
Genetic Characterization in Familial Rotator Cuff Tear: An Exome Sequencing Study
 , , , ,
, , , , 
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
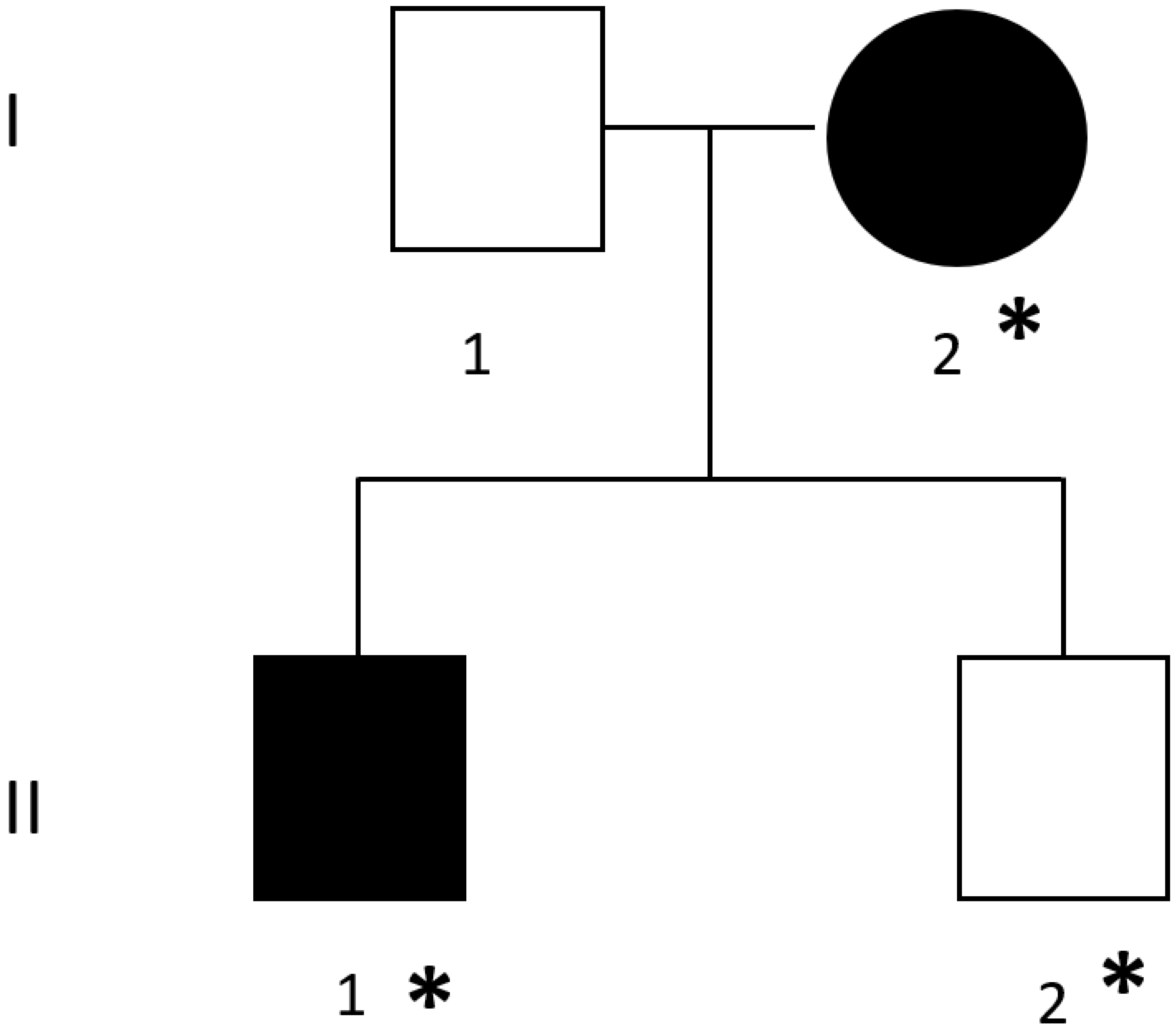
2.1. Family
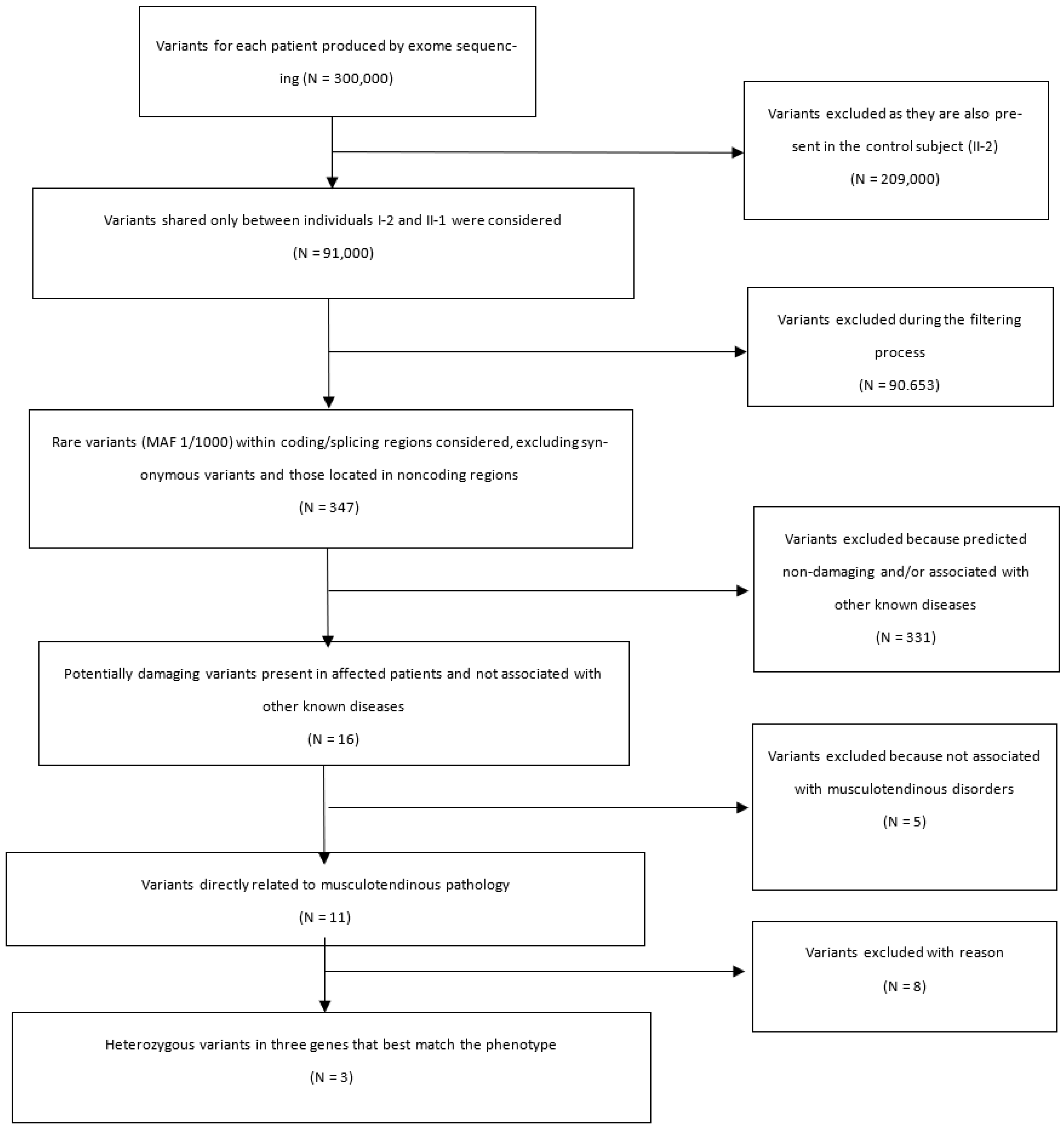
2.2. Exome Analysis by Next Generation Sequencing
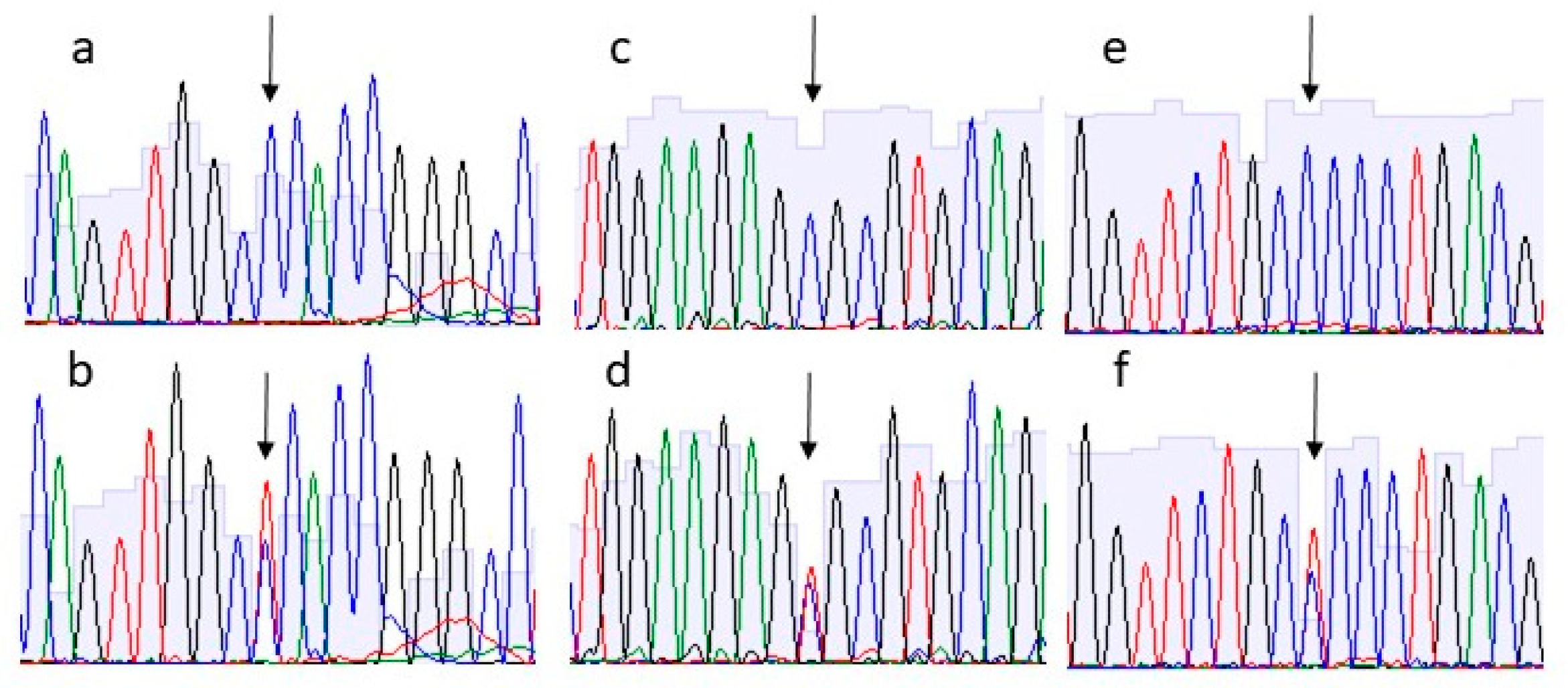
2.3. Sanger Sequencing of Variants
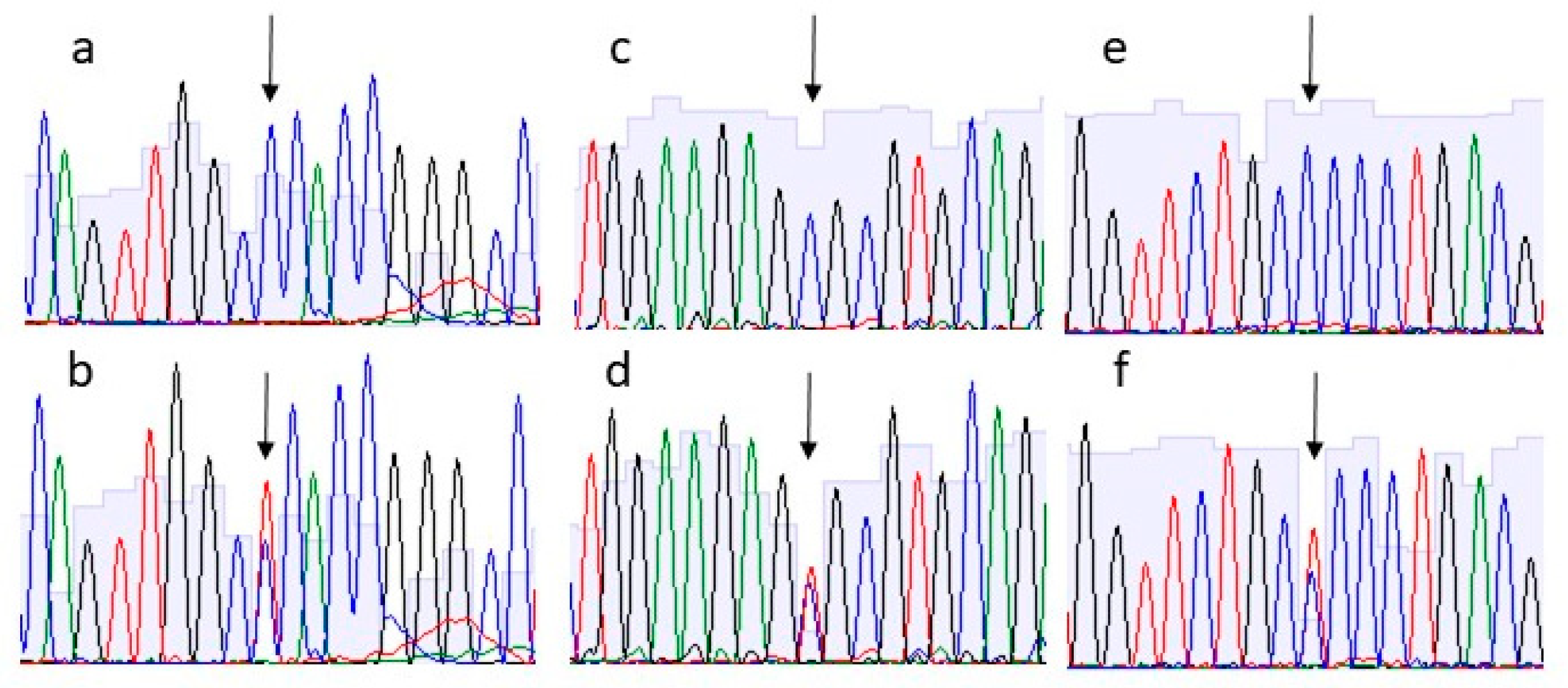
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Longo, U.G.; Salvatore, G.; Rizzello, G.; Berton, A.; Ciuffreda, M.; Candela, V.; Denaro, V. The burden of rotator cuff surgery in Italy: A nationwide registry study. Arch. Orthop. Trauma Surg. 2017, 137, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Longo, U.G.; Candela, V.; Berton, A.; Salvatore, G.; Guarnieri, A.; DeAngelis, J.; Nazarian, A.; Denaro, V. Genetic basis of rotator cuff injury: A systematic review. BMC Med. Genet. 2019, 20, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tashjian, R.Z.; Saltzman, E.G.; Granger, E.K.; Hung, M. Incidence of familial tendon dysfunction in patients with full-thickness rotator cuff tears. Open Access J. Sports Med. 2014, 5, 137–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tashjian, R.Z.; Farnham, J.M.; Albright, F.S.; Teerlink, C.C.; Cannon-Albright, L.A. Evidence for an inherited predisposition contributing to the risk for rotator cuff disease. J. Bone Jt. Surg. Am. 2009, 91, 1136–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X. Exome sequencing greatly expedites the progressive research of Mendelian diseases. Front. Med. 2014, 8, 42–57. [Google Scholar] [CrossRef] [PubMed]
