
Double and Triple Photoionization of CCl4

 ,
,  and
and
Abstract
:1. Introduction
2. Results
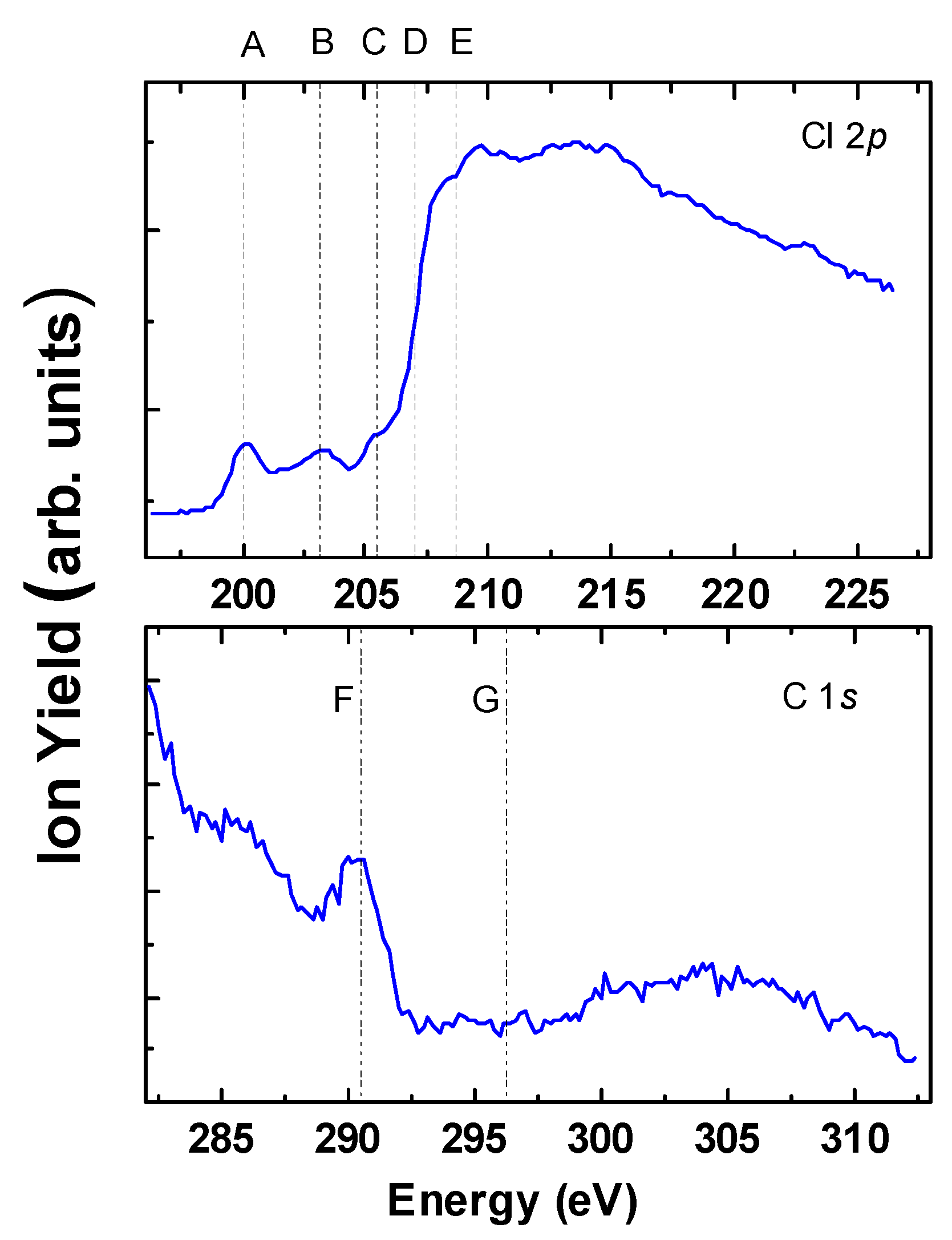
2.1. Total Ion Yield
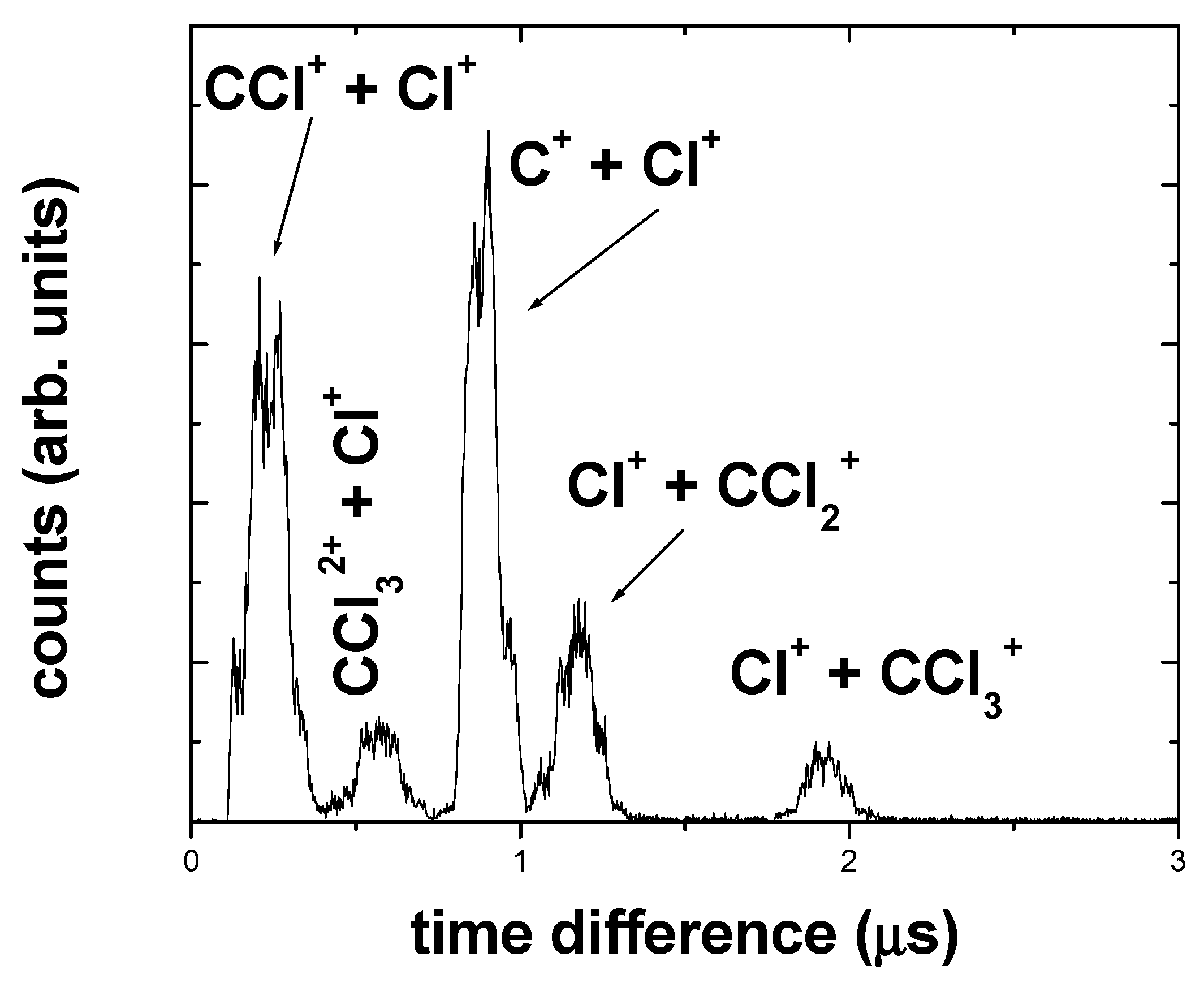
2.2. PIPICO
2.3. Kinetic Energy Release (KER)
3. Discussion
4. Materials and Methods
4.1. Experimental
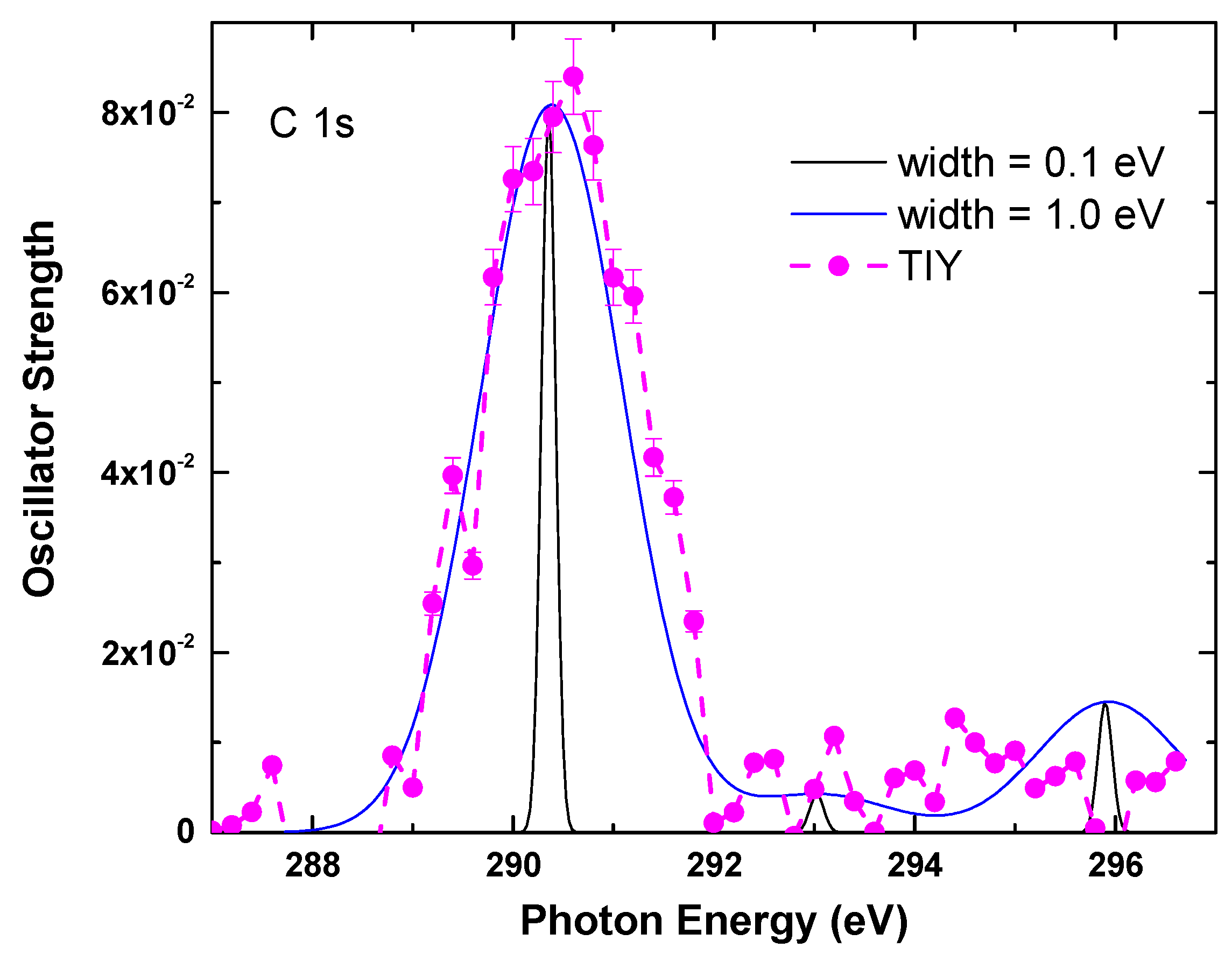
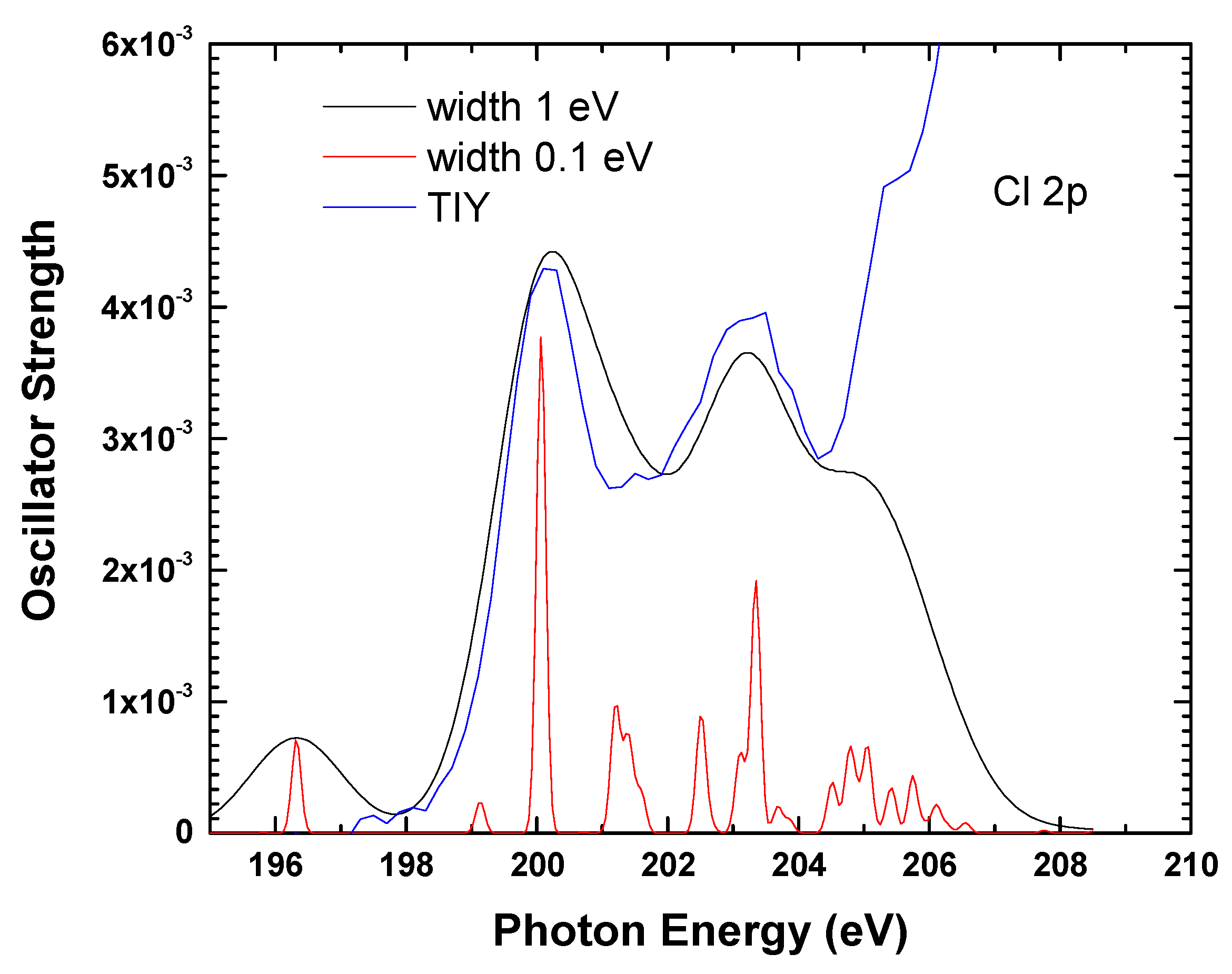
4.2. Calculations Details
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Burton, G.R.; Chan, W.F.; Cooper, G.; Brion, C.E. Valence-and inner-shell (Cl 2p, 2s; C 1s) photoabsorption and photoionization of carbon tetrachloride. Absolute oscillator strength (5–400 eV) and dipole-induced breakdown pathways. Chem. Phys. 1994, 181, 147–172. [Google Scholar] [CrossRef]
- Hitchcock, A.P.; Brion, C.E. Inner shell excitation spectroscopy of molecules using inelastic electron scattering. J. Electr. Spectr. Rel. Phen. 1978, 14, 417. [Google Scholar] [CrossRef]
- Zhang, W.; Ibuki, T.; Brion, C.E. Absolute dipole differential oscillator strengths for inner shell spectra from high resolution electron energy loss studies of the freon molecules CF4, CF3Cl, CF2Cl2, CFCl3 and CCl4. Chem. Phys. 1992, 160, 435. [Google Scholar] [CrossRef]
