
Proteome Characterization of Leaves in Common Bean


Abstract
:1. Introduction
2. Experimental Section
2.1. Plant Material and Growth Conditions
2.2. Protein Extraction
2.3. Protein Digestion
2.4. Mass Spectrometry
2.5. Data Analysis
3. Results and Discussion

3.1. Characterization of Bean Proteins
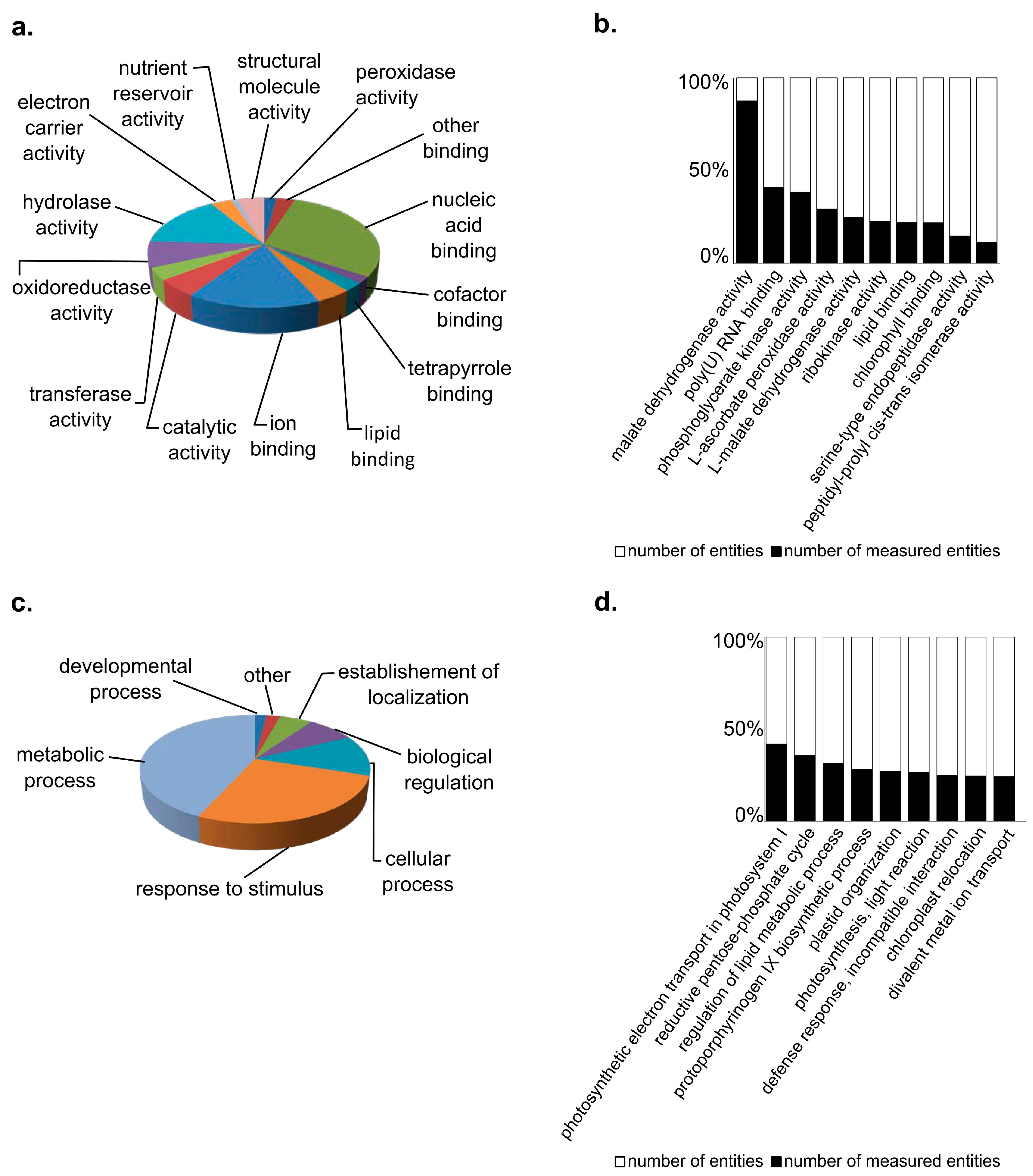

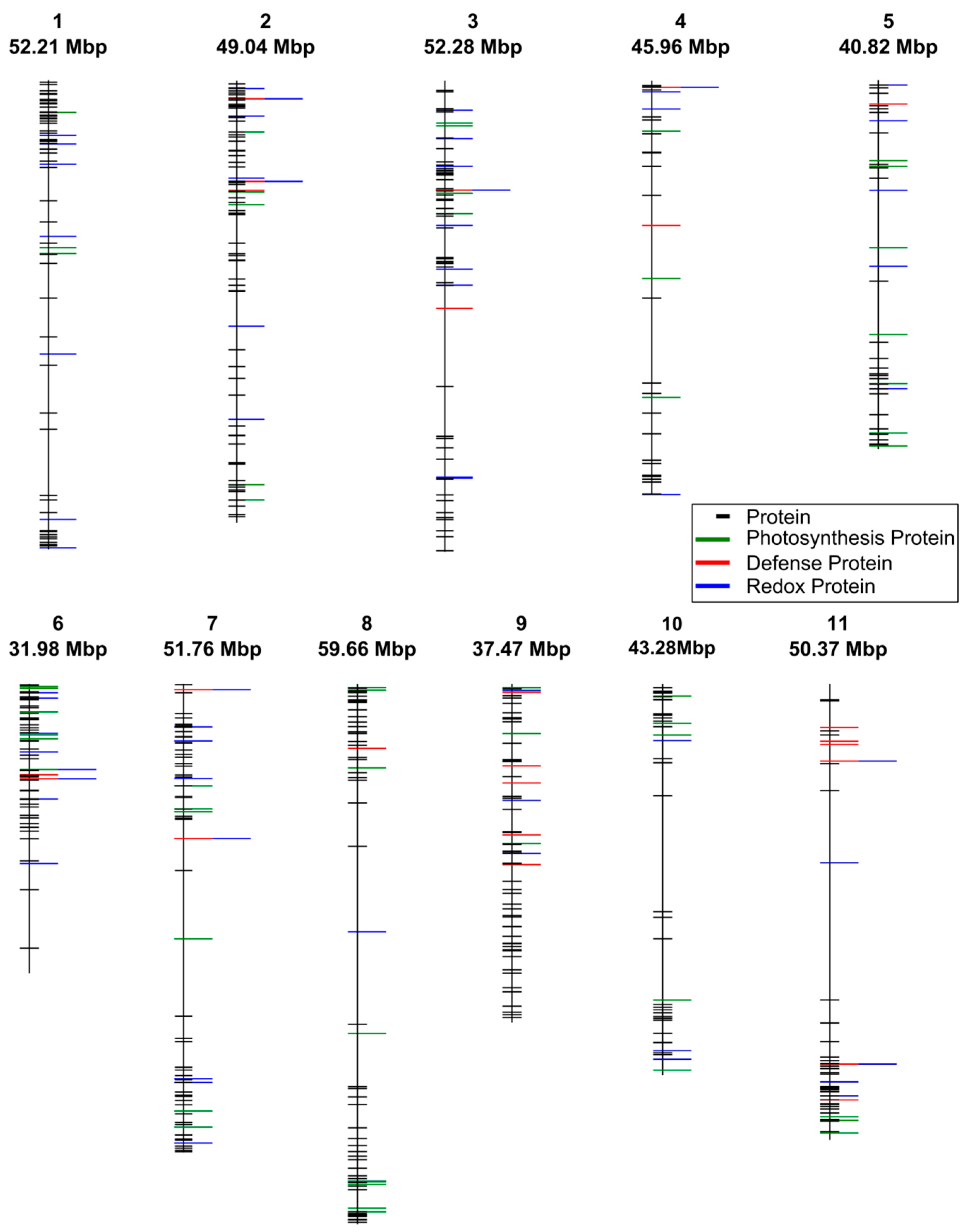
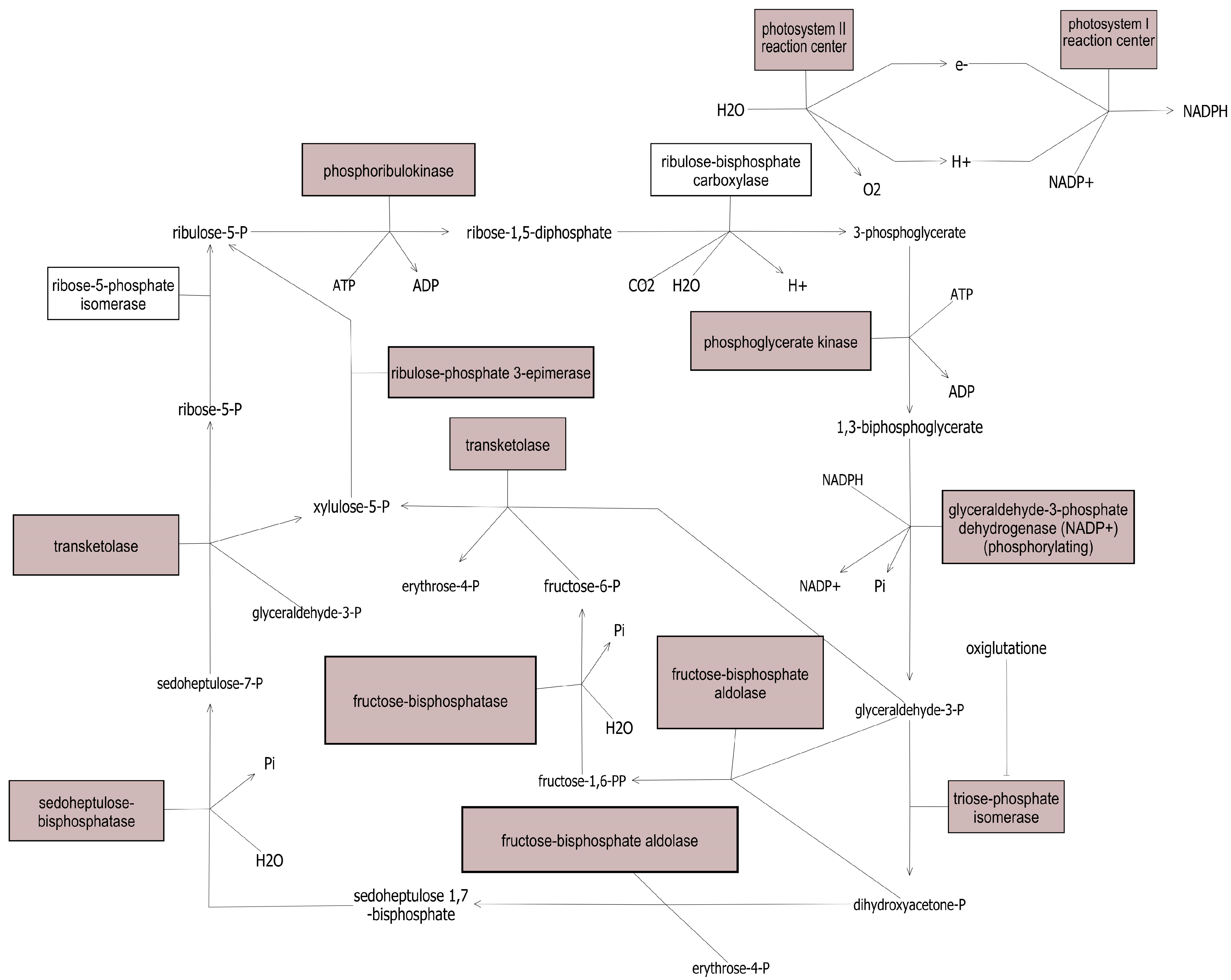
3.2. Quantitative Variation in Proteins between Bean Lines

{kind=link}
{kind=link}
{kind=link}
| Go Term | Protein Name | Accession Number | p-Value | Fold Change * |
|---|---|---|---|---|
| Photosynthesis | light harvesting complex photosystem II subunit 6 | Phvul.006G207300.1|PACid:27166485 | 0.0031 | 145.40 |
| Photosynthesis | light-harvesting chlorophyll B-binding protein 3 | Phvul.005G085800.1|PACid:27148435 | <0.00010 | 63.33 |
| Photosynthesis | light harvesting complex photosystem II subunit 6 | Phvul.002G207500.1|PACid:27170403 | <0.00010 | 53.33 |
