Wnt/β-Catenin Signaling during Cardiac Development and Repair

 ,
,
Abstract
:1. Introduction
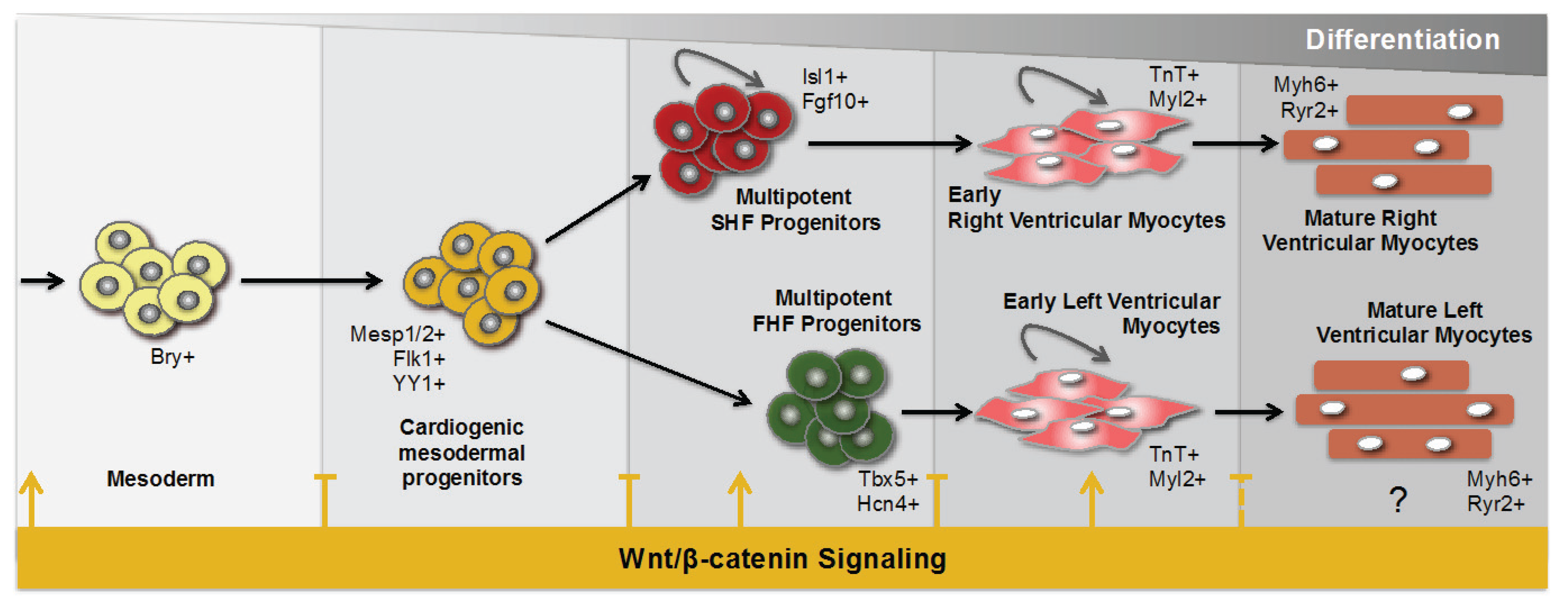
2. Wnt Signaling during Cardiogenesis
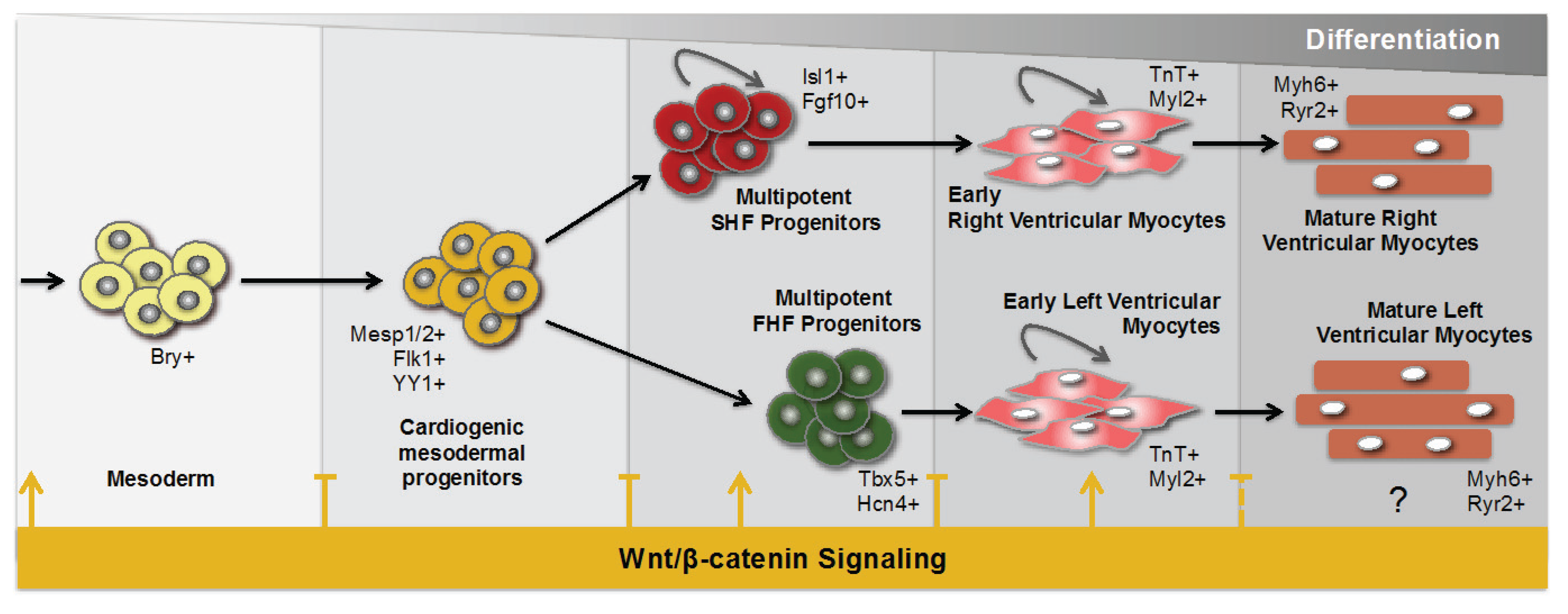
3. Wnt Signaling during Cardiac Repair


{kind=link}
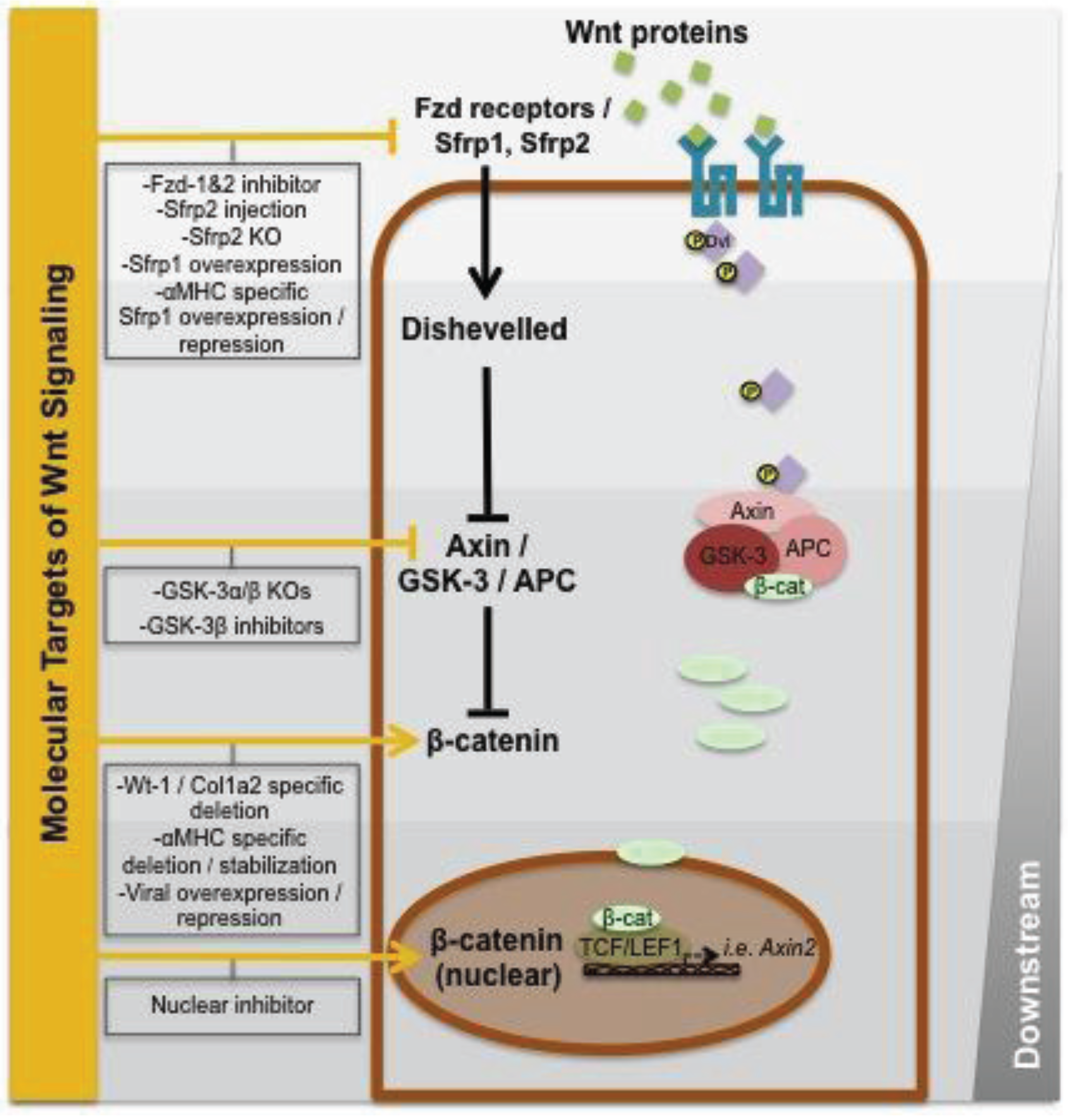{kind=link}
| References | Target | Treatment/modulation | Wnt/β-cat | Timing | Outcome |
|---|---|---|---|---|---|
| [61] | Fzd-1/Fzd-2 | Peptidergic Fzd-1/Fzd-2 antagonist UM206 | ↓ | 0 or 14d post-MI (similar results) | Reduction of infarct expansion and increased repair in infarct area |
| [59] | Sfrp2 | Recombinant Sfrp2 injection | ↓ | Injection 2d post-MI | Reduced fibrosis and improved LVEF |
| [60] | Sfrp2 | KO mice | ↑ | - | Reduced fibrosis and significantly higher LVEF compared to controls |
| [62] | Sfrp1 | αMHC-specific Sfrp1 overexpression with doxycycline inducible repression | ↓ | Overexpression/repression (1wk prior to MI) | Ischemic preconditioning caused improved outcomes after MI due to GSK-3β inhibition, but this effect was diminished in Sfrp1-overexpressing CM |
| [58] | Sfrp1 | Overexpression of Sfrp1 | ↓ | - | Reduction of infarct size, fibrosis and improved cardiac function (7 d or 30 d post-MI) |
| [64] | GSK-3α | KO mice | ↑ | - | Increased mortality (10 d post-MI), more LV dilatation, dysfunction, hypertrophy, fibrosis and heart failure (8 weeks post-MI) and increased apoptosis in border zone (2 d post-MI) in KO mice |
| [65] | GSK-3β | KO mice (tamoxifen inducible) | ↑ | KO at 3d post-MI | Improved LVEF and LV dilatation with less hypertrophy post-MI in GSK-3β KO mice (8 weeks post-MI) |
| [63] | GSK-3 | KO mice | ↑ | - | No functional difference between GSK-3 KO mice and controls |
| [66] | GSK-3β | Inhibitors (Lithium/SB216763) | ↑ | Directly after MI | GSK-3β inhibition mimicked ischemic precondition, resulting in less apoptotic cardiomyocytes and increased capillary density |
| [69] | β-catenin | Nuclear inhibitor (ICG-001) | ↓ | Directly after MI for 10d | Improved LVEF (10 d post-MI) and increased EMT in epicardial cells in treated mice |
| [6] | β-catenin | Downregulation (tamoxifen inducible) in cardiac fibroblasts | ↓ | 10d prior to MI | Increased left ventricular dilatation (8 d post-MI) and decreased cardiac fibroblast proliferation in vitro |
| [68] | β-catenin | αMHC specific depletion orstabilization of β-catenin | ↓/↑ | - | Upregulation of fetal gene program (GATA4, Tbx5) and improved MI outcomes (LVEF and mortality) in β-catenin depleted animals compared to stabilization |
