Genetic Risk Scores Associated with Baseline Lipoprotein Subfraction Concentrations Do Not Associate with Their Responses to Fenofibrate

Abstract
:1. Introduction
2. Experimental Section
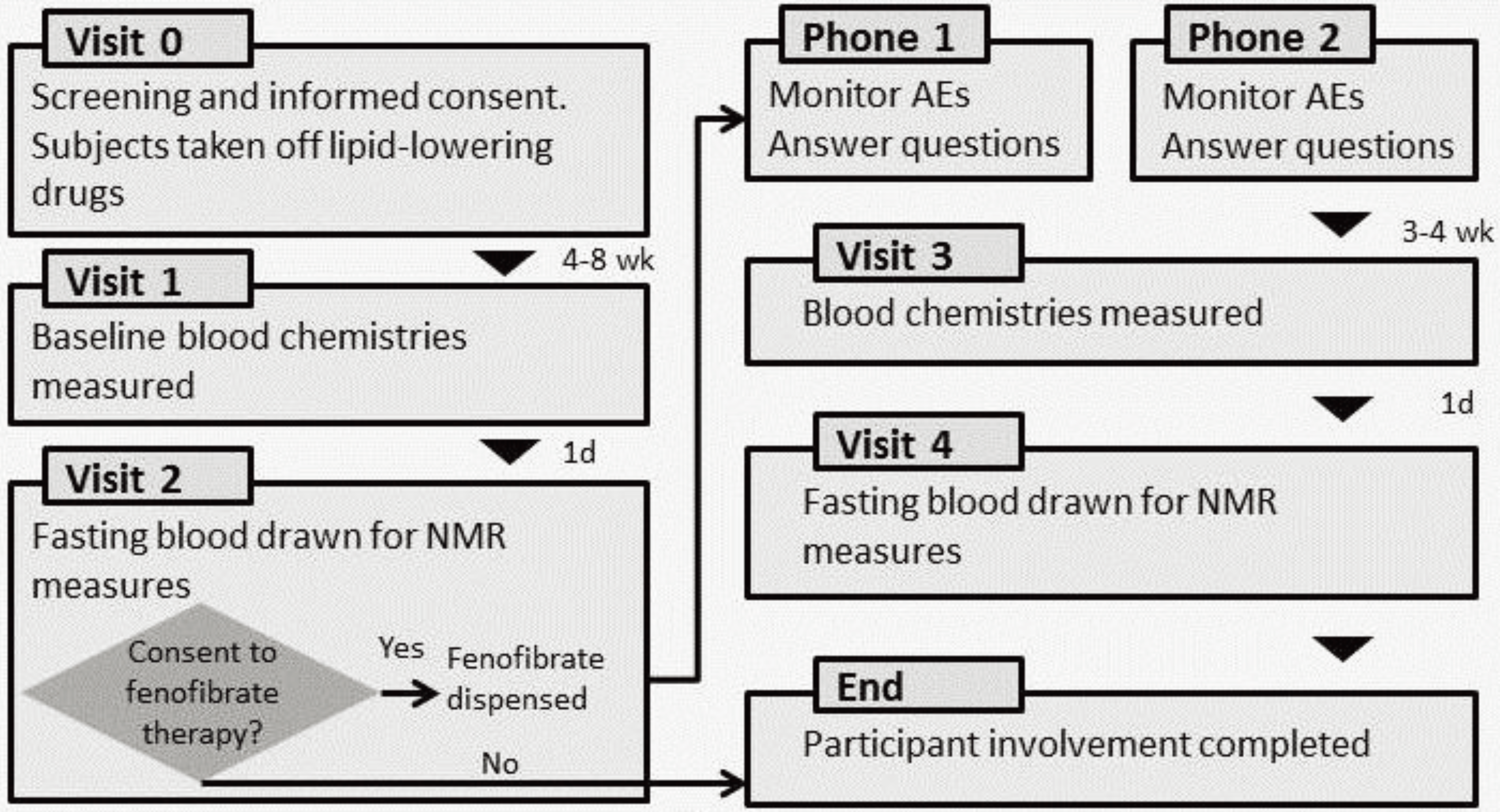
2.1. Study Population

2.2. Biochemical Measurements

{kind=link}
| NMR Lipoprotein Parameter | Diameter Range (nm) |
|---|---|
| VLDL | |
| Large VLDL/chylomicrons | >60 |
| Medium VLDL | 35–60 |
| Small VLDL | 27–35 |
| LDL | |
| Large LDL | 21.2–23 |
| Small LDL | 18–21.2 |
| HDL | |
| Large HDL | 8.8–13 |
| Medium HDL | 8.2–8.8 |
| Small HDL | 7.3–8.2 |
2.3. Genotyping
2.4. Statistical Methods
3. Results
| Age, year | 48.42 (16.34) | ||
| Gender, % male | 48.28 | ||
| Smoker, % current | 7.79 | ||
| BMI | 28.25 (5.62) | ||
| Baseline | Post-Fenofibrate | p | |
| Small VLDL concentration, nmol/L | 32.93 (21.89) | 22.83 (15.83) | <0.0001 |
| Medium VLDL concentration, nmol/L | 37.49 (36.74) | 18.46 (20.44) | <0.0001 |
| Large VLDL concentration, nmol/L | 3.93 (7.68) | 2.09 (3.24) | <0.0001 |
| VLDL total particles; nmol/L | 74.38 (50.81) | 43.41 (31.73) | <0.0001 |
| VLDL diameter; nm | 51.39 (7.86) | 51.84 (8.71) | 0.14 |
| Small LDL concentration, nmol/L | 925.64 (557.03) | 779.49 (373.04) | <0.0001 |
| Large LDL concentration, nmol/L | 407.11 (272.91) | 397.23 (201.37) | 0.19 |
| LDL total particles; nmol/L | 1375.33 (437.46) | 1209.35 (380.68) | <0.0001 |
| LDL diameter; nm | 20.81 (0.88) | 20.90 (0.58) | <0.0001 |
| Small HDL concentration, nmol/L | 21.65 (5.54) | 20.76 (6.71) | 0.003 |
| Medium HDL concentration, nmol/L | 3.00 (3.61) | 5.92 (4.80) | <0.0001 |
| Large HDL concentration, nmol/L | 6.32 (3.54) | 5.73 (3.25) | <0.0001 |
| HDL total particles; nmol/L | 30.97 (5.63) | 32.42 (5.95) | <0.0001 |
| HDL diameter; nm | 8.85 (0.45) | 8.73 (0.39) | <0.0001 |
| Baseline fasting TG < 150 mg/dL (N = 544) | |||
| Small VLDL concentration, nmol/L | 28.13 (15.67) | 18.60 (12.60) | <0.001 |
| Medium VLDL concentration, nmol/L | 21.06 (14.68) | 12.34 (11.07) | <0.001 |
| Large VLDL concentration, nmol/L | 1.39 (1.53) | 0.95 (1.22) | <0.001 |
| VLDL total particles; nmol/L | 50.62 (24.01) | 31.92 (19.76) | <0.001 |
| VLDL diameter; nm | 50.73 (8.20) | 51.12 (9.19) | 0.44 |
| Small LDL concentration, nmol/L | 705.82 (403.85) | 662.38 (261.74) | <0.001 |
| Large LDL concentration, nmol/L | 478.91 (252.68) | 398.55 (184.97) | <0.001 |
| LDL total particles; nmol/L | 1213.23 (364.60) | 1073.37 (279.02) | <0.001 |
| LDL diameter; nm | 21.14 (0.73) | 21.01 (0.53) | 0.001 |
| Small HDL concentration, nmol/L | 20.53 (5.17) | 19.25 (6.08) | <0.001 |
| Medium HDL concentration, nmol/L | 3.34 (3.54) | 6.67 (4.47) | <0.001 |
