Optimization of DNA Extraction from Individual Sand Flies for PCR Amplification



Abstract
:1. Introduction
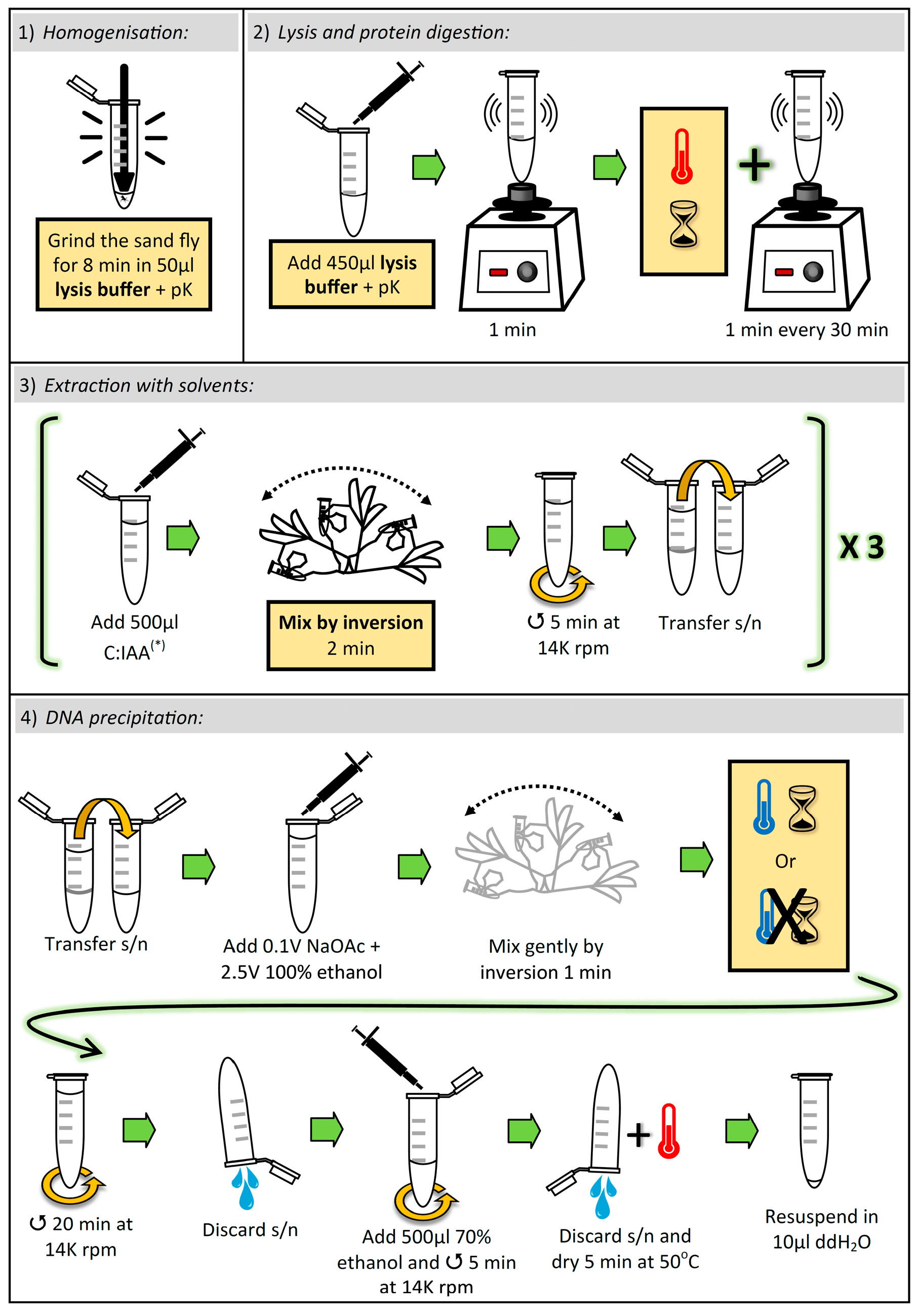
2. Experimental Design
3. Final Procedure
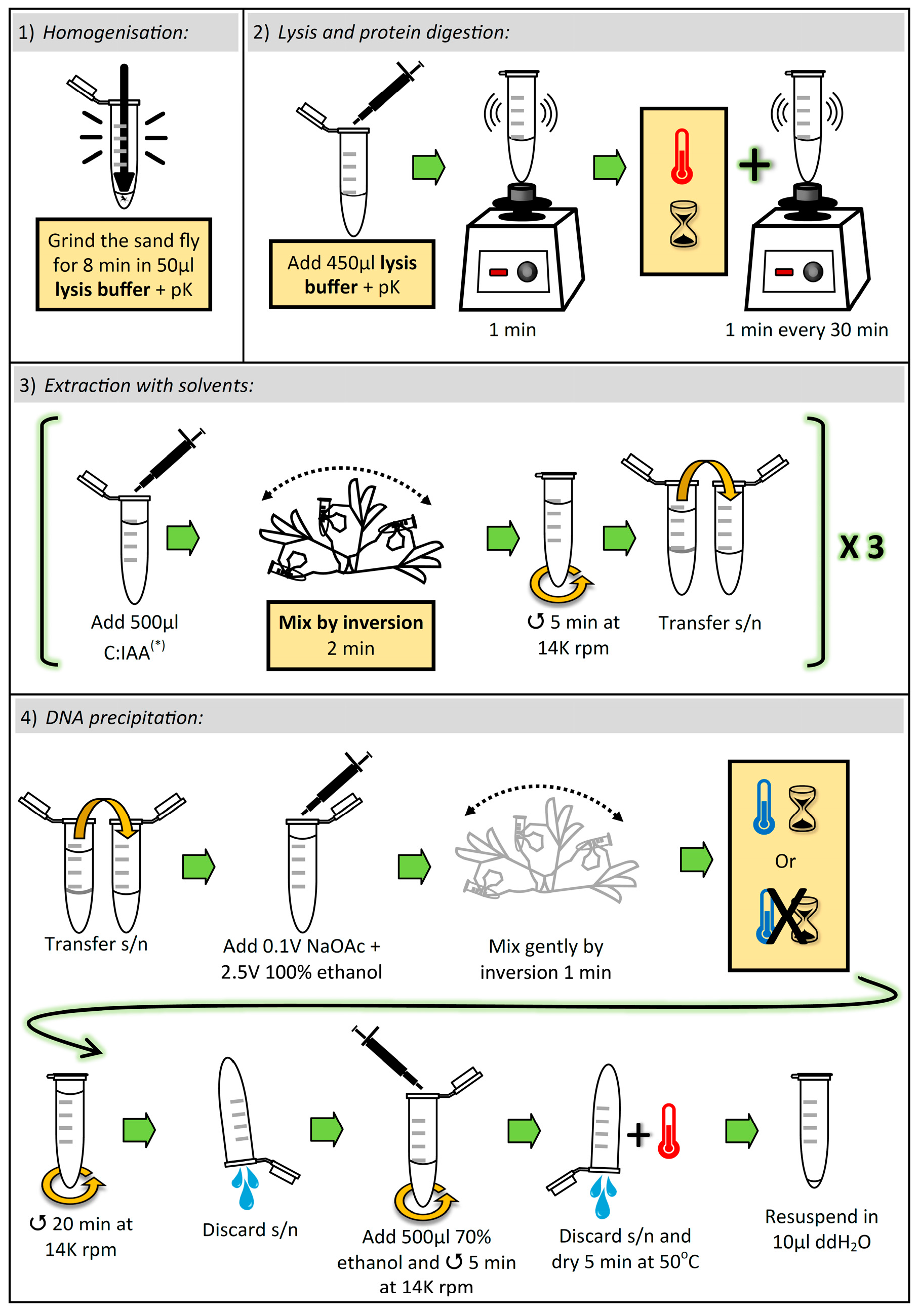
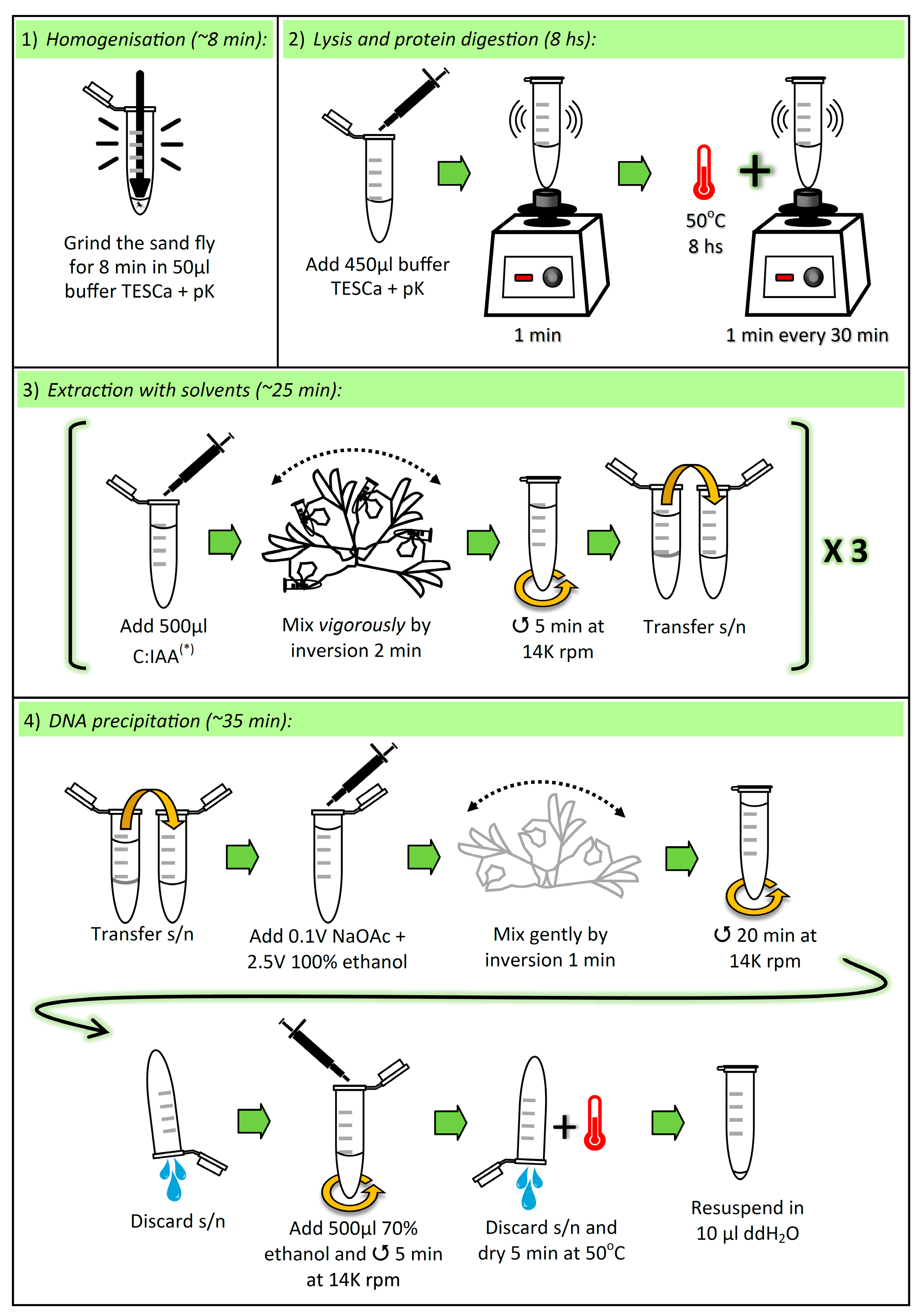
3.1. Lysis and Elimination of Proteins. Time for Completion: ~8 h, 8 min
3.1.1. Homogenization of Sand Fly
- Aliquot sufficient volume of buffer TESCa (30 mM Tris-HCl pH 8; 10 mM EDTA; 1% SDS, 5 mM CaCl2; see “Section 4.3”), i.e., 500 μL per sample, and add proteinase K (pK) (to the aliquot) to a final concentration of (0.42 μg/μL).
- Place one adult sand fly in a 1.5 mL microcentrifuge tube, and add 50 µL of buffer TESCa + pK.
