Tannic Acid-Mediated Aggregate Stabilization of Poly(N-vinylpyrrolidone)-b-poly(oligo (ethylene glycol) methyl ether methacrylate) Double Hydrophilic Block Copolymers




Abstract
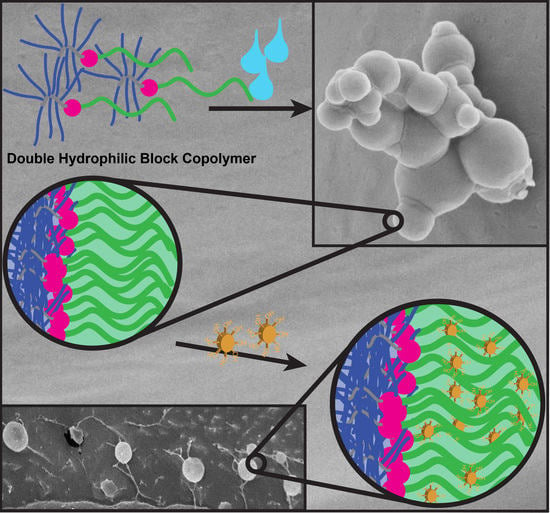
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
3. Results
3.1. Synthesis of PVP-b-P(OEGMA)
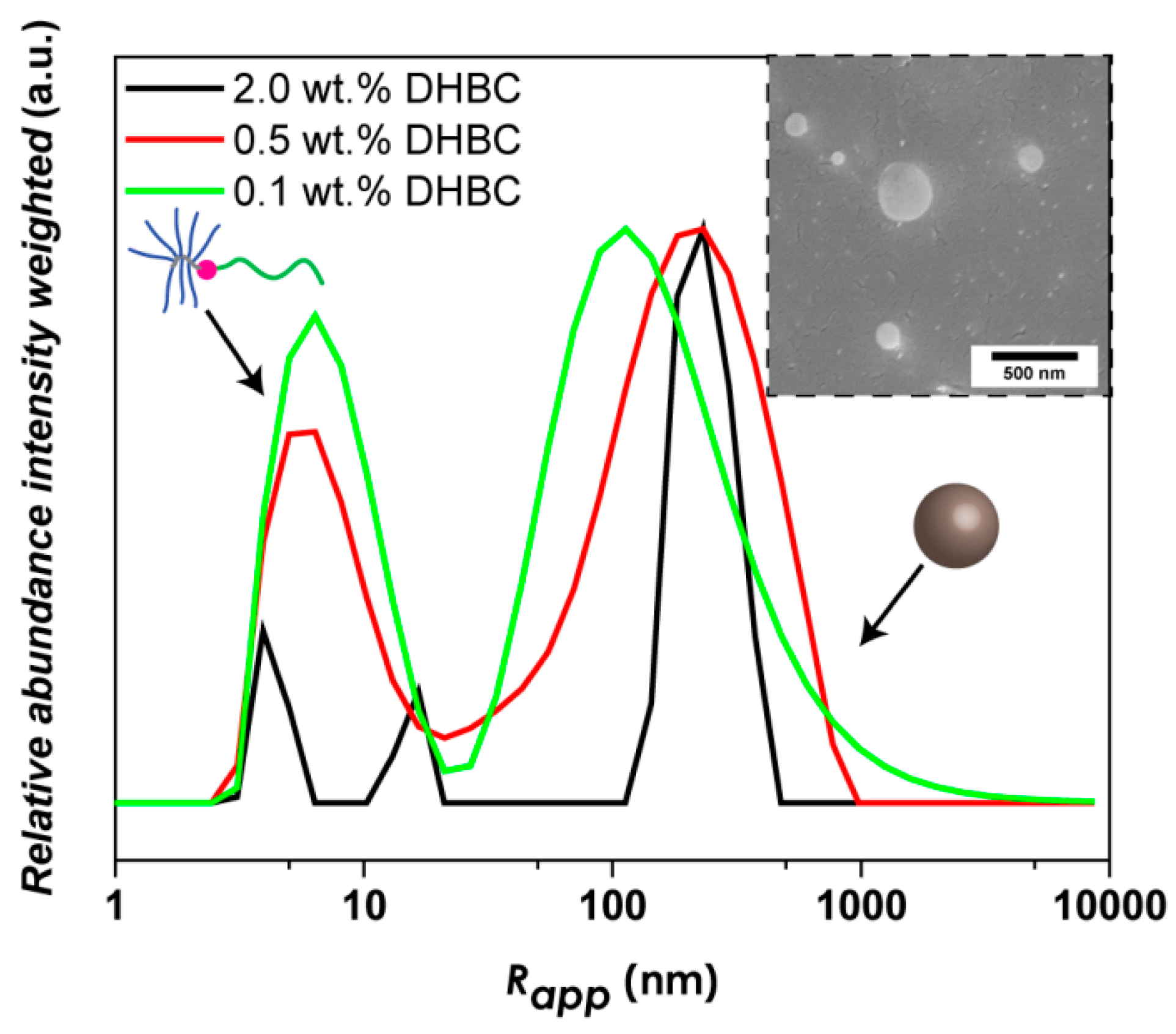
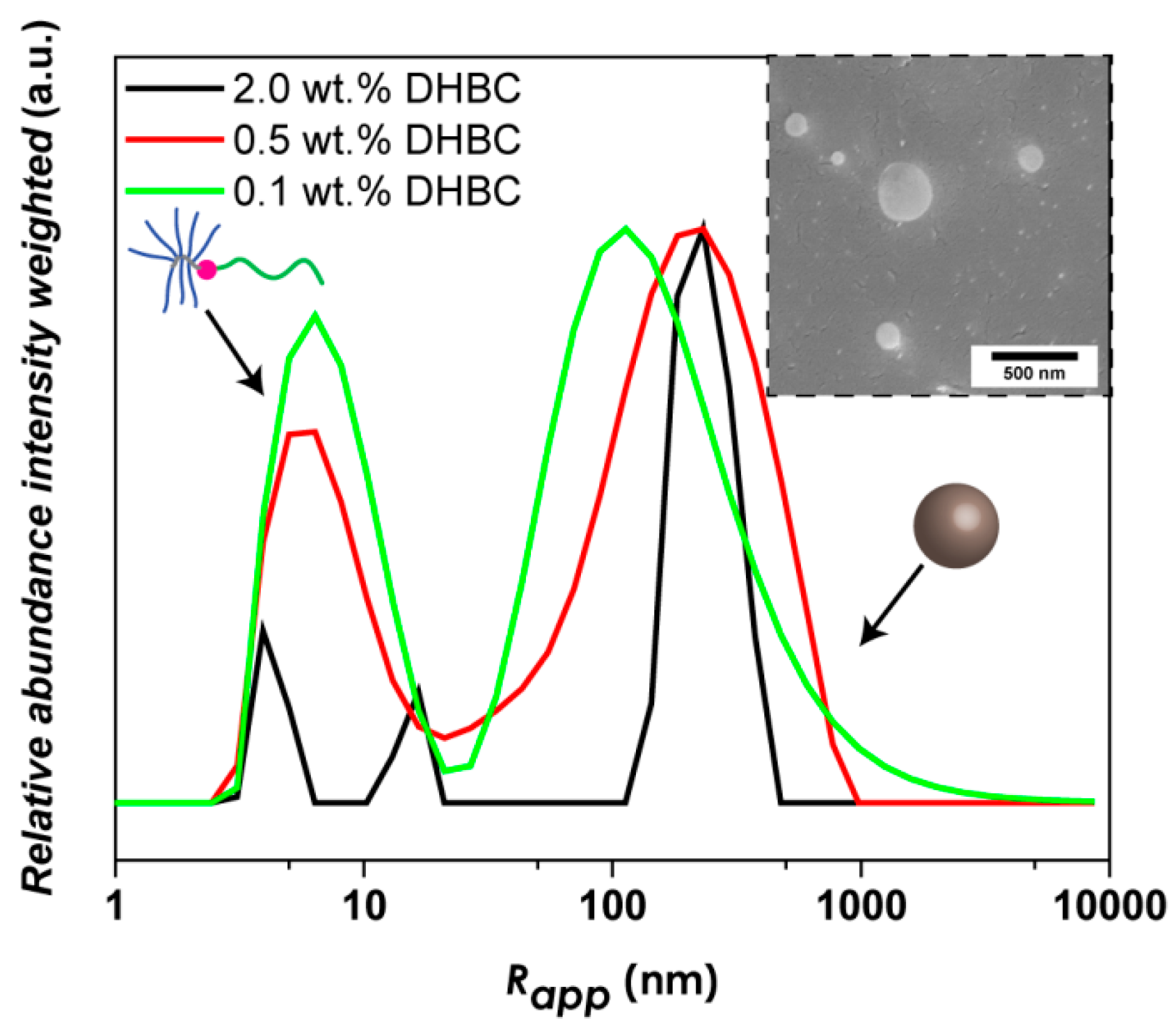
