G-Protein-Coupled Estrogen Receptor (GPER)-Specific Agonist G1 Induces ER Stress Leading to Cell Death in MCF-7 Cells

 , ,
, ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Lines and Cell Culture
2.3. Cell Treatment
2.4. Cell Cycle and Apoptosis Analysis by Flow Cytometry
2.5. Immunoblotting
2.6. Indo-1 AM Staining
2.7. Measurement of Protein Synthesis by the Surface Sensing of Translation (SUnSET) Puromycin End-Labeling Assay
2.8. Real-Time PCR
2.9. Knockdown of CHOP by Small Interfering RNA
2.10. Determination of Cell Viability
2.11. Mitochondrial Potential Measurement
2.12. Subcellular Fractionation
2.13. Statistical Analysis
3. Results
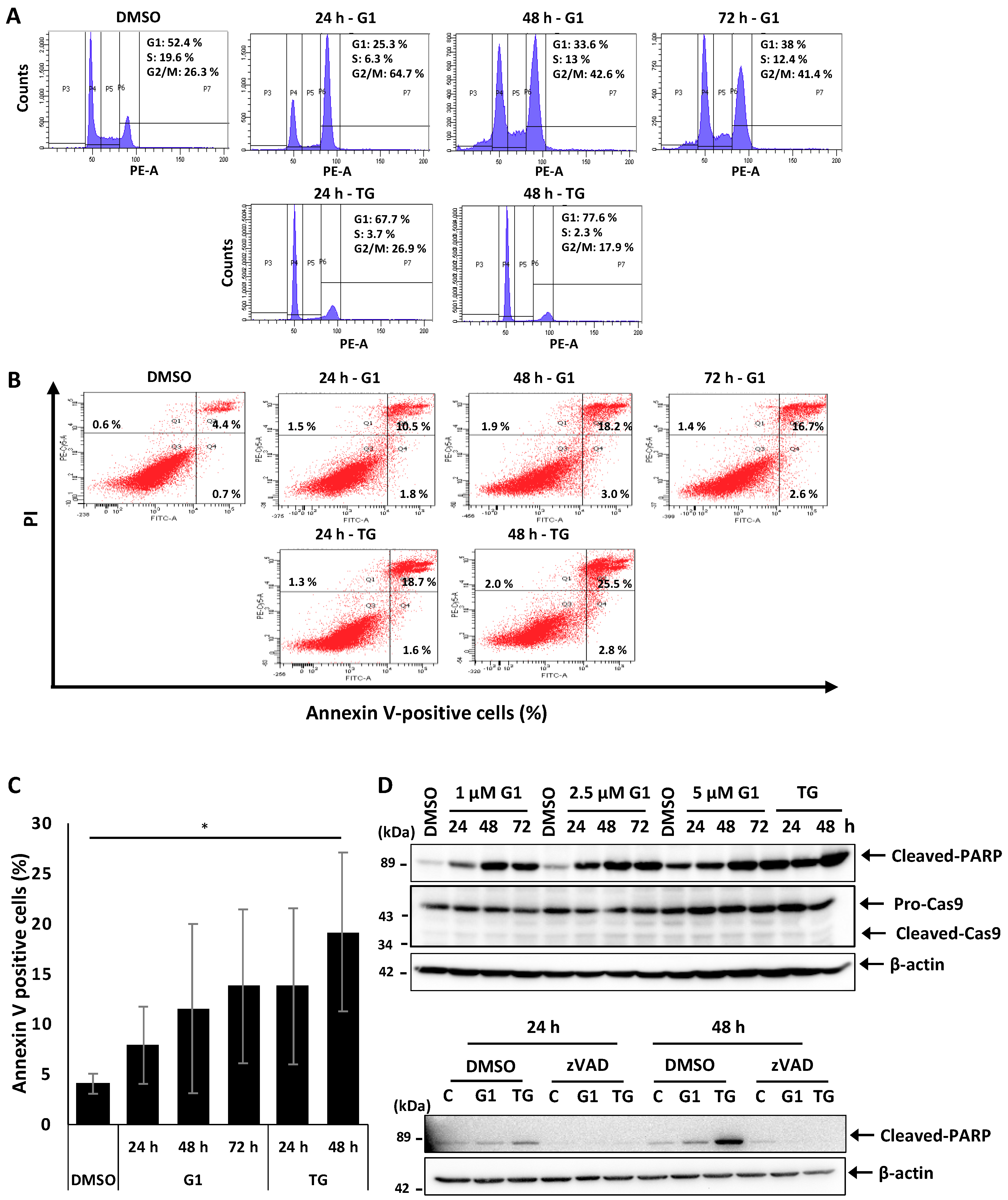
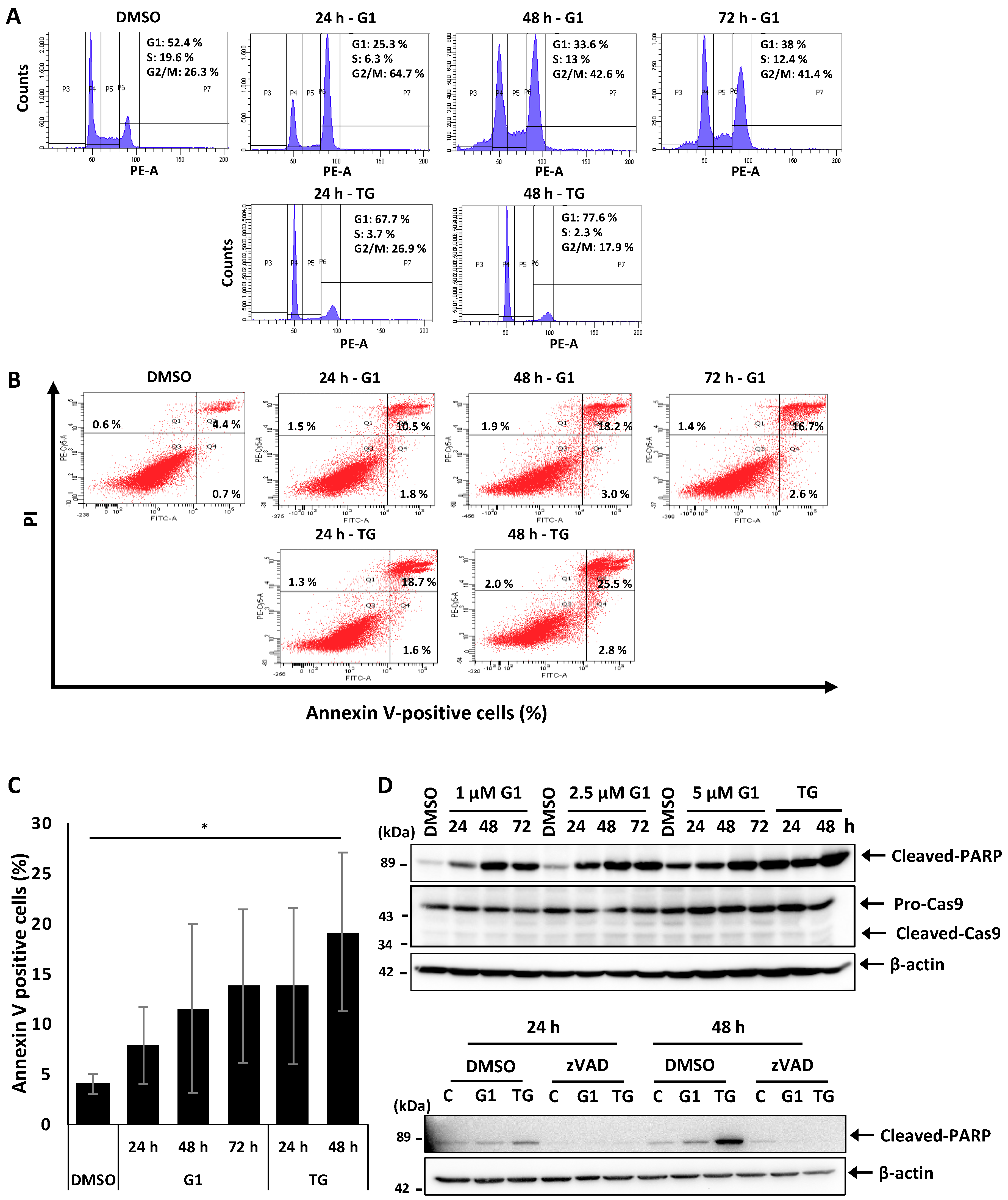
3.1. G1 and TG Induced Cell Cycle Arrest in G2/M and Apoptosis without Activating Caspase-3 and 9 in MCF-7 Cells
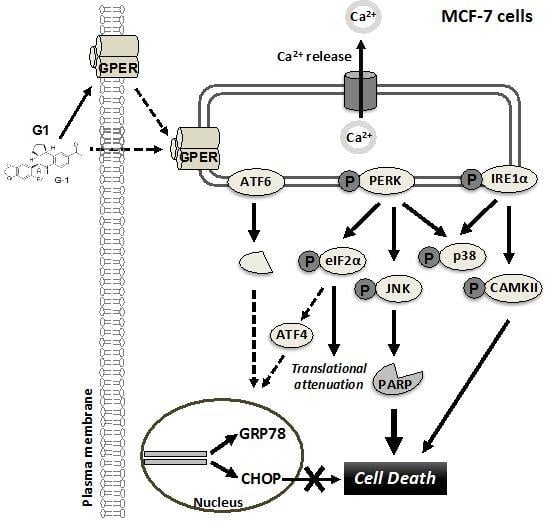
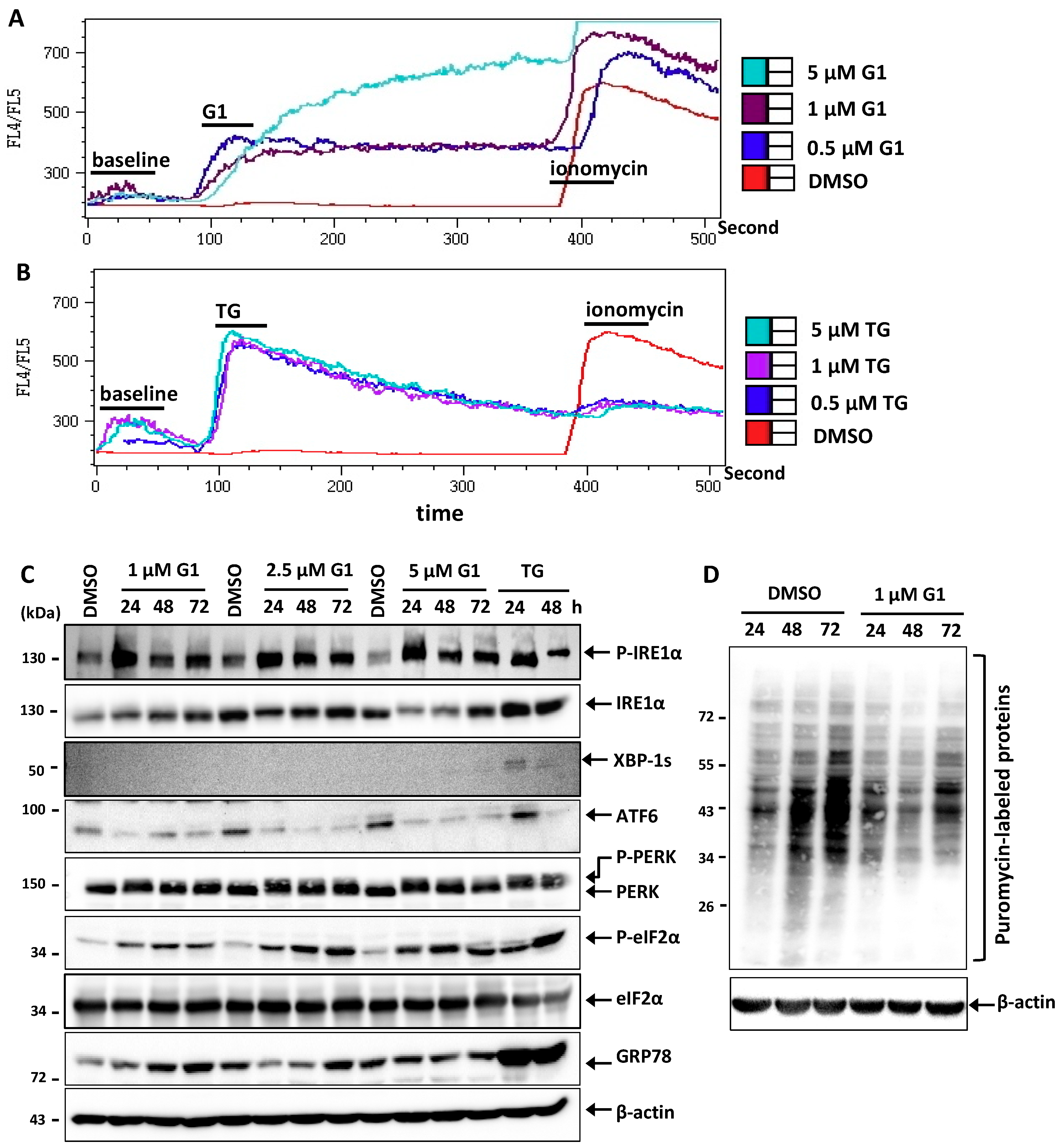
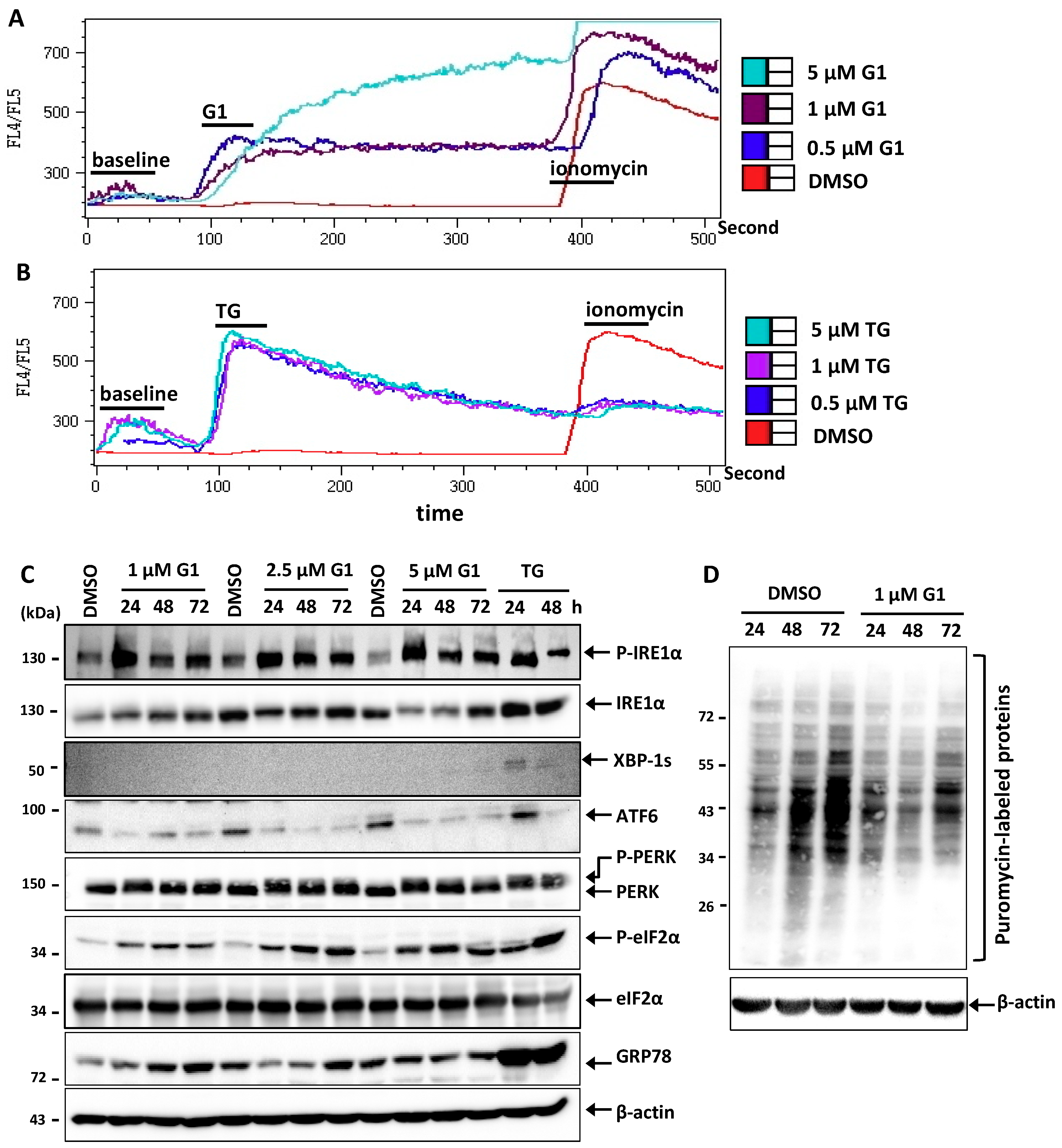
3.2. G1 and TG Induced Ca2+ Efflux and Activated the UPR in MCF-7 Cells
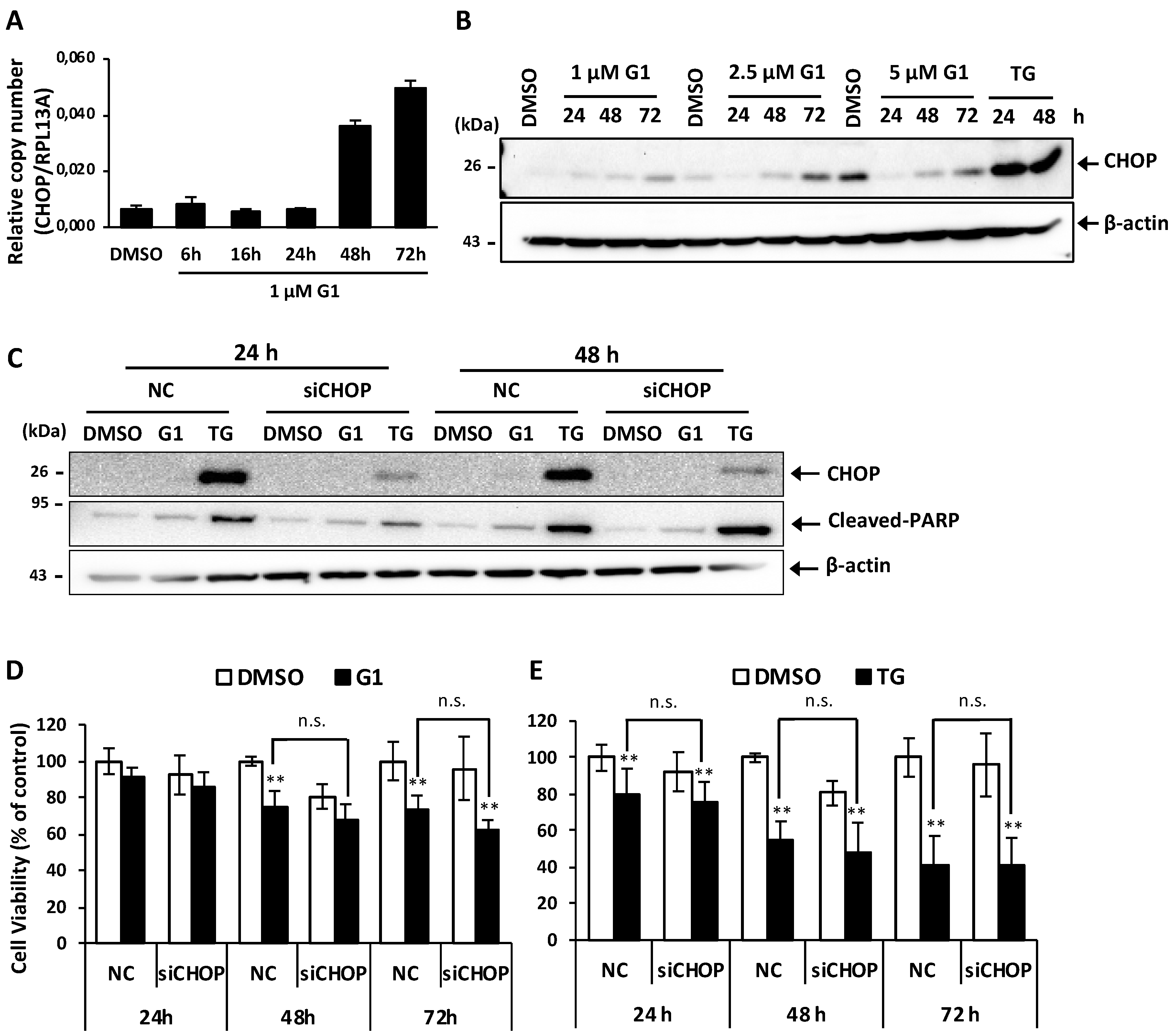
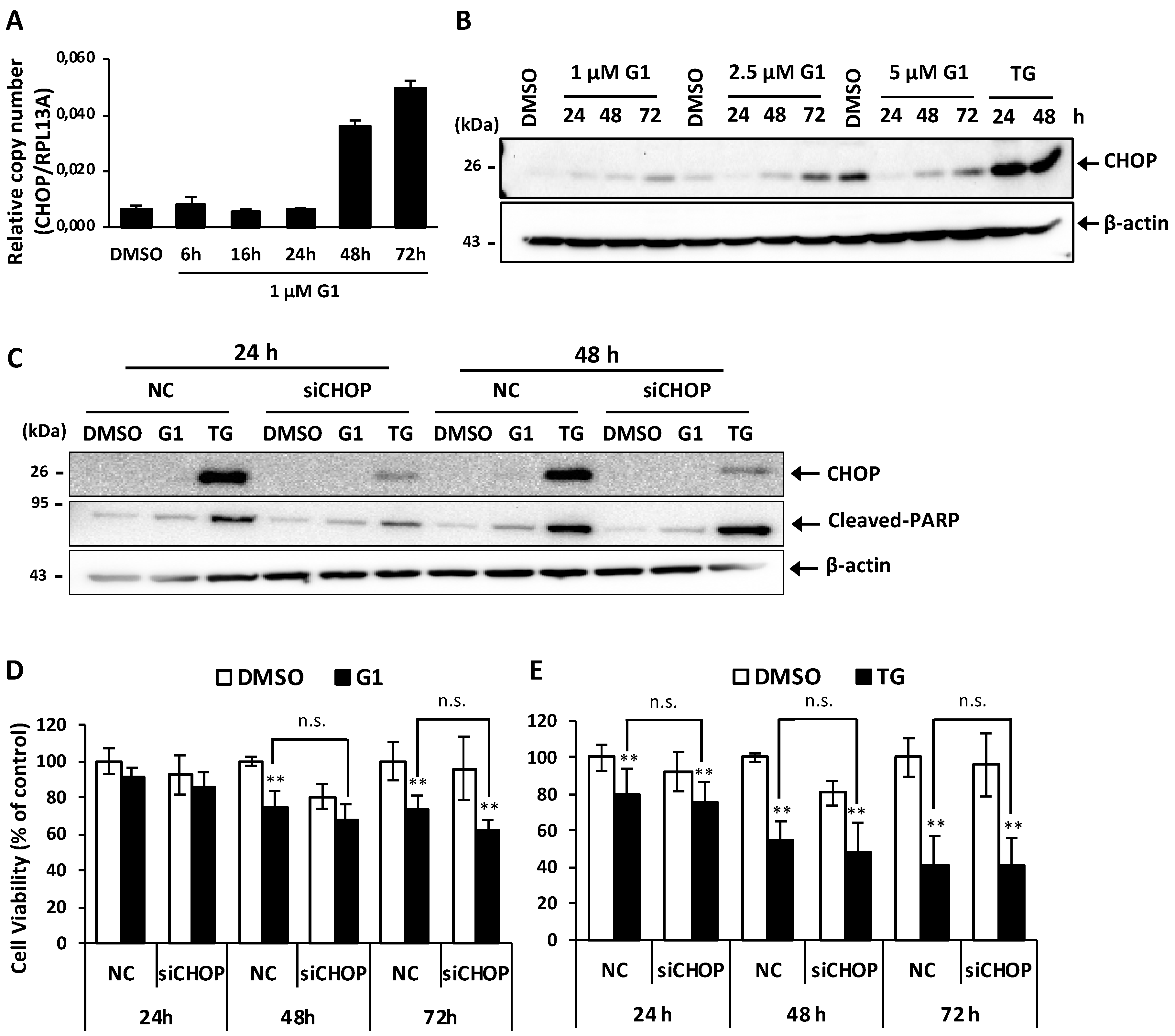
3.3. Up-Regulation of CHOP Expression Was Not Involved in G1-Induced Cell Death in MCF-7 Cells
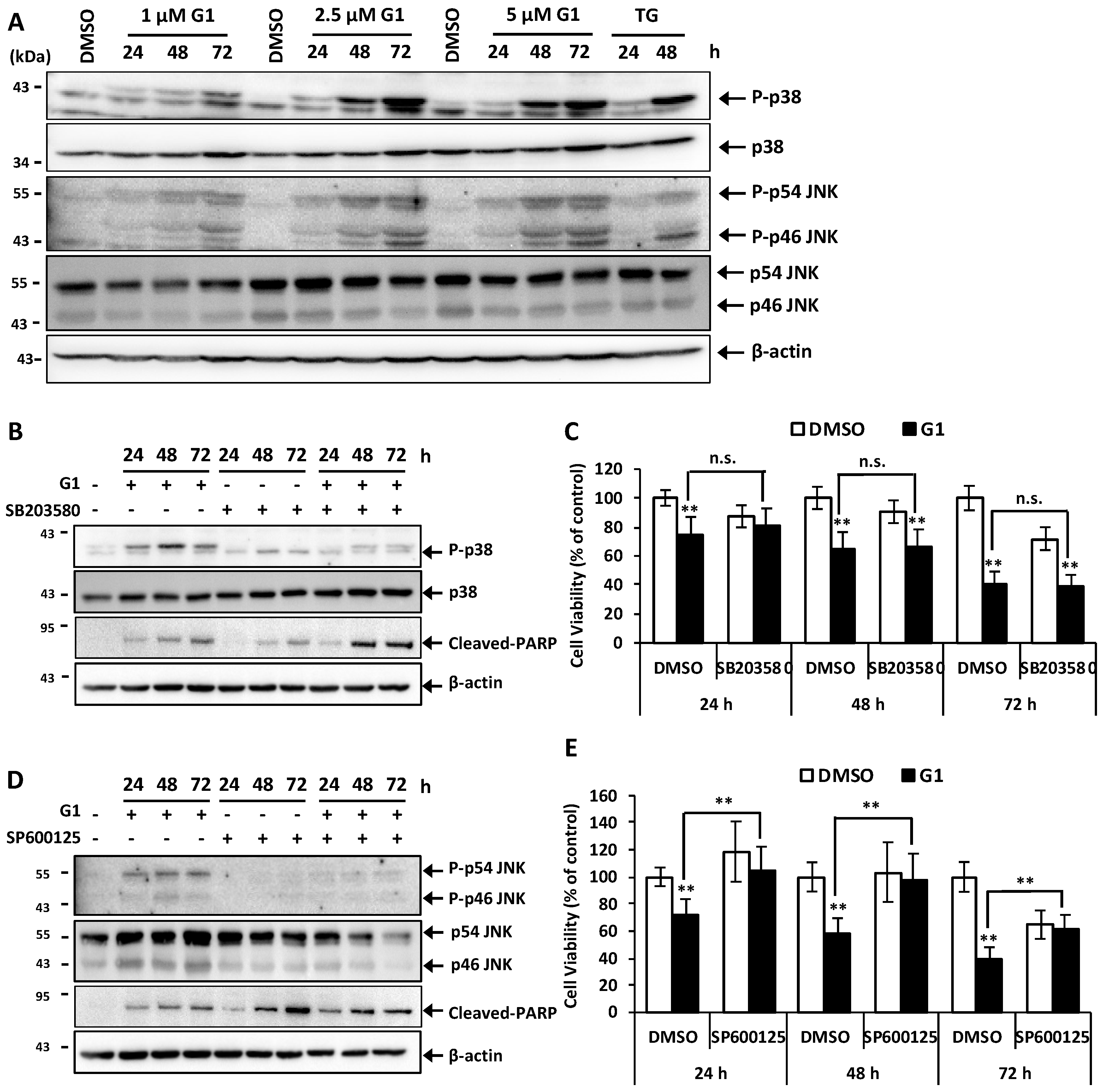
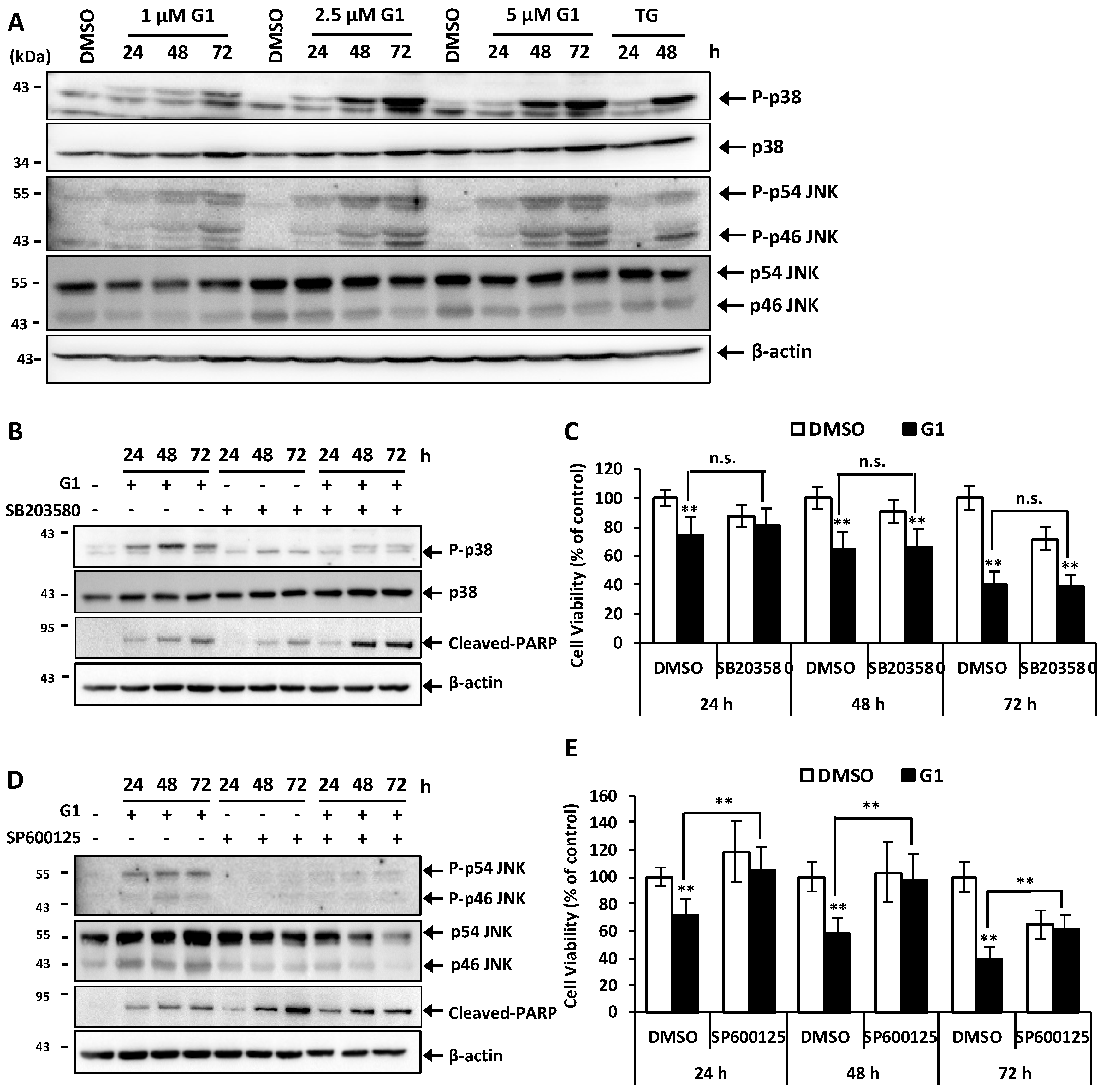
3.4. G1 Activated p38 and JNK and a Selective Inhibitor of Phosphorylation of JNK Inhibited G1-Induced Cell Death in MCF-7 Cells
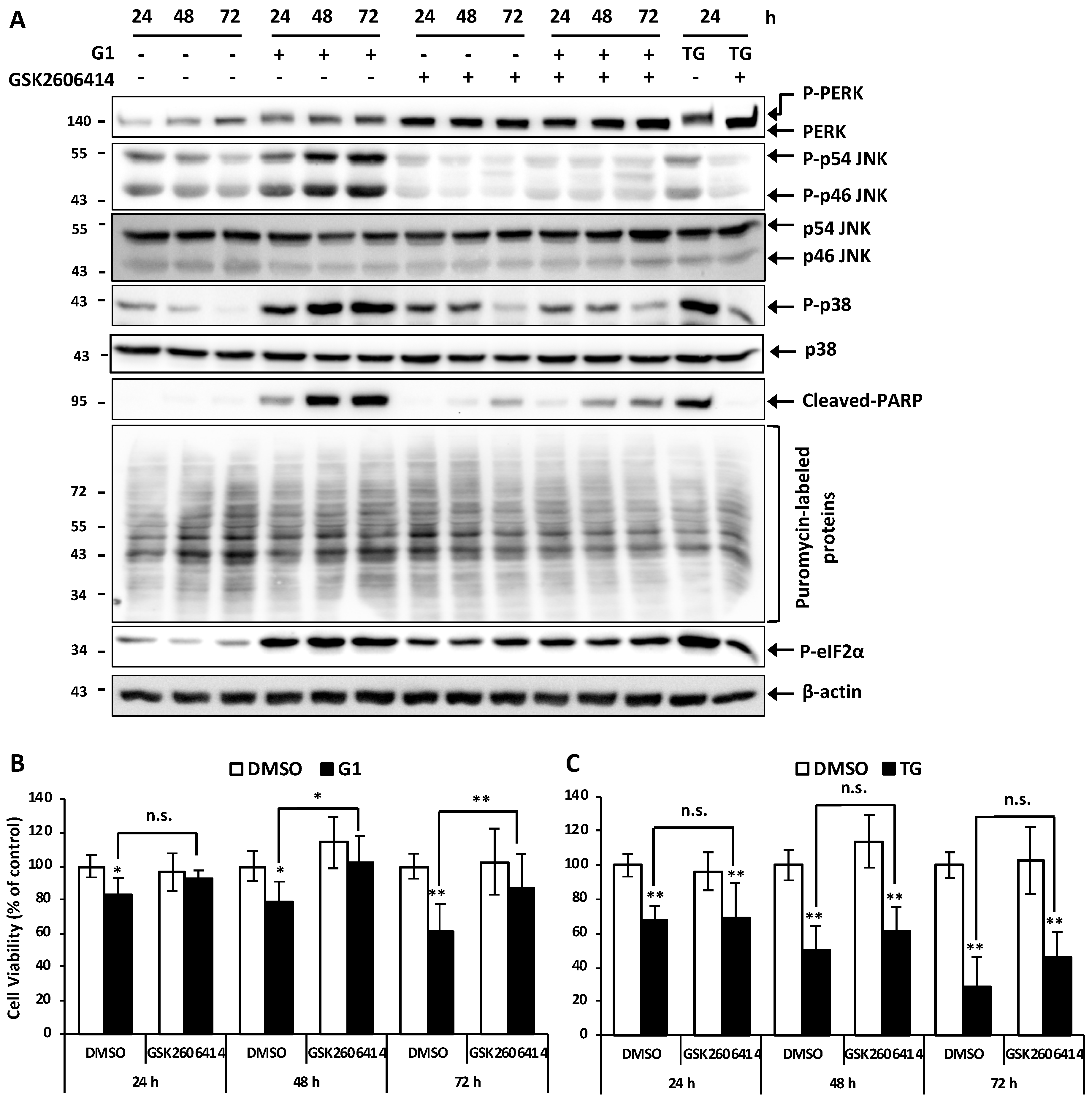
3.5. Inhibition of PERK Inhibited the Activation of JNK and Prevented G1-Induced Cell Death in MCF-7 Cells
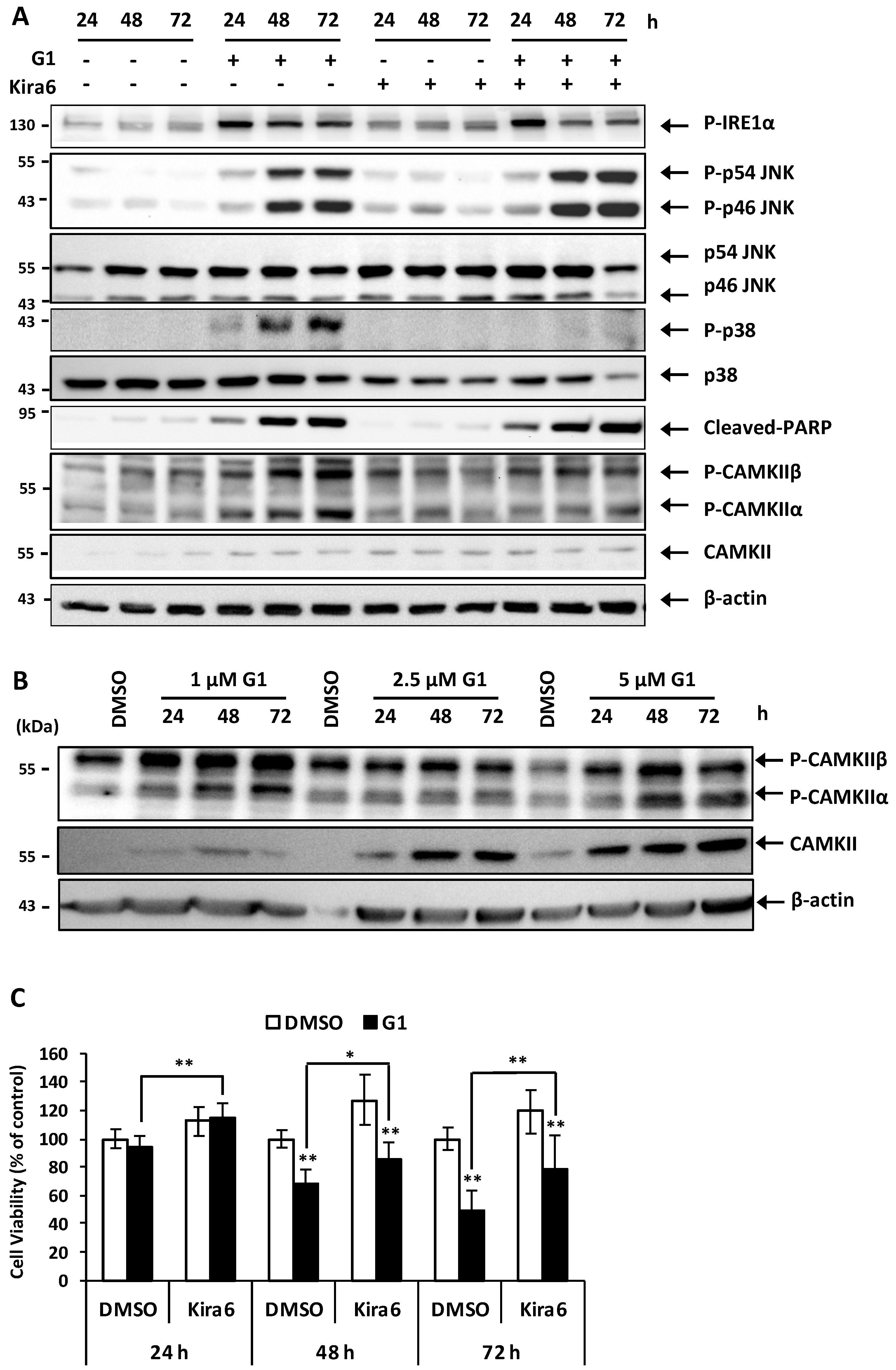
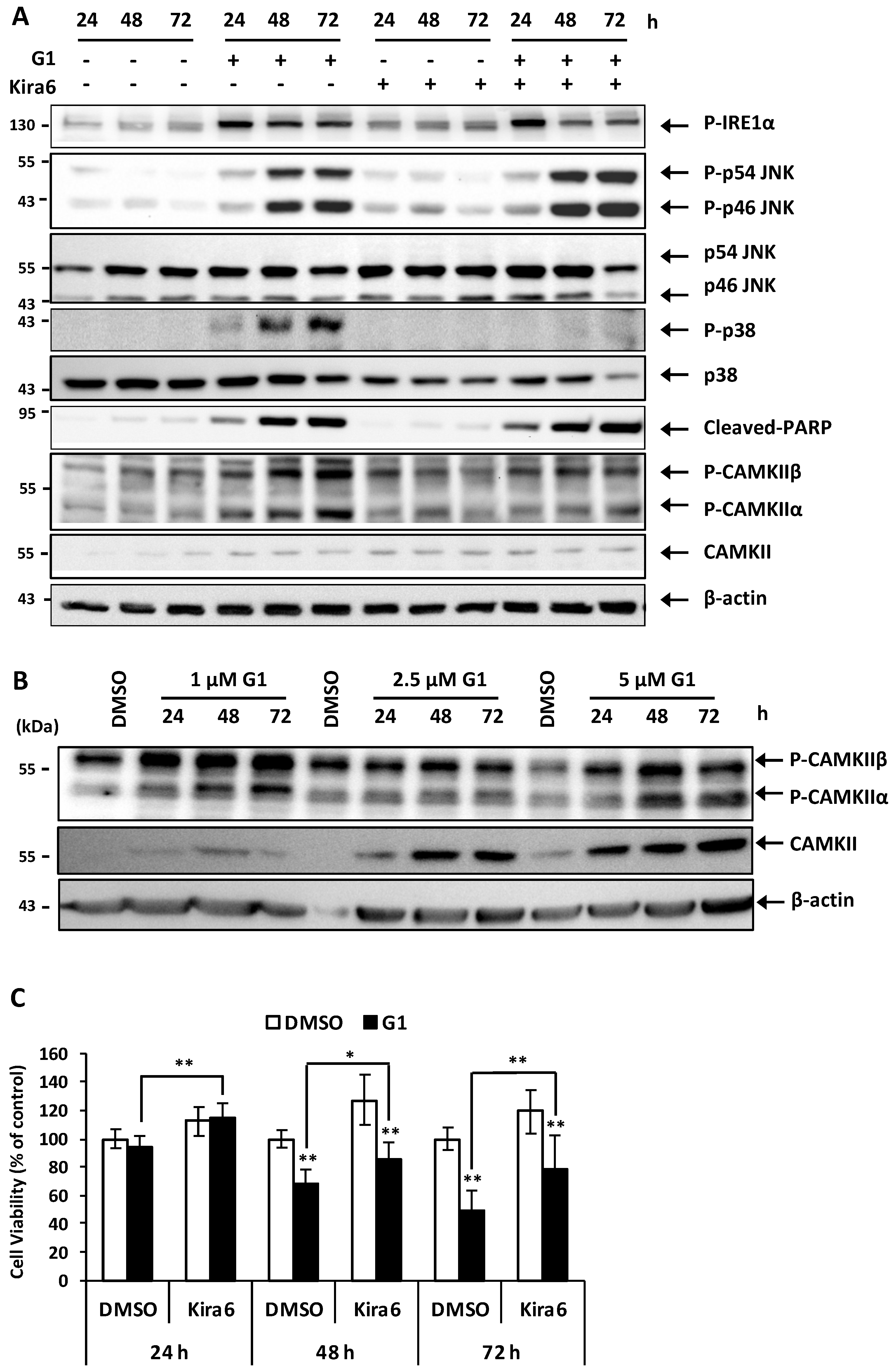
3.6. Inhibition of IRE1α Inhibited the Phosphorylation of CAMKII and Prevented G1-Induced Cell Death in MCF-7 Cells
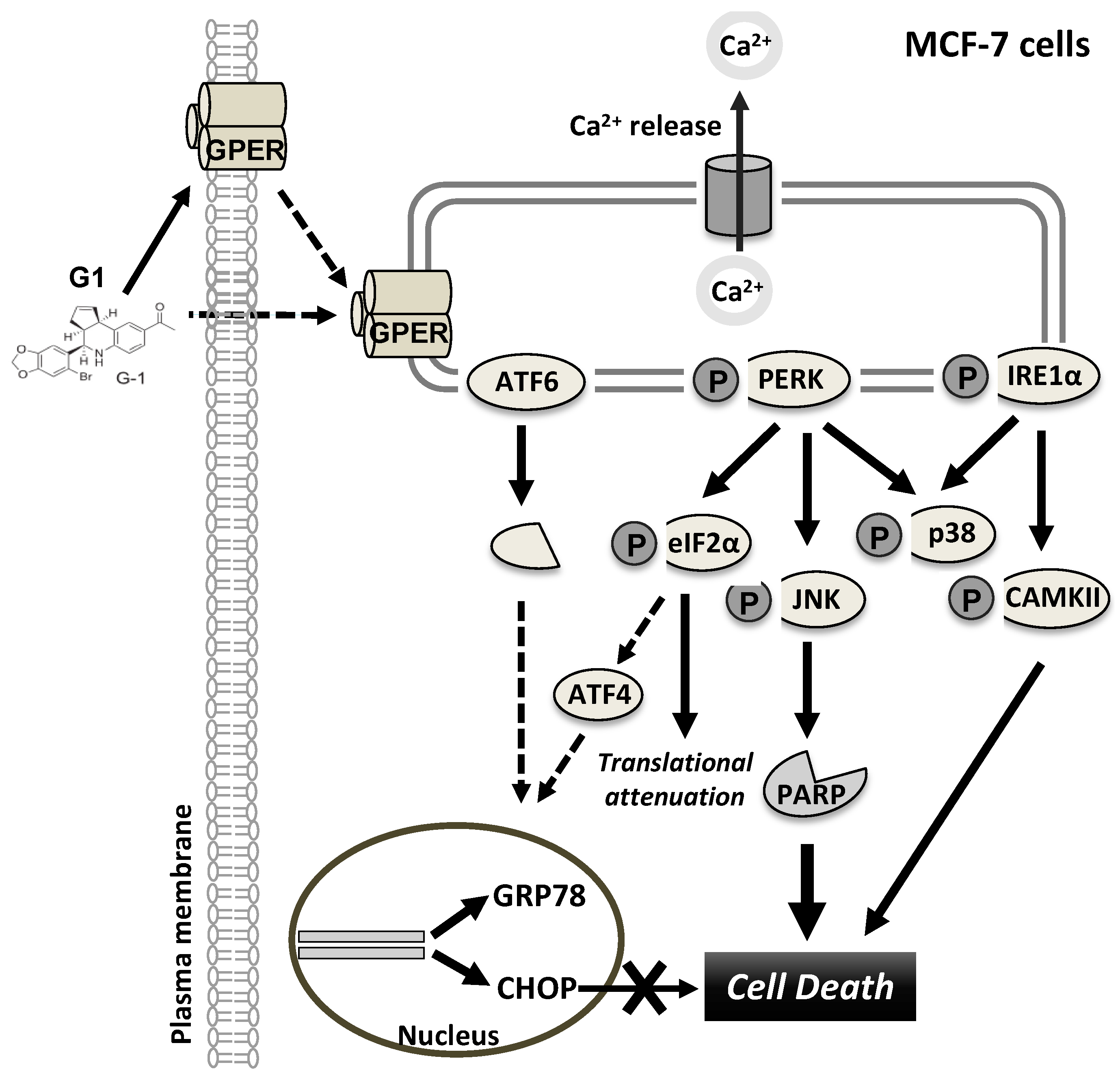
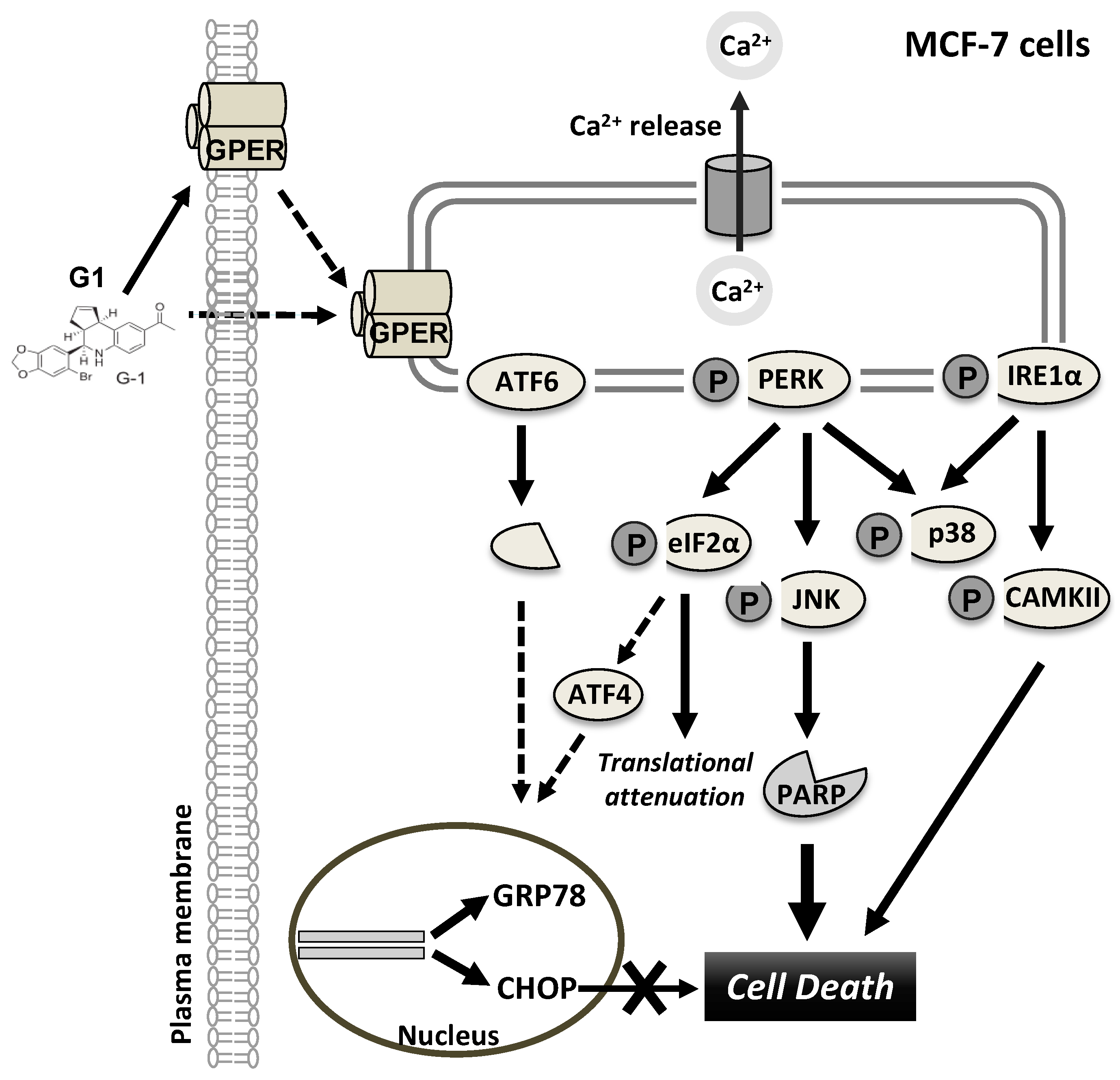