![Mps 02 00036 i001]() CRITICAL STEP: Grind the sand fly thoroughly for 8 min with a Teflon micropestle. To avoid cross-contamination between samples, the micropestle must be cleaned and autoclaved after each grinding (i.e., one micropestle per sample).
CRITICAL STEP: Grind the sand fly thoroughly for 8 min with a Teflon micropestle. To avoid cross-contamination between samples, the micropestle must be cleaned and autoclaved after each grinding (i.e., one micropestle per sample).

3.1.2. Cell Lysis, and Protein Denaturation and Digestion
- 4.
![Mps 02 00036 i001]() CRITICAL STEP: Add 450 µL buffer TESCa + pK (to a final volume of 500 µL), vortex for 1 min, and incubate at 50 °C for 8 h, vortexing for 1 min every 30 min.
CRITICAL STEP: Add 450 µL buffer TESCa + pK (to a final volume of 500 µL), vortex for 1 min, and incubate at 50 °C for 8 h, vortexing for 1 min every 30 min.
3.2. Extraction with Solvents. Time for Completion: ~25 min
3.2.1. First Extraction
- 5.
![Mps 02 00036 i001]() CRITICAL STEP: Add 500 µL chloroform:isoamyl alcohol (C:IAA) (24:1 v/v) and mix vigorously by inversion for 2 min. Immediately centrifuge at 14,000 rpm for 5 min. Transfer the supernatant (~480 µL) to a new 1.5 mL microcentrifuge tube.
CRITICAL STEP: Add 500 µL chloroform:isoamyl alcohol (C:IAA) (24:1 v/v) and mix vigorously by inversion for 2 min. Immediately centrifuge at 14,000 rpm for 5 min. Transfer the supernatant (~480 µL) to a new 1.5 mL microcentrifuge tube.
3.2.2. Second Extraction
- 6.
![Mps 02 00036 i001]() CRITICAL STEP: Add 500 µL C:IAA (24:1 v/v) and mix vigorously by inversion for 2 min. Immediately centrifuge at 14,000 rpm for 5 min. Transfer the supernatant (~460 µL) to a new 1.5 mL microcentrifuge tube.
CRITICAL STEP: Add 500 µL C:IAA (24:1 v/v) and mix vigorously by inversion for 2 min. Immediately centrifuge at 14,000 rpm for 5 min. Transfer the supernatant (~460 µL) to a new 1.5 mL microcentrifuge tube.
3.2.3. Third Extraction
- 7.
![Mps 02 00036 i001]() CRITICAL STEP: Add 700 µL C:IAA (24:1 v/v) and mix vigorously by inversion for 2 min. Immediately centrifuge at 14,000 rpm for 5 min. Transfer the supernatant (~400 µL) to a new 1.5 mL microcentrifuge tube.
CRITICAL STEP: Add 700 µL C:IAA (24:1 v/v) and mix vigorously by inversion for 2 min. Immediately centrifuge at 14,000 rpm for 5 min. Transfer the supernatant (~400 µL) to a new 1.5 mL microcentrifuge tube.
