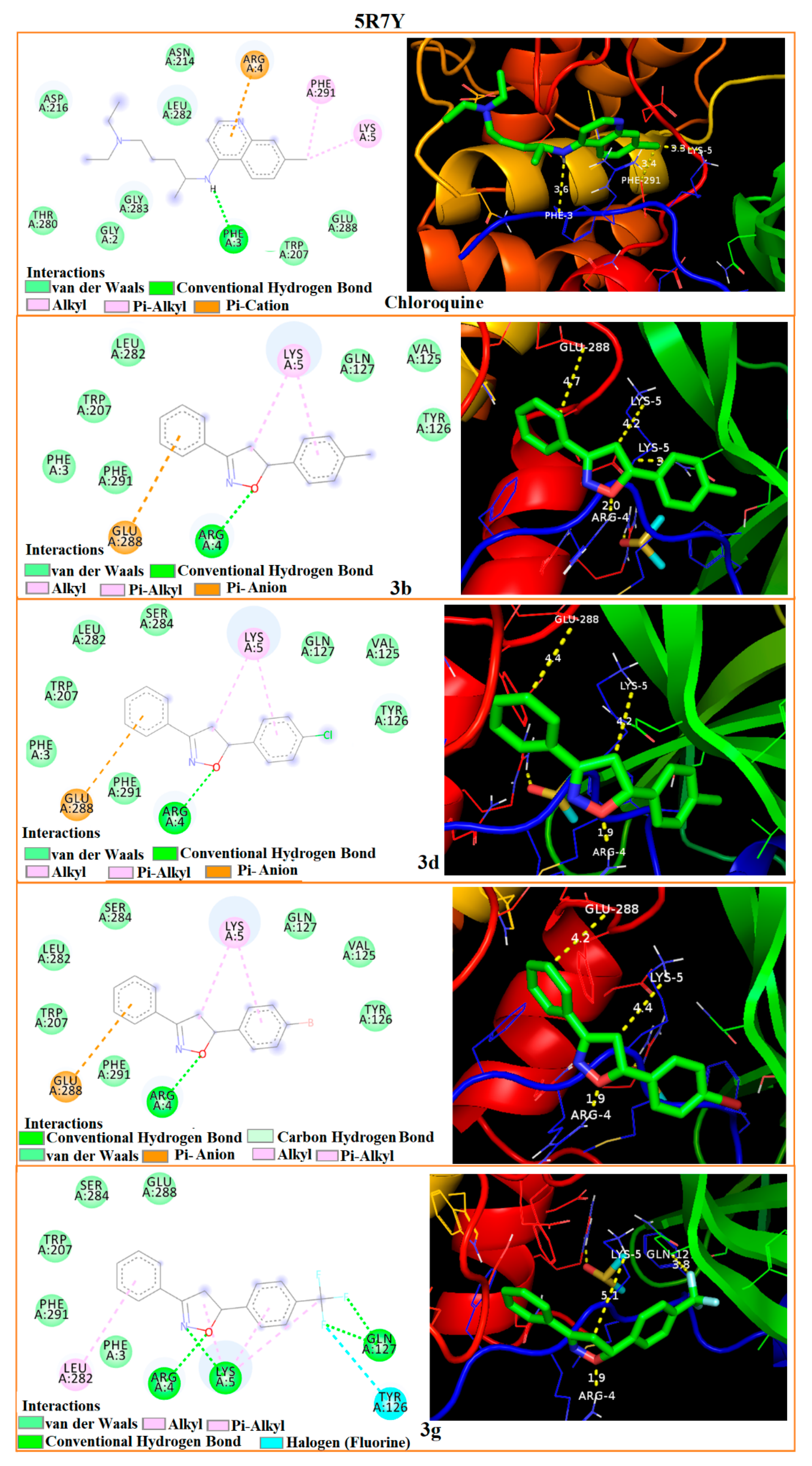
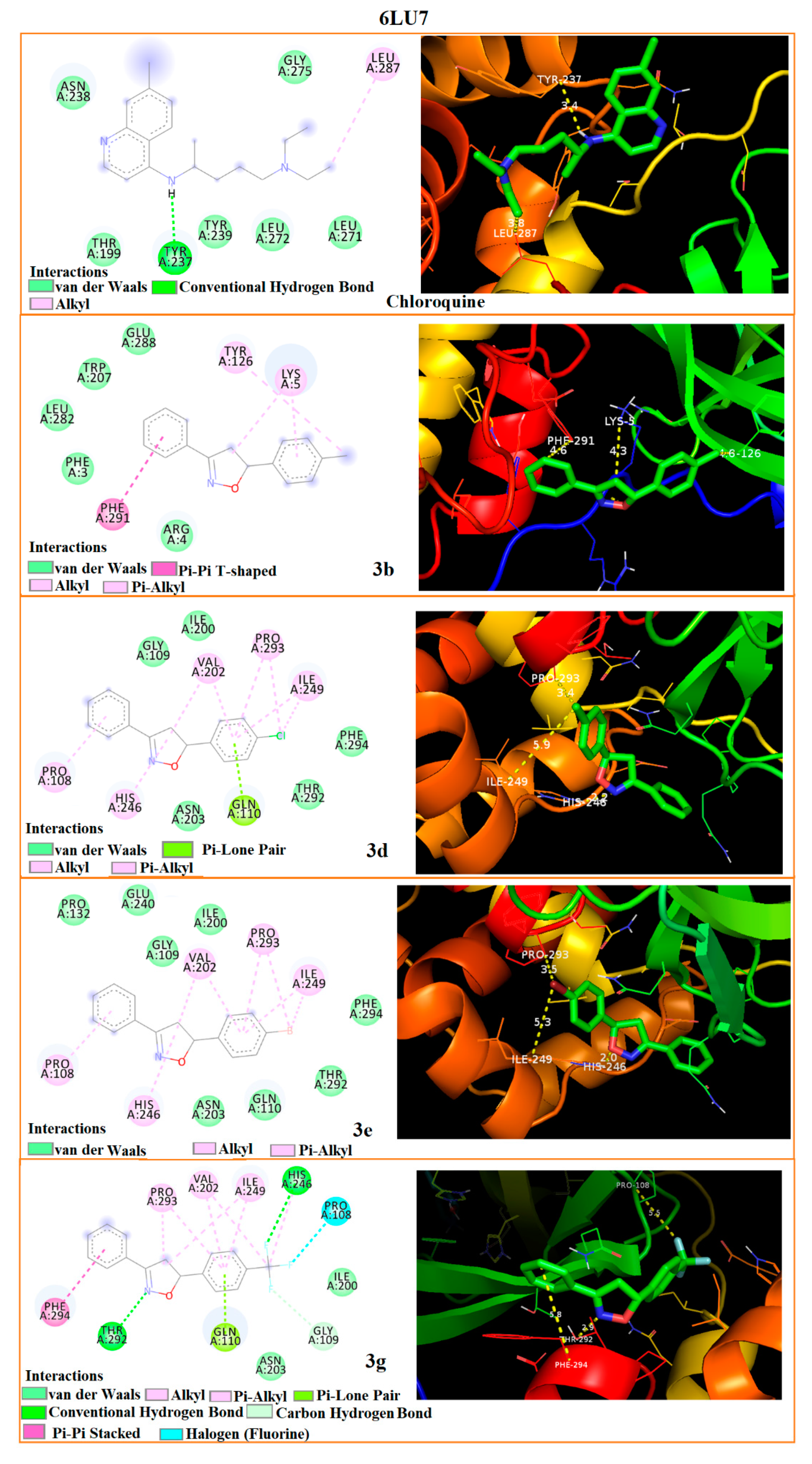
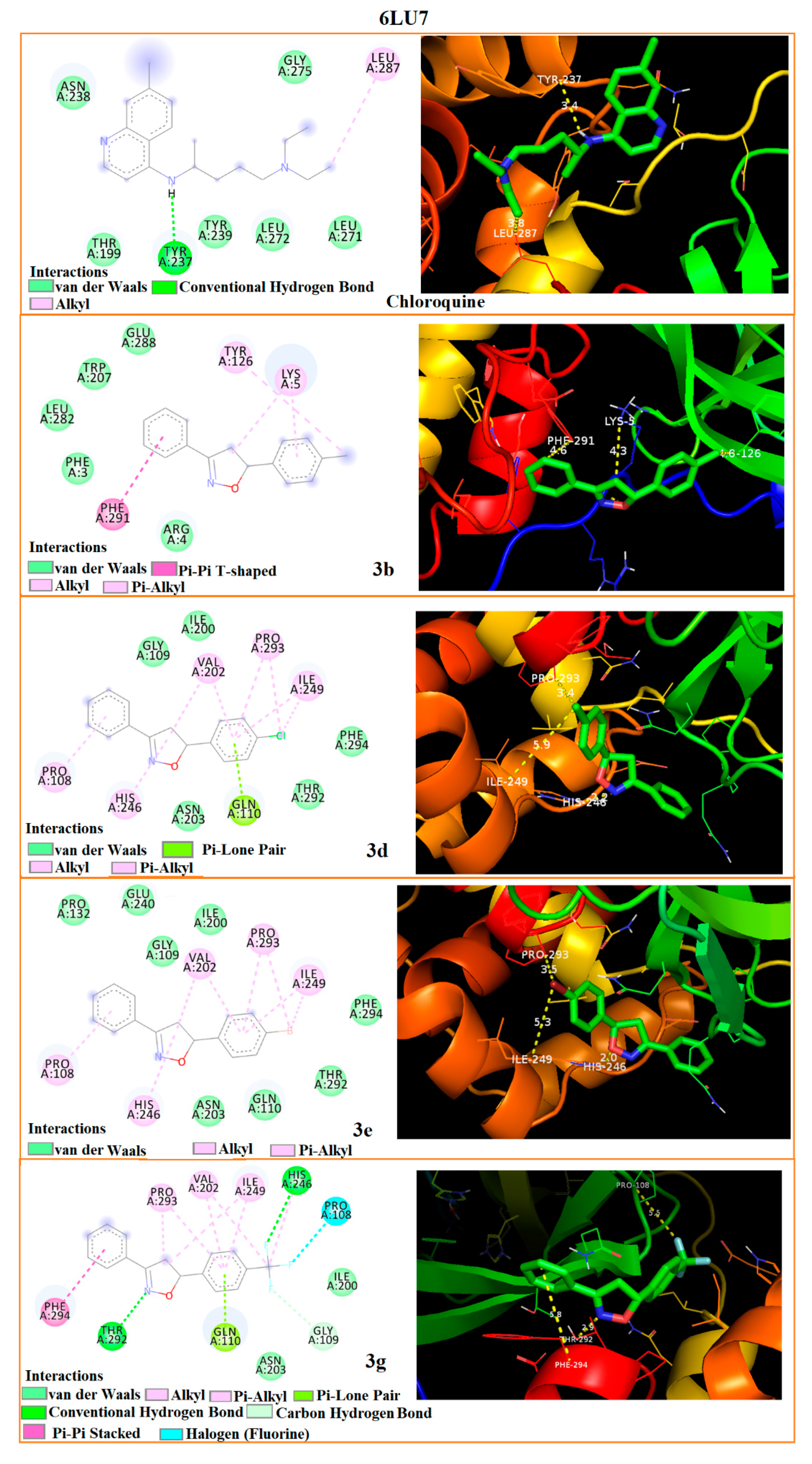
3.5. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Southon, I.W.; Buckingham, J. Dictionary of Alkaloids; Chapman & Hall: New York, NY, USA, 1989. [Google Scholar]
- Gothelf, K.V.; Jørgensen, K.A. Asymmetric 1,3-Dipolar Cycloaddition Reactions. Chem. Rev. 1998, 98, 863–910. [Google Scholar] [CrossRef]
- Huisgen, R. Kinetics and Mechanism of 1,3-Dipolar Cycloadditions. Angew. Chem. Int. Ed. 1963, 2, 633–645. [Google Scholar] [CrossRef]
- Huisgen, R. 1,3-Dipolar cycloadditions. 76. Concerted nature of 1,3-dipolar cycloadditions and the question of diradical intermediates. J. Org. Chem. 1976, 41, 403–419. [Google Scholar] [CrossRef]
- Firestone, R.A. Mechanism of 1,3-dipolar cycloadditions. J. Org. Chem. 1968, 33, 2285–2290. [Google Scholar] [CrossRef]
- Firestone, R.A. The diradical mechanism for 1,3-dipolar cycloadditions and related thermal pericyclic reactions. Tetrahedron 1977, 33, 3009–3039. [Google Scholar] [CrossRef]
- Firestone, R.A. The Low Energy of Concert in Many Symmetry Allowed Cycloadditions Supports a Stepwise Diradical Mechanism. Int. J. Chem. Kinet. 2013, 45, 415–428. [Google Scholar] [CrossRef]
- Kozikowski, A.P. The isoxazoline route to the molecules of nature. Acc. Chem. Res. 1984, 17, 410–416. [Google Scholar] [CrossRef]
- Kumar, A.B.V.K.; Sankar, A.U.R.; Kim, S.H. A Simple, Efficient One Pot Synthesis of 2-Isoxazoline Derivatives and their Antimicrobial Activity. J. Heterocycl. Chem. 2014, 51, E146–E151. [Google Scholar] [CrossRef]
- Diana, G.D.; McKinlay, M.A.; Brisson, C.J.; Zalay, E.S.; Miralles, J.V.; Salvador, U.J. Isoxazoles with antipi cornavirus activity. J. Med. Chem. 1985, 28, 748–752. [Google Scholar] [CrossRef] [PubMed]
- Antczak, C.; Bauvois, B.; Monneret, C.; Florent, J.C. A new acivicin prodrug designed for tumor-Targeted delivery. Bioorg. Med. Chem. 2001, 9, 2843–2848. [Google Scholar] [CrossRef]
- Prajapti, S.K.; Shrivastava, S.; Bihade, U.; Gupta, A.K.; Naidu, V.G.M.; Banerjee, U.C.; Babu, B.N. Synthesis and biological evaluation of novel Δ2-isoxazoline fused cyclopentane derivatives as potential antimicrobial and anticancer agents. Med. Chem. Comm. 2015, 6, 839–845. [Google Scholar] [CrossRef]
- Zhang, P.; Wei, C.; Wang, E.; Wang, W.; Liu, M.; Yin, Q.; Chen, H.; Wang, K.; Li, X.; Zhang, J. Synthesis and biological activities of novel isoxazoline-linked pseudodisaccharide derivatives. Carbohydr. Res. 2012, 351, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Abdelall, E.K.A. Synthesis and biological evaluations of novel isoxazoles and furoxan derivative as anti-inflammatory agents. Bioorg. Chem. 2019, 94, 103441. [Google Scholar] [CrossRef]