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Anchisi, L.; Dessi, S.; Pani, A.; Mandas, A. Cholesterol homeostasis: A key to prevent or slow down neurodegeneration. Front. Physiol. 2012, 3, 486. [Google Scholar] [CrossRef]
- Olde, B.; Leeb-Lundberg, L.M. GPR30/GPER1: Searching for a role in estrogen physiology. Trends Endocrin. Met. 2009, 20, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Filardo, E.J.; Quinn, J.A.; Bland, K.I.; Frackelton, A.R., Jr. Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol. Endocrinol. 2000, 14, 1649–1660. [Google Scholar] [CrossRef]
- Revankar, C.M.; Cimino, D.F.; Sklar, L.A.; Arterburn, J.B.; Prossnitz, E.R. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science 2005, 307, 1625–1630. [Google Scholar] [CrossRef] [PubMed]
- Prossnitz, E.R.; Arterburn, J.B. International Union of Basic and Clinical Pharmacology. XCVII. G Protein-Coupled Estrogen Receptor and Its Pharmacologic Modulators. Pharmacol. Rev. 2015, 67, 505–540. [Google Scholar] [CrossRef] [PubMed]
- Lappano, R.; Pisano, A.; Maggiolini, M. GPER Function in Breast Cancer: An Overview. Front. Endocrinol. 2014, 5, 66. [Google Scholar] [CrossRef] [Green Version]
- Ignatov, T.; Weißenborn, C.; Poehlmann, A.; Lemke, A.; Semczuk, A.; Roessner, A.; Costa, S.D.; Kalinski, T.; Ignatov, A. GPER-1 expression decreases during breast cancer tumorigenesis. Cancer Investig. 2013, 31, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Li, J.G.; Zheng, X.Y.; Jin, F.; Dong, H.T. Expression of CD133, PAX2, ESA, and GPR30 in invasive ductal breast carcinomas. Chin. Med. J. 2009, 122, 2763–2769. [Google Scholar] [PubMed]
- Ignatov, A.; Ignatov, T.; Weißenborn, C.; Eggemann, H.; Bischoff, J.; Semczuk, A.; Roessner, A.; Costa, S.D.; Kalinski, T. G-protein-coupled estrogen receptor GPR30 and tamoxifen resistance in breast cancer. Breast Cancer Res. Treat. 