- Longo, U.G.; Lamberti, A.; Maffulli, N.; Denaro, V. Tendon augmentation grafts: A systematic review. Br. Med. Bull. 2010, 94, 165–188. [Google Scholar] [CrossRef] [Green Version]
- Longo, U.G.; Lamberti, A.; Maffulli, N.; Denaro, V. Tissue engineered biological augmentation for tendon healing: A systematic review. Br. Med. Bull. 2011, 98, 31–59. [Google Scholar] [CrossRef] [Green Version]
- September, A.V.; Schwellnus, M.P.; Collins, M. Tendon and ligament injuries: The genetic component. Br. J. Sports Med. 2007, 41, 241–246, discussion 246. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Ditsios, K.; Middleton, W.D.; Hildebolt, C.F.; Galatz, L.M.; Teefey, S.A. The demographic and morphological features of rotator cuff disease. A comparison of asymptomatic and symptomatic shoulders. J. Bone Jt. Surg. Am. 2006, 88, 1699–1704. [Google Scholar] [CrossRef]
- Harvie, P.; Ostlere, S.J.; Teh, J.; McNally, E.G.; Clipsham, K.; Burston, B.J.; Pollard, T.C.; Carr, A.J. Genetic influences in the aetiology of tears of the rotator cuff. Sibling risk of a full-thickness tear. J. Bone Jt. Surg. Br. 2004, 86, 696–700. [Google Scholar] [CrossRef]
- Gwilym, S.E.; Watkins, B.; Cooper, C.D.; Harvie, P.; Auplish, S.; Pollard, T.C.; Rees, J.L.; Carr, A.J. Genetic influences in the progression of tears of the rotator cuff. J. Bone Jt. Surg. Br. 2009, 91, 915–917. [Google Scholar] [CrossRef] [PubMed]