| [67] | β-catenin | Overexpression of β-catenin | ↑ | Directly after MI | Decreased left ventricular dilatation, increased fractional shortening and decreased apoptosis compared to controls (7 d post-MI) |
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Senyo, S.E.; Steinhauser, M.L.; Pizzimenti, C.L.; Yang, V.K.; Cai, L.; Wang, M.; Wu, T.D.; Guerquin-Kern, J.L.; Lechene, C.P.; Lee, R.T. Mammalian heart renewal by pre-existing cardiomyocytes. Nature 2013, 493, 433–436. [Google Scholar]
- Buckingham, M.; Meilhac, S.; Zaffran, S. Building the mammalian heart from two sources of myocardial cells. Nat. Rev. Genet. 2005, 6, 826–835. [Google Scholar] [CrossRef]
- Chien, K.R.; Domian, I.J.; Parker, K.K. Cardiogenesis and the complex biology of regenerative cardiovascular medicine. Science 2008, 322, 1494–1497. [Google Scholar]
- Klaus, A.; Saga, Y.; Taketo, M.M.; Tzahor, E.; Birchmeier, W. Distinct roles of Wnt/beta-catenin and Bmp signaling during early cardiogenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 18531–18536. [Google Scholar] [CrossRef]
- Oerlemans, M.I.; Goumans, M.J.; van Middelaar, B.; Clevers, H.; Doevendans, P.A.; Sluijter, J.P. Active Wnt signaling in response to cardiac injury. Basic Res. Cardiol. 2010, 105, 631–641. [Google Scholar] [CrossRef]
- Duan, J.; Gherghe, C.; Liu, D.; Hamlett, E.; Srikantha, L.; Rodgers, L.; Regan, J.N.; Rojas, M.; Willis, M.; Leask, A.; et al. Wnt1/βcatenin injury response activates the epicardium and cardiac fibroblasts to promote cardiac repair. EMBO J. 2012, 31, 429–442. [Google Scholar] [CrossRef]
- Olson, E.N. Gene regulatory networks in the evolution and development of the heart. Science 2006, 313, 1922–1927. [Google Scholar] [CrossRef]
- Srivastava, D. Making or breaking the heart: from lineage determination to morphogenesis. Cell 2006, 126, 1037–1048. [Google Scholar] [CrossRef]
- Tzahor, E. Wnt/beta-catenin signaling and cardiogenesis: Timing does matter. Dev. Cell 2007, 13, 10–13. [Google Scholar] [CrossRef]
- Gessert, S.; Kuhl, M. The multiple phases and faces of wnt signaling during cardiac differentiation and development. Circ. Res. 2010, 107, 186–199. [Google Scholar] [CrossRef]
- Gurley, K.A.; Rink, J.C.; Sanchez Alvarado, A. Beta-catenin defines head versus tail identity during planarian regeneration and homeostasis. Science 2008, 319, 323–327. [Google Scholar] [CrossRef]
- Petersen, C.P.; Reddien, P.W. Smed-betacatenin-1 is required for anteroposterior blastema polarity in planarian regeneration. Science 2008, 319, 327–330. [Google Scholar] [CrossRef]
