Synthesis and Biomedical Potential of Azapeptide Modulators of the Cluster of Differentiation 36 Receptor (CD36)

 ,
, 

Abstract
:
1. Introduction
2. Discussion
2.1. The Cluster of Differentiation 36 Receptor (CD36) Is a Medicinally Relevant Target
2.2. Growth Hormone Releasing Peptide-6 (GHRP-6) Analogues as Lead CD36 Binding Ligands
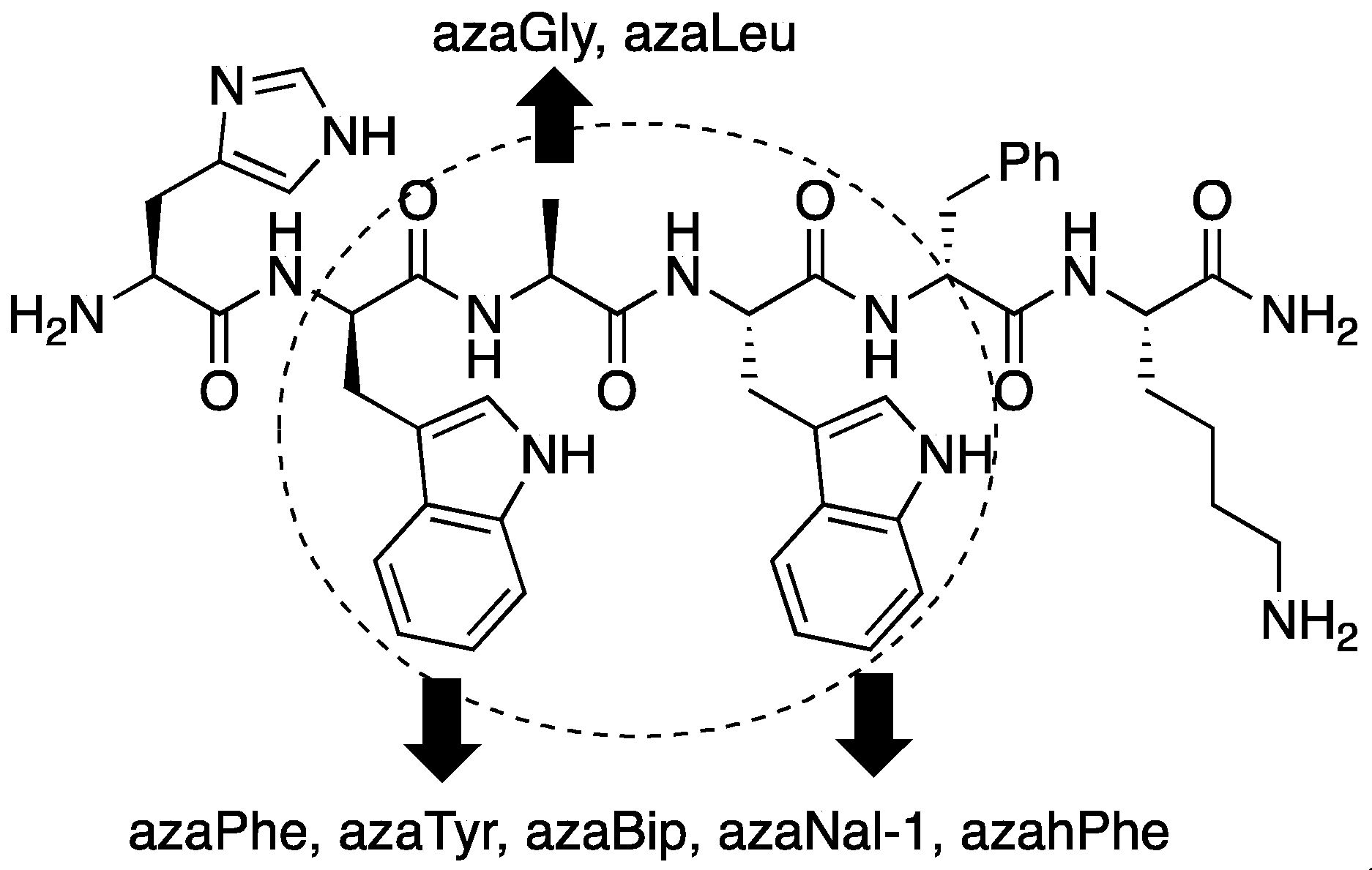
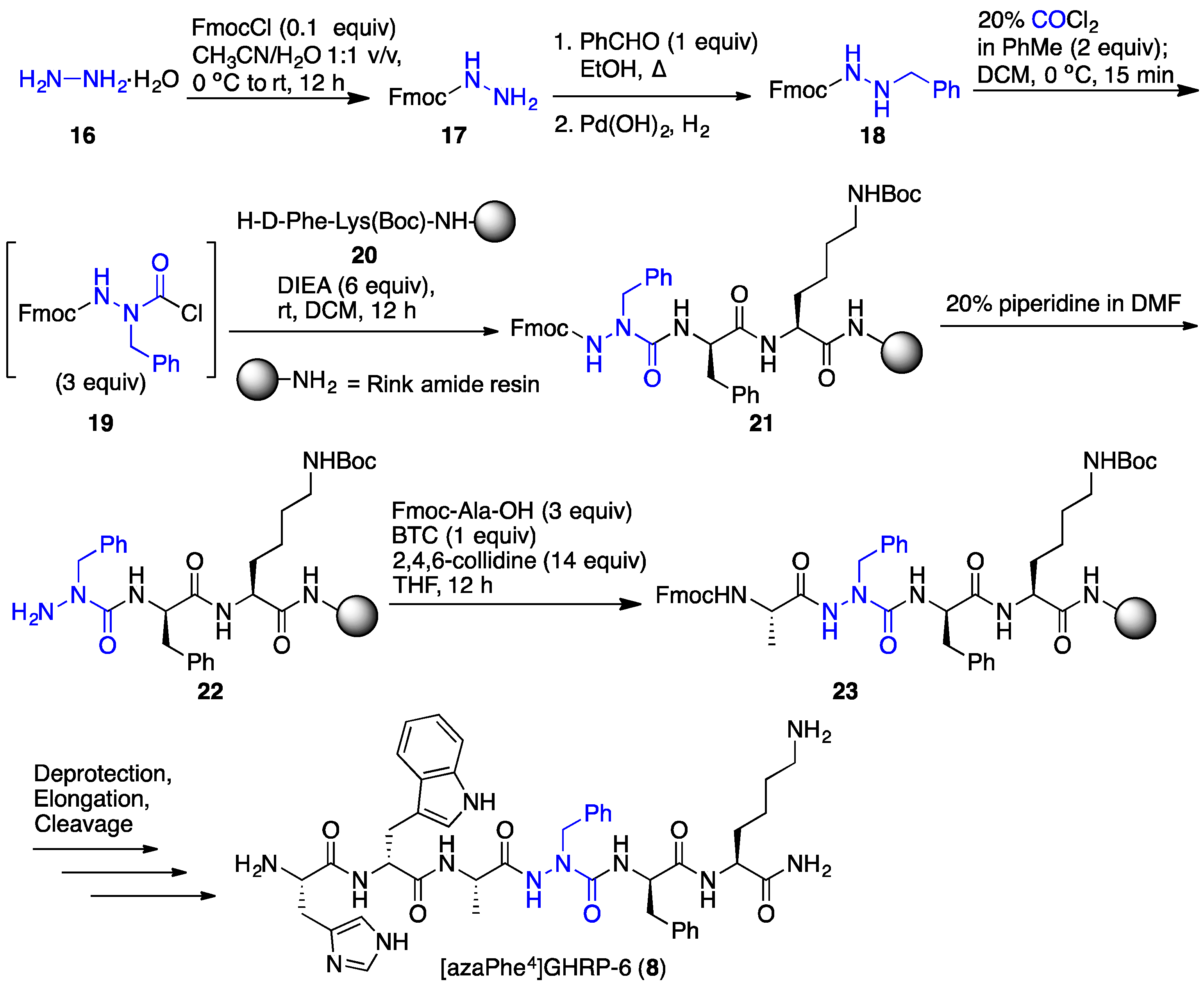
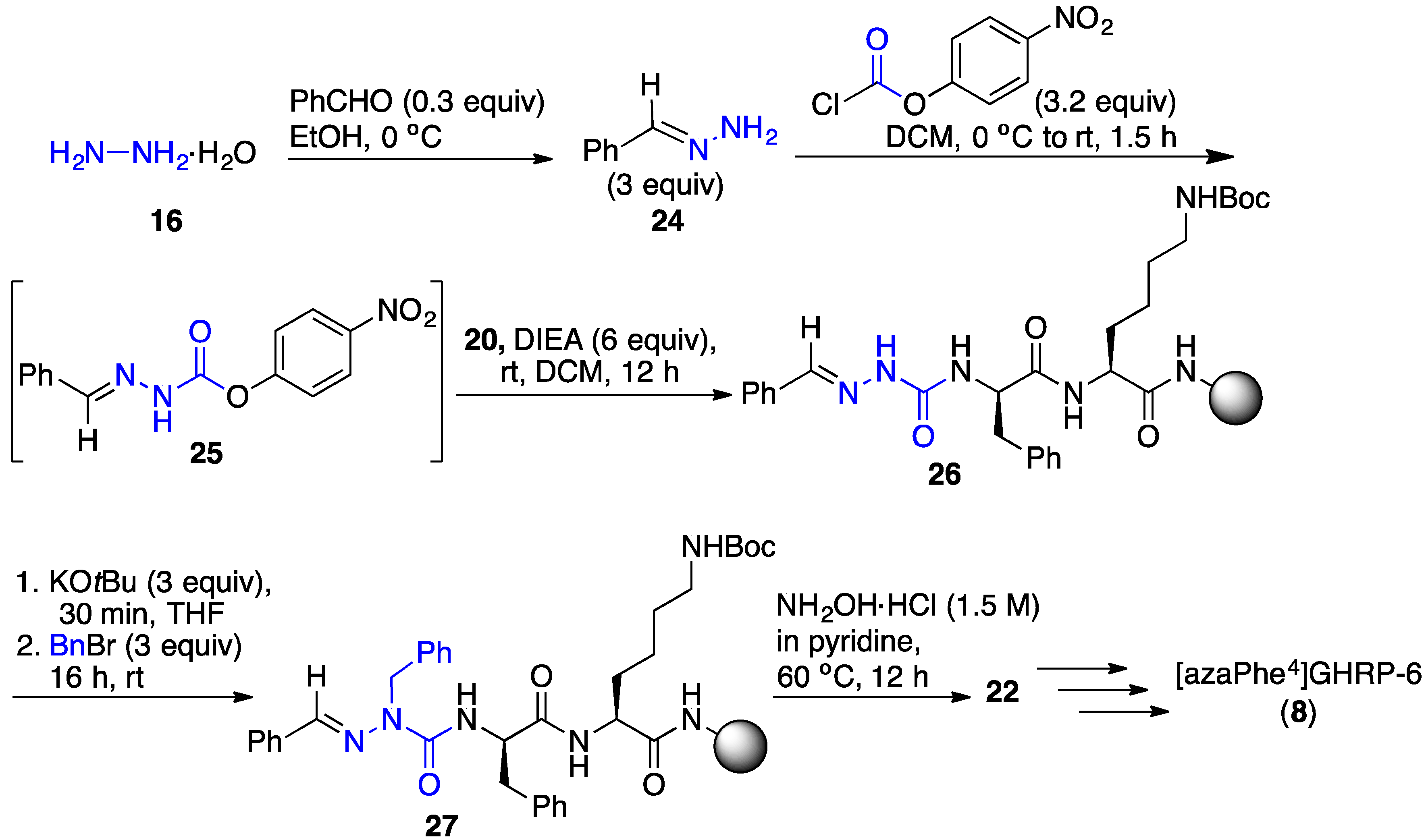
2.3. Conception of Azapeptide GHRP-6 Ligands Exhibiting CD36-Selective Binding Affinity
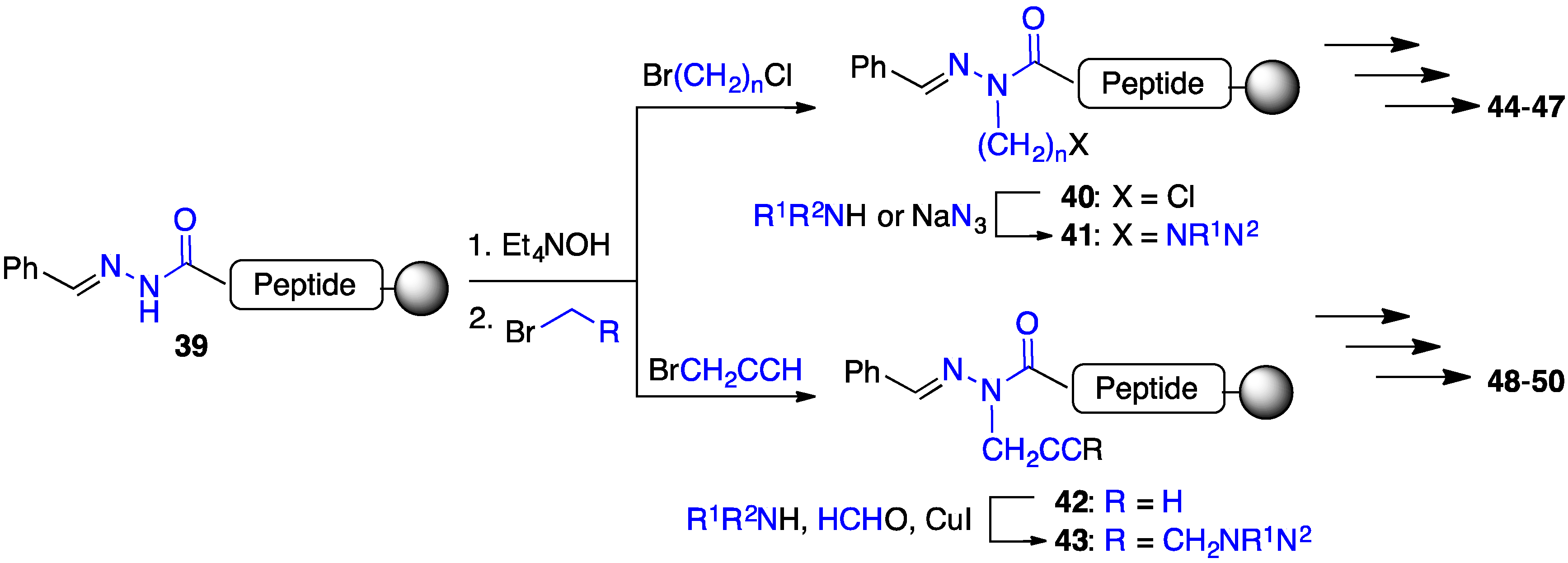
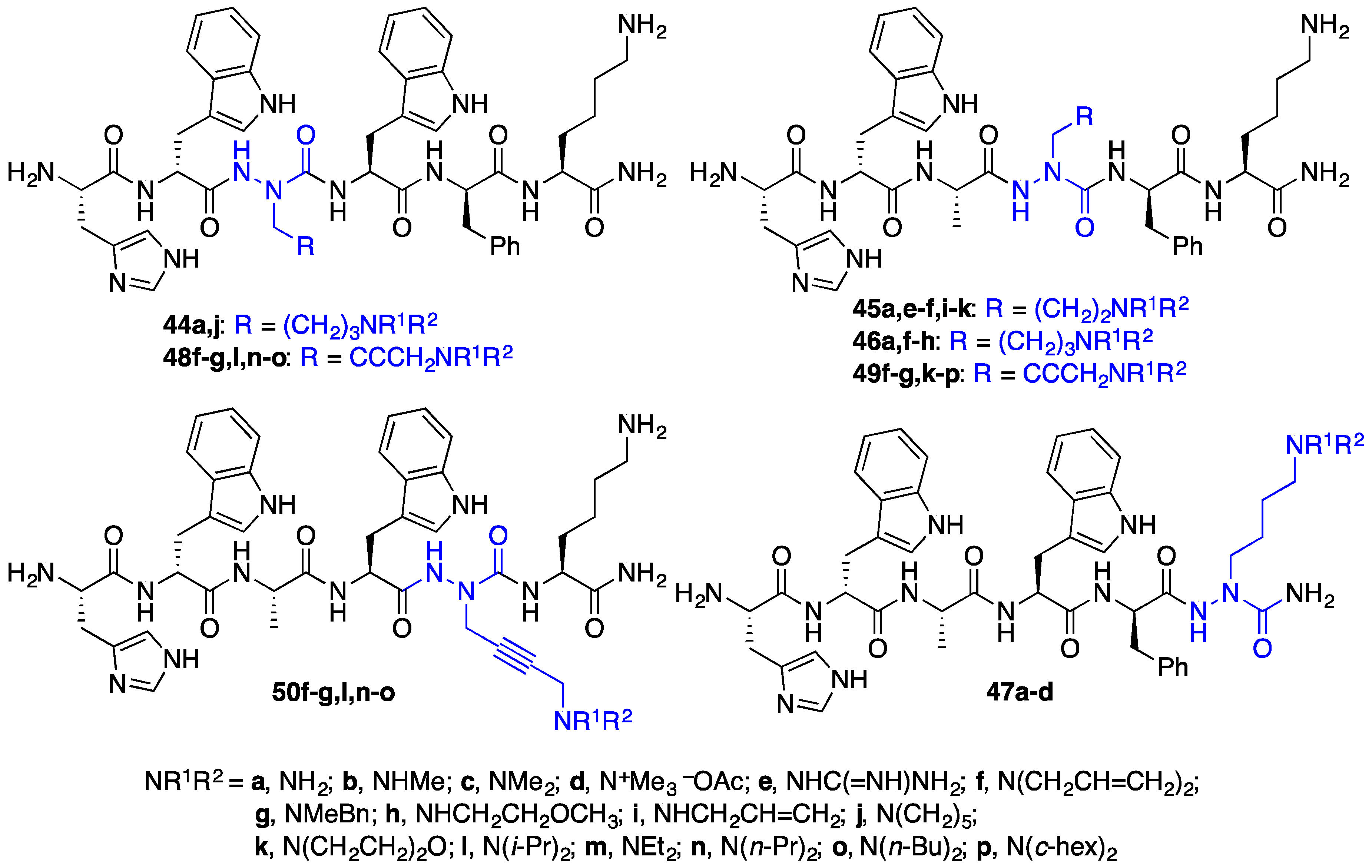
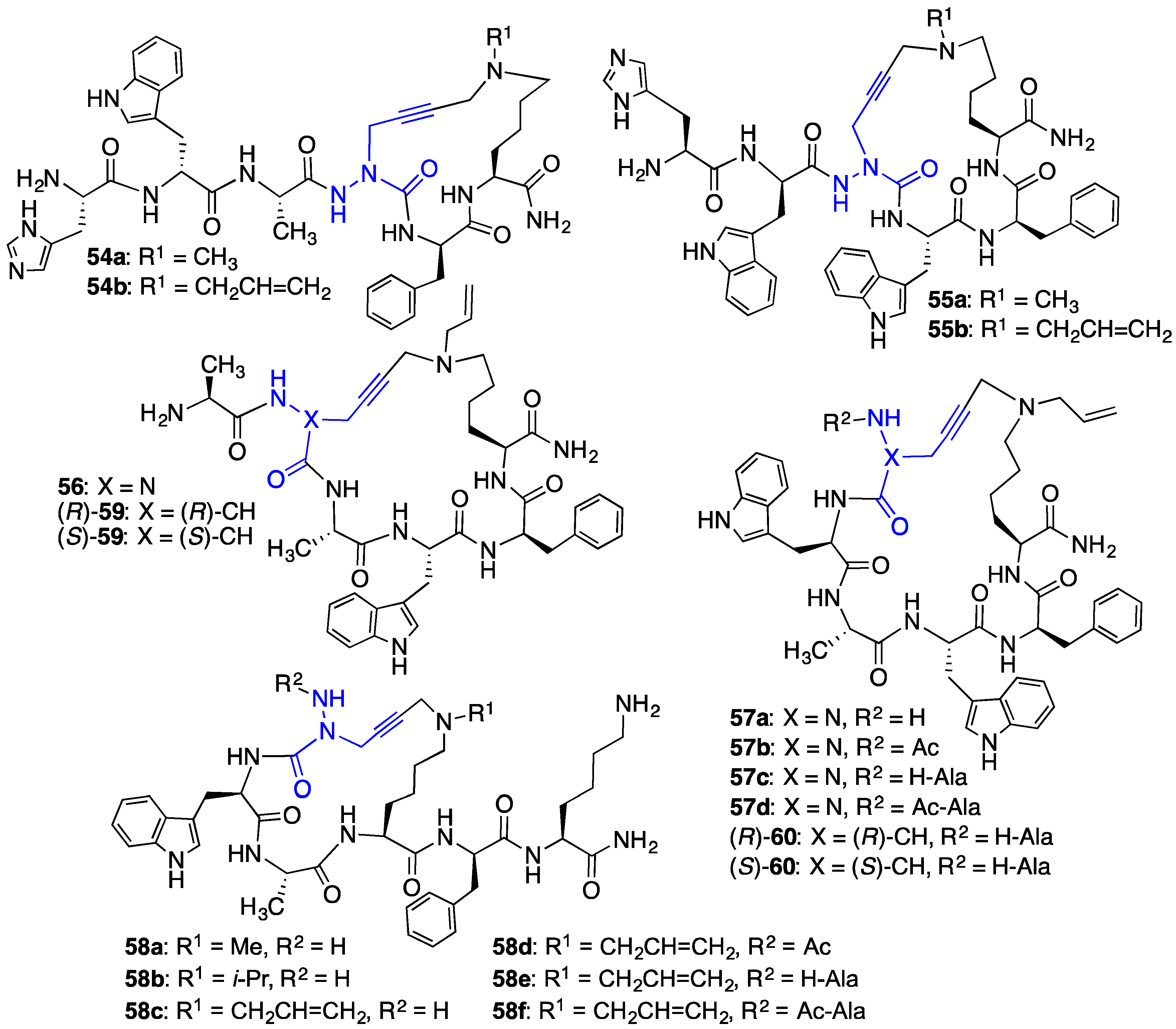
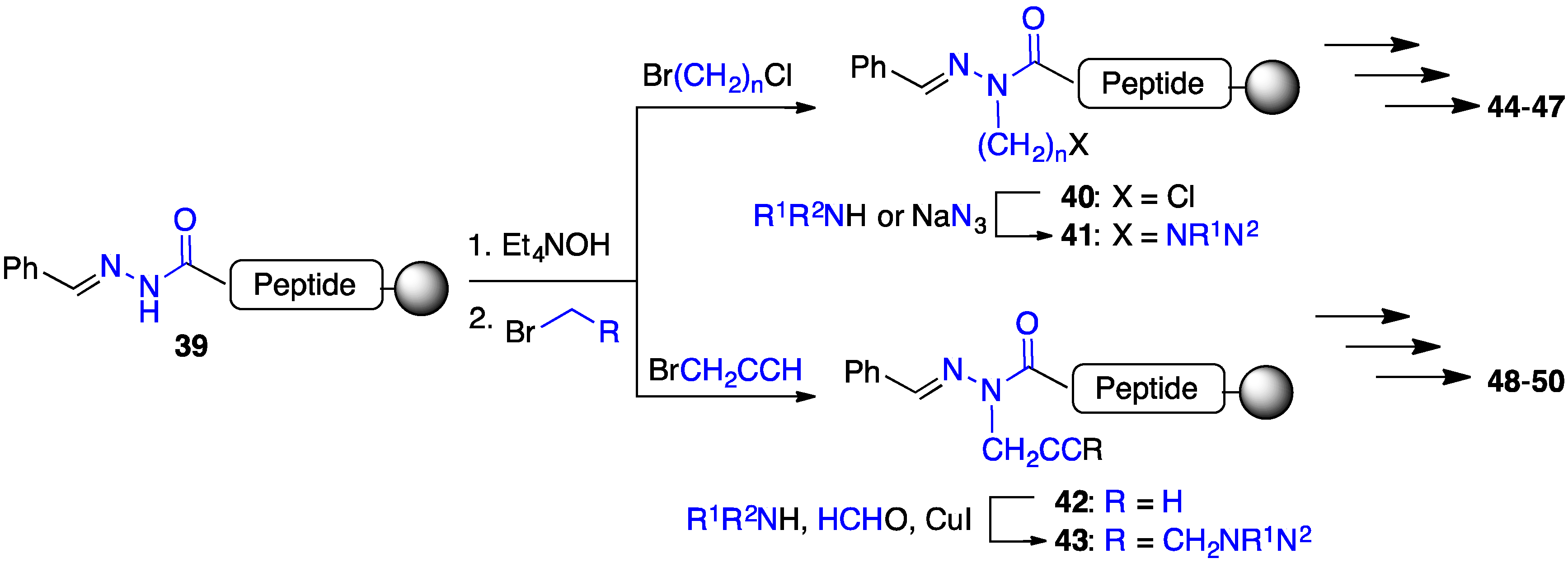
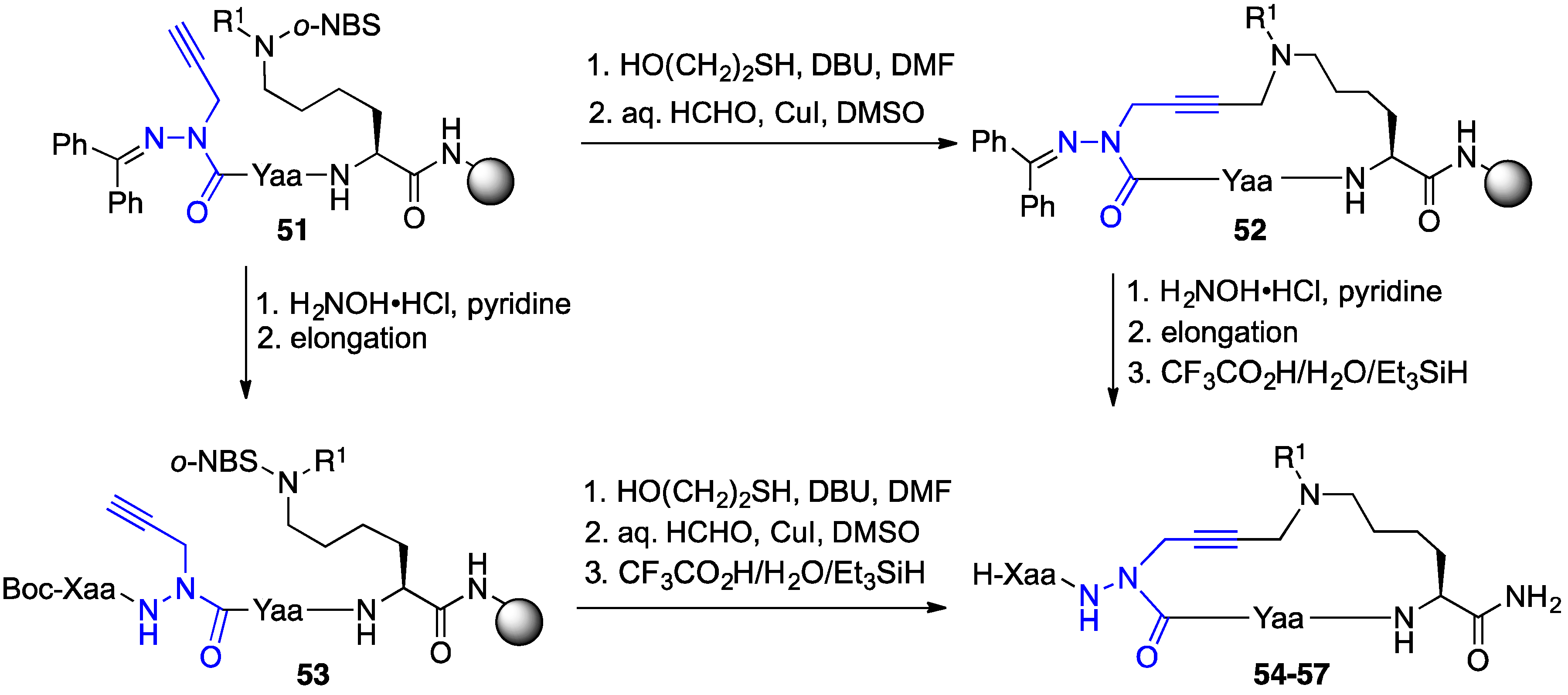
2.4. Expanding Aza-GHRP-6 Analogue Diversity by azaGly Alkylation
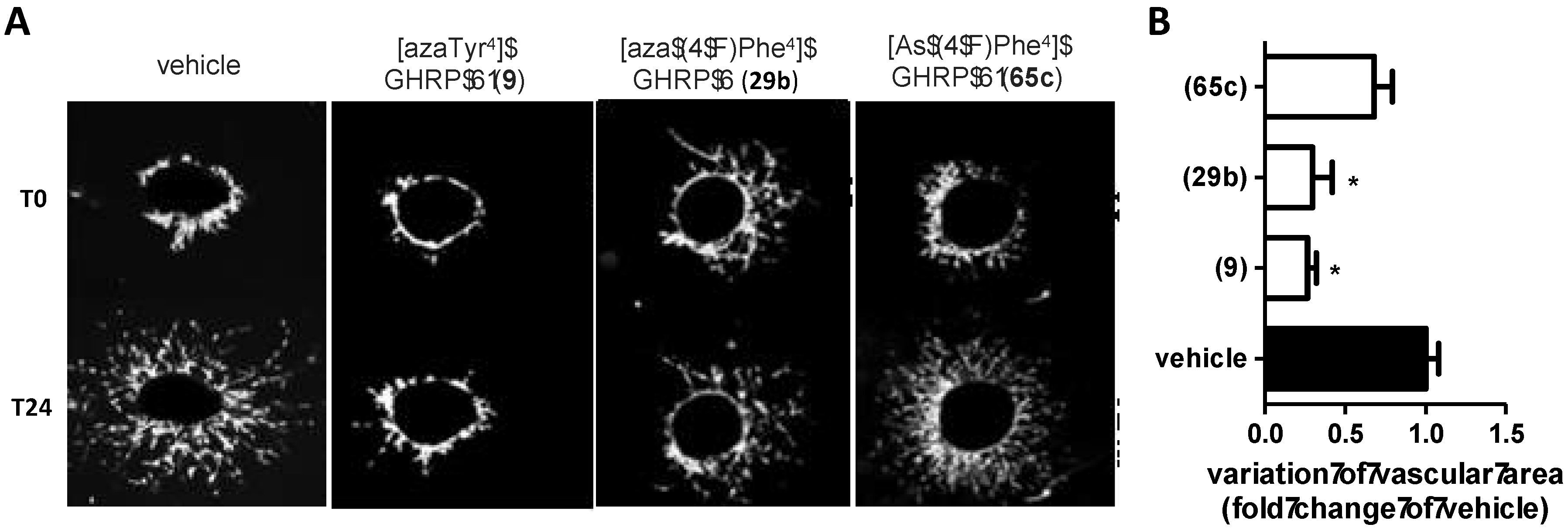
2.5. Divergent Effects of Aza-GHRP-6 Analogues on Microvascular Sprouting Reveals the Relevance of azaPhe4 Ring Substitution
2.6. Effect of His1 and azaPhe4 Substitutions on GHRP-6 Derivative Binding Affinity and Bioactivity
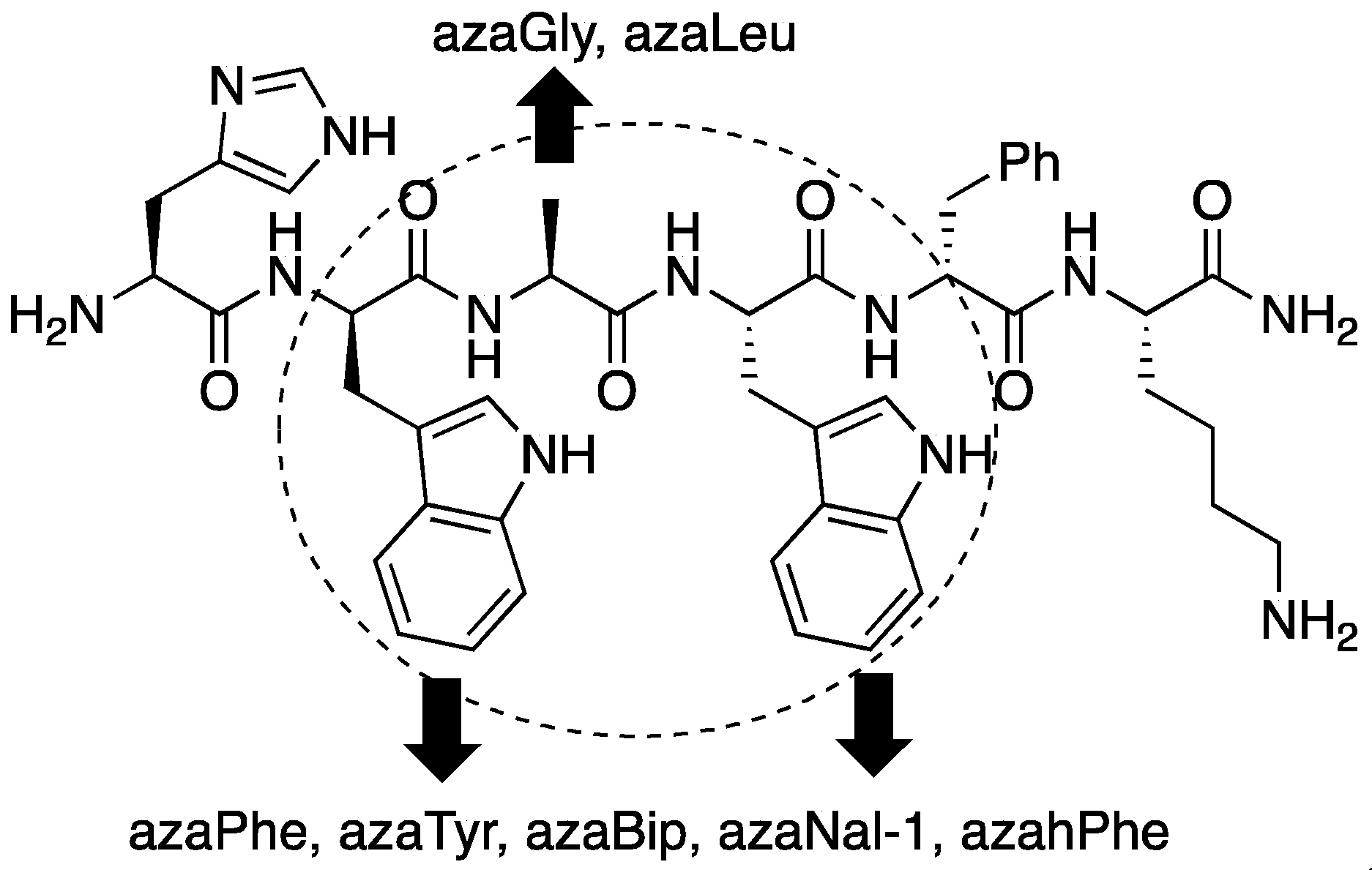
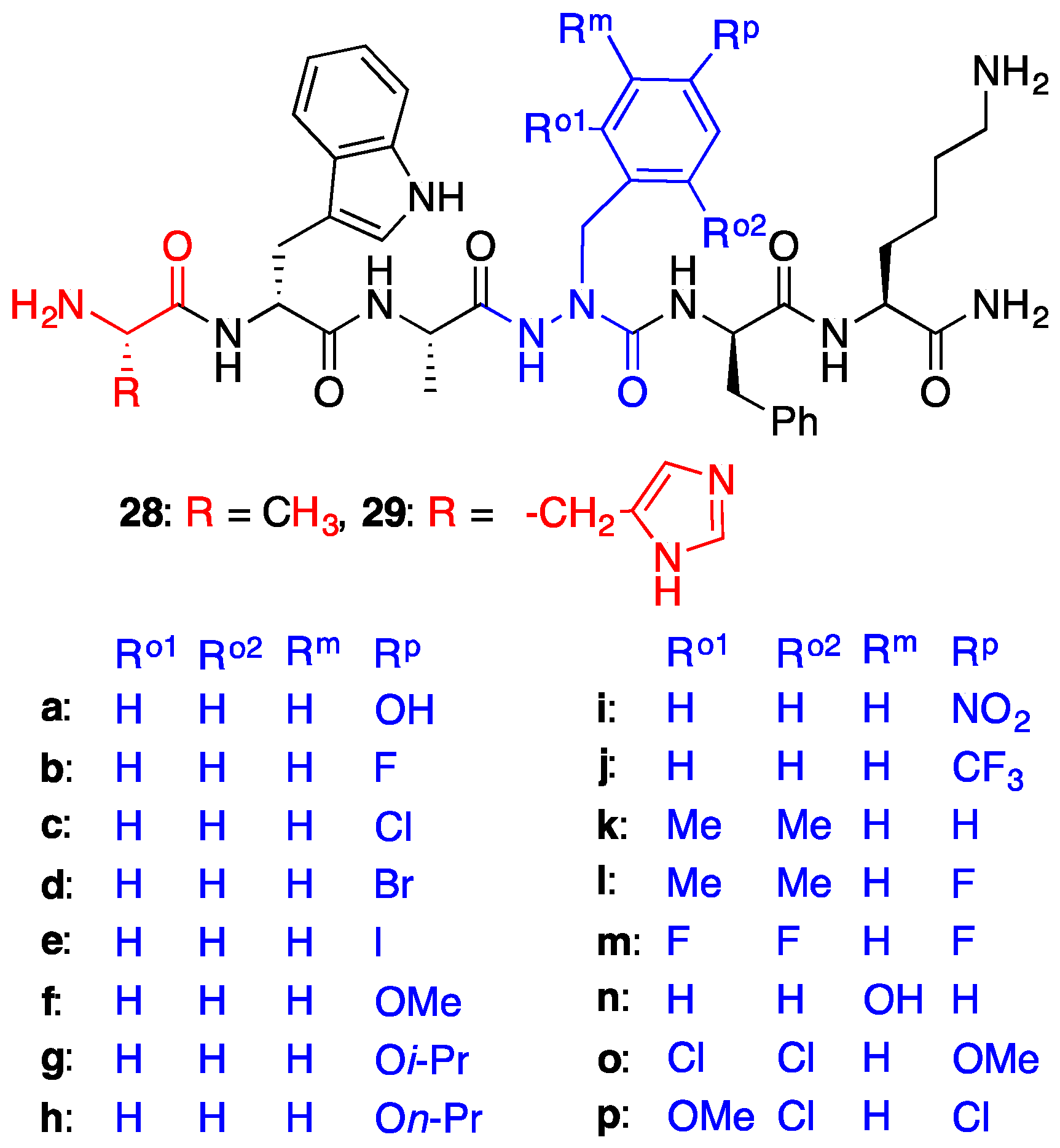
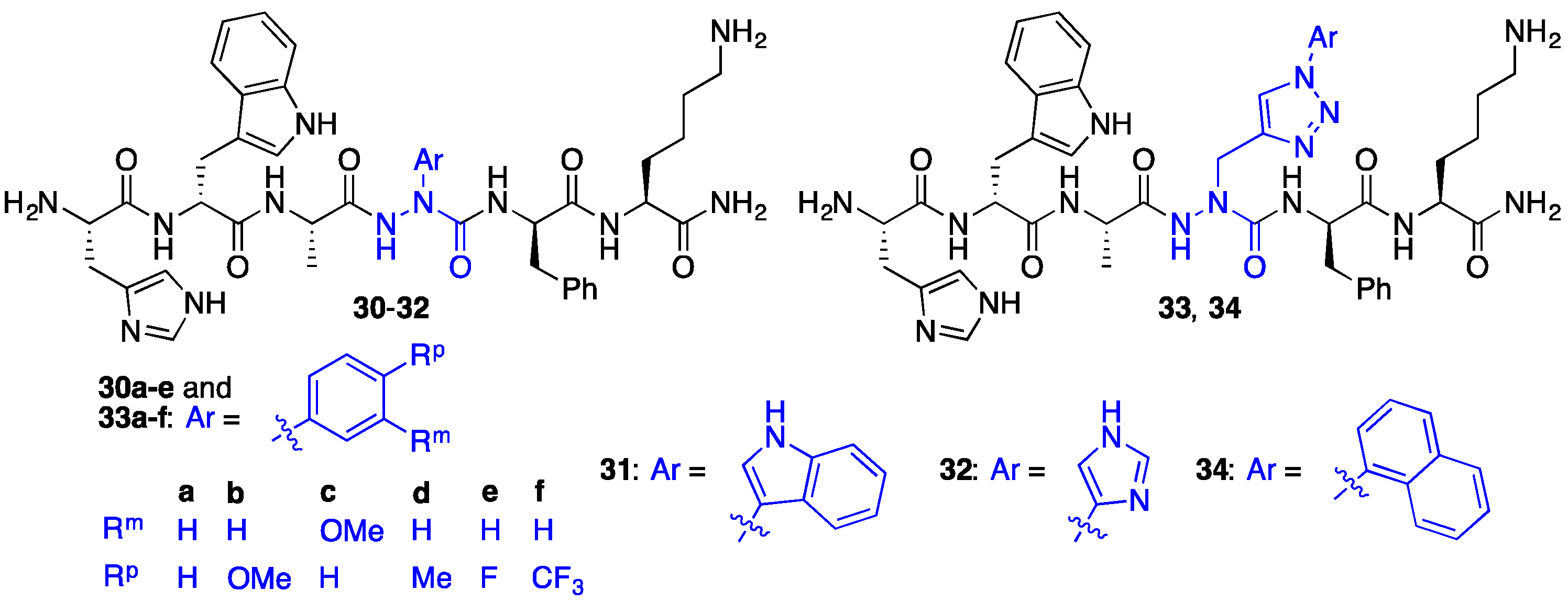
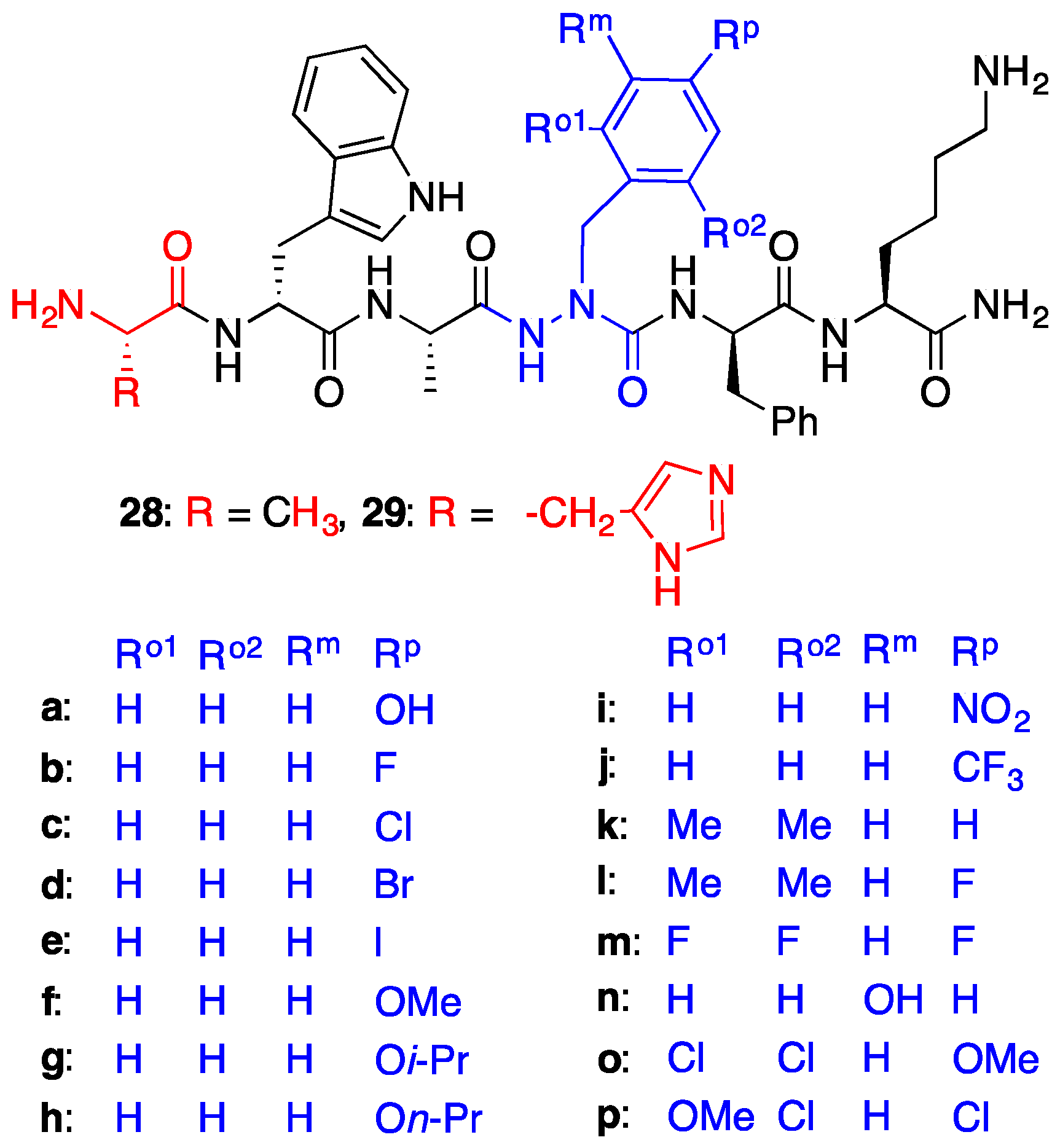
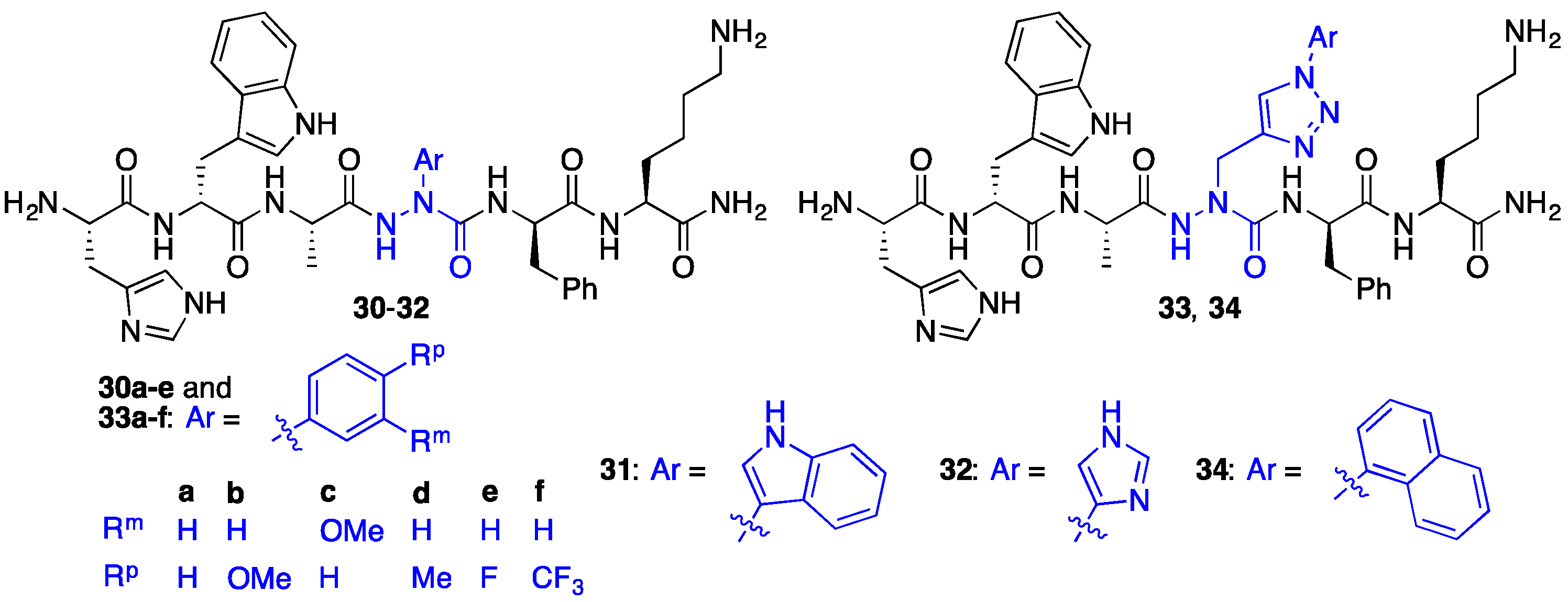
2.7. Further Probing of the azaPhe4 Residue Using aza-arylGly4- and aza-1,2,3-triazole-3-Ala4-GHRP-6 Analogues
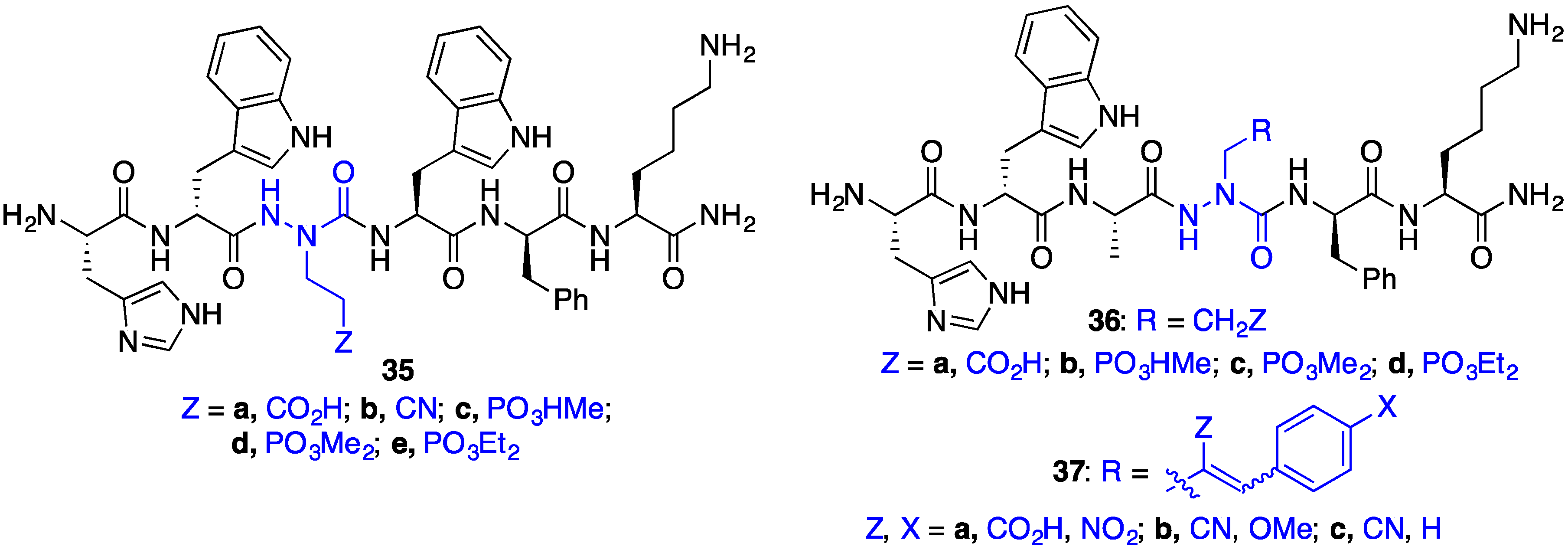
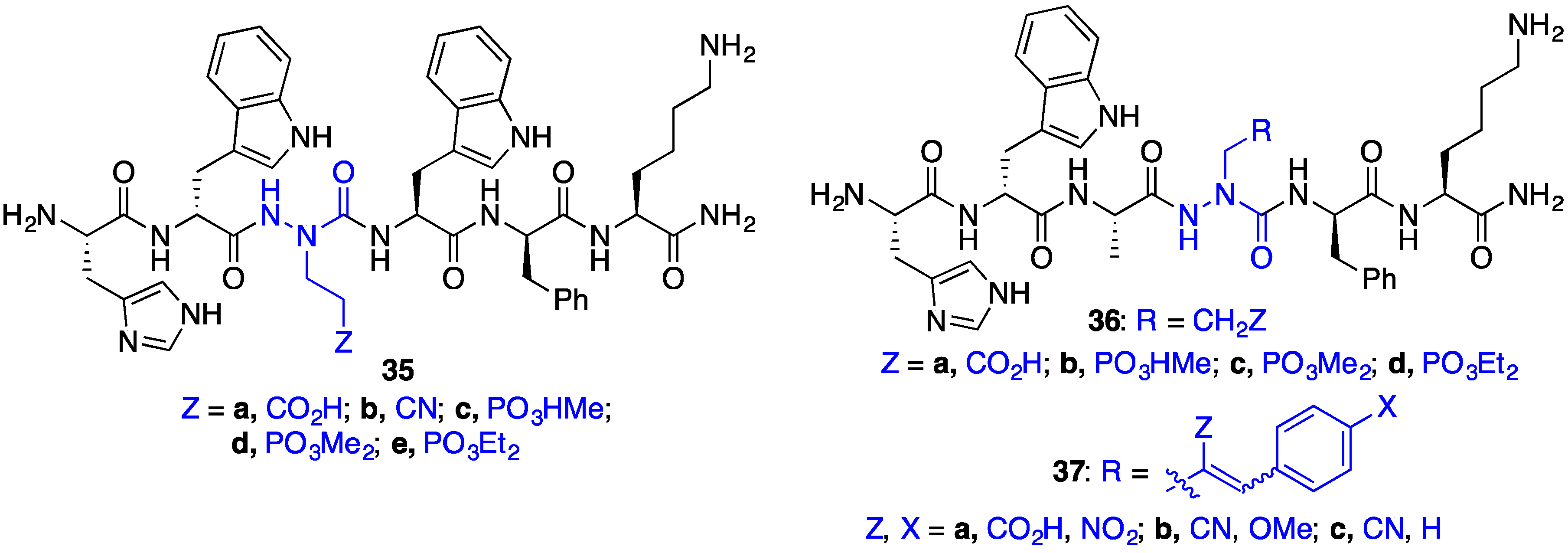
2.8. Exploring Potential Salt–Bridge Interactions Between CD36 and azaGlu-GHRP-6 Analogues
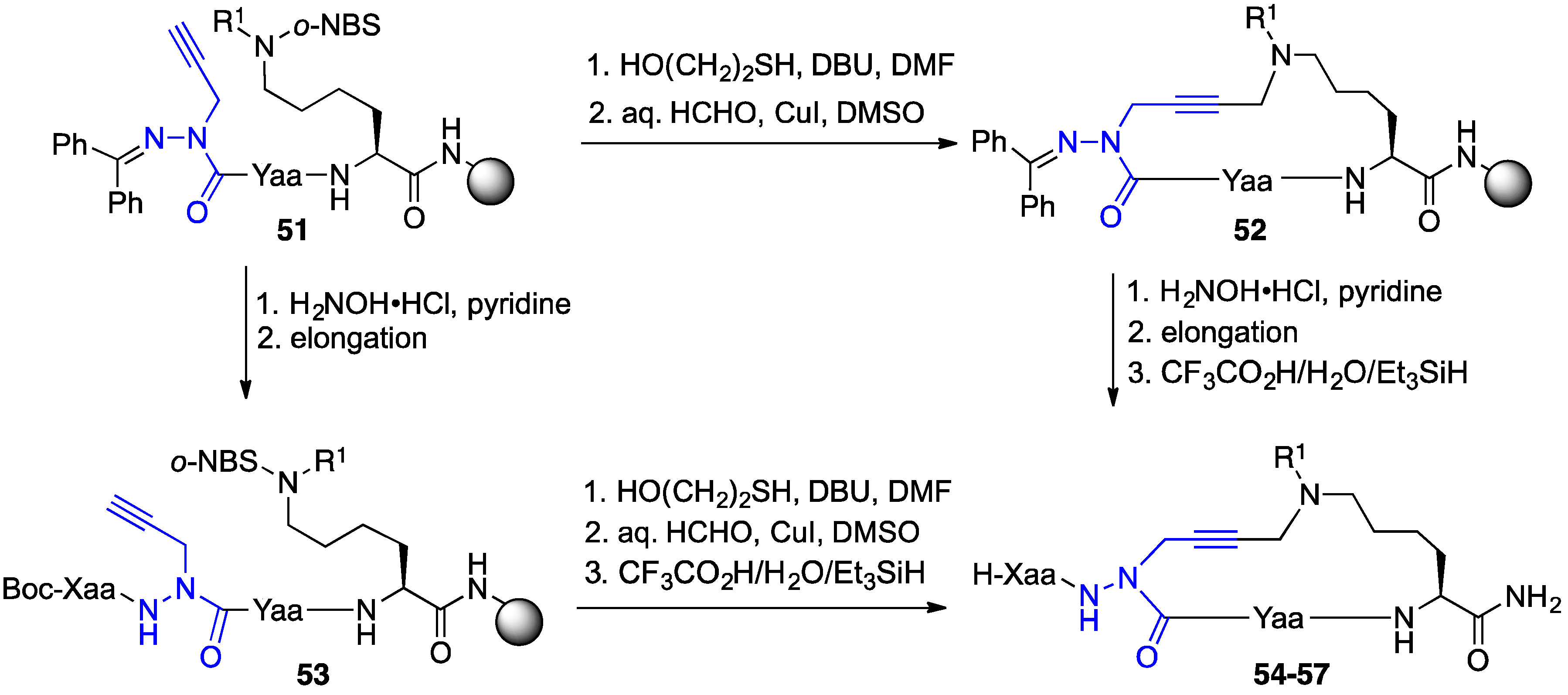
2.9. Aza-Lysine GHRP-6 Analogues
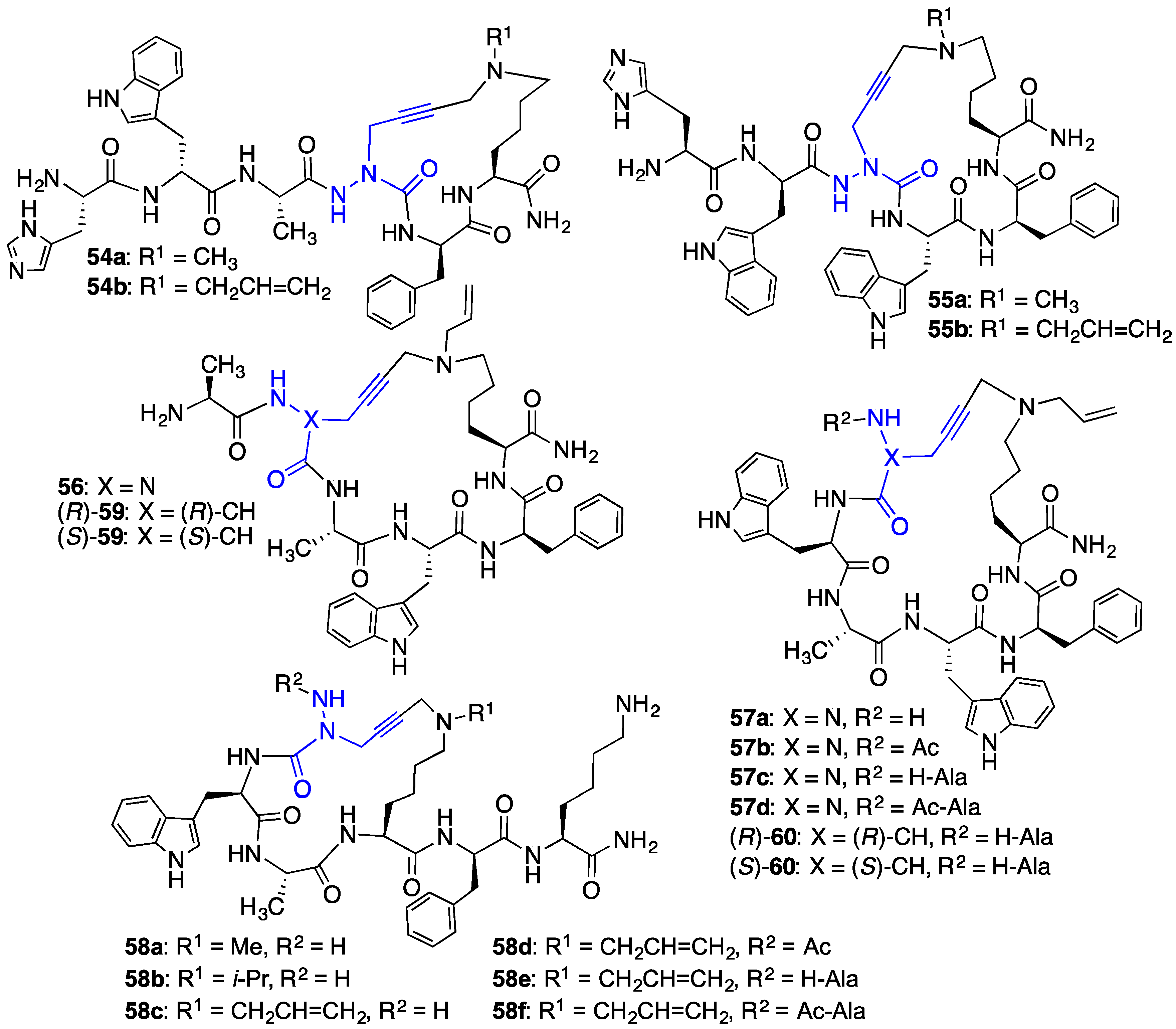
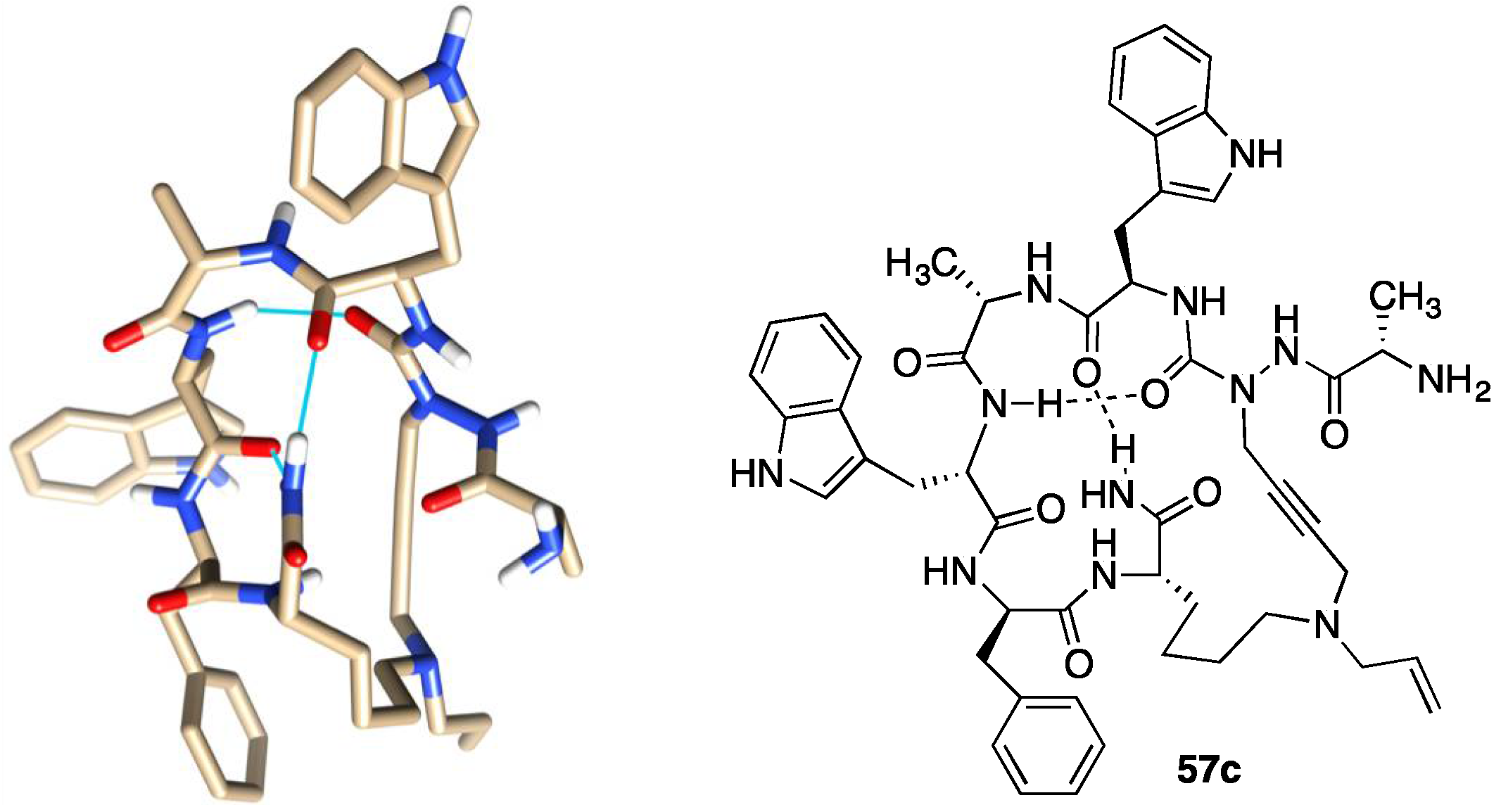
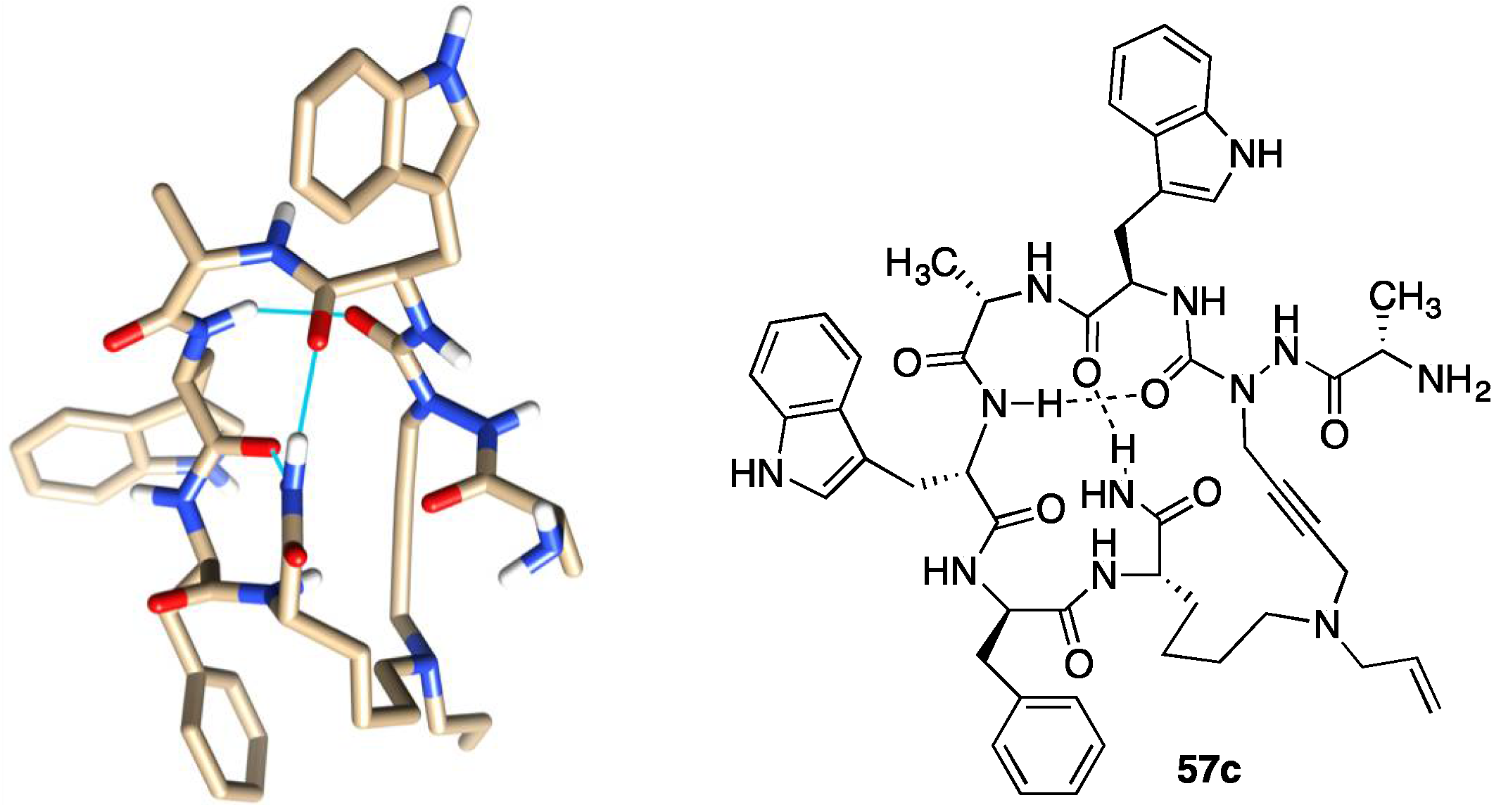
2.10. Unprecedented Binding Affinity and Activity Achieved by Azacyclopeptide-GHRP-6 Analogue Synthesis by A3-Macrocyclization
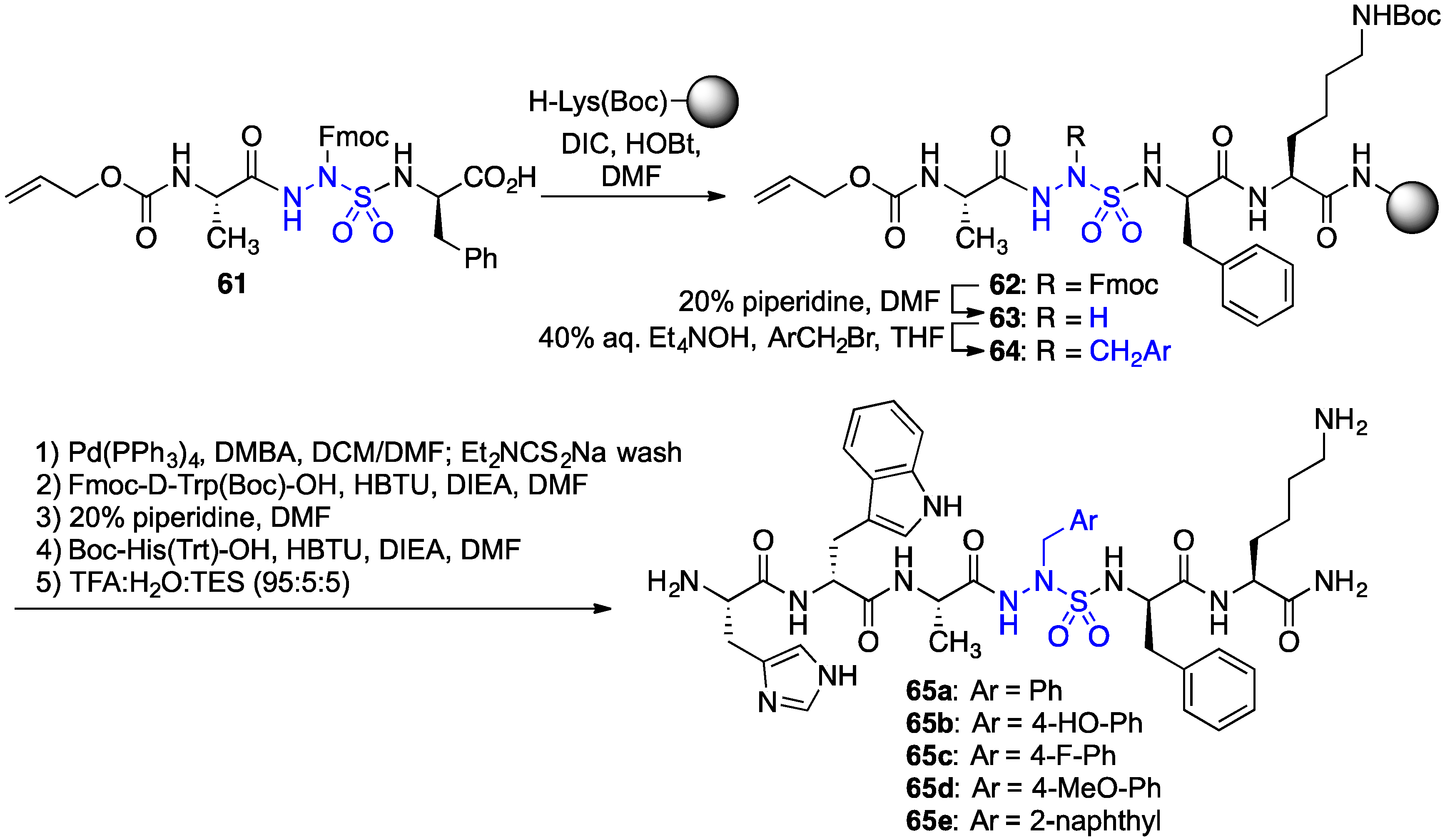
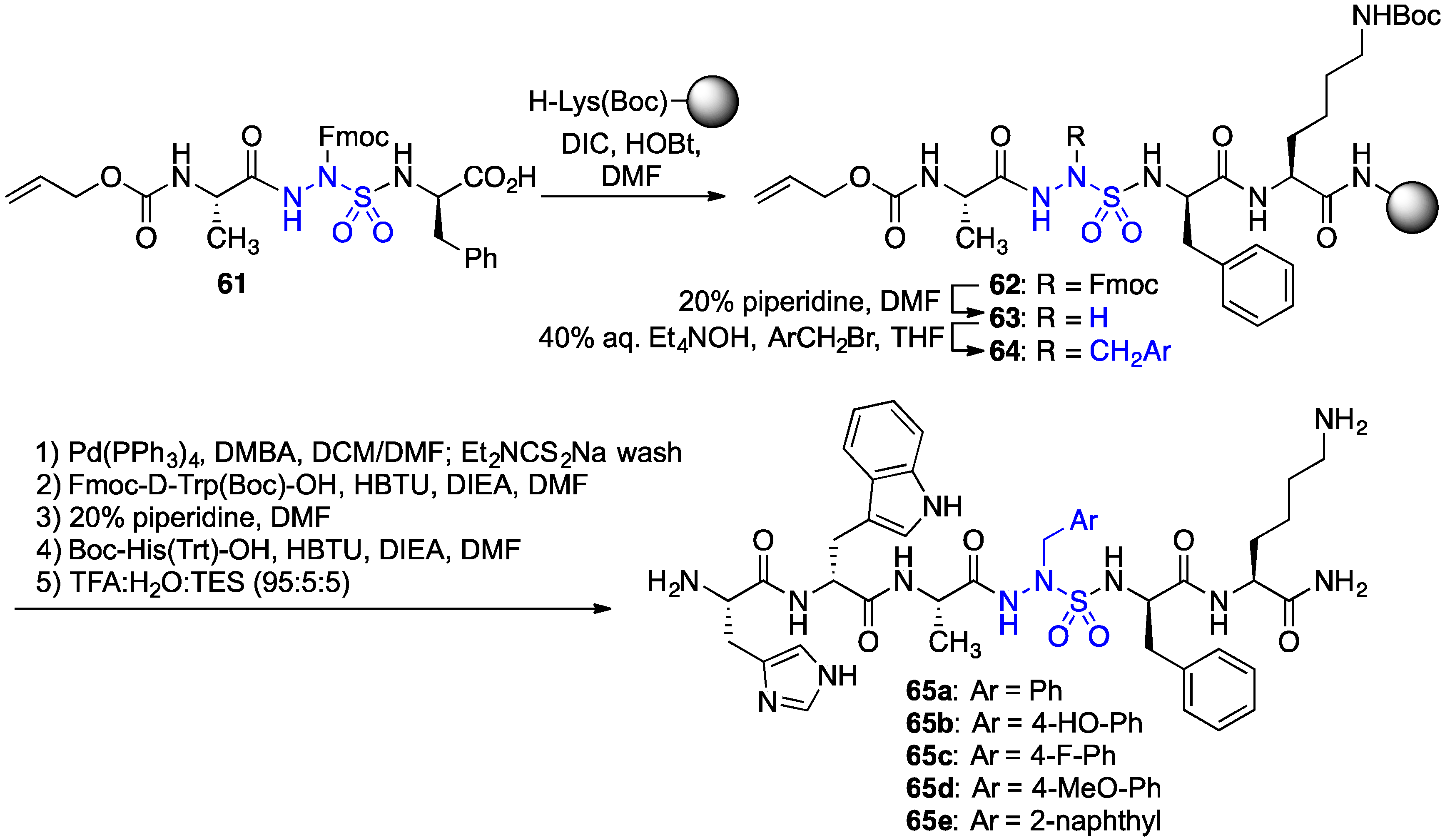
2.11. Exploration of Differences between Semicarbazides and N-Aminosulfamides Using Azasulfurylpeptides
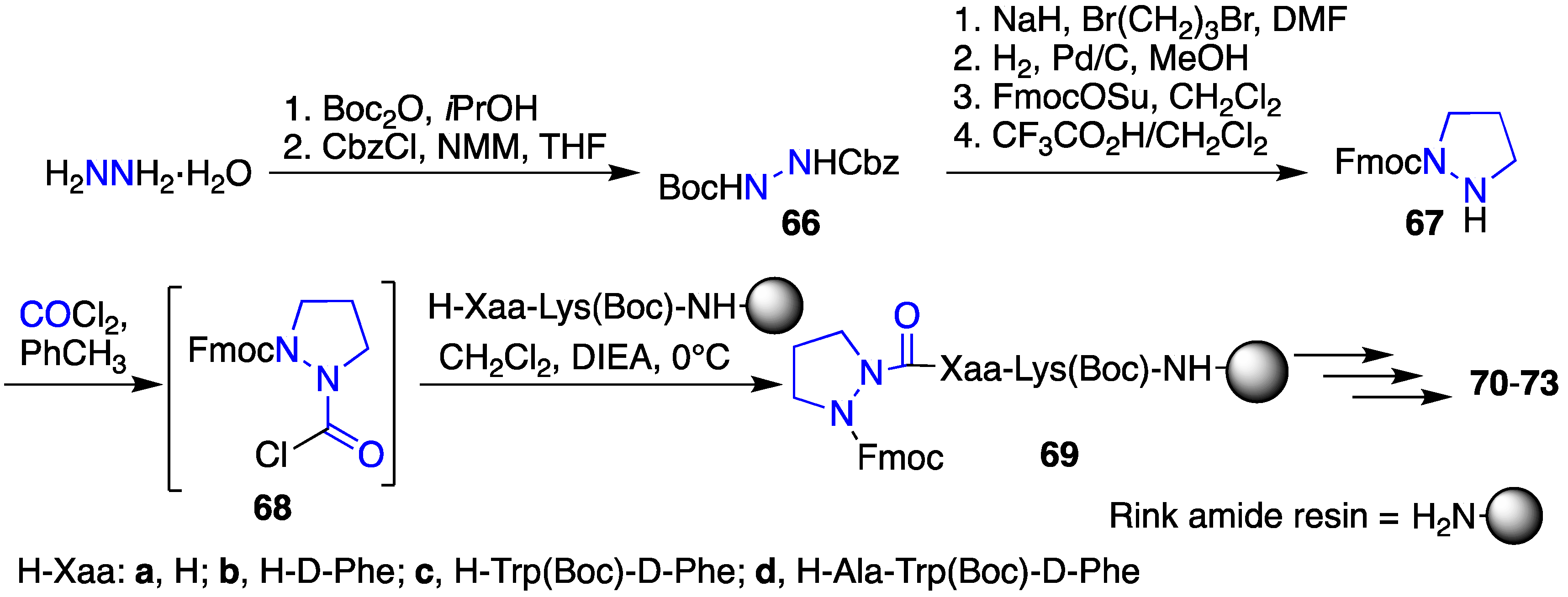
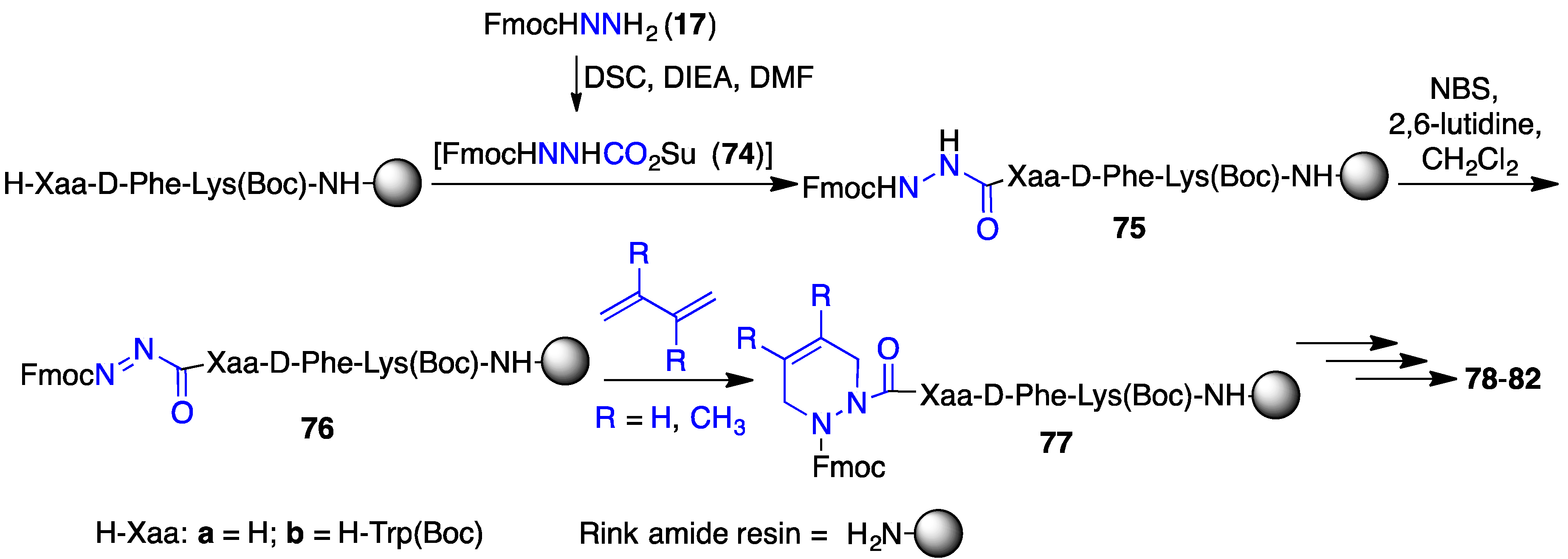
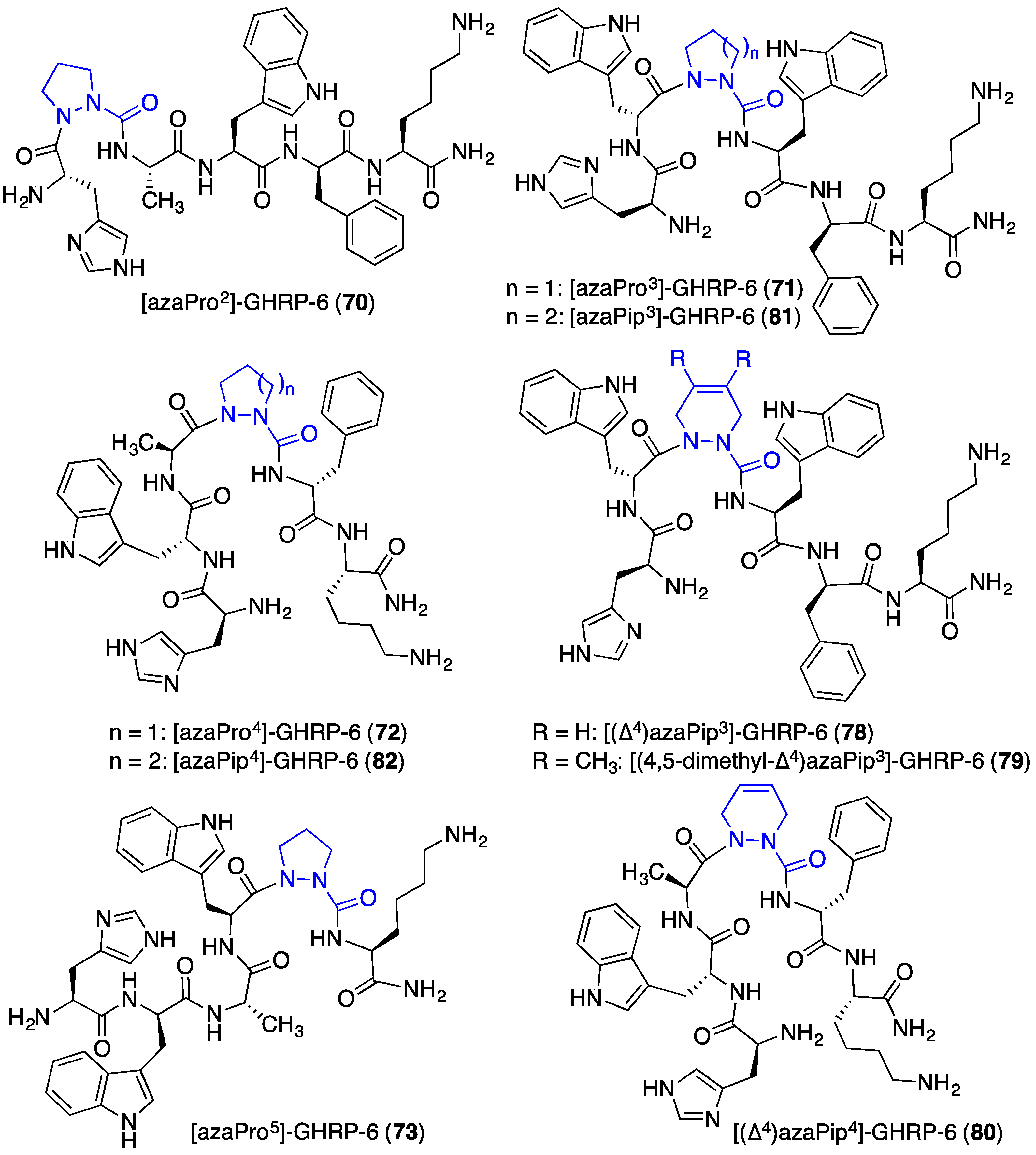
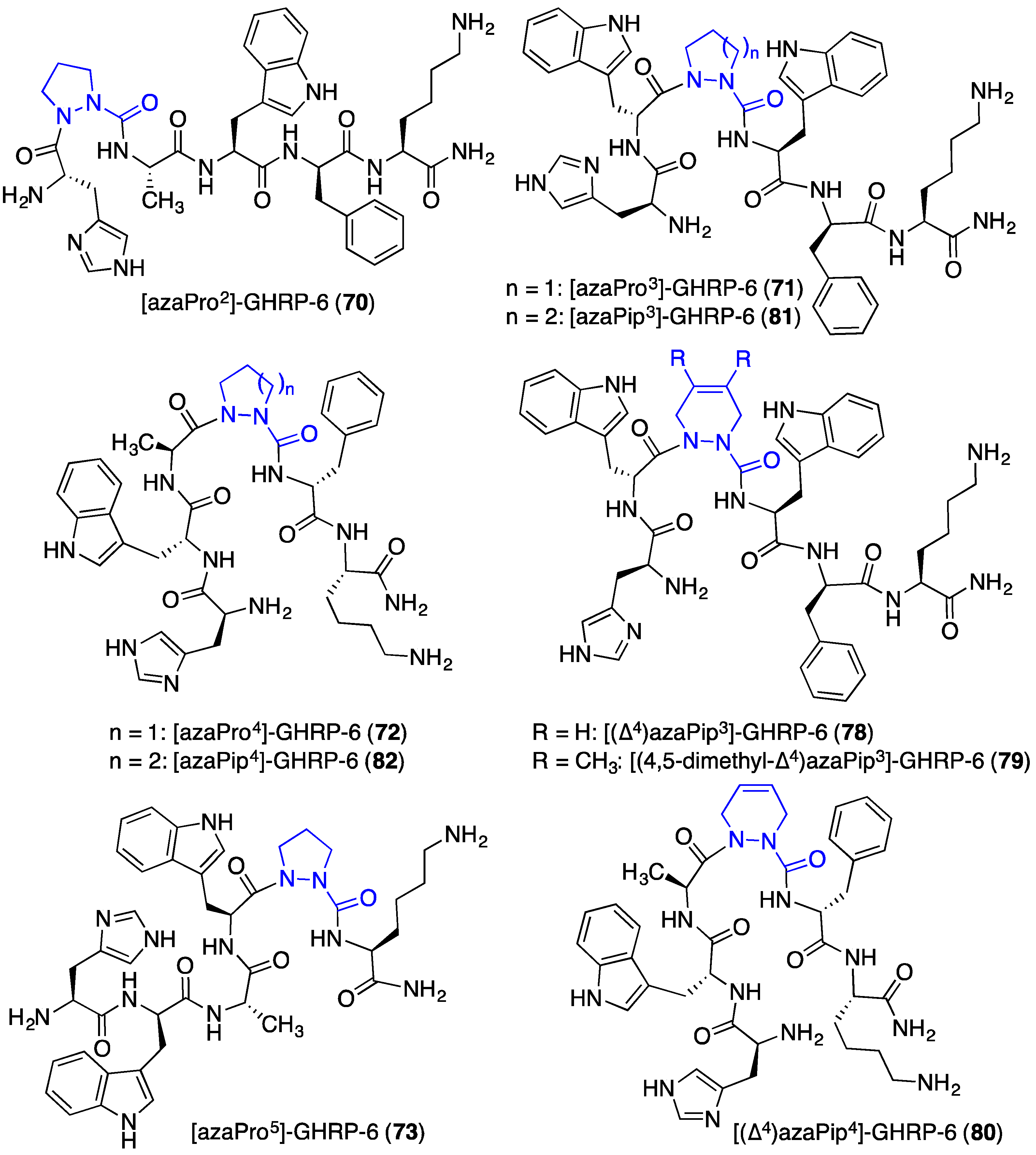
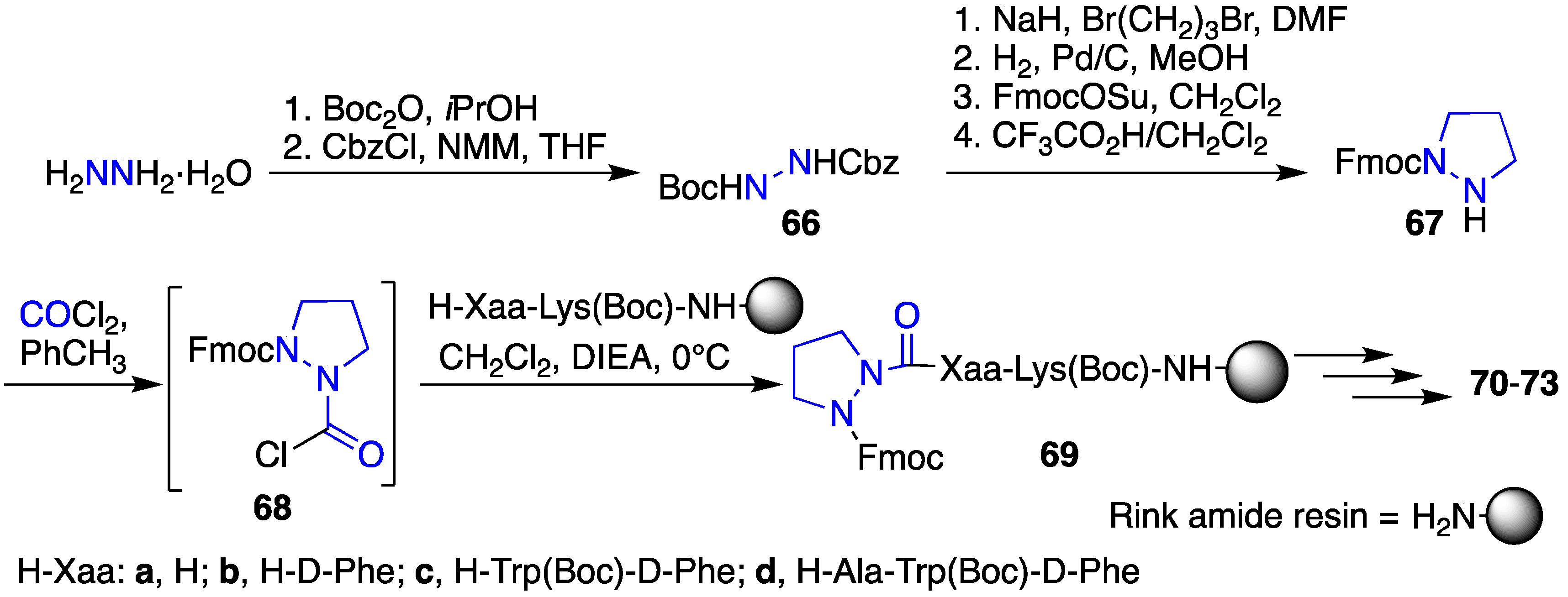
2.12. Probing for Turn Conformations Using Aza-Proline and Aza-Pipecolic Acid Mimics
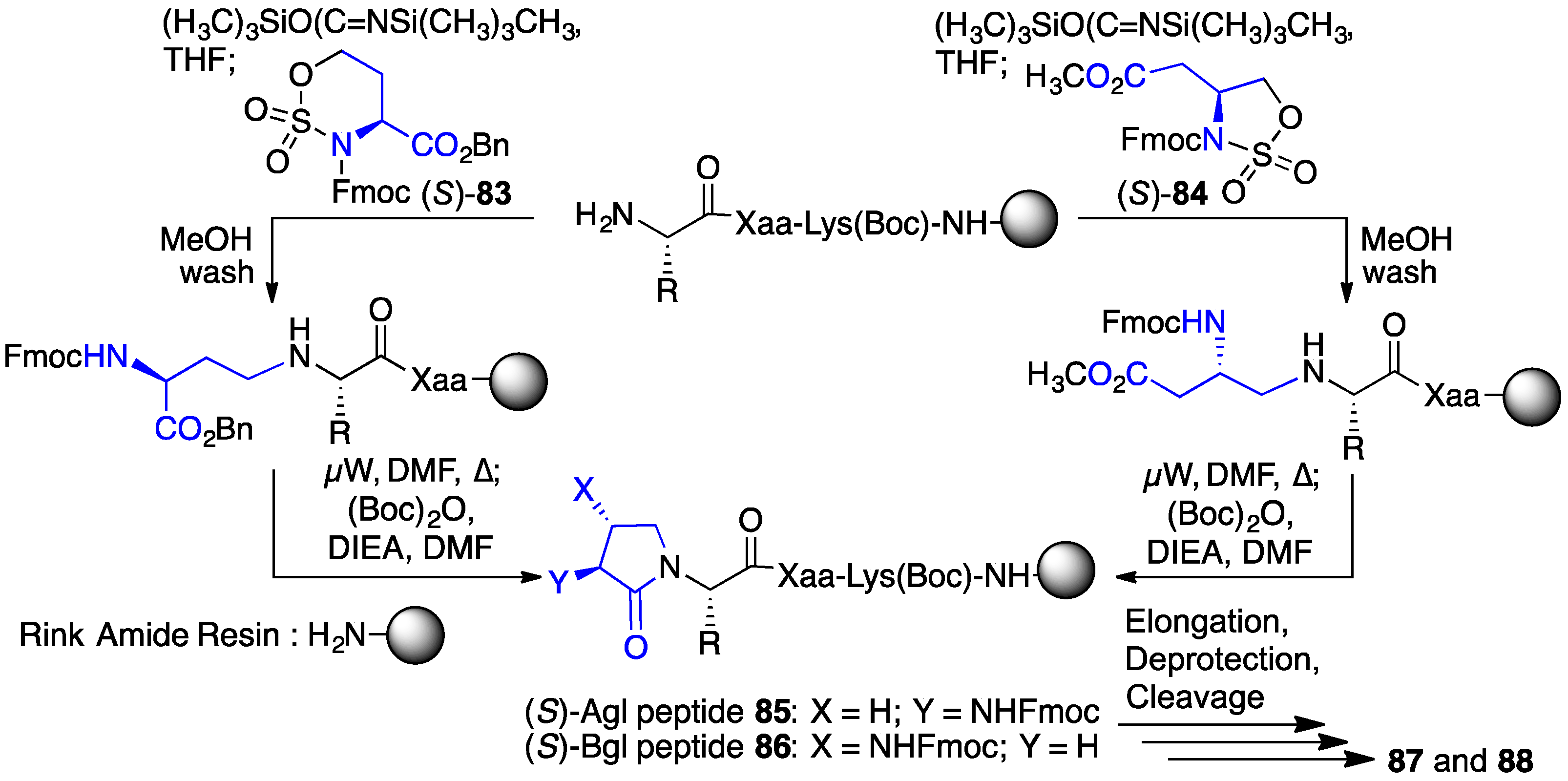
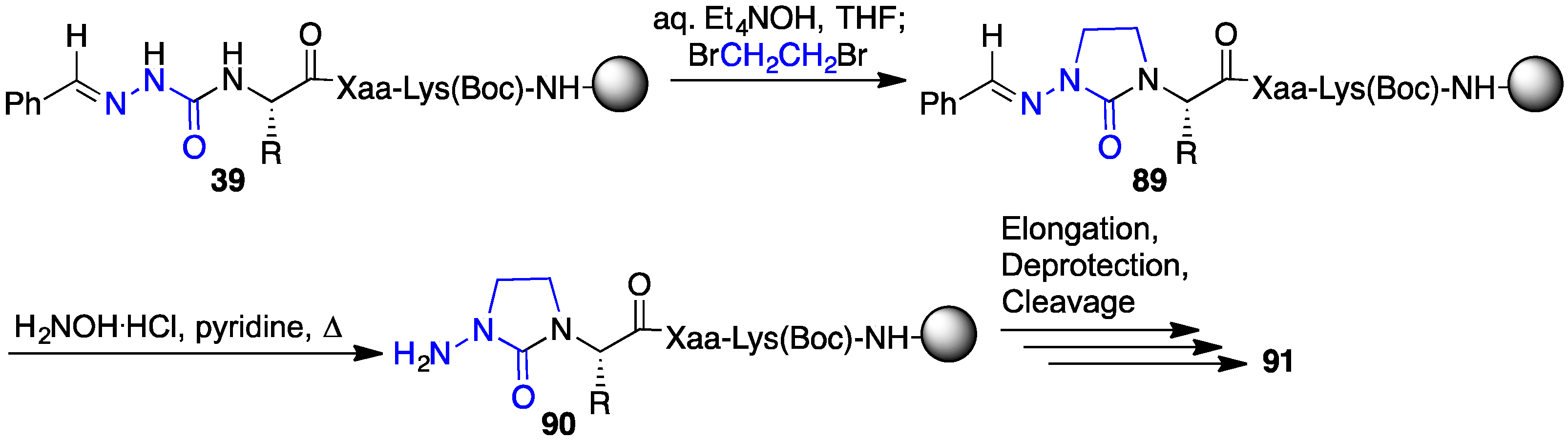
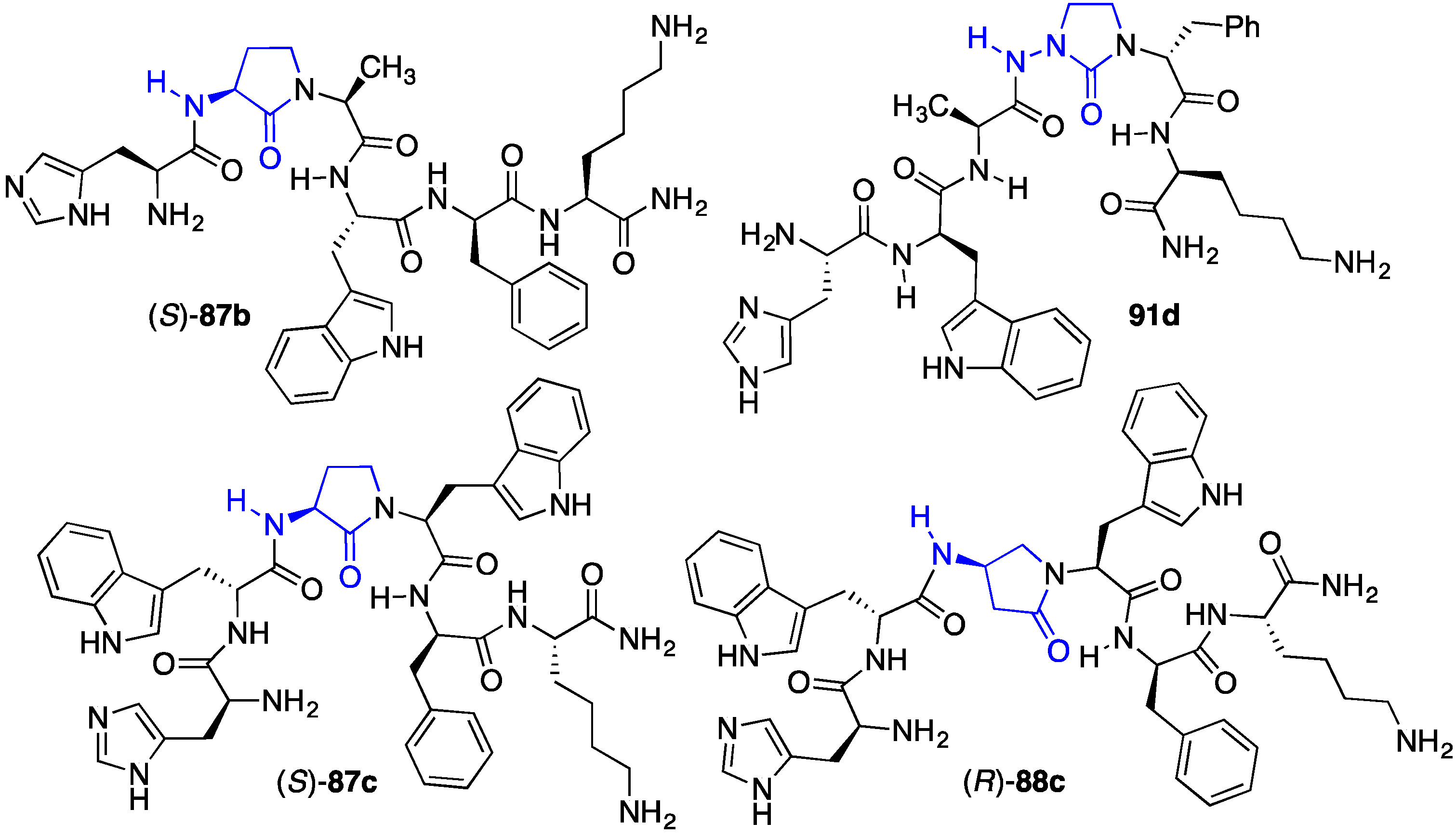
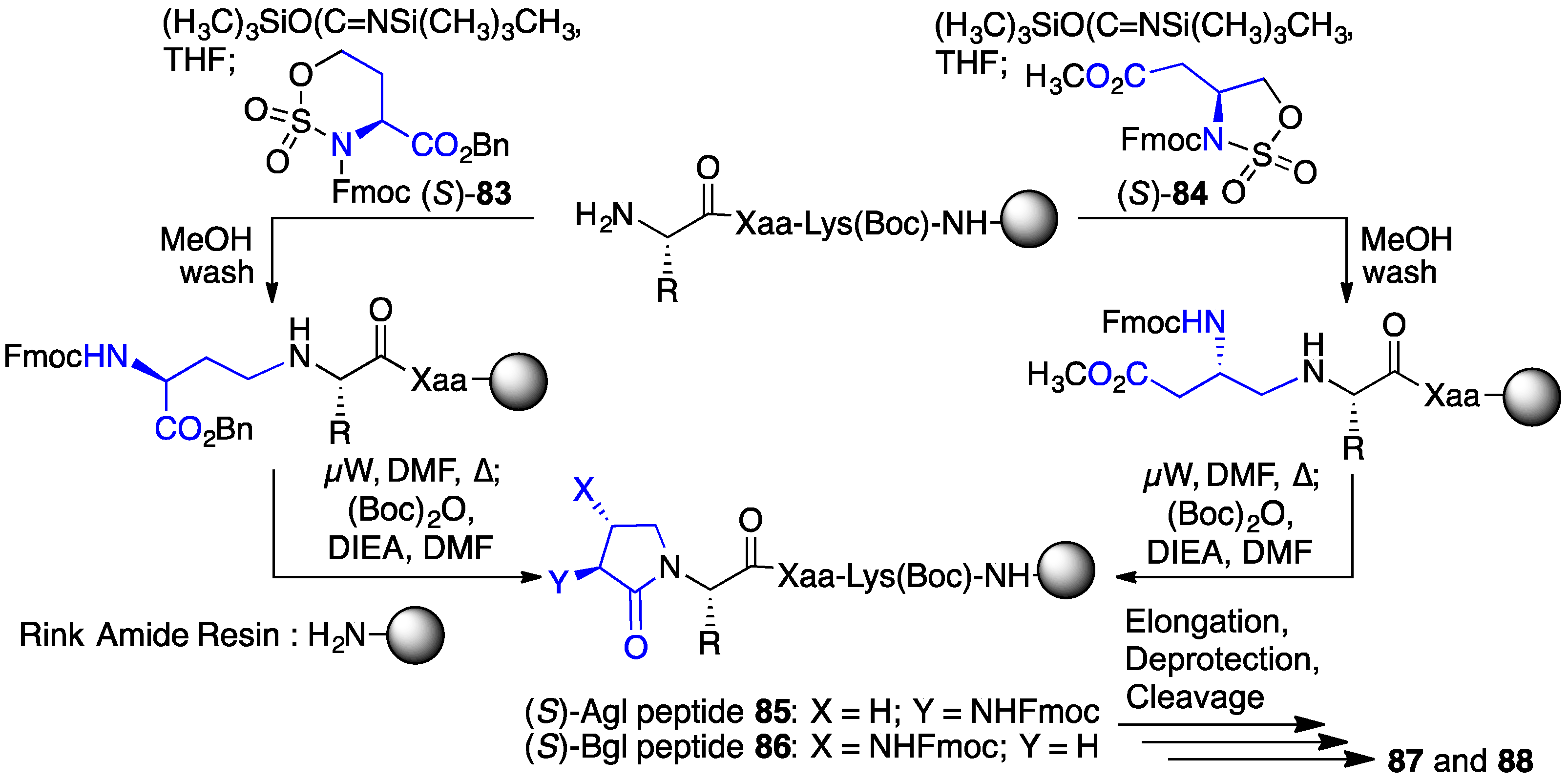
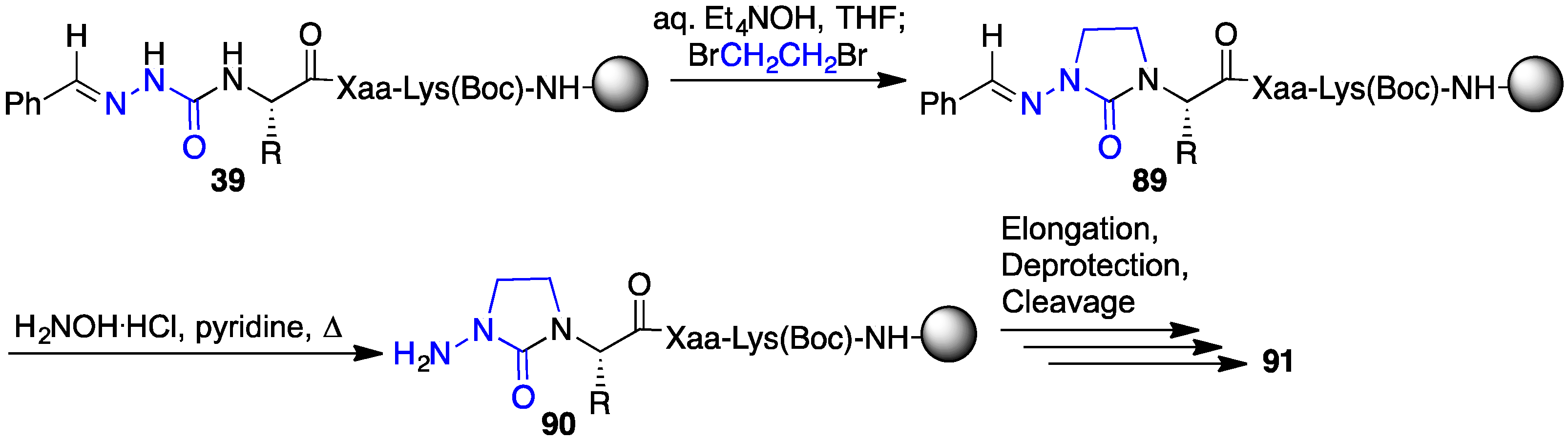