- Teerlink, C.C.; Cannon-Albright, L.A.; Tashjian, R.Z. Significant association of full-thickness rotator cuff tears and estrogen-related receptor-β (ESRRB). J. Shoulder Elb. Surg. 2015, 24, e31–e35. [Google Scholar] [CrossRef] [PubMed]
- Motta, G.a.R.; Amaral, M.V.; Rezende, E.; Pitta, R.; Vieira, T.C.; Duarte, M.E.; Vieira, A.R.; Casado, P.L. Evidence of genetic variations associated with rotator cuff disease. J. Shoulder Elb. Surg 2014, 23, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Assunção, J.H.; Godoy-Santos, A.L.; Dos Santos, M.C.L.G.; Malavolta, E.A.; Gracitelli, M.E.C.; Ferreira Neto, A.A. Matrix Metalloproteases 1 and 3 Promoter Gene Polymorphism Is Associated With Rotator Cuff Tear. Clin. Orthop. Relat. Res. 2017, 475, 1904–1910. [Google Scholar] [CrossRef] [Green Version]
- Kluger, R.; Burgstaller, J.; Vogl, C.; Brem, G.; Skultety, M.; Mueller, S. Candidate gene approach identifies six SNPs in tenascin-C (TNC) associated with degenerative rotator cuff tears. J. Orthop. Res. 2017, 35, 894–901. [Google Scholar] [CrossRef]
- Petrillo, S.; Longo, U.G.; Margiotti, K.; Candela, V.; Fusilli, C.; Rizzello, G.; De Luca, A.; Denaro, V. Genetic factors in rotator cuff pathology: Potential influence of col 5A1 polymorphism in outcomes of rotator cuff repair. BMC Med. Genet. 2020, 21, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salles, J.I.; Lopes, L.R.; Duarte, M.E.L.; Morrissey, D.; Martins, M.B.; Machado, D.E.; Guimaraes, J.A.M.; Perini, J.A. Fc receptor-like 3 (-169T>C) polymorphism increases the risk of tendinopathy in volleyball athletes: A case control study. BMC Med. Genet. 2018, 19, 119. [Google Scholar] [CrossRef]