3.3. DNA Precipitation. Time for Completion: ~35 min
3.3.1. Addition of Salt and Alcohol
- 8.
- Add 0.1 volumes (~40 µL) 3 M Sodium Acetate (NaOAc) pH 5.2 and 2.5 volumes (~1 mL) 100% ethanol, and gently mix by inversion for 1 min.
![Mps 02 00036 i002]() PAUSE STEP and OPTIONAL STEP: We found that after adding NaOAc and ethanol, results improved when the sample was immediately centrifuged (i.e., was not incubated at all). Nevertheless, due to the length of the previous stages (~9 h), the protocol can be paused here and the sample stored overnight (ON) at −20 °C.
PAUSE STEP and OPTIONAL STEP: We found that after adding NaOAc and ethanol, results improved when the sample was immediately centrifuged (i.e., was not incubated at all). Nevertheless, due to the length of the previous stages (~9 h), the protocol can be paused here and the sample stored overnight (ON) at −20 °C.

3.3.2. Centrifugation
- 9.
- Centrifuge at 14,000 rpm for 20 min and discard the supernatant by inversion.
- 10.
- Add 500 µL 70% ethanol and centrifuge at 14,000 rpm for 5 min. Discard the supernatant by inversion and dry the pellet at 50 °C for 5 min. Resuspend the pellet in 10 µL double-distilled water.
4. Materials, Equipment, and Reagents Setup
4.1. Materials
- TRIS buffer (NH2(CH2OH)3, 121.14 g/mol) (Anedra, Tigre, Argentina; Cat. no.: AN00915709)
- Hydrochloric acid (HCl, 36.46 g/mol) (Biopack, Buenos Aires, Argentina; Cat. no.: 9632.08)
- Sodium Dodecyl Sulfate (SDS, C12H25NaO4S, 288.38 g/mol) (Anedra, Tigre, Argentina; Cat. no.: AN219483180)
- EDTA (C10H16N2O8, 292.24 g/mol) (Anedra, Tigre, Argentina; Cat. no.: AN00605609)
- Calcium chloride dihydrate (CaCl2·2H2O, 147 g/mol) (Anedra, Tigre, Argentina; Cat. No.: AN6456)
- Proteinase K (Fermentas-Thermo Fisher Scientific, Waltham, MA, USA; Cat. No.: #EO0491)
- Double-distilled water (ddH2O)
- Chloroform (CHCl3, 119.38 g/mol) (Cicarelli Laboratorios, San Lorenzo, Argentina; Cat. no.: 1116110)
- Isoamyl alcohol (Anedra, Tigre, Argentina; Cat. no.: AN00659925)
- Sodium acetate (CH3COONa, 82.03 g/mol) (Anedra, Tigre, Argentina; Cat. No.: AN00651808)
- Glacial acetic acid (CH3COOH, 60,05 g/mol) (Anedra, Tigre, Argentina; Cat. No.: AN6082)
- Absolute ethanol (C2H6O, 46.07 g/mol) (Biopack, Buenos Aires, Argentina; Cat. no.: 1654.08)
4.2. Equipment
- Teflon micropestle (Eppendorf-Fisher Scientific, Leicestershire, UK; Cat. no.: 10683001)