- De Lau, W.; Barker, N.; Low, T.Y.; Koo, B.-K.; Li, V.S.W.; Teunissen, H.; Kujala, P.; Haegebarth, A.; Peters, P.J.; van de Wetering, M.; et al. Lgr5 homologues associate with Wnt receptors and mediate R-spondin signalling. Nature 2011, 476, 293–297. [Google Scholar] [CrossRef]
- Ito, M.; Yang, Z.; Andi, T.; Cui, C.; Kim, N.; Millar, S.E.; Cotsarelis, G. Wnt-dependent de novo hair follicle regeneration in adult mouse skin after wounding. Nature 2007, 447, 316–320. [Google Scholar] [CrossRef]
- Lu, C.P.; Polak, L.; Rocha, A.S.; Pasolli, H.A.; Chen, S.C.; Sharma, N.; Blanpain, C.; Fuchs, E. Identification of stem cell populations in sweat glands and ducts reveals roles in homeostasis and wound repair. Cell 2012, 150, 136–150. [Google Scholar] [CrossRef]
- Bu, L.; Polak, L.; Rocha, A.S.; Pasolli, H.A.; Chen, S.-C.; Sharma, N.; Blanpain, C.; Fuchs, E. Human ISL1 heart progenitors generate diverse multipotent cardiovascular cell lineages. Nature 2009, 460, 113–117. [Google Scholar] [CrossRef]
- Eisenberg, C.A.; Eisenberg, L.M. WNT11 promotes cardiac tissue formation of early mesoderm. Dev. Dyn. 1999, 216, 45–58. [Google Scholar] [CrossRef]
- Qyang, Y.; Martin-Puig, S.; Chiravuri, M.; Chen, S.; Xu, H.; Bu, L.; Jiang, X.; Lin, L.; Granger, A.; Moretti, A.; et al. The renewal and differentiation of Isl1+ cardiovascular progenitors are controlled by a Wnt/beta-catenin pathway. Cell Stem Cell 2007, 1, 165–179. [Google Scholar] [CrossRef]
- Haegel, H.; Larue, L.; Ohsugi, M.; Fedorov, L.; Herrenknecht, K.; Kemler, R. Lack of beta-catenin affects mouse development at gastrulation. Development 1995, 121, 3529–3537. [Google Scholar]
- Kitajima, S.; Takagi, A.; Inoue, T.; Saga, Y. MesP1 and MesP2 are essential for the development of cardiac mesoderm. Development 2000, 127, 3215–3226. [Google Scholar]
- Saga, Y.; Miyagawa-Tomita, S.; Takagi, A.; Kitajima, S.; Miyazaki, J.; Inoue, T. MesP1 is expressed in the heart precursor cells and required for the formation of a single heart tube. Development 1999, 126, 3437–3447. [Google Scholar]
- David, R.; Brenner, C.; Stieber, J.; Schwarz, F.; Brunner, S.; Vollmer, M.; Mentele, E.; Müller-Höcker, J.; Kitajima, S.; Lickert, H.; et al. MesP1 drives vertebrate cardiovascular differentiation through Dkk-1-mediated blockade of Wnt-signalling. Nat. Cell Biol. 2008, 10, 338–345. [Google Scholar] [CrossRef]
- Bondue, A.; Lapouge, G.; Paulissen, C.; Semeraro, C.; Iacovino, M.; Kyba, M.; Blanpain, C. Mesp1 acts as a master regulator of multipotent cardiovascular progenitor specification. Cell Stem Cell 2008, 3, 69–84. [Google Scholar] [CrossRef]