- Tashjian, R.Z.; Granger, E.K.; Farnham, J.M.; Cannon-Albright, L.A.; Teerlink, C.C. Genome-wide association study for rotator cuff tears identifies two significant single-nucleotide polymorphisms. J. Shoulder Elb. Surg. 2016, 25, 174–179. [Google Scholar] [CrossRef]
- Roos, T.R.; Roos, A.K.; Avins, A.L.; Ahmed, M.A.; Kleimeyer, J.P.; Fredericson, M.; Ioannidis, J.P.A.; Dragoo, J.L.; Kim, S.K. Genome-wide association study identifies a locus associated with rotator cuff injury. PLoS ONE 2017, 12, e0189317. [Google Scholar] [CrossRef] [Green Version]
- Goecks, J.; Nekrutenko, A.; Taylor, J.; Team, G. Galaxy: A comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome. Biol. 2010, 11, R86. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebbert, M.T.; Wadsworth, M.E.; Staley, L.A.; Hoyt, K.L.; Pickett, B.; Miller, J.; Duce, J.; Kauwe, J.S.; Ridge, P.G.; Initiative, A.s.D.N. Evaluating the necessity of PCR duplicate removal from next-generation sequencing data and a comparison of approaches. BMC Bioinform. 2016, 17 (Suppl. S7), 239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Stelzer, G.; Plaschkes, I.; Oz-Levi, D.; Alkelai, A.; Olendder, T.; Zimmerman, S.; Twik, M.; Belinky, F.; Fishilevich, S.; Nudel, R.; et al. VarElect: The phenotype-based variation prioritizer of the GeneCards Suite. BMC Genom. 