| Oxidation-Reduction, Defense | plastid transcriptionally active 16 | Phvul.011G061600.1|PACid:27152979 | 0.019 | 5.97 |
| ATP synthesis coupled proton transport | ATP synthase δ-subunit gene | Phvul.003G211100.1|PACid:27143051 | 0.049 | 2.15 |
| -- | uridylyltransferase-related | Phvul.005G181700.1|PACid:27149150 | 0.0052 | 1.93 |
| Oxidation-Reduction, Defense | 2-cysteine peroxiredoxin B | Phvul.001G194100.1|PACid:27163227 | 0.038 | 1.62 |
| RNA Binding | elongation factor Ts family protein | Phvul.004G168100.1|PACid:27157476 | 0.0095 | 1.62 |
| Ion Binding | copper ion binding;cobalt ion binding;zinc ion binding | Phvul.002G265700.1|PACid:27169903 | 0.035 | 1.44 |
| Oxidation-Reduction, Defense | Oxidoreductase, zinc-binding dehydrogenase family protein | Phvul.003G060300.1|PACid:27143379 | 0.018 | 0.83 |
| Oxidation-Reduction | thioredoxin M-type 4 | Phvul.005G071200.1|PACid:27149735 | 0.043 | 0.77 |
| -- | RNA-binding (RRM/RBD/RNP motifs) family protein | Phvul.005G039000.1|PACid:27150205 | 0.053 | 0.69 |