- O’Sullivan, G. Chlorine L-edge absorption in CCl4 and CCl2F2. J. Phys. B. At. Mol. Opt. Phys. 1982, 15, 2385. [Google Scholar] [CrossRef]
- Fainelli, F.; Maracci, R.; Platania, L.; Avaldi, J. The fragmentation of CCl4 dications studied by Auger electron-ion coincidence experiments. Electron. Spectrosc. Rel. Phenom. 2001, 119, 81. [Google Scholar] [CrossRef]
- Stolte, W.C.; Santos, A.C.F.; de Souza, G.G.B.; Sant’anna, M.M.; Leung, K.T. Deep-core photoabsorption and photofragmentation of tetrachloromethane near the Cl K-edge. J. Chem. Phys. 2017, 146, 214306. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-H.; Du, X.-J.; Xu, Y.-C.; Nie, Z.-W.; Wang, D.-H.; Wang, S.-X.; Zhu, L.-F. Generalized Oscillator Strengths for the Valence Shell Excitations in Carbon Tetrachloride Studied by Fast Electron Impact. J. Phys. Chem. A 2022, 126, 453–461. [Google Scholar] [CrossRef]
- Kokkonen, E.; Jänkälä, K.; Patanen, M.; Cao, W.; Hrast, M.; Bučar, K.; Žitnik, M.; Huttula, M. Role of ultrafast dissociation in the fragmentation of chlorinated methanes. J. Chem. Phys. 2018, 148, 174301. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, N.; Takahashi, M. Theoretical Study of Valence Shell Excitation by Electron Impact in CCl4. J. Phys. Chem. A 2023, 127, 1866–1873. [Google Scholar] [CrossRef] [PubMed]
- Santos, A.C.F.; Maciel, J.B.; de Souza, G.G.B. Valence and core level ionization of the CCl4 molecule. J. Elect. Spectr. 2007, 156, 236. [Google Scholar] [CrossRef]
- Fournier, P.G.; Comtet, G.; Fournier, J.; Svensson, S.; Karlsson, L.; Keane, M.P.; De Brito, A.N. Double-ionization energies of CCl 4 by double-charge-transfer and x-ray Auger-electron spectroscopies. J. Phys. Rev. A 1989, 40, 163. [Google Scholar] [CrossRef] [PubMed]
- Grant, R.P.; Harris, F.M.; Andrews, S.R.; Parry, D.E. Double ionization to singlet and triplet electronic states of CCl42+. Int. J. Mass Spectrom. Ion Process. 1995, 142, 117. [Google Scholar] [CrossRef]
- Lago, A.F.; Santos, A.C.F.; de Souza, G.G.B. Mass spectrometry study of the fragmentation of valence and core-shell (Cl2p) excited CHCl3 and CDCl3 molecules. J. Chem. Phys. 2004, 120, 9547. [Google Scholar] [CrossRef]