| Large HDL concentration, nmol/L | 7.27 (2.30) | 6.42 (3.30) | <0.001 |
| HDL total particles; nmol/L | 31.15 (5.18) | 32.35 (5.44) | <0.001 |
| HDL diameter; nm | 8.97 (0.43) | 8.83 (0.34) | <0.001 |
| Baseline fasting TG ≥ 150 mg/dL (N = 248) | |||
| Small VLDL concentration, nmol/L | 43.62 (28.93) | 31.97 (18.10) | <0.001 |
| Medium VLDL concentration, nmol/L | 74.12 (44.03) | 31.67 (28.32) | <0.001 |
| Large VLDL concentration, nmol/L | 9.60 (11.74) | 4.55 (4.60) | <0.001 |
| VLDL total particles; nmol/L | 127.38 (54.65) | 68.22 (37.96) | <0.001 |
| VLDL diameter; nm | 52.84 (6.85) | 53.39 (7.35) | 0.09 |
| Small LDL concentration, nmol/L | 1415.81 (539.35) | 1032.66 (445.66) | <0.001 |
| Large LDL concentration, nmol/L | 247.00 (247.40) | 413.84 (232.50) | <0.001 |
| LDL total particles; nmol/L | 1736.81 (488.611) | 1503.32 (404.81) | <0.001 |
| LDL diameter; nm | 20.07 (0.72) | 20.67 (0.62) | <0.001 |
| Small HDL concentration, nmol/L | 24.14 (5.54) | 24.03 (8.84) | 0.36 |
| Medium HDL concentration, nmol/L | 2.23 (3.65) | 4.30 (5.08) | <0.001 |
| Small VLDL concentration, nmol/L | 43.62 (28.93) | 31.97 (18.10) | <0.001 |
| Medium VLDL concentration, nmol/L | 74.12 (44.03) | 31.67 (28.32) | <0.001 |
| Large VLDL concentration, nmol/L | 9.60 (11.74) | 4.55 (4.60) | <0.001 |
| NMR Measure | Min | Max | Mean (SD) | Genetic Risk Score | |||||
|---|---|---|---|---|---|---|---|---|---|
| GRS-Phenotype Associations | |||||||||
| Baseline | Fenofibrate Response | ||||||||
| F-Value | δ | p | F-Value | δ | p | ||||
| Full GOLDN sample (n = 817) | |||||||||
| Small VLDL concentration | 9.44 | 22.02 | 15.75 (2.11) | 0.04 | 0.01 | 0.85 | 0.45 | 0.05 | 0.50 |
| Medium VLDL concentration | 5.26 | 13.65 | 9.79 (1.49) | 3.97 | 0.14 | 0.04 | 0.39 | 0.04 | 0.53 |
| Large VLDL concentration | 9.16 | 22.49 | 15.87 (2.34) | 0.66 | 0.06 | 0.41 | 1.77 | 0.09 | 0.18 |
| VLDL total particles | 1.01 | 8.34 | 5.15 (1.16) | 26.37 | 0.36 | <0.0001 * | 0.50 | 0.05 | 0.48 |
| VLDL diameter | 1.00 | 10.09 | 5.32 (1.61) | 2.03 | 0.10 | <0.15 | 0.01 | 0.007 | 0.93 |
| Small LDL concentration | 18.00 | 27.99 | 23.25 (1.63) | 8.90 | 0.21 | 0.003 * | 1.65 | 0.09 | 0.20 |
| Large LDL concentration | 8.37 | 24.01 | 16.29 (2.48) | 11.48 | 0.24 | 0.0007 * | 0.00 | 0 | 0.99 |
| LDL total particles | 5.96 | 19.65 | 12.39 (2.16) | 8.10 | 0.20 | 0.004 * | 0.24 | 0.03 | 0.62 |
| LDL diameter | 0.14 | 7.97 | 3.55 (1.25) | 7.04 | 0.19 | 0.008 * | 0.24 | 0.03 | 0.63 |
| Small HDL concentration | 5.00 | 18.01 | 10.64 (2.06) | 13.86 | 0.26 | 0.0002 * | 0.02 | 0.01 | 0.87 |
| Medium HDL concentration | 7.27 | 17.27 | 12.25 (1.77) | 9.11 | 0.21 | 0.0003 * | 6.38 | 0.17 | 0.01 |
| Large HDL concentration | 5.98 | 19.05 | 12.26 (1.97) | 12.14 | 0.24 | <0.0005 * | 0.19 | 0.03 | 0.67 |