2011, 128, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Arias-Pulido, H.; Royce, M.; Gong, Y.; Joste, N.; Lomo, L.; Lee, S.J.; Chaher, N.; Verschraegen, C.; Lara, J.; Prossnitz, E.R.; et al. GPR30 and estrogen receptor expression: New insights into hormone dependence of inflammatory breast cancer. Breast Cancer Res. Treat. 2010, 123, 51–58. [Google Scholar] [CrossRef]
- Ignatov, T.; Modl, S.; Thulig, M.; Weißenborn, C.; Treeck, O.; Ortmann, O.; Zenclussen, A.C.; Costa, S.D.; Kalinski, T.; Ignatov, A. GPER-1 acts as a tumor suppressor in ovarian cancer. J. Ovarian Res. 2013, 6, 51. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Chen, Z.; Jiang, G.; Zhou, Y.; Yang, X.; Huang, H.; Liu, H.; Du, J.; Wang, H. Epigenetic down regulation of G protein-coupled estrogen receptor (GPER) functions as a tumor suppressor in colorectal cancer. Mol. Cancer 2017, 16, 87. [Google Scholar] [CrossRef] [PubMed]
- Filardo, E.J.; Graeber, C.T.; Quinn, J.A.; Resnick, M.B.; Giri, D.; DeLellis, R.A.; Steinhoff, M.M.; Sabo, E. Distribution of GPR30, a seven membrane-spanning estrogen receptor, in primary breast cancer and its association with clinicopathologic determinants of tumor progression. Clin. Cancer Res. 2006, 12, 6359–6366. [Google Scholar] [CrossRef] [PubMed]
- Ignatov, T.; Claus, M.; Nass, N.; Haybaeck, J.; Seifert, B.; Kalinski, T.; Ortmann, O.; Ignatov, A. G-protein-coupled estrogen receptor GPER-1 expression in hormone receptor-positive breast cancer is associated with poor benefit of tamoxifen. Breast Cancer Res. Treat. 2019, 174, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Ignatov, A.; Ignatov, T.; Roessner, A.; Costa, S.D.; Kalinski, T. Role of GPR30 in the mechanisms of tamoxifen resistance in breast cancer MCF-7 cells. Breast Cancer Res. Treat. 2010, 123, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Mo, Z.; Liu, M.; Yang, F.; Luo, H.; Li, Z.; Tu, G.; Yang, G. GPR30 as an initiator of tamoxifen resistance in hormone-dependent breast cancer. Breast Cancer Res. 2013, 15, R114. [Google Scholar] [CrossRef] [PubMed]