| -- | CP12 domain-containing protein 2 | Phvul.001G212900.1|PACid:27164757 | 0.0072 | 0.67 |
| -- | protochlorophyllide oxidoreductase A | Phvul.005G083700.1|PACid:27149633 | 0.048 | 0.67 |
| Translation | Ribosomal L5P family protein | Phvul.008G059200.1|PACid:27153603 | 0.022 | 0.60 |
| Photosynthesis | cold, circadian rhythm, and rna binding 2 | Phvul.009G023700.1|PACid:27146551 | 0.027 | 0.53 |
| -- | FASCICLIN-like arabinogalactan 2 | Phvul.008G288800.1|PACid:27153618 | 0.036 | 0.51 |
| Defense | metacaspase 5 | Phvul.011G180300.1|PACid:27150868 | 0.015 | 0.45 |
| Photosynthesis | 2Fe-2S ferredoxin-like superfamily protein | Phvul.002G045400.1|PACid:27168785 | 0.017 | 0.33 |
| Protein Binding | DnaJ/Hsp4Unknown cysteine-rich domain superfamily protein | Phvul.003G164500.1|PACid:27144762 | 0.03 | 0.30 |
| -- | Ribosomal protein S19e family protein | Phvul.005G018600.1|PACid:27149117 | 0.089 | 0.30 |
| -- | FASCICLIN-like arabinogalactan-protein 1 Unknown | Phvul.008G075000.1|PACid:27155369 | 0.079 | 0.22 |