| HDL total particles | 1.19 | 10.88 | 5.91 (1.66) | 2.56 | 0.11 | 0.11 | 0.35 | 0.04 | 0.56 |
| HDL diameter | 6.80 | 20.46 | 13.50 (2.23) | 10.05 | 0.22 | <0.002 * | 0.54 | 0.05 | 0.46 |
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jeyarajah, E.J.; Cromwell, W.C.; Otvos, J.D. Lipoprotein particle analysis by nuclear magnetic resonance spectroscopy. Clin. Lab. Med. 2006, 26, 847–870. [Google Scholar] [CrossRef] [PubMed]
- Gray, R.S.; Robbins, D.C.; Wang, W.; Yeh, J.L.; Fabsitz, R.R.; Cowan, L.D.; Welty, T.K.; Lee, E.T.; Krauss, R.M.; Howard, B.V. Relation of LDL size to the insulin resistance syndrome and coronary heart disease in American Indians. The Strong Heart Study. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 2713–2720. [Google Scholar] [CrossRef]
- Mykkanen, L.; Haffner, S.M.; Rainwater, D.L.; Karhapaa, P.; Miettinen, H.; Laakso, M. Relationship of LDL size to insulin sensitivity in normoglycemic men. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 1447–1453. [Google Scholar] [CrossRef] [PubMed]
- Mora, S.; Szklo, M.; Otvos, J.D.; Greenland, P.; Psaty, B.M.; Goff, D.C., Jr.; O’Leary, D.H.; Saad, M.F.; Tsai, M.Y.; Sharrett, A.R. LDL particle subclasses, LDL particle size, and carotid atherosclerosis in the Multi-Ethnic Study of Atherosclerosis (MESA). Atherosclerosis 2007, 192, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Hulthe, J.; Bokemark, L.; Wikstrand, J.; Fagerberg, B. The metabolic syndrome, LDL particle size, and atherosclerosis: The atherosclerosis and insulin resistance (AIR) study. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 2140–2147. [Google Scholar] [CrossRef] [PubMed]
- Vakkilainen, J.; Steiner, G.; Ansquer, J.C.; Aubin, F.; Rattier, S.; Foucher, C.; Hamsten, A.; Taskinen, M.R. Relationships between low-density lipoprotein particle size, plasma lipoproteins, and progression of coronary artery disease: The diabetes atherosclerosis intervention study (DAIS). Circulation 2003, 107, 1733–1737. [Google Scholar] [CrossRef] [PubMed]
- Frazier-Wood, A.C.; Glasser, S.; Garvey, W.T.; Kabagambe, E.K.; Borecki, I.B.; Tiwari, H.K.; Tsai, M.Y.; Hopkins, P.N.; Ordovas, J.M.; Arnett, D.K. A clustering analysis of lipoprotein diameters in the metabolic syndrome. Lipids Health Dis. 2011, 10. [Google Scholar] [CrossRef]
- Sacks, F.M.; Campos, H. Low-density lipoprotein size and cardiovascular disease: A reappraisal. J. Clin. Endocrinol. Metab. 2003, 88, 4525–4532. [Google Scholar] [CrossRef] [PubMed]
- Chasman, D.I.; Paré, G.; Mora, S.; Hopewell, J.C.; Peloso, G.; Clarke, R.; Cupples, L.A.; Hamsten, A.; Kathiresan, S.; Mälarstig, A.; et al. Forty-three loci associated with plasma lipoprotein size, concentration, and cholesterol content in genome-wide analysis. PLoS Genet. 2009, 5, e1000730. [Google Scholar] [CrossRef] [PubMed]