- Bruneau, B.G.; Nemer, G.; Schmitt, J.P.; Charron, F.; Robitaille, L.; Caron, S.; Conner, D.A.; Gessler, M.; Nemer, M.; Seidman, C.E.; Seidman, J.G. A murine model of Holt-Oram syndrome defines roles of the T-box transcription factor Tbx5 in cardiogenesis and disease. Cell 2001, 106, 709–721. [Google Scholar] [CrossRef]
- Spater, D.; Nemer, G.; Schmitt, J.P.; Charron, F.; Robitaille, L.; Caron, S.; Conner, D.A.; Gessler, M.; Nemer, M.; Seidman, C.E.; et al. A HCN4+ cardiomyogenic progenitor derived from the first heart field and human pluripotent stem cells. Nat. Cell Biol. 2013, 15, 1098–1106. [Google Scholar] [CrossRef]
- Takeuchi, J.K.; Ohgi, M.; Koshiba-Takeuchi, K.; Shiratori, H.; Sakaki, I.; Ogura, K.; Saijoh, Y.; Ogura, T. Tbx5 specifies the left/right ventricles and ventricular septum position during cardiogenesis. Development 2003, 130, 5953–5964. [Google Scholar] [CrossRef]
- Liang, X.; Wang, G.; Lin, L.; Lowe, J.; Zhang, Q.; Bu, L.; Chen, Y.; Chen, J.; Sun, Y.; Evans, S.M. HCN4 Dynamically Marks the First Heart Field and Conduction System Precursors. Circ. Res. 2013, 113, 399–407. [Google Scholar] [CrossRef]
- Cai, C.L.; Liang, X.; Shi, Y.; Chu, P.H.; Pfaff, S.L.; Chen, J.; Evans, S. Isl1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Dev. Cell 2003, 5, 877–889. [Google Scholar] [CrossRef]
- Kelly, R.G.; Brown, N.A.; Buckingham, M.E. The arterial pole of the mouse heart forms from Fgf10-expressing cells in pharyngeal mesoderm. Dev. Cell 2001, 1, 435–440. [Google Scholar] [CrossRef]
- Verzi, M.P.; McCulley, D.J.; de Val, S.; Dodou, E.; Black, B.L. The right ventricle, outflow tract, and ventricular septum comprise a restricted expression domain within the secondary/anterior heart field. Dev. Biol. 2005, 287, 134–145. [Google Scholar] [CrossRef]
- Zaffran, S.; Kelly, R.G.; Meilhac, S.M.; Buckingham, M.E.; Brown, N.A. Right ventricular myocardium derives from the anterior heart field. Circ. Res. 2004, 95, 261–268. [Google Scholar] [CrossRef]
- Vincent, S.D.; Buckingham, M.E. How to make a heart: The origin and regulation of cardiac progenitor cells. Current Top. Dev. Biol. 2010, 90, 1–41. [Google Scholar] [CrossRef]
- Wu, S.M.; Fujiwara, Y.; Cibulsky, S.M.; Clapham, D.E.; Lien, C.L.; Schultheiss, T.M.; Orkin, S.H. Developmental origin of a bipotential myocardial and smooth muscle cell precursor in the mammalian heart. Cell 2006, 127, 1137–1150. [Google Scholar] [CrossRef]
- Moretti, A.; Caron, L.; Nakano, A.; Lam, J.T.; Bernshausen, A.; Chen, Y.; Qyang, Y.; Bu, L.; Sasaki, M.; Martin-Puig, S.; et al. Multipotent embryonic isl1+ progenitor cells lead to cardiac, smooth muscle, and endothelial cell diversification. Cell 2006, 127, 1151–1165. [Google Scholar] [CrossRef]