- Bologa, C.G.; Revankar, C.M.; Young, S.M.; Edwards, B.S.; Arterburn, J.B.; Kiselyov, A.S.; Parker, M.A.; Tkachenko, S.E.; Savchuck, N.P.; Sklar, L.A.; et al. Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat. Chem. Biol. 2006, 2, 207–212. [Google Scholar] [CrossRef]
- Ariazi, E.A.; Brailoiu, E.; Yerrum, S.; Shupp, H.A.; Slifker, M.J.; Cunliffe, H.E.; Black, M.A.; Donato, A.L.; Arterburn, J.B.; Oprea, T.I.; et al. The G protein-coupled receptor GPR30 inhibits proliferation of estrogen receptor-positive breast cancer cells. Cancer Res. 2010, 70, 1184–1194. [Google Scholar] [CrossRef] [PubMed]
- Chan, Q.K.; Lam, H.M.; Ng, C.F.; Lee, A.Y.; Chan, E.S.; Ng, H.K.; Ho, S.M.; Lau, K.M. Activation of GPR30 inhibits the growth of prostate cancer cells through sustained activation of Erk1/2, c-jun/c-fos-dependent upregulation of p21, and induction of G(2) cell-cycle arrest. Cell Death Differ. 2010, 17, 1511–1523. [Google Scholar] [CrossRef] [PubMed]
- Chimento, A.; Casaburi, I.; Rosano, C.; Avena, P.; De Luca, A.; Campana, C.; Martire, E.; Santolla, M.F.; Maggiolini, M.; Pezzi, V.; et al. Oleuropein and hydroxytyrosol activate GPER/ GPR30-dependent pathways leading to apoptosis of ER-negative SKBR3 breast cancer cells. Mol. Nutr. Food Res. 2014, 58, 478–489. [Google Scholar] [CrossRef] [PubMed]
- Weissenborn, C.; Ignatov, T.; Ochel, H.J.; Costa, S.D.; Zenclussen, A.C.; Ignatova, Z.; Ignatov, A. GPER functions as a tumor suppressor in triple-negative breast cancer cells. J. Cancer Res. Clin. 2014, 140, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Weissenborn, C.; Ignatov, T.; Poehlmann, A.; Wege, A.K.; Costa, S.D.; Zenclussen, A.C.; Ignatov, A. GPER functions as a tumor suppressor in MCF-7 and SK-BR-3 breast cancer cells. J. Cancer Res. Clin. 2014, 140, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Chen, Z.J.; Zhang, K.S.; Yang, X.L.; Wu, Y.M.; Chen, X.H.; Huang, H.B.; Liu, H.L.; Cai, S.H.; Du, J.; et al. The activation of G protein-coupled receptor 30 (GPR30) inhibits proliferation of estrogen receptor-negative breast cancer cells in vitro and in vivo. Cell Ceath Dis. 2014, 5, e1428. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Han, Z.; Shao, L.; Zhao, Y. Ultrasound-targeted microbubble destruction of calcium channel subunit alpha 1D siRNA inhibits breast cancer via G protein-coupled receptor 30. Oncol. Rep. 2016, 36, 1886–1892. [Google Scholar] [CrossRef] [PubMed]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Bio. 2013, 13, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galluzzi, L.; Brenner, C. Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 2007, 87, 99–163. [Google Scholar] [CrossRef] [PubMed]
- Urano, Y.; Vo, H.D.K.; Hirofumi, A.; Noguchi, N. 24(S)-Hydroxycholesterol induces ER dysfunction-mediated unconventional cell death. Cell Ceath Discov. 2019, 5, 113. [Google Scholar] [CrossRef]
- Ron, D.; Hubbard, S.R. How IRE1 reacts to ER stress. Cell 2008, 132, 24–26. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Wang, H.G. CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J. Biol. Chem. 2004, 279, 45495–45502. [Google Scholar] [CrossRef]
- Li, Y.; Guo, Y.; Tang, J.; Jiang, J.; Chen, Z. New insights into the roles of CHOP-induced apoptosis in ER stress. Acta Biochim. Biophys. Sin. 2014, 46, 629–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denmeade, S.R.; Jakobsen, C.M.; Janssen, S.; Khan, S.R.; Garrett, E.S.; Lilja, H.; Christensen, S.B.; Isaacs, J.T. Prostate-specific antigen-activated thapsigargin prodrug as targeted therapy for prostate cancer. J. Natl. Cancer Inst. 2003, 95, 990–1000. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.; Sundaram, C.; Jhurani, S.; Kunnumakkara, A.B.; Aggarwal, B.B. Curcumin and cancer: An “old-age” disease with an “age-old” solution. Cancer Lett. 2008, 267, 133–164. [Google Scholar] [CrossRef] [PubMed]