- Petersen, A.K.; Stark, K.; Musameh, M.D.; Nelson, C.P.; Römisch-Margl, W.; Kremer, W.; Raffler, J.; Krug, S.; Skurk, T.; Rist, M.J.; et al. Genetic associations with lipoprotein subfractions provide information on their biological nature. Hum. Mol. Genet. 2012, 21, 1433–1443. [Google Scholar]
- Tukiainen, T.; Kettunen, J.; Kangas, A.J.; Lyytikäinen, L.P.; Soininen, P.; Sarin, A.P.; Tikkanen, E.; O’Reilly, P.F.; Savolainen, M.J.; Kaski, K.; et al. Detailed metabolic and genetic characterization reveals new associations for 30 known lipid loci. Hum. Mol. Genet. 2012, 21, 1444–1455. [Google Scholar] [CrossRef] [PubMed]
- Knopp, R.H.; Walden, C.E.; Warnick, G.R.; Albers, J.J.; Ginsberg, J.; McGinnis, B.M. Effect of fenofibrate treatment on plasma lipoprotein lipids, high-density lipoprotein cholesterol subfractions, and apolipoproteins B, AI, AII, and E. Am. J. Med. 1987, 83, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Lemieux, I.; Laperrière, L.; Dzavik, V.; Tremblay, G.; Bourgeois, J.; Després, J.P. A 16-week fenofibrate treatment increases LDL particle size in type IIA dyslipidemic patients. Atherosclerosis 2002, 162, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Melenovsky, V.; Malik, J.; Wichterle, D.; Simek, J.; Pisarikova, A.; Skrha, J.; Poledne, R.; Stavek, P.; Ceska, R. Comparison of the effects of atorvastatin or fenofibrate on nonlipid biochemical risk factors and the LDL particle size in subjects with combined hyperlipidemia. Am. Heart J. 2002, 144, E11–E18. [Google Scholar] [CrossRef]
- Williams, P.T.; Krauss, R.M.; Vranizan, K.M.; Albers, J.J.; Wood, P.D. Effects of weight-loss by exercise and by diet on apolipoproteins A-I and A-II and the particle-size distribution of high-density lipoproteins in men. Metab. Clin. Exp. 1992, 41, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Mauger, J.F.; Lichtenstein, A.H.; Ausman, L.M.; Jalbert, S.M.; Jauhiainen, M.; Ehnholm, C.; Lamarche, B. Effect of different forms of dietary hydrogenated fats on LDL particle size. Am. J. Clin. Nutr. 2003, 78, 370–375. [Google Scholar] [PubMed]
- Okopien, B.; Krysiak, R.; Herman, Z.S. Effects of short-term fenofibrate treatment on circulating markers of inflammation and hemostasis in patients with impaired glucose tolerance. J. Clin. Endocrinol. Metab. 2006, 91, 1770–1778. [Google Scholar] [CrossRef] [PubMed]