- Cohen, E.D.; Wang, Z.; Lepore, J.J.; Lu, M.M.; Taketo, M.M.; Epstein, D.J.; Morrisey, E.E. Wnt/beta-catenin signaling promotes expansion of Isl-1-positive cardiac progenitor cells through regulation of FGF signaling. J. Clin. Invest. 2007, 117, 1794–1804. [Google Scholar] [CrossRef]
- Lin, L.; Cui, L.; Zhou, W.; Dufort, D.; Zhang, X.; Cai, C.L.; Bu, L.; Yang, L.; Martin, J.; Kemler, R.; et al. Beta-catenin directly regulates Islet1 expression in cardiovascular progenitors and is required for multiple aspects of cardiogenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 9313–9318. [Google Scholar] [CrossRef]
- Jeter, J.R., Jr.; Cameron, I.L. Cell proliferation patterns during cytodifferentiation in embryonic chick tissues: Liver, heart and erythrocytes. J. Embryol. Exp. Morphol. 1971, 25, 405–422. [Google Scholar]
- Luxan, G.; Casanova, J.C.; Martínez-Poveda, B.; Prados, B.; D’Amato, G.; MacGrogan, D.; Gonzalez-Rajal, A.; Dobarro, D.; Torroja, C.; et al. Mutations in the NOTCH pathway regulator MIB1 cause left ventricular noncompaction cardiomyopathy. Nat. Med. 2013, 19, 193–201. [Google Scholar] [CrossRef]
- Buikema, J.W.; Mady, A.S.; Mittal, N.V.; Atmanli, A.; Caron, L.; Doevendans, P.A.; Sluijter, J.P.; Domian, I.J. Wnt/beta-catenin signaling directs the regional expansion of first and second heart field-derived ventricular cardiomyocytes. Development 2013, 140, 4165–4176. [Google Scholar] [CrossRef]
- Buikema, J.W.; Zwetsloot, P.P.; Doevendans, P.A.; Sluijter, J.P.; Domian, I.J. Expanding mouse ventricular cardiomyocytes through GSK-3 inhibition. Curr. Protoc. Cell Biol. 2013, 61. [Google Scholar] [CrossRef]
- Oka, T.; Xu, J.; Molkentin, J.D. Re-employment of developmental transcription factors in adult heart disease. Semin. Cell Dev. Biol. 2007, 18, 117–131. [Google Scholar] [CrossRef]
- Lindsley, R.C.; Gill, J.G.; Kyba, M.; Murphy, T.L.; Murphy, K.M. Canonical Wnt signaling is required for development of embryonic stem cell-derived mesoderm. Development 2006, 133, 3787–3796. [Google Scholar] [CrossRef]
- Palpant, N.J.; Pabon, L.; Rabinowitz, J.S.; Hadland, B.K.; Stoick-Cooper, C.L.; Paige, S.L.; Bernstein, I.D.; Moon, R.T.; Murry, C.E. Transmembrane protein 88: A Wnt regulatory protein that specifies cardiomyocyte development. Development 2013, 140, 3799–3808. [Google Scholar]
- Hsieh, P.C.; Segers, V.F.; Davis, M.E.; MacGillivray, C.; Gannon, J.; Molkentin, J.D.; Robbins, J.; Lee, R.T. Evidence from a genetic fate-mapping study that stem cells refresh adult mammalian cardiomyocytes after injury. Nat. Med. 2007, 13, 970–974. [Google Scholar] [CrossRef]