- Zhu, J.; Mo, J.; Lin, H.-Z.; Chen, Y.; Sun, H.-P. The recent progress of isoxazole in medicinal chemistry. Bioorg. Med. Chem. 2018, 26, 3065–3075. [Google Scholar] [CrossRef]
- Zadrozna, I.; Kurkowska, J.; Kruszewska, H.; Makuch, I. 4,5-Dihydroisoxazoles: Testing of antimicrobial activity. Il Farmaco. 2000, 55, 499–501. [Google Scholar] [CrossRef]
- Liu, X.; Lu, D.; Wu, J.-H.; Tan, J.-P.; Jiang, C.; Gao, G.; Wang, T. Stereoselective Synthesis of CF3-Containing Spiro oxindoles via 1,3-Dipolar Cycloaddition by Dipeptide Based Phosphonium Salt Catalysis. Adv. Synth. Catal. 2020, 362, 1490–1495. [Google Scholar] [CrossRef]
- Gui, H.-Z.; Wei, Y.; Shi, M. Recent Advances in the Construction of Trifluoromethyl Containing Spiro oxindoles through Cycloaddition Reactions. Chem. Asian J. 2020, 15, 1225–1233. [Google Scholar] [CrossRef]
- Uno, H.; Imai, T.; Harada, K.; Shibata, N. Synthesis of Highly Functionalized 12-Membered Trifluoromethyl Heterocycles via a Nonde carboxyl ative Pd-Catalyzed [6 + 6] Annulation. ACS Catal. 2020, 10, 1454–1459. [Google Scholar] [CrossRef]
- Bigelow, L.A. The Action of Elementary Fluorine upon Organic Compounds. Chem. Rev. 1947, 40, 51–115. [Google Scholar] [CrossRef]
- Yoshimura, A.; Zhdankin, V.V. Advances in Synthetic Applications of Hypervalent Iodine Compounds. Chem. Rev. 2016, 116, 3328–3435. [Google Scholar] [CrossRef]
- Muller, K.; Faeh, C.; Diederich, F. Fluorine in Pharmaceuticals: Looking Beyond Intuition. Science 2007, 317, 1881–1886. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, R.; Bansal, A.; Mittal, A. Synthesis and antimicrobial activity of 3-(2-thienyl)-4-arylazo-5-hydroxy-5-trifluoromethyl-Δ2-isoxazolines and 3-(2-thienyl)-4-arylazo-5-trifluoromethylisoxazoles. J. Fluor.Chem. 2013, 145, 95–101. [Google Scholar] [CrossRef]
- Filler, R.; Kobayashi, Y. Biomedicinal Aspects of Fluorine Chemistry; Elsevier: New York, NY, USA, 1982. [Google Scholar]
- Giovannoni, M.P.; Vergelli, C.; Ghelardini, C.; Galeotti, N.; Bartolini, A.; Piaz, V.D. [(3-Chlorophenyl)piperazinylpropyl]pyridazinones and Analogues as Potent Antinociceptive Agents. J. Med. Chem. 2003, 46, 1055–1059. [Google Scholar] [CrossRef]
- Karthikeyan, K.; Seelan, T.V.; Lalitha, K.G.; Perumal, P.T. Synthesis and antinociceptive activity of pyrazolyl isoxazolines and pyrazolyl isoxazoles. Bioorg. Med. Chem. Lett. 2009, 19, 3370–3373. [Google Scholar] [CrossRef]