- Lemieux, I.; Salomon, H.; Després, J.P. Contribution of apo CIII reduction to the greater effect of 12-week micronized fenofibrate than atorvastatin therapy on triglyceride levels and LDL size in dyslipidemic patients. Ann. Med. 2003, 35, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Kon, K.K.; Yeal, A.J.; Hwan, H.S.; Kyu, J.D.; Sik, K.H.; Cheon, L.K.; Kyun, S.E.; Sakuma, I. Effects of fenofibrate on lipoproteins, vasomotor function, and serological markers of inflammation, plaque stabilization, and hemostasis. Atherosclerosis 2004, 174, 379–383. [Google Scholar] [CrossRef] [PubMed]
- Rosenson, R.S.; Huskin, A.L.; Wolff, D.A.; Helenowski, I.B.; Rademaker, A.W. Fenofibrate reduces fasting and postprandial inflammatory responses among hypertriglyceridemia patients with the metabolic syndrome. Atherosclerosis 2008, 198, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Belfort, R.; Berria, R.; Cornell, J.; Cusi, K. Fenofibrate reduces systemic inflammation markers independent of its effects on lipid and glucose metabolism in patients with the metabolic syndrome. J. Clin. Endocrinol. Metab. 2010, 95, 829–836. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.Q.; Arnett, D.K.; Corella, D.; Straka, R.J.; Tsai, M.Y.; Peacock, J.M.; Adiconis, X.; Parnell, L.D.; Hixson, J.E.; Province, M.A.; et al. Fenofibrate effect on triglyceride and postprandial response of apolipoprotein A5 variants: The GOLDN study. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1417–1425. [Google Scholar] [CrossRef] [PubMed]
- Vakkilainen, J.; Steiner, G.; Ansquer, J.C.; Perttunen-Nio, H.; Taskinen, M.R. Fenofibrate lowers plasma triglycerides and increases LDL particle diameter in subjects with type 2 diabetes. Diabetes Care 2002, 25, 627–628. [Google Scholar] [CrossRef] [PubMed]
- Guerin, M.; Bruckert, E.; Dolphin, P.J.; Turpin, G.; Chapman, M.J. Fenofibrate reduces plasma cholesteryl ester transfer from HDL to VLDL and normalizes the atherogenic, dense LDL profile in combined hyperlipidemia. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Hiukka, A.; Leinonen, E.; Jauhiainen, M.; Sundvall, J.; Ehnholm, C.; Keech, A.C.; Taskinen, M.R. Long-term effects of fenofibrate on VLDL and HDL subspecies in participants with type 2 diabetes mellitus. Diabetologia 2007, 50, 2067–2075. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.Y.; Ordovas, J.M.; Li, N.; Straka, R.J.; Hanson, N.Q.; Arends, V.L.; Arnett, D. Effect of fenofibrate therapy and ABCA1 polymorphisms on high-density lipoprotein subclasses in the genetics of lipid lowering drugs and diet network. Mol. Genet. Metab. 