- Porrello, E.R.; Mahmoud, A.I.; Simpson, E.; Hill, J.A.; Richardson, J.A.; Olson, E.N.; Sadek, H.A. Transient regenerative potential of the neonatal mouse heart. Science 2011, 331, 1078–1080. [Google Scholar] [CrossRef]
- Bergmann, O.; Bhardwaj, R.D.; Bernard, S.; Zdunek, S.; Barnabé-Heider, F.; Walsh, S.; Zupicich, J.; Alkass, K.; Buchholz, B.A.; Druid, H.; et al. Evidence for cardiomyocyte renewal in humans. Science 2009, 324, 98–102. [Google Scholar] [CrossRef]
- Huch, M.; Dorrell, C.; Boj, S.F.; van Es, J.H.; Li, V.S.; van de Wetering, M.; Sato, T.; Hamer, K.; Sasaki, N.; Finegold, M.J.; et al. In vitro expansion of single Lgr5+ liver stem cells induced by Wnt-driven regeneration. Nature 2013, 494, 247–250. [Google Scholar] [CrossRef]
- O'Gara, P.T.; Kushner, F.G.; Ascheim, D.D.; Casey, D.E., Jr.; Chung, M.K.; de Lemos, J.A.; Ettinger, S.M.; Fang, J.C.; Fesmire, F.M.; Franklin, B.A.; et al. 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2013, 127, e362–e425. [Google Scholar] [CrossRef]
- Buikema, J.; van der Meer, P.; Sluijter, J.P.; Domian, I.J. Engineering Myocardial Tissue: The Convergence of Stem Cells Biology and Tissue Engineering Technology. Stem Cells 2013, 31, 2587–2589. [Google Scholar] [CrossRef]
- Bhanot, P.; Brink, M.; Samos, C.H.; Hsieh, J.C.; Wang, Y.; Macke, J.P.; Andrew, D.; Nathans, J.; Nusse, R. A new member of the frizzled family from Drosophila functions as a Wingless receptor. Nature 1996, 382, 225–230. [Google Scholar] [CrossRef]
- Pinson, K.I.; Brennan, J.; Monkley, S.; Avery, B.J.; Skarnes, W.C. An LDL-receptor-related protein mediates Wnt signalling in mice. Nature 2000, 407, 535–538. [Google Scholar] [CrossRef]
- Ikeda, S.; Kishida, S.; Yamamoto, H.; Murai, H.; Koyama, S.; Kikuchi, A. Axin, a negative regulator of the Wnt signaling pathway, forms a complex with GSK-3beta and beta-catenin and promotes GSK-3beta-dependent phosphorylation of beta-catenin. EMBO J. 1998, 17, 1371–1384. [Google Scholar] [CrossRef]
- Behrens, J.; von Kries, J.P.; Kühl, M.; Bruhn, L.; Wedlich, D.; Grosschedl, R.; Birchmeier, W. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature 1996, 382, 638–642. [Google Scholar] [CrossRef]
- Van de Wetering, M.; Cavallo, R.; Dooijes, D.; van Beest, M.; van Es, J.; Loureiro, J.; Ypma, A.; Hursh, D.; Jones, T.; Bejsovec, A.; et al. Armadillo coactivates transcription driven by the product of the Drosophila segment polarity gene dTCF. Cell 1997, 88, 789–799. [Google Scholar] [CrossRef]