| Lipid Metabolic Process | GDSL-like Lipase/Acylhydrolase superfamily protein | Phvul.006G169100.1|PACid:27166530 | 0.045 | 0.20 |
| -- | FKBP-like peptidyl-prolyl cis-trans isomerase family protein | Phvul.011G051700.1|PACid:27151439 | 0.01 | 0.15 |
| Translation | plastid ribosomal protein l11 | Phvul.004G090600.1|PACid:27158071 | 0.0011 | 0.13 |
| Protein Binding | Embryo-specific protein 3, (ATS3) | Phvul.005G177100.1|PACid:27149941 | 0.0023 | 0.03 |
| Response to Stress | Unknown | Phvul.003G074600.1|PACid:27143731 | 0.0001 | 0.01 |
| Oxidation-Reduction, Defense | Peroxidase superfamily protein | Phvul.007G082600.1|PACid:27161387 | 0.00015 | 0.01 |
| Ion Binding | allantoinase | Phvul.006G186800.1|PACid:27164938 | 0.0039 | 0.01 |
4. Conclusions
Supplementary Materials
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jones, A.L. Phaseolus Bean: Post-Harvest Operations; Centro Internacional de Agricultura Tropical: Cali, Colombia, 1999; pp. 1–24. Available online: http://www.fao.org/3/a-av015e.pdf (accessed on 12 August 2015).