- Kamal, A.; Bharathi, E.V.; Reddy, J.S.; Ramaiah, M.J.; Dastagiri, D.; Reddy, M.K.; Viswanath, A.; Reddy, T.L.; Shaik, T.B.; Pushpavalli, S.N.C.V.L.; et al. Synthesis and biological evaluation of 3,5-diaryl isoxazoline/isoxazole linked 2,3-dihydroquinazolinone hybrids as anticancer agents. Eur. J. Med. Chem. 2011, 46, 691–703. [Google Scholar] [CrossRef]
- Patrizia, D.; Carbone, A.; Barraja, P.; Kelter, G.; Fiebig, H.H.; Cirrincione, G. Synthesis and antitumor activity of 2,5-bis(3′-indolyl)-furans and 3,5-bis(3′-indolyl)-isoxazoles, nortopsentin analogues. Bioorg. Med. Chem. 2010, 18, 4524–4529. [Google Scholar]
- Isanbor, C.; O’Hagan, D. Fluorine in medicinal chemistry: A review of anti-cancer agents. J. Fluor. Chem. 2006, 127, 303–319. [Google Scholar] [CrossRef]
- De Weerd, N.J.; Bukovsky, E.V.; Castro, P.K.; Kuvychko, I.V.; Popovb, A.A.; Strauss, S.H.; Olga Boltalina, V. Steric and electronic effects of CF3 conformations in acene(CF3)n derivatives. J. Fluor. Chem. 2019, 221, 1–7. [Google Scholar] [CrossRef]
- Haranahalli, K.; Honda, T.; Ojima, I. Recent progress in the strategic incorporation of fluorine into medicinally active compounds. J. Fluor. Chem. 2019, 217, 29–40. [Google Scholar] [CrossRef]
- Agbaje, C.O.; Fadeyi, O.O.; Okoro, O.C. Lewis acid mediated diastereoselective synthesis of fused fluorinated spiroketal as potential biologically active compounds. Tetrahedron Lett. 2011, 52, 5297–5300. [Google Scholar] [CrossRef]
- Jbeily, M.; Kressler, J. Fluorophilicity and lipophilicity of fluorinated rhodamines determined by their partition coefficients in biphasic solvent systems. J. Fluor. Chem. 2017, 193, 67–72. [Google Scholar] [CrossRef]
- Ojima, I. Fluorine in Medicinal Chemistry and Chemical Biology; Wiley-Blackwell: Chichester, UK, 2009. [Google Scholar]
- Wang, J.; Sánchez-Roselló, M.; Aceña, J.L.; del Pozo, C.; Sorochinsky, A.E.; Fustero, S.; Soloshonok, V.A.; Liu, H. Fluorine in Pharmaceutical Industry: Fluorine-Containing Drugs Introduced to the Market in the Last Decade (2001–2011). Chem. Rev. 2014, 114, 2432–2506. [Google Scholar] [CrossRef] [PubMed]
- Koldobskii, A.B.; Shilova, O.S.; Artyushin, O.I.; Kagramanov, N.D.; Moiseev, S.K. Polyfluorinated esters of 4-chloro-2-oxobut-3-ynoic acid. Cycloaddition reactions of hexafluoro isopropyl 4-chloro-2-oxobut-3-ynoate, an incredibly electrophilic alkyne. J. Fluor. Chem. 2020, 231, 109463. [Google Scholar] [CrossRef]