2.13. Probing for Turn Conformations Using Lactam and Aza-Lactam Residues
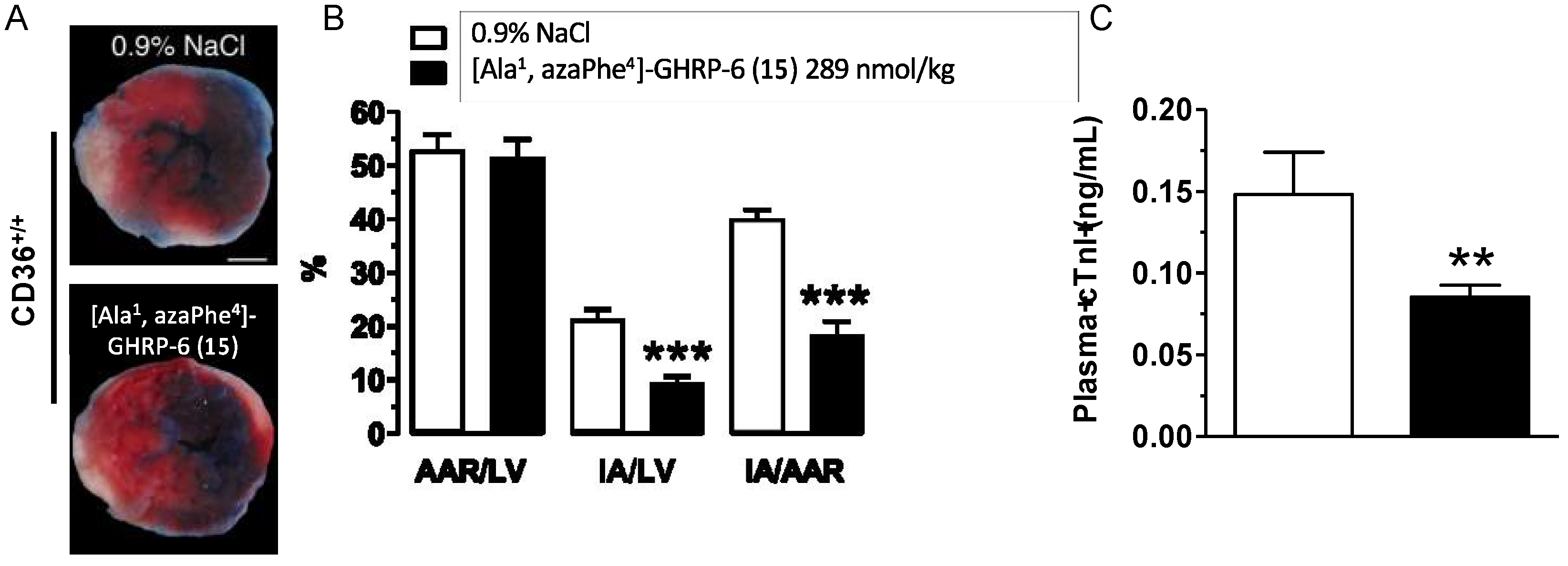
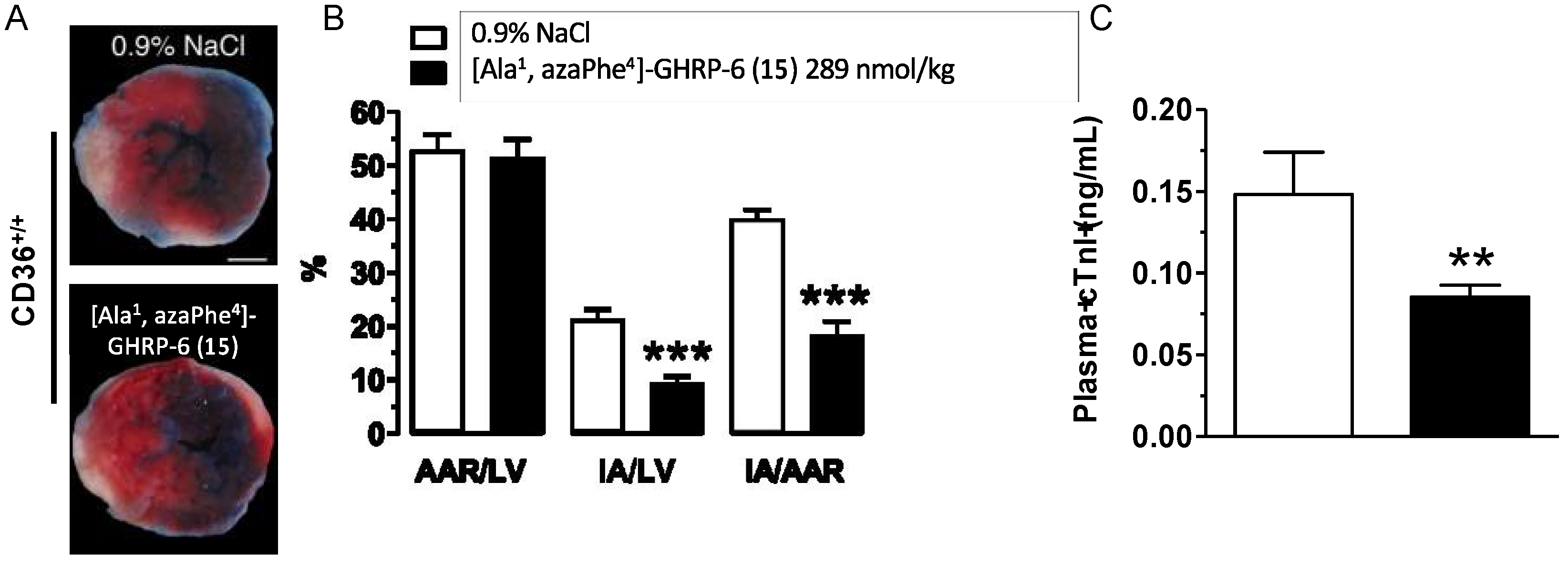
2.14. Cardioprotective Effects of [Ala1, azaPhe4]-GHRP-6 (15)
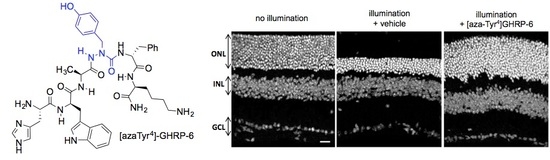
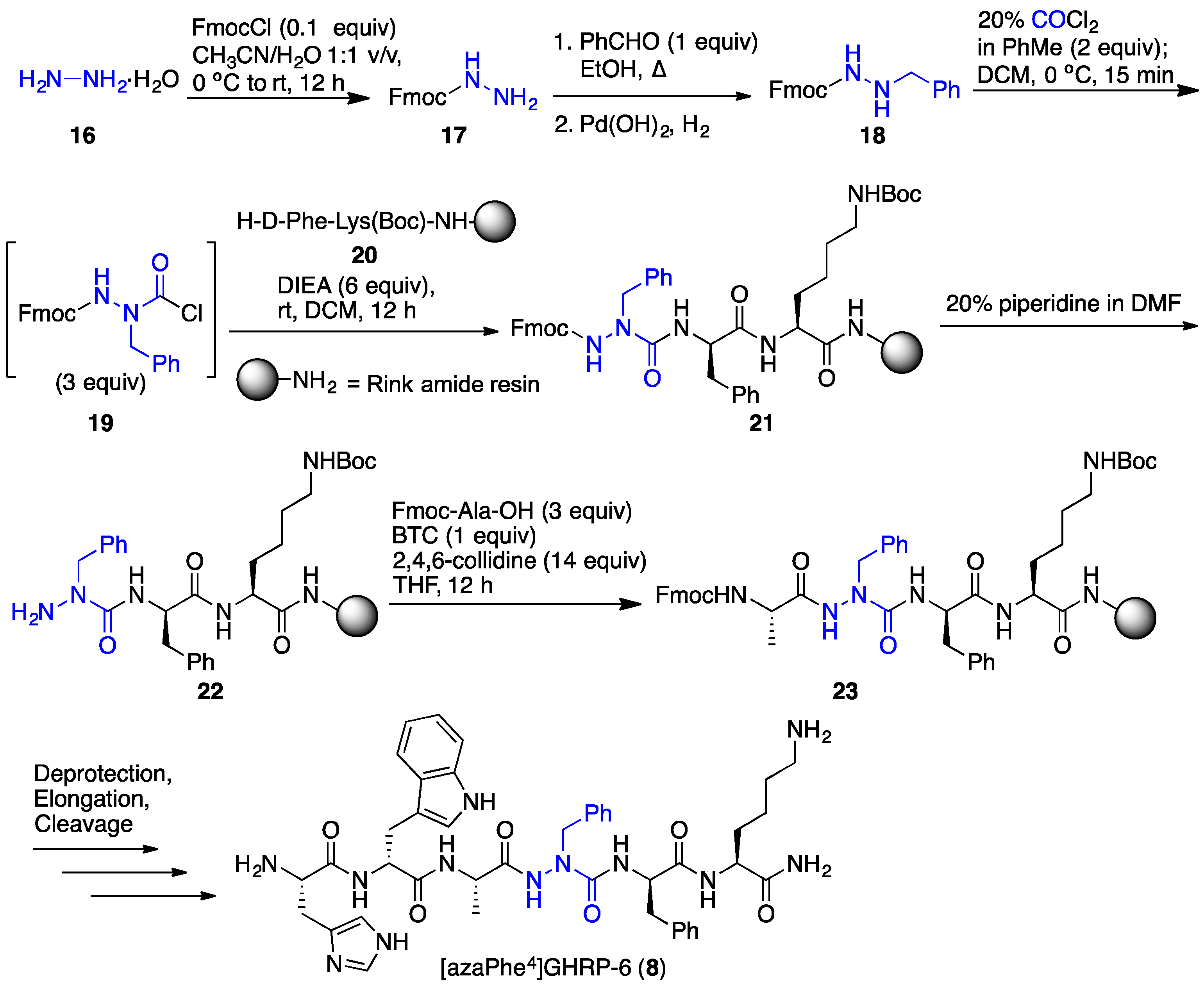
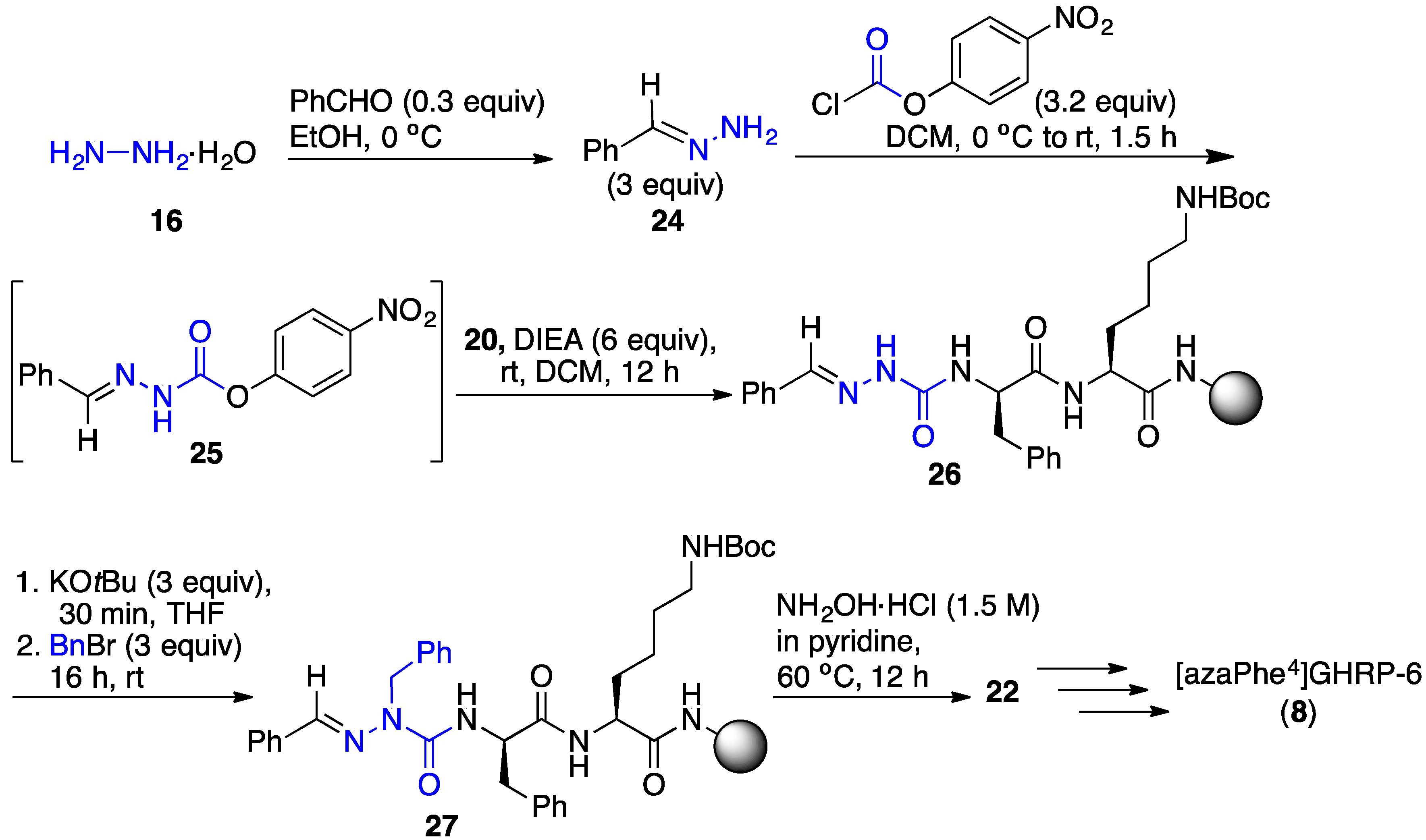
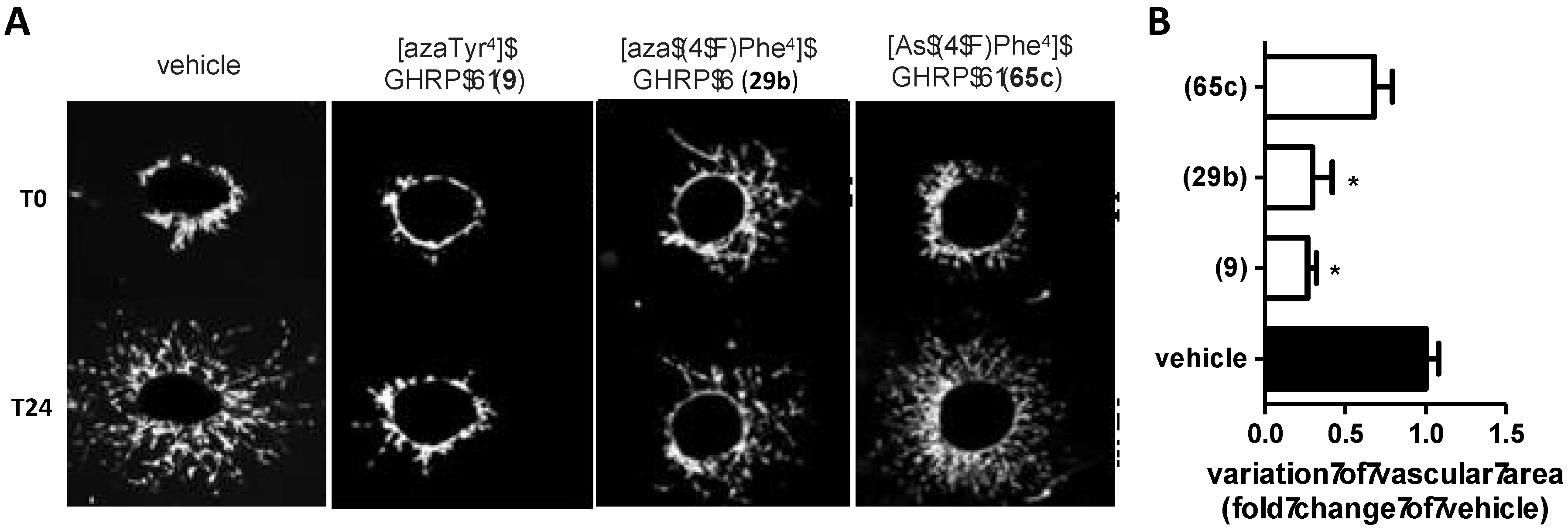
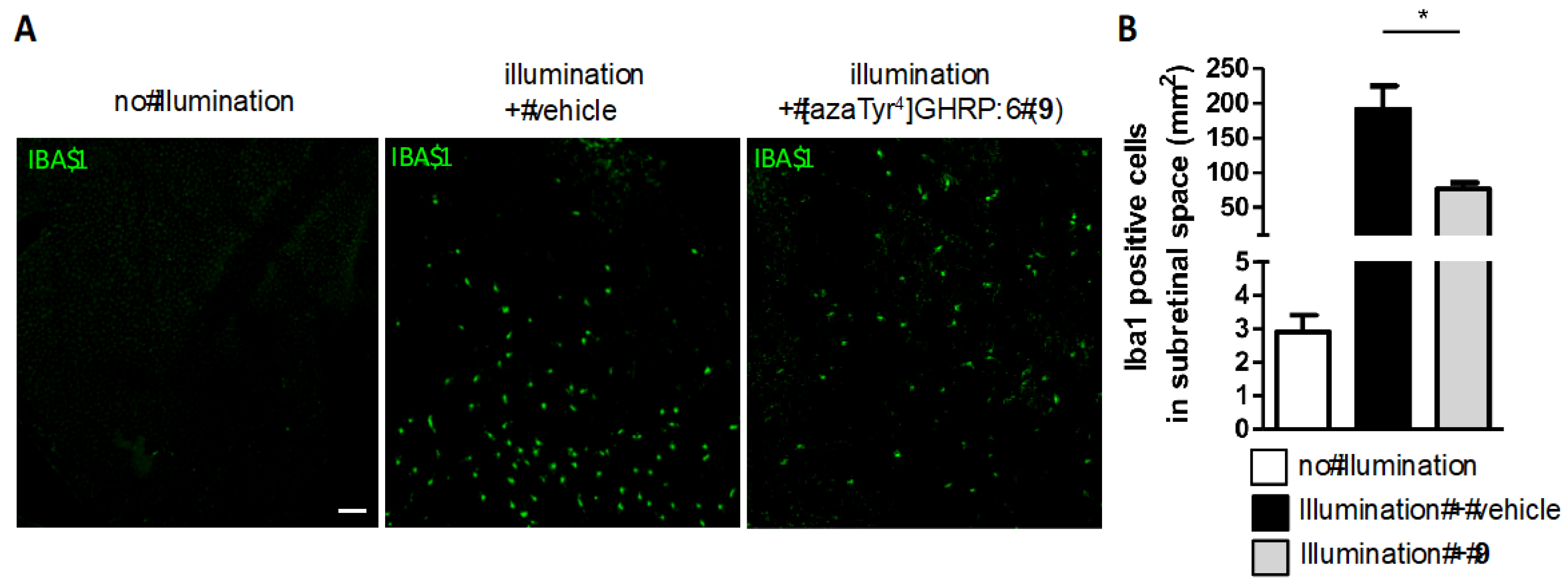
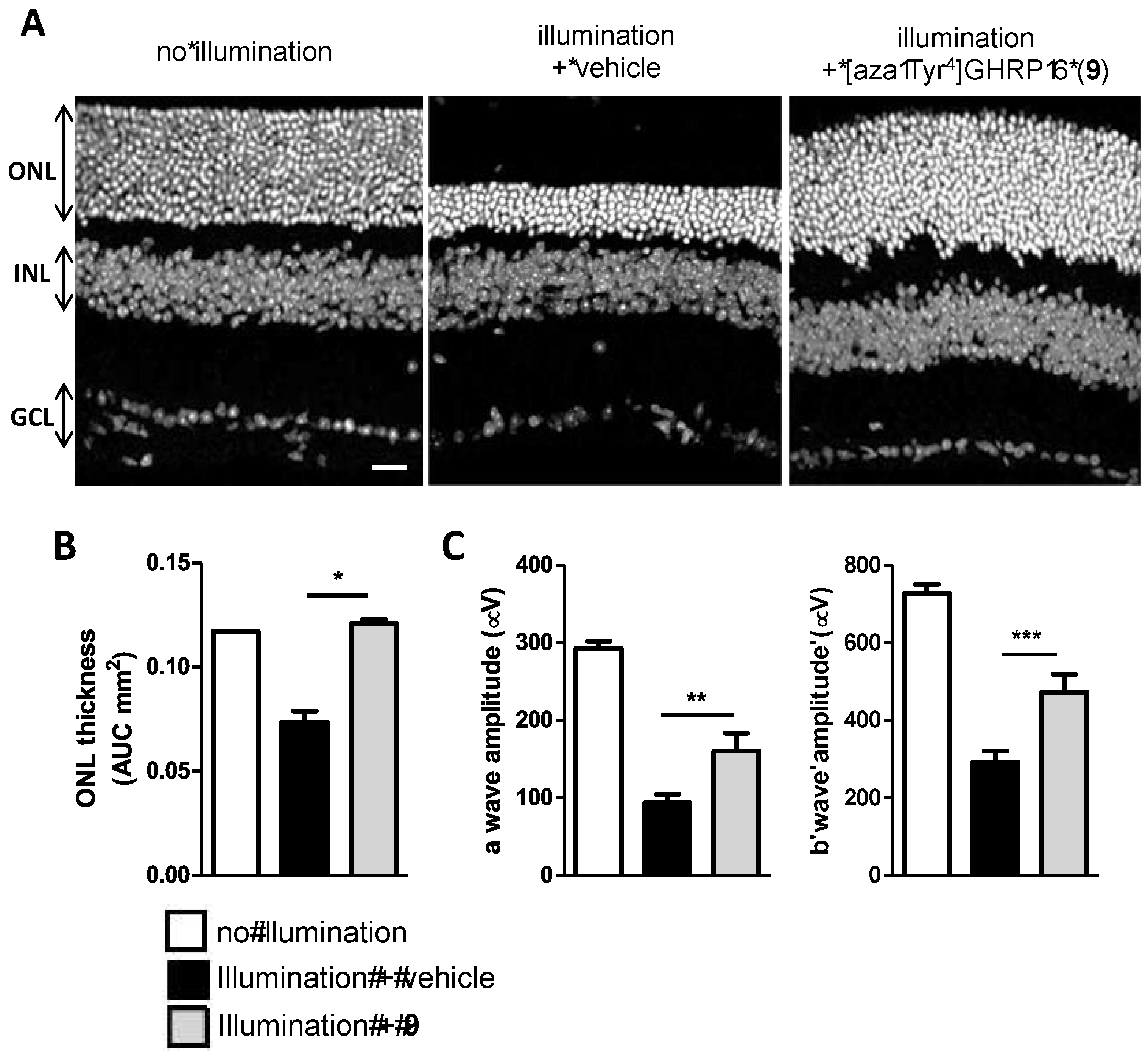
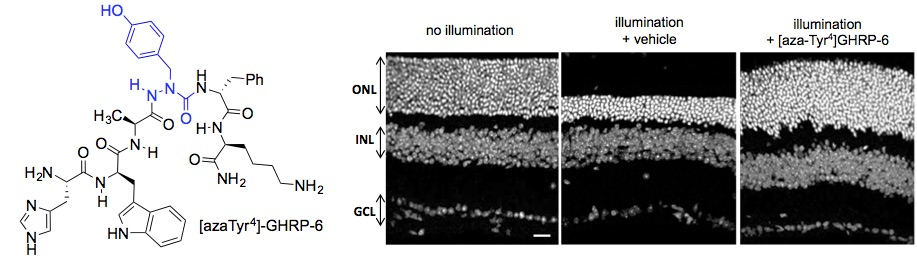
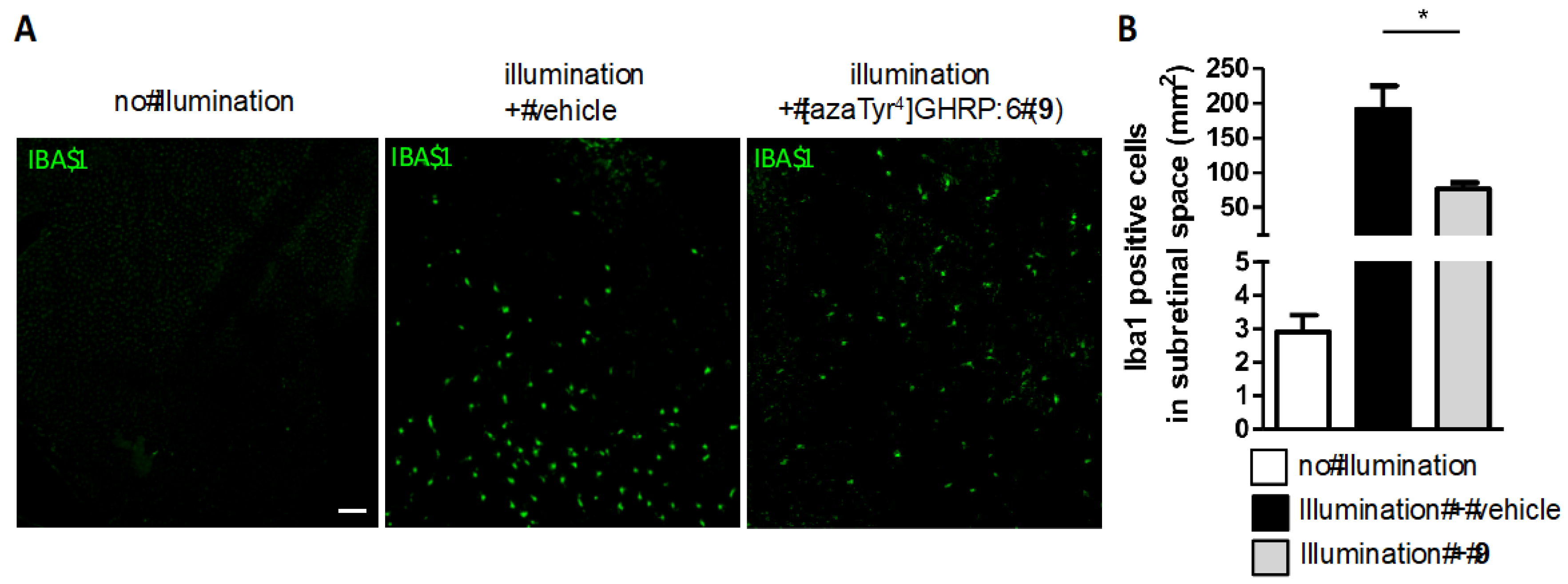
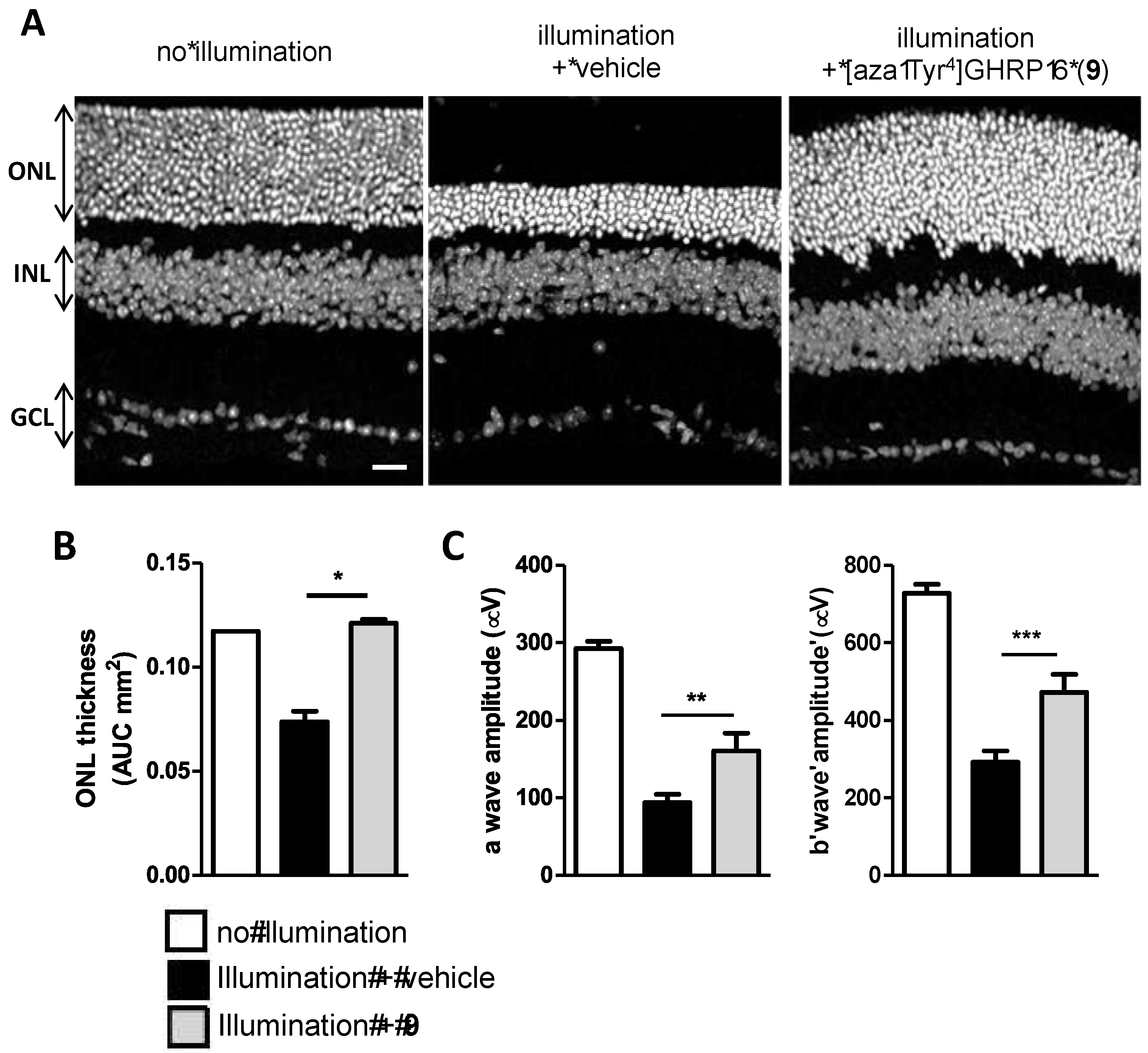
2.15. Mitigation of Subretinal Macrophage-Driven Inflammation Using [azaTyr4]GHRP-6 (9)
2.16. Implications of aza-GHRP-6 Conformation on Activity
3. Conclusions and Future Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Endemann, G.; Stanton, L.W.; Madden, K.S.; Bryant, C.M.; White, R.T.; Protter, A.A. CD36 is a receptor for oxidized low density lipoproteins. J. Biol. Chem. 1993, 268, 11811–11816. [Google Scholar] [PubMed]
- Febbraio, M.; Hajjar, D.P.; Silverstein, R.L.J. CD36: A class B scavenger receptor involved in angiogenesis, athersosclerosis, inflammation and lipid metabolism. Clin. Investig. 2001, 108, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Avallone, R.; Demers, A.; Rodrigue-Way, A.; Bujold, K.; Harb, D.; Anghel, S.; Wahli, W.; Marleau, S.; Ong, H.; Tremblay, A. A growth hormone-releasing peptide that binds scavenger receptor CD36 and ghrelin receptor up-regulates sterol transporters and cholesterol efflux in macrophages through a peroxisome proliferator-activated receptor gamma-dependent pathway. Mol. Endocrinol. 2006, 20, 3165–3178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoosdally, S.J.; Andress, E.J.; Wooding, C.; Martin, C.A.; Linton, K.J. The Human Scavenger Receptor CD36. J. Biol. Chem. 2009, 284, 16277–16288. [Google Scholar] [CrossRef] [Green Version]
- Febbraio, M.; Silverstein, R.L. CD36: Implications in Cardiovascular Disease. Int. J. Biochem. Cell Biol. 2007, 39, 2012–2030. [Google Scholar] [CrossRef] [Green Version]
- Jimenez, B.; Volpert, O.V.; Crawford, S.E.; Febbraio, M.; Silverstein, R.L.; Bouck, N. Signals leading to apoptosis-dependent inhibition of neovascularization by thrombospondin-1. Nat. Med. 2000, 6, 41–48. [Google Scholar] [CrossRef]
- Sun, J.; Hopkins, B.D.; Tsujikawa, K.; Perruzi, C.; Adini, I.; Swerlick, R.; Bornstein, P.; Lawler, J.; Benjamin, L.E. Thrombospondin-1 modulates VEGF-A mediated Akt signaling and capillary survival in the developing retina. Am. J. Physiol. 2009, 296, H1344–H1351. [Google Scholar] [CrossRef] [Green Version]