- Blankesteijn, W.; van Gijn, M.; Essers-Janssen, Y.; Daemen, M.; Smits, J. Beta-catenin, an inducer of uncontrolled cell proliferation and migration in malignancies, is localized in the cytoplasm of vascular endothelium during neovascularization after myocardial infarction. Am. J. Pathol. 2000, 157, 877–883. [Google Scholar] [CrossRef]
- Chen, L.; Wu, Q.; Guo, F.; Xia, B.; Zuo, J. Expression of Dishevelled-1 in wound healing after acute myocardial infarction: Possible involvement in myofibroblast proliferation and migration. J. Cell. Mol. Med. 2004, 8, 257–264. [Google Scholar] [CrossRef]
- Aisagbonhi, O.; Rai, M.; Ryzhov, S.; Atria, N.; Feoktistov, I.; Hatzopoulos, A.K. Experimental myocardial infarction triggers canonical Wnt signaling and endothelial-to-mesenchymal transition. Dis. Models Mech. 2011, 4, 469–483. [Google Scholar] [CrossRef]
- Barandon, L.; Couffinhal, T.; Ezan, J.; Dufourcq, P.; Costet, P.; Alzieu, P.; Leroux, L.; Moreau, C.; Dare, D.; Duplàa, C. Reduction of infarct size and prevention of cardiac rupture in transgenic mice overexpressing FrzA. Circulation 2003, 108, 2282–2289. [Google Scholar] [CrossRef]
- He, W.; Zhang, L.; Ni, A.; Zhang, Z.; Mirotsou, M.; Mao, L.; Pratt, R.E.; Dzau, V.J. Exogenously administered secreted frizzled related protein 2 (Sfrp2) reduces fibrosis and improves cardiac function in a rat model of myocardial infarction. Proc. Natl. Acad. Sci. USA 2010, 107, 21110–21115. [Google Scholar]
- Kobayashi, K.; Luo, M.; Zhang, Y.; Wilkes, D.C.; Ge, G.; Grieskamp, T.; Yamada, C.; Liu, T.-C.; Huang, G.; Basson, C.T.; et al. Secreted Frizzled-related protein 2 is a procollagen C proteinase enhancer with a role in fibrosis associated with myocardial infarction. Nat. Cell Biol. 2009, 11, 46–55. [Google Scholar] [CrossRef]
- Laeremans, H.; Hackeng, T.M.; van Zandvoort, M.A.; Thijssen, V.L.; Janssen, B.J.; Ottenheijm, H.C.; Smits, J.F.; Blankesteijn, W.M. Blocking of frizzled signaling with a homologous peptide fragment of wnt3a/wnt5a reduces infarct expansion and prevents the development of heart failure after myocardial infarction. Circulation 2011, 124, 1626–1635. [Google Scholar] [CrossRef]
- Barandon, L.; Dufourcq, P.; Costet, P.; Moreau, C.; Allières, C.; Daret, D.; Dos Santos, P.; Daniel Lamazière, J.M.; Couffinhal, T.; Duplàa, C. Involvement of FrzA/sFRP-1 and the Wnt/frizzled pathway in ischemic preconditioning. Circ. Res. 2005, 96, 1299–1306. [Google Scholar] [CrossRef]
- Webb, I.; Sicard, P.; Clark, J.; Redwood, S.; Marber, M. Myocardial stress remodelling after regional infarction is independent of glycogen synthase kinase-3 inactivation. J. Mol. Cell. Cardiol. 2010, 49, 897–900. [Google Scholar] [CrossRef]