- Bigotti, S.; Malpezzi, L.; Molteni, M.; Mele, A.; Panzeri, W.; Zanda, M. Functionalized fluoroalkyl heterocycles by 1,3-dipolar cycloadditions with γ-fluoro-α-nitroalkenes. Tetrahedron Lett. 2009, 50, 2540–2542. [Google Scholar] [CrossRef]
- O’Hagan, D. Fluorine in health care: Organofluorine containing blockbuster drugs. J. Fluor. Chem. 2010, 131, 1071–1081. [Google Scholar] [CrossRef]
- McGrath, N.A.; Brichacek, M.; Njardarson, J.T.A. Graphical Journey of Innovative Organic Architectures That Have Improved Our Lives. J. Chem. Educ. 2010, 87, 1348–1349. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceňa, J.L.; Soloshonok, V.A.; Izawa, K.; Liu, H. Next Generation of Fluorine-Containing Pharmaceuticals, Compounds Currently in Phase II–III Clinical Trials of Major Pharmaceutical Companies: New Structural Trends and Therapeutic Areas. Chem. Rev. 2016, 116, 422–518. [Google Scholar] [CrossRef]
- Domingo, L.R. Molecular Electron Density Theory: A Modern View of Reactivity in Organic Chemistry. Molecules 2016, 21, 1319. [Google Scholar] [CrossRef]
- Ríos-Gutiérrez, M.; Domingo, L.R. Unravelling the Mysteries of the [3 + 2] Cycloaddition Reactions. Eur. J. Org. Chem. 2019, 2019, 267–282. [Google Scholar] [CrossRef]
- Domingo, L.R.; Aurell, M.J.; Pérez, P. A DFT analysis of the participation of zwitterionic TACs in polar [3 + 2] cycloaddition reactions. Tetrahedron 2014, 70, 4519–4525. [Google Scholar] [CrossRef]
- Zeroual, A.; Ríos-Gutiérrez, M.; El Idrissi, M.; El Alaoui El Abdallaoui, H.; Domingo, L.R. An MEDT study of the mechanism and selectivities of the [3 + 2] cycloaddition reaction of tomentosin with benzonitrile oxide. Int. J. Quantum Chem. 2019, 119, e25980. [Google Scholar] [CrossRef]
- Zeroual, A.; Ríos-Gutiérrez, M.; Salah, M.; El Alaoui El Abdallaoui, H.; Domingo, L.R. An investigation of the molecular mechanism, chemoselectivity and regioselectivity of cycloaddition reaction between acetonitrile N-Oxide and 2,5-dimethyl-2H-[1,2,3]diazaphosphole: A MEDT study. J. Chem. Sci. 2019, 131, 75. [Google Scholar] [CrossRef] [Green Version]
- Yoshimura, A.; Middleton, K.R.; Todora, A.D.; Kastern, B.J.; Koski, S.R.; Maskaev, A.V.; Zhdankin, V.V. Hypervalent Iodine Catalyzed Generation of Nitrile Oxides from Oximes and their Cycloaddition with Alkenes or Alkynes. Org. Lett. 2013, 15, 4010–4013. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Hehre, W.J.; Radom, L.; Schleyer, P.R.; Pople, J.A. Ab Initio Molecular Orbital Theory; Wiley: New York, NY, USA, 1986. [Google Scholar]
- Fisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision A.02; Fox, Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Schlegel, H.B. Optimization of equilibrium geometries and transition structures. J. Comput. Chem. 1982, 3, 214–218. [Google Scholar] [CrossRef]
- Fukui, K. Formulation of the reaction coordinate. J. Phys. Chem. 1970, 74, 4161–4163. [Google Scholar] [CrossRef]
- Gonzalez, C.; Schlegel, H.B. Improved algorithms for reaction path following: Higher order implicit algorithms. J. Chem. Phys. 1991, 95, 5853–5860. [Google Scholar] [CrossRef] [Green Version]
- Tomasi, J.; Persico, M. Molecular Interactions in Solution: An Overview of Methods Based on Continuous Distributions of the Solvent. Chem. Rev. 1994, 94, 2027–2094. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M.; Tomasi, J. Geometry optimization of molecular structures in solution by the polarizable continuum model. J. Comput. Chem. 1998, 19, 404–417. [Google Scholar] [CrossRef]
- Domingo, L.R. A new C–C bond formation model based on the quantum chemical topology of electron density. RSC Adv. 2014, 4, 32415–32428. [Google Scholar] [CrossRef] [Green Version]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. Applications of the Conceptual Density Functional Theory Indices to Organic Chemistry Reactivity. Molecules 2016, 21, 748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Noury, S.; Krokidis, X.; Fuster, F.; Silvi, B. Computational tools for the electron localization function topological analysis. Comput. Chem. 1999, 23, 597–604. [Google Scholar] [CrossRef]
- Krokidis, X.; Noury, S.; Silvi, B. Characterization of Elementary Chemical Processes by Catastrophe Theory. J. Phys. Chem. A 1997, 101, 7277–7282. [Google Scholar] [CrossRef]
- Lefebre, C.; Khartabil, H.; Boisson, J.C.; Garcia, J.C.; Piquemal, J.P.; Hénon, E. The Independent Gradient Model: A New Approach for Probing Strong and Weak Interactions in Molecules from Wave Function Calculations. Chem. Phys. Chem. 2018, 19, 724–735. [Google Scholar] [CrossRef]
- Görling, A. Density-functional theory beyond the Hohenberg-Kohn theorem. Phys. Rev. A 1999, 59, 3359–3374. [Google Scholar] [CrossRef]