- Nagendran, J.; Pulinilkunnil, T.; Kienesberger, P.C.; Sung, M.M.; Fung, D.; Febbraio, M.; Dyck, J.R. Cardiomyocyte-specific ablation of CD36 improves post-ischemic functional recovery. J. Mol. Cell. Cardiol. 2013, 63, 180–188. [Google Scholar] [CrossRef]
- Bessi, V.L.; Labbe, S.M.; Huynh, D.N.; Menard, L.; Jossart, C.; Febbraio, M.; Guerin, B.; Bentourkia, M.; Lecomte, R.; Carpentier, A.C.; et al. EP 80317, a selective CD36 ligand, shows cardioprotective effects against post-ischaemic myocardial damage in mice. Cardiovasc. Res. 2012, 96, 99–108. [Google Scholar] [CrossRef] [Green Version]
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.; Jaswal, J.S.; Stanley, W.C. Myocardial fatty acid metabolism in health and disease. Physiol. Rev. 2010, 90, 207–258. [Google Scholar] [CrossRef]
- Hajri, T.; Hall, A.M.; Jensen, D.R.; Pietka, T.A.; Drover, V.A.; Tao, H.; Eckel, R.; Abumrad, N.A. CD36-facilitated fatty acid uptake inhibits leptin production and signaling in adipose tissue. Diabetes 2007, 56, 1872–1880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, L.; Kinney, B.; Schaack, J.; Shao, J. Adiponectin inhibits lipolysis in mouse adipocytes. Diabetes 2011, 60, 1519–1527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silverstein, R.L.; Febbraio, M. CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci. Signal. 2009, 2, re3. [Google Scholar] [CrossRef] [Green Version]
- Reis, A.; Mateus, C.; Melo, P.; Figueira, J.; Cunha-Vaz, J.; Castelo-Branco, M. Neuroretinal dysfunction with intact blood-retinal barrier and absent vasculopathy in type 1 diabetes. Diabetes 2014, 63, 3926–3937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, T.; Shimamura, M.; Jackman, K.; Kurinami, H.; Anrather, J.; Zhou, P.; Iadecola, C. Key role of CD36 in Toll-like receptor 2 signaling in cerebral ischemia. Stroke 2010, 41, 898–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beutler, B.; Jiang, Z.; Georgel, P.; Crozat, K.; Croker, B.; Rutschmann, S.; Du, X.; Hoebe, K. Genetic analysis of host resistance: Toll-like receptor signaling and immunity at large. Annu. Rev. Immunol. 2006, 24, 353–389. [Google Scholar] [CrossRef] [Green Version]
- Shibata, R.; Sato, K.; Pimentel, D.R.; Takemura, Y.; Kihara, S.; Ohashi, K.; Funahashi, T.; Ouchi, N.; Walsh, K. Adiponectin protects against myocardial ischemia-reperfusion injury through AMPK- and COX-2-dependent mechanisms. Nat. Med. 2005, 11, 1096–1103. [Google Scholar] [CrossRef]
- Proulx, C.; Picard, E.; Boeglin, D.; Pohankova, P.; Chemtob, S.; Ong, H.; Lubell, W.D. Azapeptide analogues of the growth hormone releasing peptide-6 as cluster of differentiation 36 receptor ligands with reduced affinity for the growth hormone secretagogue receptor 1a. J. Med. Chem. 2012, 55, 6502–6511. [Google Scholar] [CrossRef]