- Vo, D.K.; Urano, Y.; Takabe, W.; Saito, Y.; Noguchi, N. 24(S)-Hydroxycholesterol induces RIPK1-dependent but MLKL-independent cell death in the absence of caspase-8. Steroids 2015, 99, 230–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, M.; Millot, J.M.; Sebille, S.; Manfait, M. Inhibitory effects of extracellular Mg2+ on intracellular Ca2+ dynamic changes and thapsigargin-induced apoptosis in human cancer MCF7 cells. Mol. Cell. Biochem. 2002, 229, 163–171. [Google Scholar] [CrossRef]
- Van Noorden, C.J. The history of Z-VAD-FMK, a tool for understanding the significance of caspase inhibition. Acta Histochem. 2001, 103, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.K.; Clavarino, G.; Ceppi, M.; Pierre, P. SUnSET, a nonradioactive method to monitor protein synthesis. Nat. Methods 2009, 6, 275–277. [Google Scholar] [CrossRef]
- Lim, M.L.; Minamikawa, T.; Nagley, P. The protonophore CCCP induces mitochondrial permeability transition without cytochrome c release in human osteosarcoma cells. FEBS Lett. 2001, 503, 69–74. [Google Scholar] [CrossRef]
- Scaling, A.L.; Prossnitz, E.R.; Hathaway, H.J. GPER mediates estrogen-induced signaling and proliferation in human breast epithelial cells and normal and malignant breast. Horm. Cancer 2014, 5, 146–160. [Google Scholar] [CrossRef]
- Luo, H.; Yang, G.; Yu, T.; Luo, S.; Wu, C.; Sun, Y.; Liu, M.; Tu, G. GPER-mediated proliferation and estradiol production in breast cancer-associated fibroblasts. Endocr. Relat. Cancer 2014, 21, 355–369. [Google Scholar] [CrossRef] [Green Version]