- Lago, A.F.; Santos, A.C.F.; de Souza, G.G.B. Charge separation mass spectrometry study of the chloroform molecule following valence and Cl 2p photoionization. Int. J. Mass Spectrom. 2007, 262, 187. [Google Scholar] [CrossRef]
- KAlcantara, K.F.; Wolff, W.; Gomes, A.H.A.; Sigaud, L.; Soriano, S.; Oliveira, V.; Rocha, A.B.; Santos, A.C.F. Fragmentation of the CH2Cl2 molecule by proton impact and VUV photons. J. Phys. B At. Mol. Opt. Phys. 2011, 44, 165205. [Google Scholar] [CrossRef]
- Alcantara, K.F.; Gomes, A.H.A.; Wolff, W.; Sigaud, L.; Santos, A.C.F. Strong electronic selectivity in the shallow core excitation of the CH2Cl2 molecule. J. Phys. Chem. A 2015, 119, 8822–8831. [Google Scholar] [CrossRef] [PubMed]
- Lindgren, A.; Gisselbrecht, M.; Burmeister, F.; Naves de Brito, A.; Kivimäki, A.; Sorensen, S.L. Molecular alignment of ammonia studied by electron-ion-ion coincidence spectroscopy. J. Chem. Phys. 2005, 122, 114306. [Google Scholar] [CrossRef] [PubMed]
- Burmeister, F.; Coutinho, L.H.; Marinho, R.R.T.; Homem, M.G.P.; De Morais, M.A.A.; Mocellin, A.; Björneholm, O.; Sorensen, S.L.; Fonseca, P.D.T.; Lindgren, A.; et al. Description and performance of an electron-ion coincidence TOF spectrometer used at the Brazilian synchrotron facility LNLS. J. Electron. Spectrosc. Relat. Phenom. 2010, 180, 6. [Google Scholar] [CrossRef]