- Vortex (Denville Scientific, Metuchen, NJ, USA; Cat. no.: Vortexer S7030)
- Water bath (Jiangsu Jinyi Instrument Technology Company Limited, Shanghai, China; Cat. no.: SHZ-88)
- High-speed bench-top centrifuge (Heal Force, Shanghai, China; Cat. no.: Neofuge 15)
- Micropipettes p1000, p200, p20 (Gilson, Middleton, WI, USA; Cat. nos.: F144566, F144565, and F144563)
4.3. Reagents Setup
Buffer TESCa
5. Results
5.1. Assayed Modifications
5.2. Conclusions
- (1)
- an 8-h incubation with proteinase K in buffer TESCa at 50 °C;
- (2)
- vigorous mixing by inversion during the extraction with solvents step; and
- (3)
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A. Internal PCR Control Conditions
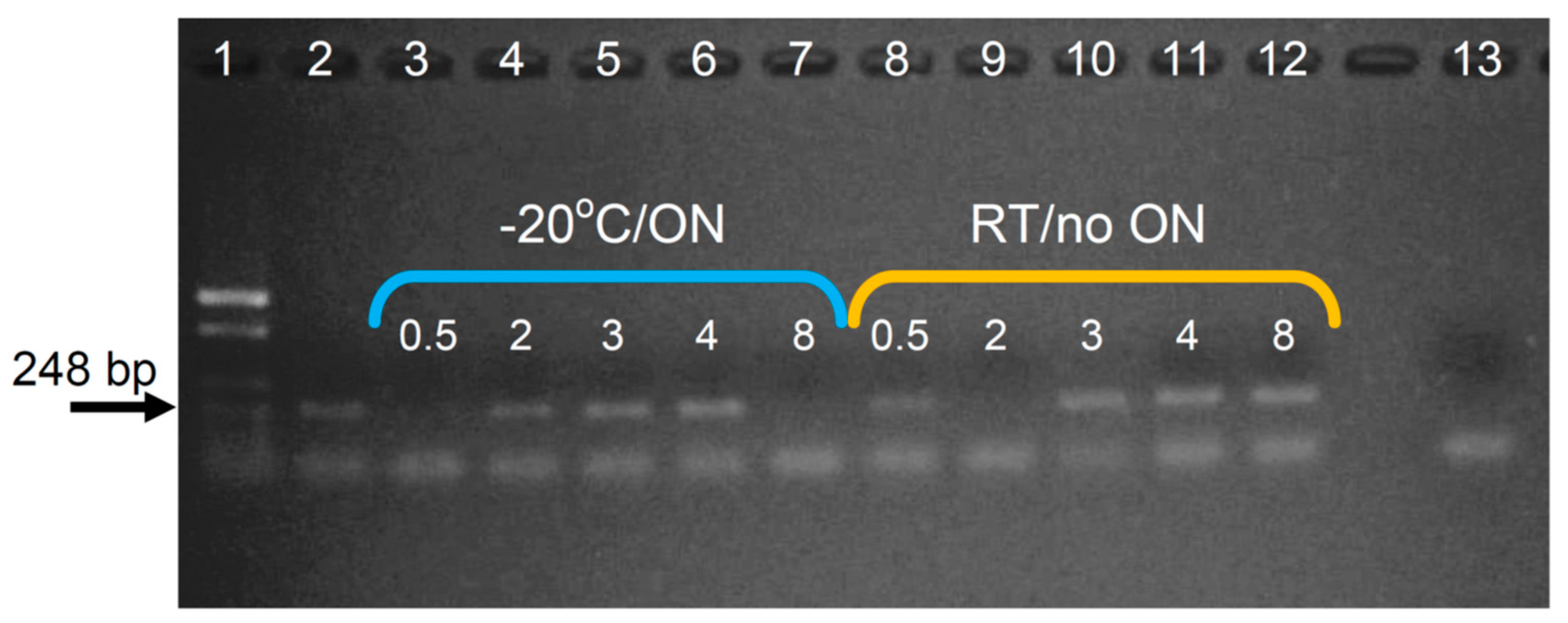
Appendix B. Assaying Different Incubation Periods with pK in Buffer pAC, and ON or No Incubation at −20 °C (in the DNA Precipitation Step)

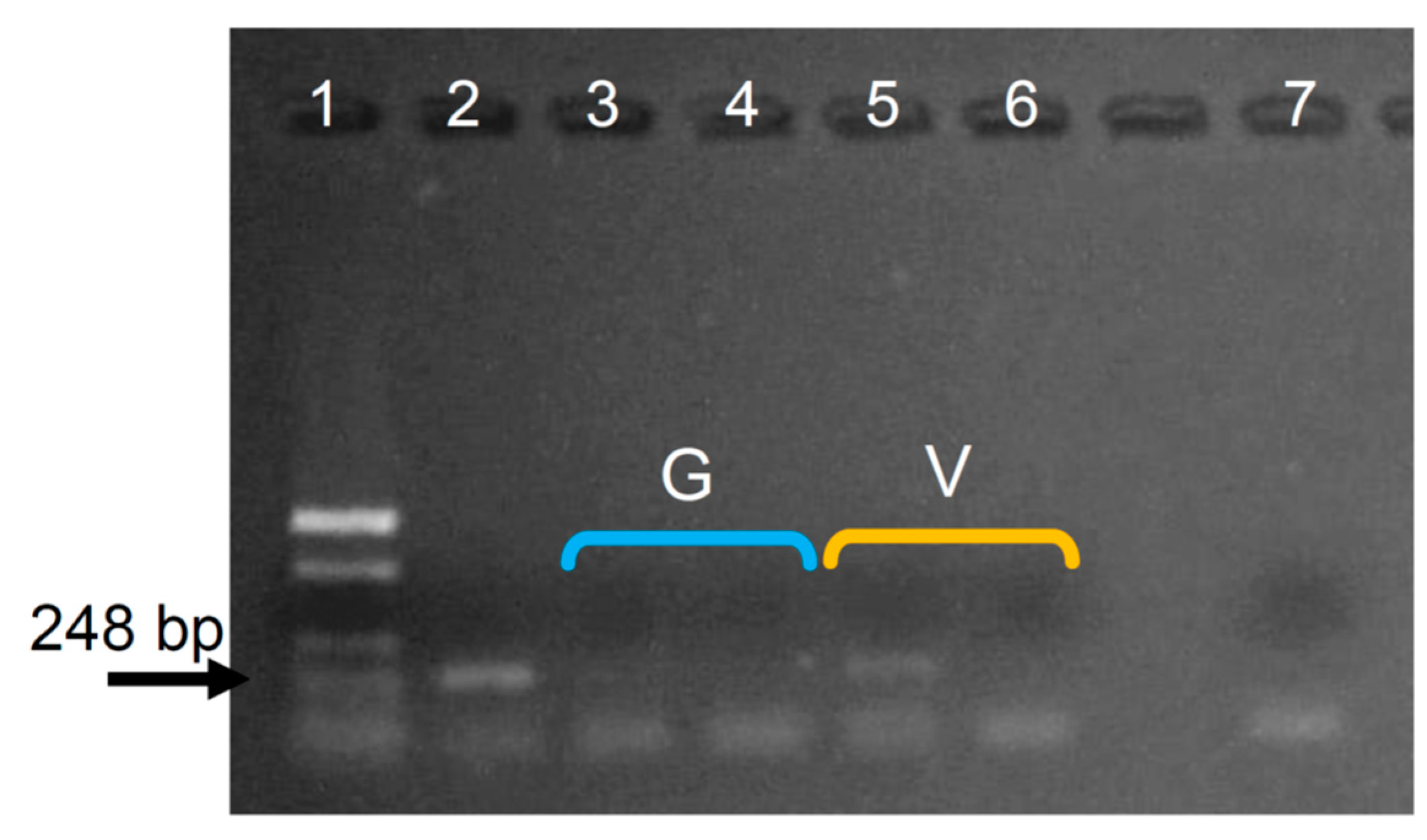
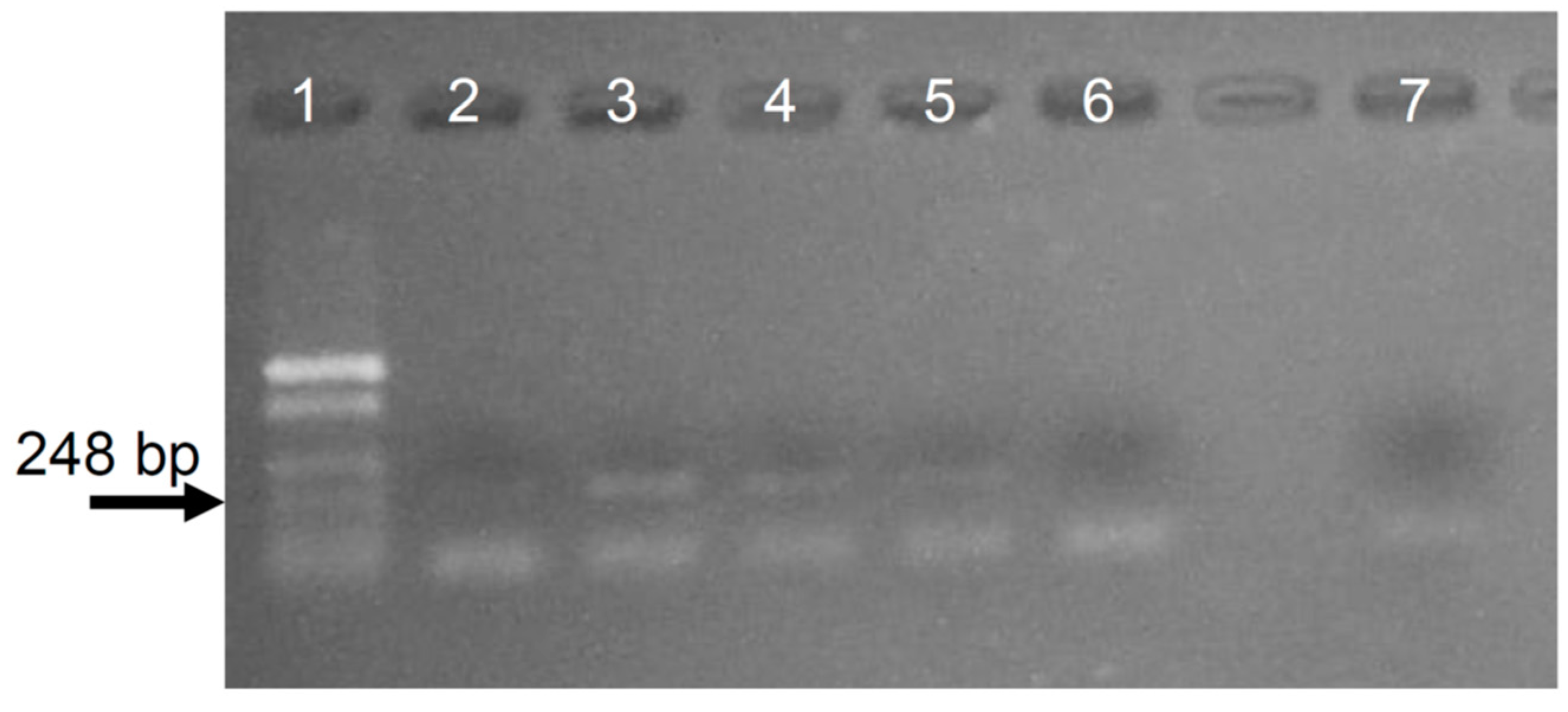
Appendix C. Assaying the Intensity When Mixing by Inversion in the Extraction with Solvents Step

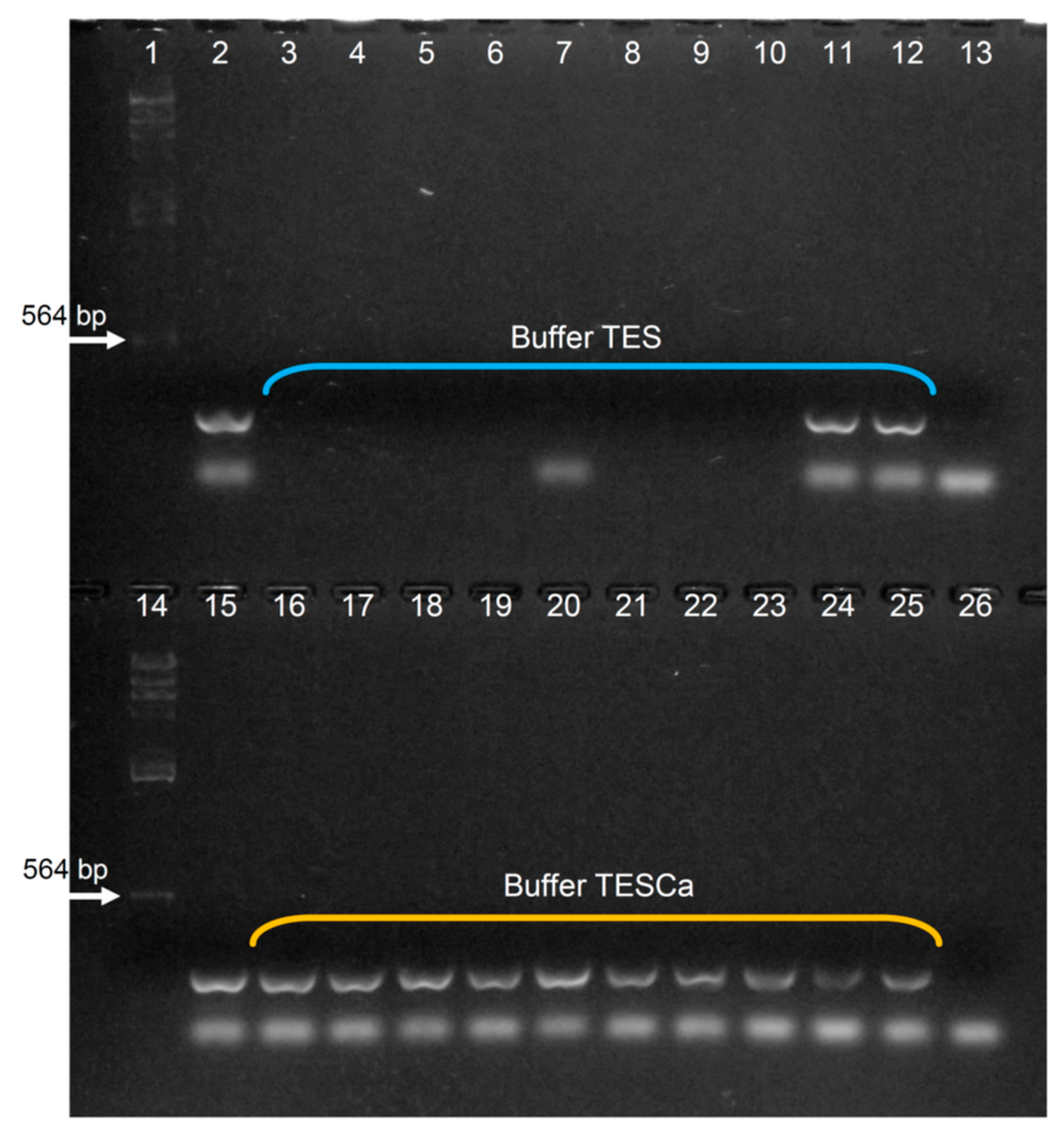
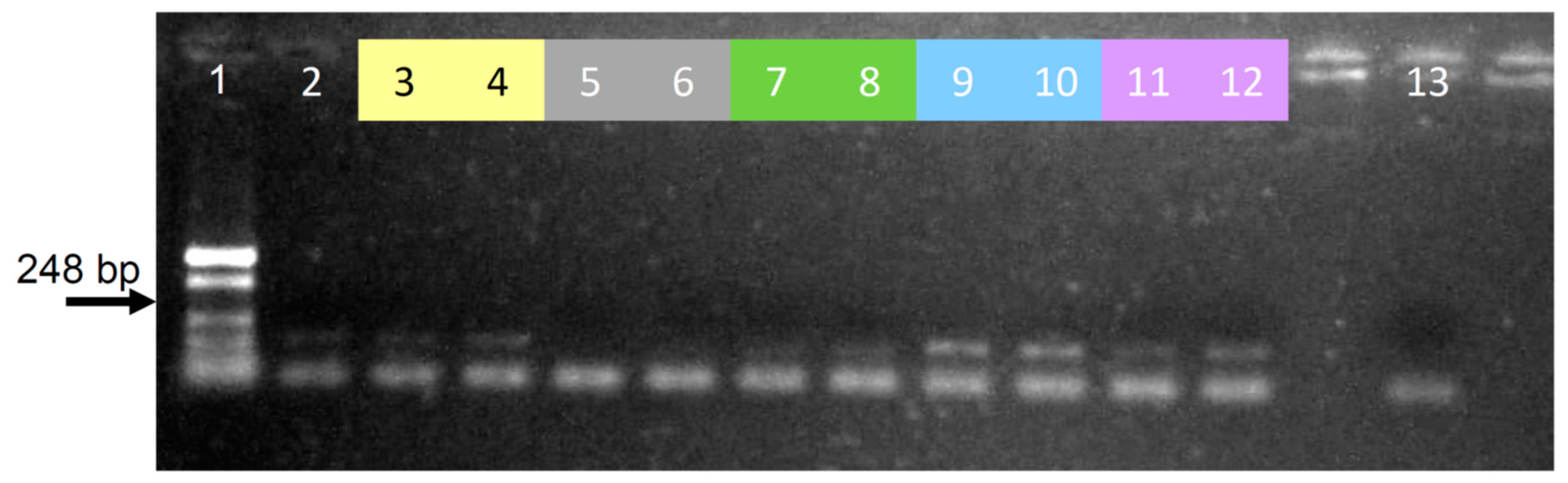
Appendix D. Assaying Different Lysis Buffers

Appendix E. Assaying Different Incubation Periods with pK in Buffer TESCa, and ON or No Incubation at −20 °C (in the DNA Precipitation Step)

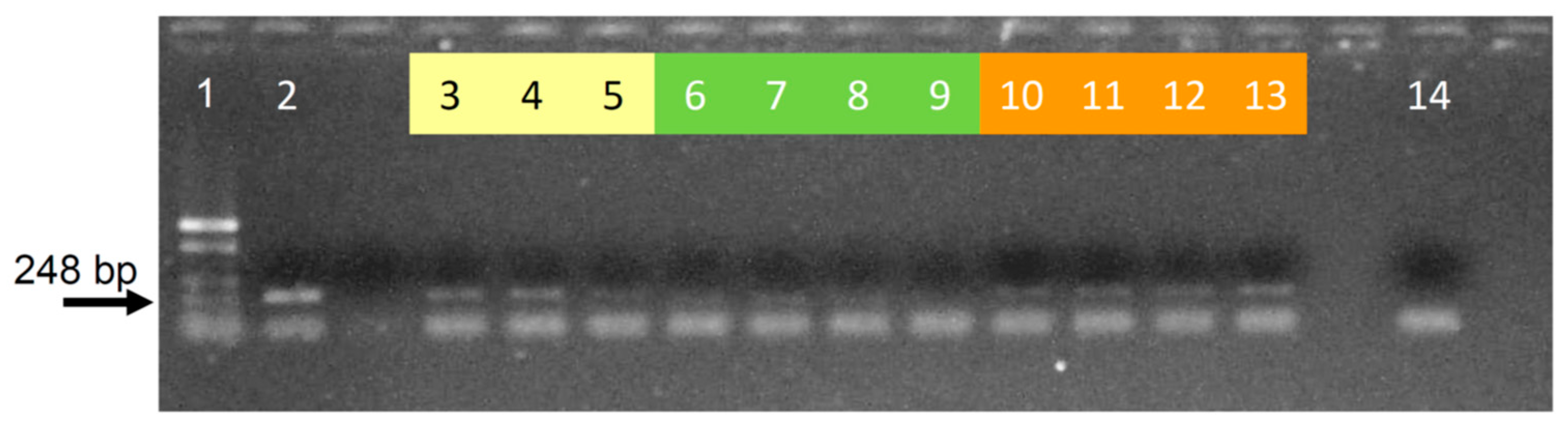
Appendix F. Results Using the Optimized Protocol with Different Lutzomyia Spp.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species 1 | City | State/Province | Country | Figure |
|---|---|---|---|---|
| L. umbratilis | Presidente Figueiredo | Amazonas | Brazil | A5, A6 |
| L. migonei | Baturite | Ceara | Brazil | A5 |
| L. renei | Lagoa Santa | Minas Gerais | Brazil | A6 |
| L. intermedia | Tancredo Neves | Bahia | Brazil | A7 |
| L. longipalpis (cavunge strain) | Cavunge | Bahia | Brazil | A5, A6 |
| L. longipalpis (jacobina strain) | Jacobina | Bahia | Brazil | A5 |
| L. longipalpis (lapinha strain) | Lagoa Santa | Minas Gerais | Brazil | A5 |
| L. longipalpis | Posadas | Misiones | Argentina | A7 |



References
- Ready, P.D.; Lainson, R.; Shaw, J.J.; Souza, A.A. DNA probes for distinguishing Psychodopygus wellcomei from Psychodopygus complexus (Diptera: Psychodidae). Mem. Inst. Oswaldo Cruz 1991, 86, 41–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parvizi, P.; Amirkhani, A. Mitochondrial DNA Characterization of Sergentomyia Sintoni Populations and Finding Mammalian Leishmania Infections in This Sandfly by Using ITS-RDNA Gene. Iran. J. Vet. Res. 2008, 9, 9–18. [Google Scholar]