2010, 100, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Brisson, D.; Ledoux, K.; Bosse, Y.; St. Pierre, J.; Julien, P.; Perron, P.; Hudson, T.J.; Vohl, M.C.; Gaudet, D. Effect of apolipoprotein E, peroxisome proliferator-activated receptor alpha and lipoprotein lipase gene mutations on the ability of fenofibrate to improve lipid profiles and reach clinical guideline targets among hypertriglyceridemic patients. Pharmacogenetics 2002, 12, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Foucher, C.; Rattier, S.; Flavell, D.M.; Talmud, P.J.; Humphries, S.E.; Kastelein, J.J.; Ayyobi, A.; Pimstone, S.; Frohlich, J.; Ansquer, J.C.; et al. Response to micronized fenofibrate treatment is associated with the peroxisome-proliferator-activated receptors alpha G/C intron7 polymorphism in subjects with type 2 diabetes. Pharmacogenetics 2004, 14, 823–829. [Google Scholar] [CrossRef] [PubMed]
- Frazier-Wood, A.C.; Ordovas, J.M.; Straka, R.J.; Hixson, J.E.; Borecki, I.B.; Tiwari, H.K.; Arnett, D.K. The PPAR alpha gene is associated with triglyceride, low-density cholesterol and inflammation marker response to fenofibrate intervention: The GOLDN study. Pharmacogenomics J. 2013, 13, 312–317. [Google Scholar]
- Frazier-Wood, A.C.; Aslibekyan, S.; Straka, R.J.; Borecki, I.B.; Tiwari, H.K.; Lai, C.Q.; Hopkins, P.N.; Ordovas, J.M.; Arnett, D.K. Genome-wide association study indicates variants associated with insulin signaling and inflammation mediate lipoprotein responses to fenofibrate. Pharmacogenetics Genomics 2012, 22, 750–757. [Google Scholar]
- Pankow, J.S.; Province, M.A.; Hunt, S.C.; Arnett, D.K. Regarding Testing for population subdivision and association in four case-control studies. Am. J. Hum. Genet. 2002, 71, 1478–1480. [Google Scholar] [CrossRef] [PubMed]
- Otvos, J.D.; Jeyarajah, E.J.; Bennett, D.W.; Krauss, R.M. Development of a proton nuclear magnetic resonance spectroscopic method for determining plasma lipoprotein concentrations and subspecies distributions from a single, rapid measurement. Clin. Chem. 1992, 38, 1632–1638. [Google Scholar] [PubMed]
- Liu, Y.; Ordovas, J.M.; Gao, G.; Province, M.; Straka, R.J.; Tsai, M.Y.; Lai, C.Q.; Zhang, K.; Borecki, I.; Hixson, J.E.; et al. The SCARB1 gene is associated with lipid response to dietary and pharmacological interventions. J. Hum. Genet. 2008, 53, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Straka, R.J.; Burkhardt, R.T.; Fisher, J.E. Determination of fenofibric acid concentrations by HPLC after anion exchange solid-phase extraction from human serum. Ther. Drug. Monit. 2007, 29, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Korn, J.M.; Kuruvilla, F.G.; McCarroll, S.A.; Wysoker, A.; Nemesh, J.; Cawley, S.; Hubbell, E.; Veitch, J.; Collins, P.J.; Darvishi, K.; et al. Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs. Nat. Genet. 2008, 40, 1253–1260. [Google Scholar] [CrossRef] [PubMed]
- Center for statistical genetics MACH 1.0. Available online: http://www.sph.umich.edu/csg/abecasis/MACH/index.html (accessed online: 20 July 2014).