- Demers, A.; McNicoll, N.; Febbraio, M.; Servant, M.; Marleau, S.; Silverstein, R.; Ong, H. Identification of the growth hormone releasing peptide binding site in CD36: A photoaffinity cross-linking study. Biochem. J. 2004, 382, 417–424. [Google Scholar] [CrossRef] [Green Version]
- Kar, N.S.; Ashraf, M.Z.; Valiyaveettil, M.; Podrez, E.A. Mapping and characterization of the binding site for specific oxidized phospholipids and oxidized low density lipoproteins of scavenger receptor CD36. J. Biol. Chem. 2008, 283, 8765–8771. [Google Scholar] [CrossRef] [Green Version]
- Sabatino, D.; Proulx, C.; Pohankova, P.; Ong, H.; Lubell, W.D. Structure-activity relationships of GHRP-6 azapeptide ligands of the CD36 scavenger receptor by solid phase submonomer azapeptide synthesis. J. Am. Chem. Soc. 2011, 133, 12493–12506. [Google Scholar] [CrossRef] [PubMed]
- Bolduc, O.R.; Lambert-Lanteigne, P.; Colin, D.Y.; Zhao, S.S.; Proulx, C.; Boeglin, D.; Lubell, W.D.; Pelletier, J.N.; Féthière, J.; Ong, H.; et al. Modified peptide monolayer binding His-tagged biomolecules for small ligand screening with SPR biosensors. Analyst 2011, 136, 3142–3148. [Google Scholar] [CrossRef] [PubMed]
- Lubell, W.D.; Beauregard, K.S.; Polyak, F. Peptides and chirality effects on the conformation and the synthesis of medicinally relevant peptides. In Comprehensive Chirality; Carreira, E.M., Yamamoto, H., Eds.; Elsevier: Amsterdam, The Netherlands, 2014; pp. 86–104. [Google Scholar]
- Proulx, C.; Sabatino, D.; Hopewell, R.; Spiegel, J.; Ramos, Y.G.; Lubell, W.D. Azapeptides and their therapeutic potential. Future Med. Chem. 2011, 3, 1139–1164. [Google Scholar] [CrossRef] [PubMed]
- Boeglin, D.; Lubell, W.D. Aza-amino acid scanning of secondary structure suited for solid-phase peptide synthesis with Fmoc chemistry and aza-amino acids with heteroatomic side chains. J. Comb. Chem. 2005, 7, 864–878. [Google Scholar] [CrossRef]
- Weinberger, H.; Lichte, E.; Griesinger, C.; Kutscher, B. Small peptide libraries: Combinatorial split-mix synthesis followed by combinatorial amino acid analysis of selected variants. Arch. Pharm. 1997, 330, 109–111. [Google Scholar] [CrossRef] [PubMed]
- Sabatino, D.; Proulx, C.; Bourguet, C.; Klocek, S.; Boeglin, D.; Ong, H.; Lubell, W.D. Exploring side chain diversity by submonomer solid phase azapeptide synthesis. Org. Lett. 2009, 11, 3650–3653. [Google Scholar] [CrossRef]