2016, 17 (Suppl. S2), 444. [Google Scholar] [CrossRef] [Green Version]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.11–11.10.33. [Google Scholar] [CrossRef] [Green Version]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- Koch, M.; Veit, G.; Stricker, S.; Bhatt, P.; Kutsch, S.; Zhou, P.; Reinders, E.; Hahn, R.A.; Song, R.; Burgeson, R.E.; et al. Expression of type XXIII collagen mRNA and protein. J. Biol. Chem. 2006, 281, 21546–21557. [Google Scholar] [CrossRef] [Green Version]
- Banyard, J.; Bao, L.; Zetter, B.R. Type XXIII collagen, a new transmembrane collagen identified in metastatic tumor cells. J. Biol. Chem. 2003, 278, 20989–20994. [Google Scholar] [CrossRef] [Green Version]
- Xu, F.; Chang, K.; Ma, J.; Qu, Y.; Xie, H.; Dai, B.; Gan, H.; Zhang, H.; Shi, G.; Zhu, Y.; et al. The Oncogenic Role of COL23A1 in Clear Cell Renal Cell Carcinoma. Sci. Rep. 2017, 7, 9846. [Google Scholar] [CrossRef]
- Leimeister, C.; Steidl, C.; Schumacher, N.; Erhard, S.; Gessler, M. Developmental expression and biochemical characterization of Emu family members. Dev. Biol. 2002, 249, 204–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doi, M.; Nagano, A.; Nakamura, Y. Molecular cloning and characterization of a novel gene, EMILIN-5, and its possible involvement in skeletal development. Biochem. Biophys Res. Commun. 2004, 313, 888–893. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.J.; Liu, J.; Bertos, N.R.; Yang, X.J. Identification of HDAC10, a novel class II human histone deacetylase containing a leucine-rich domain. Nucleic Acids Res. 2002, 30, 1114–1123. [Google Scholar] [CrossRef] [Green Version]