- Rocha, A.B. Potential curves for inner-shell states of CO calculated at multiconfigurational self-consistent field level. J. Chem. Phys. 2011, 134, 024107. [Google Scholar] [CrossRef] [PubMed]
- Rocha, A.B. Spin-orbit splitting for inner-shell 2p states. J. Mol. Model. 2014, 20, 2355. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Energy (eV) | CCl+ + Cl+ | CCl32+ + Cl+ | C+ + Cl+ | CCl2+ + Cl+ | CCl3+ + Cl+ |
|---|---|---|---|---|---|
| 35.0 | 12.7 | -- | 3.7 | 37.5 | 46.0 |
| 147.5 | 42.7 | 4.6 | 25.6 | 18.8 | 8.3 |
| 201.3 | 43.0 | 4.0 | 31.4 | 15.1 | 6.4 |
| 204.5 | 44.8 | 3.9 | 31.1 | 13.9 | 6.3 |
| 206.7 | 39.6 | 4.0 | 32.3 | 16.0 | 8.1 |
| 209.5 | 36.5 | 3.4 | 25.5 | 24.4 | 10.2 |
| 210.9 | 39.7 | 3.3 | 24.8 | 23.5 | 8.7 |
| 216.1 | 37.1 | 3.5 | 24.6 | 25.1 | 9.8 |
| 224,0 | 39.7 | 3.4 | 27.4 | 21.4 | 8.0 |
| 237.5 | 39.0 | 4.1 | 31.2 | 19.0 | 6.6 |
| 283.5 | 35.7 | 7.4 | 38.5 | 13.7 | 4.7 |
| 290.5 | 36.8 | 7.5 | 38.7 | 13.0 | 4.1 |