- Chingle, R.; Proulx, C.; Lubell, W.D. Azapeptide Synthesis Methods for Expanding Side-Chain Diversity for Biomedical Applications. Acc. Chem. Res. 2017, 50, 1541–1556. [Google Scholar] [CrossRef]
- Possi, K.C.; Mulumba, M.; Omri, S.; Garcia-Ramos, Y.; Tahiri, H.; Chemtob, S.; Ong, H.; Lubell, W.D. Influences of Histidine-1 and Azaphenylalanine-4 on the Affinity, Anti-inflammatory, and Antiangiogenic Activities of Azapeptide Cluster of Differentiation 36 Receptor Modulators. J. Med. Chem. 2017, 60, 9263–9274. [Google Scholar] [CrossRef]
- Möller, M.N.; Rios, N.; Trujillo, M.; Radi, R.; Denicola, A.; Alvarez, B. Detection and quantification of nitric oxide-derived oxidants in biological systems. J. Biol. Chem. 2019, 294, 14776–14802. [Google Scholar] [CrossRef] [Green Version]
- Proulx, C.; Lubell, W.D. Copper-catalyzed N-arylation of semicarbazones for the synthesis of aza-arylglycine-containing aza-peptides. Org. Lett. 2010, 12, 2916–2919. [Google Scholar] [CrossRef]
- Proulx, C.; Lubell, W.D. Aza-1,2,3-triazole-3-alanine synthesis via copper-catalyzed 1,3-dipolar cycloaddition on aza-progargylglycine. J. Org. Chem. 2010, 75, 5385–5387. [Google Scholar] [CrossRef] [PubMed]
- Traoré, M.; Doan, N.-D.; Lubell, W.D. Diversity-Oriented Synthesis of Azapeptides with Basic Amino Acid Residues: Aza-Lysine, Aza-Ornithine and Aza-Arginine. Org. Lett. 2014, 16, 3588–3591. [Google Scholar] [CrossRef] [PubMed]
- Doan, N.; Zhang, J.; Traoré, M.; Kamdem, W.; Lubell, W.D. Solid-phase synthesis of C-terminal azapeptides. J. Pept. Sci. 2015, 21, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Proulx, C.; Tomberg, A.; Lubell, W.D. Multicomponent diversity-oriented synthesis of aza-lysine-peptide mimics. Org. Lett. 2014, 16, 298–301. [Google Scholar] [CrossRef]
- Wei, C.; Li, C.-J. Enantioselective Direct-Addition of Terminal Alkynes to Imines Catalyzed by Copper(I)pybox Complex in Water and in Toluene. J. Am. Chem. Soc. 2002, 124, 5638–5639. [Google Scholar] [CrossRef]
- Zhang, J.; Mulumba, M.; Ong, H.; Lubell, W.D. Diversity-Oriented Synthesis of Cyclic Azapeptides by A3-Macrocyclization Provides High-Affinity CD36-Modulating Peptidomimetics. Angew. Chem. Int. Ed. 2017, 56, 6284–6288. [Google Scholar] [CrossRef]
- Ahsanullah; Chingle, R.; Ohm, R.G.; Chauhan, P.S.; Lubell, W.D. Aza-propargylglycine installation by aza-amino acylation: Synthesis and Ala-scan of an azacyclopeptide CD36 modulator. Pept. Sci. 2019, 111, e24102. [Google Scholar]
- Danelius, E.; Ohm, R.G.; Ahsanullah, A.; Mulumba, M.; Ong, H.; Chemtob, S.; Erdelyi, M.; Lubell, W.D. Dynamic chirality in the mechanism of action of allosteric CD36 modulators of macrophage driven inflammation. J. Med. Chem. 2019, 62, 11071–11079. [Google Scholar] [CrossRef]