- Nass, N.; Walter, S.; Jechorek, D.; Weissenborn, C.; Ignatov, A.; Haybaeck, J.; Sel, S.; Kalinski, T. High neuronatin (NNAT) expression is associated with poor outcome in breast cancer. Virchows Arch. 2017, 471, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Merritt, R.; Fu, L.; Pan, Z. Targeting calcium signaling in cancer therapy. Acta Pharm. Sin. B 2017, 7, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Holm, A.; Baldetorp, B.; Olde, B.; Leeb-Lundberg, L.M.; Nilsson, B.O. The GPER1 agonist G-1 attenuates endothelial cell proliferation by inhibiting DNA synthesis and accumulating cells in the S and G2 phases of the cell cycle. J. Vasc. Res. 2011, 48, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Kim, T.W.; Park, G.L.; Hwang, Y.S.; Cho, H.J.; Kim, J.T.; Lee, H.G. G protein-coupled estrogen receptor-1 agonist induces chemotherapeutic via ER stress signaling in gastric cancer. BMB Rep. 2019. [Google Scholar]
- Alvarez-Gonzalez, R.; Spring, H.; Müller, M.; Bürkle, A. Selective loss of Poly(ADP-ribose) and the 85-kDa Fragment of Poly(ADP-ribose) Polymerase in Nucleoli during Alkylation-induced apoptosis of Hela cells. J. Biol. Chem. 1999, 274, 32122–32126. [Google Scholar] [CrossRef] [PubMed]
- Chaitanya, G.V.; Steven, A.J.; Babu, P.P. PARP-1 cleavage fragemnts: Signatures of cell-death proteases in neurodegeneration. Cell. Commun. Signal. 2010, 8, 31. [Google Scholar] [CrossRef]
- Szegezdi, E.; Logue, S.E.; Gorman, A.M.; Samali, A. Mediatora of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006, 7, 800–995. [Google Scholar] [CrossRef]
- Orrenius, S.; Zhivotovsky, B.; Nicotera, P. Regulation of cell death: The calcium-apoptosis link. Nat. Rev. Mol. Cell Biol. 2003, 4, 552–565. [Google Scholar] [CrossRef]
- Demay, Y.; Perochon, J.; Szuplewski, S.; Mignotte, B.; Gaumer, S. The PERK pathway independently triggers apoptosis amd a Rac1/Slpr/JNKJ/Dilp8 signaling favoring tissue homeostasis in a chronic ER stress Drosophila model. Cell Death Dis. 2014, 5, e1454. [Google Scholar] [CrossRef]