| 302.4 | 35.8 | 8.2 | 39.3 | 12.5 | 4.3 |
| Coincidence | Photon Energy (eV) | Columb Model | ||
|---|---|---|---|---|
| 209.5 | 282.5 | 302.5 | ||
| Cl+ + CCl3+ | 12.7 | -- | 3.7 | 8.2 |
| Cl + + CCl2+ | 42.7 | 4.6 | 25.6 | 8.2 |
| Cl+ + C+ | 43.0 | 4.0 | 31.4 | 8.2 |
| Cl+ + CCl32+ | 44.8 | 3.9 | 31.1 | 16.4 |
| Cl+ + CCl+ | 39.6 | 4.0 | 32.3 | 8.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
dos Santos, A.C.F.; Maciel, J.B.; Rocha, A.B.; de Souza, G.G.B. Double and Triple Photoionization of CCl4. Atoms 2024, 12, 74. https://doi.org/10.3390/atoms12120074
dos Santos ACF, Maciel JB, Rocha AB, de Souza GGB. Double and Triple Photoionization of CCl4. Atoms. 2024; 12(12):74. https://doi.org/10.3390/atoms12120074
Chicago/Turabian Styledos Santos, Antônio Carlos Fontes, Joselito Barbosa Maciel, Alexandre Braga Rocha, and Gerardo Gerson Bezerra de Souza. 2024. "Double and Triple Photoionization of CCl4" Atoms 12, no. 12: 74. https://doi.org/10.3390/atoms12120074
APA Styledos Santos, A. C. F., Maciel, J. B., Rocha, A. B., & de Souza, G. G. B. (2024). Double and Triple Photoionization of CCl4. Atoms, 12(12), 74. https://doi.org/10.3390/atoms12120074