- Salah, M.; Belghiti, M.E.; Aitouna, A.O.; Zeroual, A.; Jorio, S.; El Alaoui, A.H.; El hadki, H.; Marakchi, K.; Komiha, N. MEDT study of the 1,3-DC reaction of diazomethane with Psilostachyin and investigation about the interactions of some pyrazoline derivatives with protease (Mpro) of nCoV-2. J. Mol. Graph. Model. 2021, 102, 107763. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wave function analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Silvi, B.; Savin, A. Classification of chemical bonds based on topological analysis of electron localization functions. Nature 1994, 371, 683–686. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Parr, R.G.; Szentpaly, L.V.; Liu, S. Electrophilicity Index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Domingo, L.R.; Chamorro, E.; Pérez, P. Understanding the Reactivity of Captodative Ethylenes in Polar Cycloaddition Reactions. A Theoretical Study. J. Org. Chem. 2008, 73, 4615–4624. [Google Scholar] [CrossRef] [PubMed]
- Aurell, M.J.; Domingo, L.R.; Pérez, P.; Contreras, R. A theoretical study on the regioselectivity of 1,3-dipolar cycloadditions using DFT-based reactivity indexes. Tetrahedron 2004, 60, 11503–11509. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Castellanos Soriano, J. Understanding the Origin of the Regioselectivity in Non-Polar [3 + 2] Cycloaddition Reactions through the Molecular Electron Density Theory. Org. Chem. 2020, 1, 3. [Google Scholar] [CrossRef]
- Domingo, L.R.; Pérez, P.; Sáez, J.A. Understanding the local reactivity in polar organic reactions through electrophilic and nucleophilic Parr functions. RSC Adv. 2013, 3, 1486–1494. [Google Scholar] [CrossRef]
- Zeroual, A.; Ríos-Gutiérrez, M.; El Ghozlani, M.; El Idrissi, M.; OuledAitouna, A.; Salah, M.; El Abdallaoui, H.E.; Domingo, L.R. A molecular electron density theory investigation of the molecular mechanism, regioselectivity, stereoselectivity and chemoselectivity of cycloaddition reaction between acetonitrile N-oxide and 2,5-dimethyl-2H-[1,2,3]diazarsole. Theor. Chem. Acc. 2020, 139, 37. [Google Scholar] [CrossRef]
- Domingo, L.R.; Sáez, J.A.; Zaragozá, R.J.; Arnó, M. Understanding the Participation of Quadricyclane as Nucleophile in Polar [2σ + 2σ + 2π] Cycloadditions toward Electrophilic π Molecules. J. Org. Chem. 2008, 73, 8791–8799. [Google Scholar] [CrossRef]
- Didelot, C.; Lanneau, D.; Brunet, M.; Joly, A.L.; Thonel, A.D.; Chiosis, G.; Garrido, C. Anti-Cancer Therapeutic Approaches Based on Intracellular and Extracellular Heat Shock Proteins. Curr. Med. Chem. 2007, 14, 2839–2847. [Google Scholar] [CrossRef]
- Tumma, R.; Vamaraju, H.B. Design, Synthesis, Molecular Docking, and Biological Evaluation of Pyrazole 1-Carbothiamide incorporated Isoxazole Derivatives. Asian J. Pharm. Clin. Res. 2019, 12, 245–250. [Google Scholar] [CrossRef]