- Heller, F.R.; Desager, J.P.; Harvengt, C. Changes in plasma activities of lipolytic enzymes and lipids of normolipidemic subjects given phenobarbital, a strong microsomal inducer, alone or in combination with fenofibrate. Int. J. Clin. Pharm. Ther. Toxicol. 1988, 26, 138–142. [Google Scholar]
- Krysiak, R.; Labuzek, K.; Okopień, B. Effect of atorvastatin and fenofibric acid on adipokine release from visceral and subcutaneous adipose tissue of patients with mixed dyslipidemia and normolipidemic subjects. Pharmacol. Rep. 2009, 61, 1134–1145. [Google Scholar] [CrossRef] [PubMed]
- Dudbridge, F. Power and predictive accuracy of polygenic risk scores. PLoS Genet. 2013. [Google Scholar] [CrossRef]
- Frazier-Wood, A.C.; Garvey, W.T.; Dall, T.; Honigberg, R.; Pourfarzib, R. Opportunities for using lipoprotein subclass profile by nuclear magnetic resonance spectroscopy in assessing insulin resistance and diabetes prediction. Metab. Syndr. Relat. Disord. 2012, 10, 244–251. [Google Scholar]
- Berneis, K.K.; Krauss, R.M. Metabolic origins and clinical significance of LDL heterogeneity. J. Lipid. Res. 2002, 43, 1363–1379. [Google Scholar] [CrossRef] [PubMed]
- Mora, S.; Otvos, J.D.; Rosenson, R.S.; Pradhan, A.; Buring, J.E.; Ridker, P.M. Lipoprotein particle size and concentration by nuclear magnetic resonance and incident type 2 diabetes in women. Diabetes 2010, 59, 1153–1160. [Google Scholar] [CrossRef] [PubMed]
- Garvey, W.T.; Kwon, S.; Zheng, D.; Shaughnessy, S.; Wallace, P.; Hutto, A.; Pugh, K.; Jenkins, A.J.; Klein, R.L.; Liao, Y. Effects of insulin resistance and type 2 diabetes on lipoprotein subclass particle size and concentration determined by nuclear magnetic resonance. Diabetes 2003, 52, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Kuller, L.; Arnold, A.; Tracy, R.; Otvos, J.; Burke, G.; Psaty, B.; Siscovick, D.; Freedman, D.S.; Kronmal, R. Nuclear magnetic resonance spectroscopy of lipoproteins and risk of coronary heart disease in the cardiovascular health study. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1175–1180. [Google Scholar] [CrossRef] [PubMed]
- Grundy, S.M.; Vega, G.L. Two different views of the relationship of hypertriglyceridemia to coronary heart disease. Implications for treatment. Arch. Intern. Med. 1992, 152, 28–34. [Google Scholar] [CrossRef]
- Aslibekyan, S.; Goodarzi, M.O.; Frazier-Wood, A.C.; Yan, X.; Irvin, M.R.; Kim, E.; Tiwari, H.K.; Guo, X.; Straka, R.J.; Taylor, K.D.; et al. Variants identified in a GWAS meta-analysis for blood lipids are associated with lipid response to fenofibrate. PLoS One 2012, 7, e48663. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Frazier-Wood, A.C.; Wojczynski, M.K.; Borecki, I.B.; Hopkins, P.N.; Lai, C.-Q.; Ordovas, J.M.; Straka, R.J.; Tsai, M.Y.; Tiwari, H.K.; Arnett, D.K. Genetic Risk Scores Associated with Baseline Lipoprotein Subfraction Concentrations Do Not Associate with Their Responses to Fenofibrate. Biology 2014, 3, 536-550. https://doi.org/10.3390/biology3030536
Frazier-Wood AC, Wojczynski MK, Borecki IB, Hopkins PN, Lai C-Q, Ordovas JM, Straka RJ, Tsai MY, Tiwari HK, Arnett DK. Genetic Risk Scores Associated with Baseline Lipoprotein Subfraction Concentrations Do Not Associate with Their Responses to Fenofibrate. Biology. 2014; 3(3):536-550. https://doi.org/10.3390/biology3030536
Chicago/Turabian StyleFrazier-Wood, Alexis C., Mary K. Wojczynski, Ingrid B. Borecki, Paul N. Hopkins, Chao-Qiang Lai, Jose M. Ordovas, Robert J. Straka, Micheal Y. Tsai, Hemant K. Tiwari, and Donna K. Arnett. 2014. "Genetic Risk Scores Associated with Baseline Lipoprotein Subfraction Concentrations Do Not Associate with Their Responses to Fenofibrate" Biology 3, no. 3: 536-550. https://doi.org/10.3390/biology3030536