- Beaver, J.S.; Osorno, J.M. Achievements and limitations of contemporary common bean breeding using conventional and molecular approaches. Euphytica 2009, 168, 145–175. [Google Scholar] [CrossRef]
- Yang, D.F.; Du, X.H.; Yang, Z.Q.; Liang, Z.S.; Guo, Z.X.; Liu, Y. Transcriptomics, proteomics, and metabolomics to reveal mechanisms underlying plant secondary metabolism. Eng. Life Sci. 2014, 14, 456–466. [Google Scholar] [CrossRef]
- Kushalappa, A.C.; Gunnaiah, R. Metabolo-proteomics to discover plant biotic stress resistance genes. Trends Plant Sci. 2013, 18, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Badowiec, A.; Weidner, S. Proteomic changes in the roots of germinating Phaseolus vulgaris seeds in response to chilling stress and post-stress recovery. J. Plant Physiol. 2014, 171, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Zadraznik, T.; Hollung, K.; Egge-Jacobsen, W.; Meglic, V.; Sustar-Vozlic, J. Differential proteomic analysis of drought stress response in leaves of common bean (Phaseolus vulgaris L.). J. Proteom. 2013, 78, 254–272. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.B.; Eticha, D.; Fuhrs, H.; Heintz, D.; Ayoub, D.; van Dorsselaer, A.; Schlingmann, B.; Rao, I.M.; Braun, H.P.; Horst, W.J. Proteomic and phosphoproteomic analysis of polyethylene glycol-induced osmotic stress in root tips of common bean (Phaseolus vulgaris L.). J. Exp. Bot. 2013, 64, 5569–5586. [Google Scholar] [CrossRef] [PubMed]
- De La Fuente, M.; Borrajo, A.; Bermudez, J.; Lores, M.; Alonso, J.; Lopez, M.; Santalla, M.; de Ron, A.M.; Zapata, C.; Alvarez, G. 2-DE-based proteomic analysis of common bean (Phaseolus vulgaris L.) seeds. J. Proteom. 2011, 74, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Du, X.Q.; Wu, F.; Li, L.; Li, C.Y.; Meng, Z. Proteomics insights into the basis of interspecific facilitation for maize (Zea mays) in faba bean (Vicia faba)/maize intercropping. J. Proteom. 2014, 109, 111–124. [Google Scholar] [CrossRef] [PubMed]
- Demartini, D.R.; Carlini, C.R.; Thelen, J.J. Global and targeted proteomics in developing jack bean (Canavalia ensiformis) seedlings: An investigation of urease isoforms mobilization in early stages of development. Plant Mol. Biol. 2011, 75, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Torres, N.L.; Cho, K.; Shibato, J.; Hirano, M.; Kubo, A.; Masuo, Y.; Iwahashi, H.; Jwa, N.S.; Agarwal, G.K.; Rakwal, R. Gel-based proteomics reveals potential novel protein markers of ozone stress in leaves of cultivated bean and maize species of panama. Electrophoresis 2007, 28, 4369–4381. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Chu, C.H.; Chen, S.L.; Juan, H.F.; Chen, Y.M. A proteomics study of the mung bean epicotyl regulated by brassinosteroids under conditions of chilling stress. Cell. Mol. Biol. Lett. 2006, 11, 264–278. [Google Scholar] [CrossRef] [PubMed]
- Katam, R.; Basha, S.M.; Suravajhala, P.; Pechan, T. Analysis of peanut leaf proteome. J. Proteome Res. 2010, 9, 2236–2254. [Google Scholar] [CrossRef] [PubMed]
- Salavati, A.; Taleei, A.; Bushehri, A.A.; Komatsu, S. Analysis of the proteome of common bean (Phaseolus vulgaris L.) roots after inoculation with rhizobium etli. Protein Pept. Lett. 2012, 19, 880–889. [Google Scholar] [CrossRef] [PubMed]
- Vanderschuren, H.; Nyaboga, E.; Poon, J.S.; Baerenfaller, K.; Grossmann, J.; Hirsch-Hoffmann, M.; Kirchgessner, N.; Nanni, P.; Gruissem, W. Large-scale proteomics of the cassava storage root and identification of a target gene to reduce postharvest deterioration. Plant cell 2014, 26, 1913–1924. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.R.; Teran, H.; Lema, M.; Schwartz, H.F.; Miklas, P.N. Registration of white mold resistant dry bean germplasm line a 195. J. Plant Regist. 2007, 1, 62–63. [Google Scholar] [CrossRef]
- Shanahan, J.F.; Schwartz, H.F.; Brick, M.A.; Schweissing, F.C.; Meyer, R.F.; Pearson, C.H.; Hain, J.P.; Johnson, C.L.; Barry Ogg, J. Colorado Dry Bean Variety Performance Trials; Technical Report TR93-8. Colorado Agricultural Experiment Station: Fort Collins, CO, USA, December 1993. Available online: http://cospl.coalliance.org/fedora/repository/co%3A9650/ucsu20211tr938internet.pdf/ (accessed on 12 August 2015).