- Loh, B.; Vozzolo, L.; Mok, B.J.; Lee, C.C.; Fitzmaurice, R.J.; Caddick, S.; Fassati, A. Inhibition of HIV-1 Replication by Isoxazolidine and Isoxazole Sulfonamides. Chem. Biol. Drug. Des. 2010, 75, 461–474. [Google Scholar] [CrossRef]
- Panda, S.S.; Chowdary, P.V.; Jayashree, B.S. Synthesis, Antiinflammatory and Antibacterial Activity of Novel Indolyl-isoxazoles. Indian J. Pharm. Sci. 2009, 71, 684–687. [Google Scholar] [CrossRef] [Green Version]
- Durgamma, S.; Reddy, P.R.; Padmavathi, V.; Padmaja, A. Synthesis and Antioxidant Activity of Amido-Linked Benzoxazolyl/Benzothiazolyl/BenzimidazolylPyrazoles and Isoxazoles. J. Heterocycl. Chem. 2016, 53, 738–747. [Google Scholar] [CrossRef]
- Makarov, V.A.; Riabova, O.B.; Granik, V.G.; Wutzler, P.; Schmidtke, M. Novel [(biphenyloxy)propyl]isoxazole derivatives for inhibition of human rhinovirus 2 and coxsackie virus B3 replication. J. Antimicrob. Chemother. 2005, 55, 483–488. [Google Scholar] [CrossRef]
- Basha, S.S.; Divya, K.; Padmaja, A.; Padmavathi, V. Synthesis and antimicrobial activity of thiazolyl pyrazoles and isoxazoles. Res. Chem. Int. 2015, 41, 10067–10083. [Google Scholar] [CrossRef]
- Arrieta, A.; Carrillo, J.R.; Cossio, F.P.; Diaz-Ortiz, A.; Gomez-Escalonilla, M.J.; Hoz, A.D.L.; Langa, F.; Moreno, A. Efficient tautomerization hydrazone-azomethine imine under microwave irradiation. Synthesis of [4,3′] and [5,3′]bipyrazoles. Tetrahedron 1998, 54, 13167–31180. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Burley, S.K.; Berman, H.M.; Bhikadiya, C.; Bi, C.; Chen, L.; Costanzo, L.D.; Feng, Z. RCSB Protein Data Bank: Biological macromolecular structures enabling research and education in fundamental biology, biomedicine, biotechnology and energy. Nucleic Acids Res. 2019, 47, D464–D474. [Google Scholar] [CrossRef] [Green Version]
- Goodsell, D.S.; Olson, A.J. Automated Docking of Substrates to Proteins by Simulated Annealing. Proteins Struct. Funct. Bioinform. 1990, 8, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef] [Green Version]
- Katritzky, A.R.; Mu, L.; Lobanov, V.S.; Karelson, M. Correlation of Boiling Points with Molecular Structure. 1. A Training Set of 298 Diverse Organics and a Test Set of 9 Simple Inorganics. J. Phys. Chem. 1996, 100, 10400–10407. [Google Scholar] [CrossRef]
- Solis, F.J.; Wets, R.J.B. Minimization by Random Search Techniques. Math. Oper. Res. 1981, 6, 19–30. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | η | µ | ω | N |
|---|---|---|---|---|
| BrVB 1 | 4.97 | −3.63 | 1.32 | 3.41 |
| ClVB 2 | 5.02 | −3.64 | 1.32 | 3.36 |
| BzNO 3 | 5.02 | −3.82 | 1.45 | 3.19 |