- Timmins, J.M.; Ozcan, L.; Seimon, T.A.; Li, G.; Malagelada, C.; Backs, J.; Backs, T.; Bassel-Duby, R.; Olson, E.N.; Anderson, M.E.; et al. Calcium/calmodulin-dependent protein kinase II links ER stress with Fas and mitochondrial apoptosis pathways. J. Clin. Investig. 2009, 119, 2925–2941. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Mongillo, M.; Chin, K.T.; Harding, H.; Ron, D.; Marks, A.R.; Tabas, I. Role of ERO1α-mediated stimulation of inositol 1, 4, 5-triphosphate receptor activity in endoplasmic reticulum stress-induced apoptosis. J. Cell. Biol. 2009, 186, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Chen, J.; Ai, Y.; Gu, X.; Li, L.; Che, D.; Jiang, Z.; Li, L.; Chen, S.; Huang, H.; et al. Estrogen-Related Hormones Induce Apoptosis by Stabilizing Schlafen-12 Protein Turnover. Mol. Cell. 2019. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Jin, Y.; Zhao, Y.; Shan, A.; Fang, H.; Shen, J.; Zhou, C.; Yu, H.; Zhou, Y.F.; Wang, X.; et al. Zearalenone induces apoptosis in bovine mammary epithelial cells by activating endoplasmic reticulum stress. J. Dairy Sci. 2019. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vo, D.-K.H.; Hartig, R.; Weinert, S.; Haybaeck, J.; Nass, N. G-Protein-Coupled Estrogen Receptor (GPER)-Specific Agonist G1 Induces ER Stress Leading to Cell Death in MCF-7 Cells. Biomolecules 2019, 9, 503. https://doi.org/10.3390/biom9090503
Vo D-KH, Hartig R, Weinert S, Haybaeck J, Nass N. G-Protein-Coupled Estrogen Receptor (GPER)-Specific Agonist G1 Induces ER Stress Leading to Cell Death in MCF-7 Cells. Biomolecules. 2019; 9(9):503. https://doi.org/10.3390/biom9090503
Chicago/Turabian StyleVo, Diep-Khanh Ho, Roland Hartig, Sönke Weinert, Johannes Haybaeck, and Norbert Nass. 2019. "G-Protein-Coupled Estrogen Receptor (GPER)-Specific Agonist G1 Induces ER Stress Leading to Cell Death in MCF-7 Cells" Biomolecules 9, no. 9: 503. https://doi.org/10.3390/biom9090503