- Golczer, G.; Arrivillaga, J. Modification of a Standard Protocol for DNA Extraction from Smaller Sandflies (Phlebotominae: Lutzomyia). Rev. Colomb. Entomol. 2008, 34, 199–200. [Google Scholar]
- Torgerson, D.G.; Velázquez, Y.; Woo, P.T.K.; Lampo, M. Genetic relationships among some species groups within the genus lutzomyia (Diptera: Psychodidae). Am. J. Trop. Med. Hyg. 2003, 69, 484–493. [Google Scholar] [CrossRef] [PubMed]
- Michalsky, É.M.; Pimenta, P.F.; Secundino, N.F.; Fortes-Dias, C.L.; Dias, E.S. Assessment of PCR in the detection of Leishmania spp in experimentally infected individual phlebotomine sandflies (Diptera: Psychodidae: Phlebotominae). Rev. Inst. Med. Trop. São Paulo 2002, 44, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Uezato, H.; Katakura, K.; Calvopiña, M.; Marco, J.D.; Barroso, P.A.; Gomez, E.A.; Mimori, T.; Korenaga, M.; Iwata, H.; Nonaka, S.; Hashiguchi, Y. Detection and Identification of Leishmania Species within Naturally Infected Sand Flies in the Andean Areas of Ecuador by a Polymerase Chain Reaction. Am. J. Trop. Med. Hyg. 2005, 71, 87–93. [Google Scholar] [CrossRef]
- Beati, L.; Cáceres, A.G.; Lee, J.A.; Munstermann, L.E. Systematic relationships among Lutzomyia sand flies (Diptera: Psychodidae) of Peru and Colombia based on the analysis of 12S and 28S ribosomal DNA sequences. Int. J. Parasitol. 2004, 34, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Giantsis, I.A.; Chaskopoulou, A.; Bon, M.C. Mild-Vectolysis: A Nondestructive DNA Extraction Method for Vouchering Sand Flies and Mosquitoes. J. Med. Entomol. 2016, 53, 692–695. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, O.L.; Munstermann, L.E.; Cardenas, R.; Ferro, C. PCR as a Tool in Confirming the Experimental Transmission of Leishmania Chagasi to Hamsters by Lutzomyia Longipalpis (Diptera:Psychodidae). Biomedica 2003, 23, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Jorquera, A.; González, R.; Marchán-Marcano, E.; Oviedo, M.; Matos, M. Multiplex-PCR for Detection of Natural Leishmania Infection in Lutzomyia Spp. Captured in an Endemic Region for Cutaneous Leishmaniasis in State of Sucre, Venezuela. Mem. Inst. Oswaldo Cruz 2005, 100, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Caligiuri, L.G.; Acardi, S.A.; Soledad