- Vinogradov, A.A.; Yin, Y.; Suga, H. Macrocyclic Peptides as Drug Candidates: Recent Progress and Remaining Challenges. J. Am. Chem. Soc. 2019, 141, 4167–4181. [Google Scholar] [CrossRef]
- Turcotte, S.; Lubell, W.D. Crystal structure analyses of azasulfuryltripeptides reveal potential for γ-turn mimicry. Pept. Sci. 2015, 104, 622–628. [Google Scholar] [CrossRef]
- Turcotte, S.; Mellal, K.; Chingle, R.; Mulumba, M.; Omri, S.; Dif-Yaiche, L.; Chemtob, S.; Ong, H.; Lubell, W.D. Azasulfurylpeptide Modulation of CD-36 Mediated Inflammation Without Effect on Neovascularization. Biomedicines 2018, 6, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chingle, R.; Mulumba, M.; Chung, N.N.; Nguyen, T.M.; Ong, H.; Ballet, S.; Schiller, P.W.; Lubell, W.D. Solid-Phase Azopeptide Diels–Alder Chemistry for Aza-pipecolyl Residue Synthesis to Study Peptide Conformation. J. Org. Chem. 2019, 84, 6006–6016. [Google Scholar] [CrossRef] [PubMed]
- Che, Y.; Marshall, G.R. Engineering cyclic tetrapeptides containing chimeric amino acids as preferred reverse-turn scaffolds. J. Med. Chem. 2006, 49, 111–124. [Google Scholar] [CrossRef] [PubMed]
- Che, Y.; Marshall, G.R. Impact of azaproline on peptide conformation. J. Org. Chem. 2004, 69, 9030–9042. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.-J.; Berglund, A.; Kao, J.L.-F.; Couty, J.-P.; Gershengorn, M.C.; Marshall, G.R. Impact of azaproline on amide cis−trans isomerism: Conformational analyses and NMR studies of model peptides including TRH analogues. J. Am. Chem. Soc. 2003, 125, 1221–1235. [Google Scholar] [CrossRef] [PubMed]
- Hemmerlin, C.; Cung, M.T.; Boussard, G. Synthesis and conformational preferences in solution and crystalline states of an aza-tripeptide. Tetrahedron Lett. 2001, 42, 5009–5012. [Google Scholar] [CrossRef]
- Didierjean, C.; Aubry, A.; Wyckaert, F.; Boussard, G. Structural features of the Pip/AzPip couple in the crystalline state: Influence of the relative AzPip location in an aza dipeptide sequence upon the induced chirality and conformational characteristics. J. Pept. Res. 2000, 55, 308–317. [Google Scholar] [CrossRef]
- Lecoq, A.; Boussard, G.; Marraud, M.; Aubry, A. Crystal state conformation of three azapeptides containing the azaproline residue, a β-turn regulator. Biopolymers 1993, 33, 1051–1059. [Google Scholar] [CrossRef]
- Lecoq, A.; Boussard, G.; Marraud, M.; Aubry, A. The couple Pro/AzaPro: A means of β-turn formation control synthesis and conformation of two azapro-containing dipeptides. Tetrahedron Lett. 1992, 33, 5209–5212. [Google Scholar] [CrossRef]