- Hart, L.P.; Saettler, A.W.; Correa, F. Angular leaf spot of dry beans; Extension Bulletin, E-1753. Michigan State University: East Lansing, MI, USA, February 1984. Available online: http://archive.lib.msu.edu/DMC/Ag.%20Ext.%202007-Chelsie/PDF/e1753-1984.pdf (accessed on 12 August 2015).
- Bello, M.H.; Moghaddam, S.M.; Massoudi, M.; McClean, P.E.; Cregan, P.B.; Miklas, P.N. Application of in silico bulked segregant analysis for rapid development of markers linked to bean common mosaic virus resistance in common bean. BMC Genom. 2014, 15, 13. [Google Scholar] [CrossRef] [PubMed]
- Keller, A.; Nesvizhskii, A.I.; Kolker, E.; Aebersold, R. Empirical statistical model to estimate the accuracy of peptide identifications made by ms/ms and database search. Anal. Chem. 2002, 74, 5383–5392. [Google Scholar] [CrossRef] [PubMed]
- Searle, B.C.; Turner, M.; Nesvizhskii, A.I. Improving sensitivity by probabilistically combining results from multiple ms/ms search methodologies. J. Proteome Res. 2008, 7, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Kall, L.; Storey, J.D.; MacCoss, M.J.; Noble, W.S. Assigning significance to peptides identified by tandem mass spectrometry using decoy databases. J. Proteome Res. 2008, 7, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Rudnick, P.A.; Clauser, K.R.; Kilpatrick, L.E.; Tchekhovskoi, D.V.; Neta, P.; Blonder, N.; Billheimer, D.D.; Blackman, R.K.; Bunk, D.M.; Cardasis, H.L.; et al. Performance metrics for liquid chromatography-tandem mass spectrometry systems in proteomics analyses. Mol. Cell. Proteom. 2010, 9, 225–241. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Sadygov, R.G.; Yates, J.R., III. A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal. Chem. 2004, 76, 4193–4201. [Google Scholar] [CrossRef] [PubMed]
- Freund, D.M.; Prenni, J.E. Improved detection of quantitative differences using a combination of spectral counting and MS/MS total ion current. J. Proteome Res. 2013, 12, 1996–2004. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Goodstein, D.M.; Shu, S.Q.; Howson, R.; Neupane, R.; Hayes, R.D.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N.; et al. Phytozome: A comparative platform for green plant genomics. Nucleic Acids Res. 2012, 40, D1178–D1186. [Google Scholar] [CrossRef] [PubMed]
- Van der Hoorn, R.A.L.; Jones, J.D. The plant proteolytic machinery and its role in defence. Curr. Opin. Plant Biol. 2004, 7, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, N.; Lam, E. Recent advance in the study of caspase-like proteases and bax inhibitor-1 in plants: Their possible roles as regulator of programmed cell death. Mol. Plant Pathol. 2004, 5, 65–70. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Robison, F.M.; Heuberger, A.L.; Brick, M.A.; Prenni, J.E. Proteome Characterization of Leaves in Common Bean. Proteomes 2015, 3, 236-248. https://doi.org/10.3390/proteomes3030236
Robison FM, Heuberger AL, Brick MA, Prenni JE. Proteome Characterization of Leaves in Common Bean. Proteomes. 2015; 3(3):236-248. https://doi.org/10.3390/proteomes3030236
Chicago/Turabian StyleRobison, Faith M., Adam L. Heuberger, Mark A Brick, and Jessica E. Prenni. 2015. "Proteome Characterization of Leaves in Common Bean" Proteomes 3, no. 3: 236-248. https://doi.org/10.3390/proteomes3030236