| Name | 5-(p-methylphenyl)-3-phenyl-2-isoxazoline (D-m) | 5-(p-chlorophenyl)-3-phenyl-2-isoxazoline (C-m) | 5-(p-bromophenyl)-3-phenyl-2-isoxazoline (B-m) | 3-phenyl-5-(p-trifluoromethyl-phenyl)-2-isoxazoline (A-m) | Chloroquine | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2D Compound Structure |  X: CH3 | 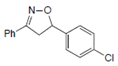 X: Cl |  X: Br |  X: CF3 |  | |||||||||||
| 3D Compound Structure |  |  |  |  |  | |||||||||||
| Binding Energy (kcal·mol−1) | 5R7Y | −6.91 | −6.98 | −7.03 | −6.63 | −6.28 | ||||||||||
| 6LU7 | −5.59 | −6.21 | −6.41 | −5.99 | −4.36 | |||||||||||
| Inhibition constant (μM) | 5R7Y | 8.62 | 7.64 | 7.03 | 13.79 | 24.95 | ||||||||||
| 6LU7 | 79.42 | 28.07 | 19.97 | 41.01 | 634.41 | |||||||||||
| İntermolecular energy (kcal·mol−1) | 5R7Y | −7.51 | −7.58 | −7.63 | −7.53 | −8.67 | ||||||||||
| 6LU7 | −6.19 | −6.81 | −7.01 | −6.88 | −6.75 | |||||||||||
| Interaction Residues-Bond distances (Å) | 5R7Y | ARG-4 2.0 | LYS-5 4.2 | GLU-288 4.7 | ARG-4 1.9 | LYS-5 4.2 | GLU-288 4.4 | ARG-4 1.9 | LYS-5 4.4 | GLU-288 4.2 | ARG-4 1.9 | LYS-5 5.1 | TYR-126 2.9 | PHE-3 3.6 | LYS-5 3.3 | PHE-291 3.4 |
| 6LU7 | TYR-126 4.6 | LYS-5 4.3 | PHE-291 4.6 | HIS-246 2.2 | PRO-293 3.4 | ILE-249 5.9 | HIS-246 2.0 | PRO-293 3.5 | ILE-249 5.3 | THR-292 2.9 | PHE-294 5.8 | PRO-108 5.5 | TYR-237 3.4 | LEU-287 3.8 | - | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
El Idrissi, M.; El Ghozlani, M.; Eşme, A.; Ríos-Gutiérrez, M.; Ouled Aitouna, A.; Salah, M.; El Abdallaoui, H.E.A.; Zeroual, A.; Mazoir, N.; Domingo, L.R. Mpro-SARS-CoV-2 Inhibitors and Various Chemical Reactivity of 1-Bromo- and 1-Chloro-4-vinylbenzene in [3 + 2] Cycloaddition Reactions. Organics 2021, 2, 1-16. https://doi.org/10.3390/org2010001
El Idrissi M, El Ghozlani M, Eşme A, Ríos-Gutiérrez M, Ouled Aitouna A, Salah M, El Abdallaoui HEA, Zeroual A, Mazoir N, Domingo LR. Mpro-SARS-CoV-2 Inhibitors and Various Chemical Reactivity of 1-Bromo- and 1-Chloro-4-vinylbenzene in [3 + 2] Cycloaddition Reactions. Organics. 2021; 2(1):1-16. https://doi.org/10.3390/org2010001
Chicago/Turabian StyleEl Idrissi, Mohammed, Mohamed El Ghozlani, Asli Eşme, Mar Ríos-Gutiérrez, Anas Ouled Aitouna, Mohammed Salah, Habib El Alaoui El Abdallaoui, Abdellah Zeroual, Noureddine Mazoir, and Luis R. Domingo. 2021. "Mpro-SARS-CoV-2 Inhibitors and Various Chemical Reactivity of 1-Bromo- and 1-Chloro-4-vinylbenzene in [3 + 2] Cycloaddition Reactions" Organics 2, no. 1: 1-16. https://doi.org/10.3390/org2010001