- Kluger, R.; Huber, K.R.; Seely, P.G.; Berger, C.E.; Frommlet, F. Novel Tenascin-C Haplotype Modifies the Risk for a Failure to Heal After Rotator Cuff Repair. Am. J. Sports Med. 2017, 45, 2955–2964. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.; Sun, J.; Liu, W.; Li, W.; Jia, J.; Ou, F.; Su, K.; Zheng, Y.; Zhang, Z.; Sun, Y. HDAC10 upregulation contributes to interleukin 1β-mediated inflammatory activation of synovium-derived mesenchymal stem cells in temporomandibular joint. J. Cell. Physiol. 2019, 234, 12646–12662. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Varshney, R.R.; Ren, L.; Cai, D.; Wang, D.A. Synovium-derived mesenchymal stem cells: A new cell source for musculoskeletal regeneration. Tissue Eng. Part B Rev. 2009, 15, 75–86. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azzarà, A.; Risi Ambrogioni, L.; Cassano, I.; Lintas, C.; Longo, U.G.; Denaro, V.; Gurrieri, F. Genetic Characterization in Familial Rotator Cuff Tear: An Exome Sequencing Study. Biology 2022, 11, 1565. https://doi.org/10.3390/biology11111565
Azzarà A, Risi Ambrogioni L, Cassano I, Lintas C, Longo UG, Denaro V, Gurrieri F. Genetic Characterization in Familial Rotator Cuff Tear: An Exome Sequencing Study. Biology. 2022; 11(11):1565. https://doi.org/10.3390/biology11111565
Chicago/Turabian StyleAzzarà, Alessia, Laura Risi Ambrogioni, Ilaria Cassano, Carla Lintas, Umile Giuseppe Longo, Vincenzo Denaro, and Fiorella Gurrieri. 2022. "Genetic Characterization in Familial Rotator Cuff Tear: An Exome Sequencing Study" Biology 11, no. 11: 1565. https://doi.org/10.3390/biology11111565