- Zouikri, M.; Vicherat, A.; Aubry, A.; Marraud, M.; Boussard, G. Azaproline as a β-turn-inducer residue opposed to proline. J. Pept. Res. 1998, 52, 19–26. [Google Scholar] [CrossRef]
- Proulx, C.; Lubell, W.D. Solid-Phase Synthesis of Aza-Proline Analogues of GHRP-6. In Peptides: Breaking Away, Proceedings of the 21st American Peptide Symposium, Bloomington, IN, USA, 7–12 June 2009; Lebl, M., Ed.; Prompt Scientific Publishing: San Diego, CA, USA, 2009; pp. 56–57. [Google Scholar]
- Freidinger, R.M.; Veber, D.F.; Perlow, D.S.; Brooks, J.R.; Saperstein, R. Bioactive conformation of luteinizing hormone-releasing hormone: Evidence from a conformationally constrained analog. Science 1980, 210, 656–658. [Google Scholar] [CrossRef] [PubMed]
- Freidinger, R.M. Design and synthesis of novel bioactive peptides and peptidomimetics. J. Med. Chem. 2003, 46, 5553–5566. [Google Scholar] [CrossRef] [PubMed]
- St-Cyr, D.J.; García-Ramos, Y.; Doan, N.D.; Lubell, W.D. Aminolactam, N-Aminoimidazolone, and N-Aminoimdazolidinone Peptide Mimics. In Peptidomimetics I; Springer: Cham, Switzerland, 2017; pp. 125–175. [Google Scholar]
- Boutard, N.; Jamieson, A.G.; Ong, H.; Lubell, W.D. Structure-Activity Analysis of the Growth Hormone Secretagogue GHRP-6 by α− and β−Amino γ−Lactam Positional Scanning. Chem. Biol. Drug Des. 2010, 75, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Doan, N.-D.; Hopewell, R.; Lubell, W.D. N-Aminoimidazolidin-2-one Peptidomimetics. Org. Lett. 2014, 16, 2232–2235. [Google Scholar] [CrossRef] [PubMed]
- Doan, N.-D.; Lubell, W.D. X-ray Structure Analysis Reveals β-Turn Mimicry by N-amino-imidazolidin-2-ones. Biopolymers 2015, 104, 629–635. [Google Scholar] [CrossRef]
- Picard, E.; Houssier, M.; Bujold, K.; Sapieha, P.; Lubell, W.; Dorfman, A.; Racine, J.; Hardy, P.; Febbraio, M.; Lachapelle, P.; et al. CD36 plays an important role in the clearance of oxLDL and associated age-dependent sub-retinal deposits. Aging (Albany NY) 2010, 2, 981–989. [Google Scholar] [CrossRef] [Green Version]
- Huynh, D.N.; Bessi, V.L.; Ménard, L.; Piquereau, J.; Proulx, C.; Febbraio, M.; Lubell, W.D.; Carpentier, A.C.; Burelle, Y.; Ong, H.; et al. Adiponectin has a pivotal role in the cardioprotective effects of CP-3(iv), a selective CD36 azapeptide ligand, after transient coronary artery occlusion in mice. FASEB J. 2018, 32, 807–818. [Google Scholar] [CrossRef] [Green Version]
- Frégeau, G.; Sarduy, R.; Elimam, H.; Esposito, C.L.; Mellal, K.; Ménard, L.; Leitãoda Graça, S.D.; Proulx, C.; Zhang, J.; Febbraio, M.; et al. Atheroprotective and atheroregressive potential of azapeptide derivatives of GHRP-6 as selective CD36 ligands in apolipoprotein E-deficient mice. Atherosclerosis 2020, in press. [Google Scholar] [CrossRef]