- Lal, H.; Zhou, J.; Ahmad, F.; Zaka, R.; Vagnozzi, R.J.; Decaul, M.; Woodgett, J.; Gao, E.; Force, T. Glycogen synthase kinase-3α limits ischemic injury, cardiac rupture, post-myocardial infarction remodeling and death. Circulation 2012, 125, 65–75. [Google Scholar] [CrossRef]
- Woulfe, K.C.; Gao, E.; Lal, H.; Harris, D.; Fan, Q.; Vagnozzi, R.; DeCaul, M.; Shang, X.; Patel, S.; Woodgett, J.R.; et al. Glycogen synthase kinase-3beta regulates post-myocardial infarction remodeling and stress-induced cardiomyocyte proliferation in vivo. Circ. Res. 2010, 106, 1635–1645. [Google Scholar] [CrossRef]
- Kaga, S.; Zhan, L.; Altaf, E.; Maulik, N. Glycogen synthase kinase-3beta/beta-catenin promotes angiogenic and anti-apoptotic signaling through the induction of VEGF, Bcl-2 and survivin expression in rat ischemic preconditioned myocardium. J. Mol. Cell. Cardiol. 2006, 40, 138–147. [Google Scholar] [CrossRef]
- Hahn, J.-Y.; Cho, H.J.; Bae, J.W.; Yuk, H.S.; Kim, K.I.; Park, K.W.; Koo, B.K.; Chae, I.H.; Shin, C.S.; Oh, B.H.; et al. Beta-catenin overexpression reduces myocardial infarct size through differential effects on cardiomyocytes and cardiac fibroblasts. J. Biol. Chem. 2006, 281, 30979–30989. [Google Scholar] [CrossRef]
- Zelarayán, L.C.; Noack, C.; Sekkali, B.; Kmecova, J.; Gehrke, C.; Renger, A.; Zafiriou, M.P.; van der Nagel, R.; Dietz, R.; de Windt, L.J.; et al. Beta-Catenin downregulation attenuates ischemic cardiac remodeling through enhanced resident precursor cell differentiation. Proc. Natl. Acad. Sci. USA 2008, 105, 19762–19767. [Google Scholar] [CrossRef]
- Sasaki, T.; Hwang, H.; Nguyen, C.; Kloner, R.; Kahn, M. The Small Molecule Wnt Signaling Modulator ICG-001 Improves Contractile Function in Chronically Infarcted Rat Myocardium. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Buikema, J.W.; Zwetsloot, P.-P.M.; Doevendans, P.A.; Domian, I.J.; Sluijter, J.P.G. Wnt/β-Catenin Signaling during Cardiac Development and Repair. J. Cardiovasc. Dev. Dis. 2014, 1, 98-110. https://doi.org/10.3390/jcdd1010098
Buikema JW, Zwetsloot P-PM, Doevendans PA, Domian IJ, Sluijter JPG. Wnt/β-Catenin Signaling during Cardiac Development and Repair. Journal of Cardiovascular Development and Disease. 2014; 1(1):98-110. https://doi.org/10.3390/jcdd1010098
Chicago/Turabian StyleBuikema, Jan W., Peter-Paul M. Zwetsloot, Pieter A. Doevendans, Ibrahim J. Domian, and Joost P. G. Sluijter. 2014. "Wnt/β-Catenin Signaling during Cardiac Development and Repair" Journal of Cardiovascular Development and Disease 1, no. 1: 98-110. https://doi.org/10.3390/jcdd1010098