Santini, M.; Salomón, O.D.; Mccarthy, C.B. Polymerase Chain Reaction-Based Assay for the Detection and Identification of Sand Fly Gregarines in Lutzomyia Longipalpis, a Vector of Visceral Leishmaniasis. J. Vector Ecol. 2014, 39, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Peixoto, A.A.; Gomes, C.A.; de Amoretty, P.R.; Lins, R.M.; Meireles-Filho, A.C.; de Souza, N.A.; Kyriacou, C.P. New Molecular Markers for Phlebotomine Sand Flies. Int. J. Parasitol. 2001, 31, 635–639. [Google Scholar] [CrossRef]
- Lins, R.M.; Oliveira, S.G.; Souza, N.A.; de Queiroz, R.G.; Justiniano, S.C.; Ward, R.D.; Kyriacou, C.P.; Peixoto, A.A. Molecular Evolution of the Cacophony IVS6 Region in Sandflies. Insect. Mol. Biol. 2002, 11, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Acardi, S.A.; Liotta, D.J.; Santini, M.S.; Romagosa, C.M.; Salomon, O.D. Detection of Leishmania Infantum in Naturally Infected Lutzomyia Longipalpis (Diptera: Psychodidae: Phlebotominae) and Canis Familiaris in Misiones, Argentina: The First Report of a PCR-RFLP and Sequencing-Based Confirmation Assay. Mem. Inst. Oswaldo Cruz 2010, 105, 796–799. [Google Scholar] [CrossRef] [PubMed]
- Bajorath, J.; Hinrichs, W.; Saenger, W. The Enzymatic Activity of Proteinase K Is Controlled by Calcium. Eur. J. Biochem. 1988, 176, 441–447. [Google Scholar] [CrossRef]
- Bajorath, J.; Raghunathan, S.; Hinrichs, W.; Saenger, W. Long-Range Structural Changes in Proteinase K Triggered by Calcium Ion Removal. Nature 1989, 337, 481–484. [Google Scholar] [CrossRef]
- Betzel, C.; Pal, G.P.; Saenger, W. Three-dimensional Structure of Proteinase K at 0.15-nm Resolution. Eur. J. Biochem. 1988, 178, 155–171. [Google Scholar] [CrossRef]
- Müller, A.; Hinrichs, W.; Wolf, W.M.; Saenger, W. Crystal Structure of Calcium-Free Proteinase K at 1.5-A Resolution. J. Biol. Chem. 1994, 269, 23108–23111. [Google Scholar]
- McNevin, D.; Wilson-Wilde, L.; Robertson, J.; Kyd, J.; Lennard, C. Short Tandem Repeat (STR) Genotyping of Keratinised Hair Part 2. An Optimised Genomic DNA Extraction Procedure Reveals Donor Dependence of STR Profiles. Forensic Sci. Int. 2005, 153, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Farrell, R.E., Jr. Resilient Ribonucleases. In RNA Methodologies: A Laboratory Guide for Isolation and Characterization, 4th ed.; Academic Press: Cambridge, MA, USA, 2010. [Google Scholar]