- Mellal, K.; Omri, S.; Mulumba, M.; Tahiri, H.; Fortin, C.; Dorion, M.F.; Pham, H.; Garcia Ramos, Y.; Zhang, J.; Pundir, S.; et al. Immunometabolic modulation of retinal inflammation by CD36 ligand. Sci. Rep. 2019, 9, 12903. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, M.; Tsujikawa, M.; Itabe, H.; Du, Z.J.; Xie, P.; Matsumura, N.; Fu, X.; Zhang, R.; Sonoda, K.H.; Egashira, K.; et al. Chronic photo-oxidative stress and subsequent MCP-1 activation as causative factors for age-related macular degeneration. J. Cell Sci. 2012, 125, 2407–2415. [Google Scholar] [CrossRef] [Green Version]
- Bush, C.A.; Sarkar, S.K.; Kopple, K.D. Circular dichroism of β turns in peptides and proteins. Biochemistry 1978, 17, 4951–4954. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | GHS-R1a EC50 (M) | CD36 EC50 (M) |
|---|---|---|
| GHRP-6 (1) | 6.08 × 10−9 | 1.82 × 10−6 |
| [azaPhe2]GHRP-6 (4) | 1.61 × 10−5 | 7.24 × 10−5 |
| [azaTyr2]GHRP-6 (5) | 8.53 × 10−6 | 1.80 × 10−6 |
| [azaLeu3]GHRP-6 (6) | 1.20 × 10−6 | 2.89 × 10−6 |
| [azaGly3]GHRP-6 (7) | 8.08 × 10−7 | 9.61 × 10−6 |
| [azaPhe4]GHRP-6 (8) | 2.77 × 10−6 | 1.34 × 10−6 |
| [azaTyr4]GHRP-6 (9) | 1.57 × 10−5 | 2.80 × 10−5 |
| Compound | GHS-R1a EC50 (M) | CD36 EC50 (M) |
|---|---|---|
| [Ala1, azaPhe2]GHRP-6 (10) | » 10−5 | 4.27 × 10−5 |
| [Ala1, azaTyr2]GHRP-6 (11) | » 10−5 | 3.69 × 10−5 |
| [Ala1, azaLeu3]GHRP-6 (12) | 1.2×10−5 | 6.66 × 10−6 |
| [azaLeu3, Ala4]GHRP-6 (13) | » 10−5 | 2.59 × 10−5 |
| [azaLeu3, d-Ala5]GHRP-6 (14) | » 10−5 | 9.48 × 10−6 |
| [Ala1, azaPhe4]GHRP-6 (15) | » 10−5 | 7.58 × 10−6 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Proulx, C.; Zhang, J.; Sabatino, D.; Chemtob, S.; Ong, H.; Lubell, W.D. Synthesis and Biomedical Potential of Azapeptide Modulators of the Cluster of Differentiation 36 Receptor (CD36). Biomedicines 2020, 8, 241. https://doi.org/10.3390/biomedicines8080241
Proulx C, Zhang J, Sabatino D, Chemtob S, Ong H, Lubell WD. Synthesis and Biomedical Potential of Azapeptide Modulators of the Cluster of Differentiation 36 Receptor (CD36). Biomedicines. 2020; 8(8):241. https://doi.org/10.3390/biomedicines8080241
Chicago/Turabian StyleProulx, Caroline, Jinqiang Zhang, David Sabatino, Sylvain Chemtob, Huy Ong, and William D. Lubell. 2020. "Synthesis and Biomedical Potential of Azapeptide Modulators of the Cluster of Differentiation 36 Receptor (CD36)" Biomedicines 8, no. 8: 241. https://doi.org/10.3390/biomedicines8080241