- Ebeling, W.; Hennrich, N.; Klockow, M.; Metz, H.; Orth, H.D.; Lang, H. Proteinase K from Tritirachium Album Limber. Eur. J. Biochem. 1974, 47, 91–97. [Google Scholar] [CrossRef] [PubMed]

| Step | Variation A 1 | Variation B 2 | Variation C 3 | Variation D 4 |
|---|---|---|---|---|
| Homogenization | Grind with micropestle for 8 min in 50 µL buffer. | Grind with micropestle for 8 min in 50 µL buffer. | Grind with micropestle for 8 min in 50 µL buffer. | Grind with micropestle for 8 min in 50 µL buffer. |
| Lysis and protein digestion | Buffer pAC; Incubation with pK at 58 °C for 0.5, 2, 3, 4 and 8 h. | Buffer pAC; Incubation with pK at 58 °C for 8 h. | Buffers pAC, TES and TESCa; Incubation with pK at 50 °C for 8 h. | Buffer TESCa Incubation with pK at 50 °C for 1, 2, 3, 4 and 8 h. |
| Extraction with solvents | Gentle mixing by inversion with C:IAA. | Gentle and vigorous mixing by inversion with C:IAA. | Vigorous mixing by inversion with C:IAA. | Vigorous mixing by inversion with C:IAA. |
| DNA precipitation | Incubation with alcohol: ON at −20 °C, and no incubation | Incubation with alcohol at −20 °C 5 | Incubation with alcohol at −20 °C 5 | Incubation with alcohol: ON at −20 °C and no incubation |
| Final resuspension | 20 µL ddH2O | 20 µL ddH2O | 10 µL ddH2O | 10 µL ddH2O |
| Reagent | Final Concentration | Volume (Vf 1 = 12.5 mL) |
|---|---|---|
| 1 M Tris-HCl pH 8 | 30 mM | 375 µL |
| 0.5 M EDTA | 10 mM | 250 µL |
| 100 mM CaCl2 | 5 mM | 625 µL |
| dH2O 2 | - | 10.625 mL |
| 20% SDS | 1% | 625 µL |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caligiuri, L.G.; Sandoval, A.E.; Miranda, J.C.; Pessoa, F.A.; Santini, M.S.; Salomón, O.D.; Secundino, N.F.C.; McCarthy, C.B. Optimization of DNA Extraction from Individual Sand Flies for PCR Amplification. Methods Protoc. 2019, 2, 36. https://doi.org/10.3390/mps2020036
Caligiuri LG, Sandoval AE, Miranda JC, Pessoa FA, Santini MS, Salomón OD, Secundino NFC, McCarthy CB. Optimization of DNA Extraction from Individual Sand Flies for PCR Amplification. Methods and Protocols. 2019; 2(2):36. https://doi.org/10.3390/mps2020036
Chicago/Turabian StyleCaligiuri, Lorena G., Adolfo E. Sandoval, Jose C. Miranda, Felipe A. Pessoa, María S. Santini, Oscar D. Salomón, Nagila F. C. Secundino, and Christina B. McCarthy. 2019. "Optimization of DNA Extraction from Individual Sand Flies for PCR Amplification" Methods and Protocols 2, no. 2: 36. https://doi.org/10.3390/mps2020036