3.2. PVP-b-P(OEGMA) Aggregate Formation
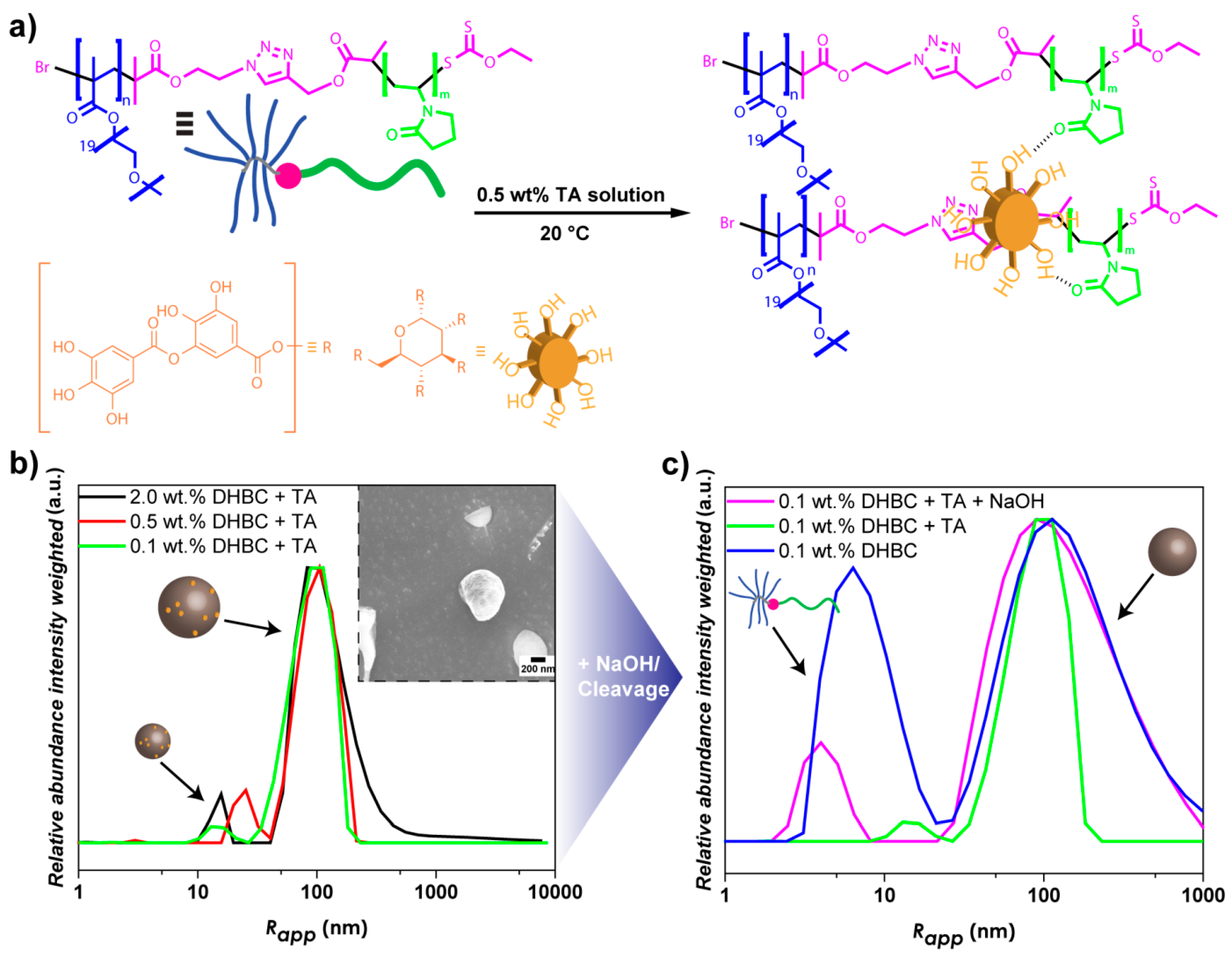
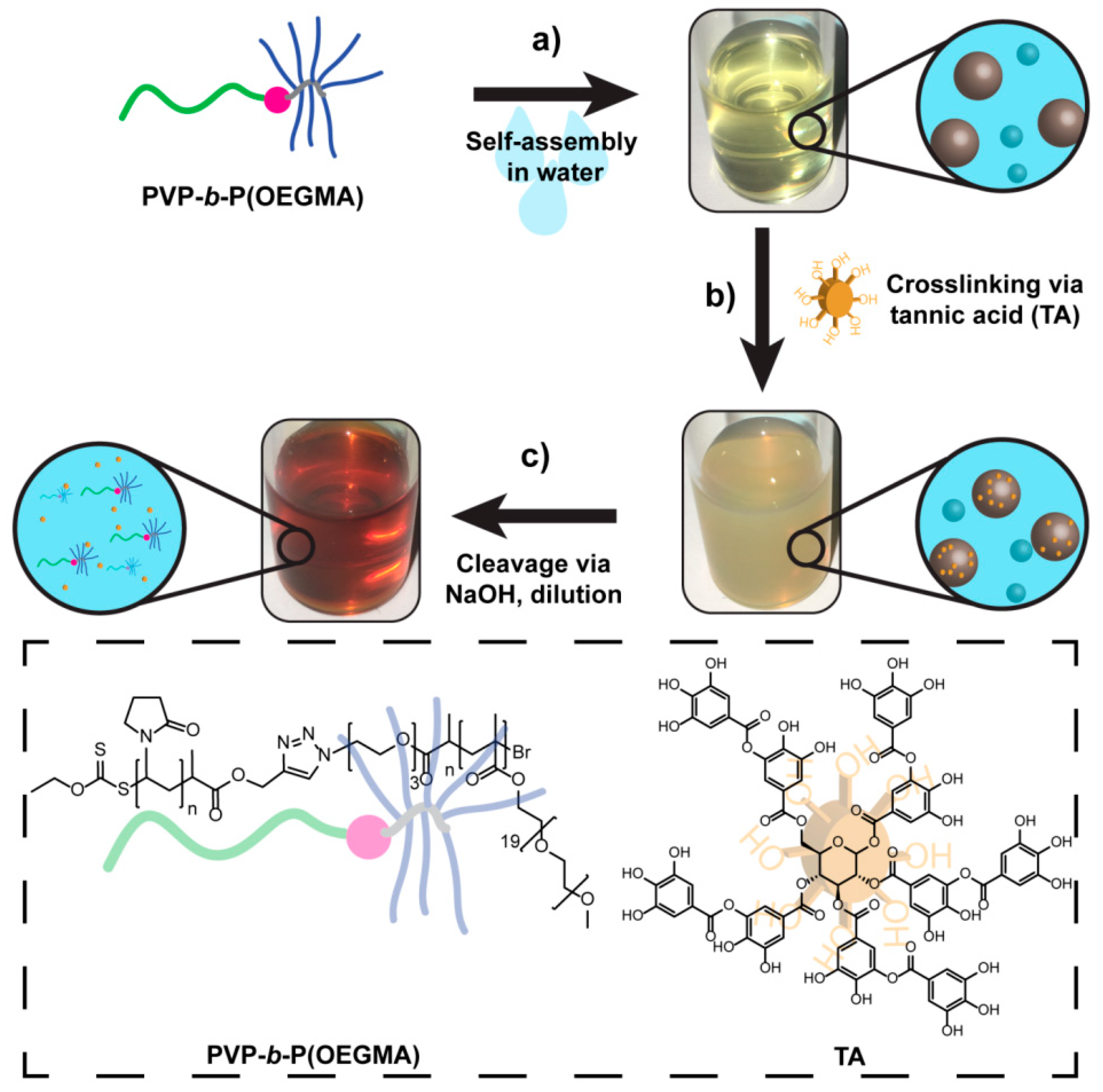
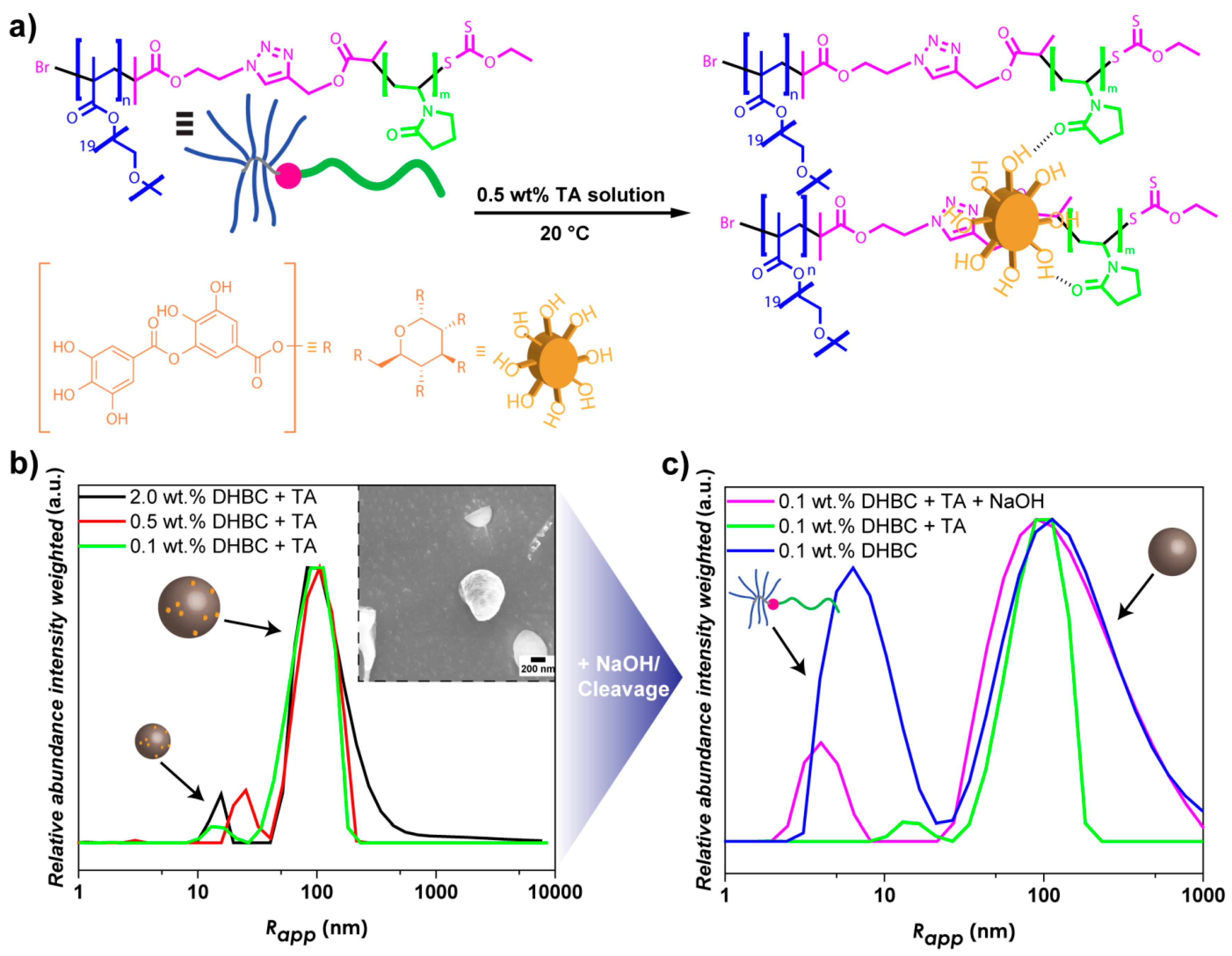
3.3. Crosslinking of DHBC Aggregates via Tannic Acid
3.4. Base-Induced Disassembly of Crosslinked PVP-b-P(OEGMA) Aggregates
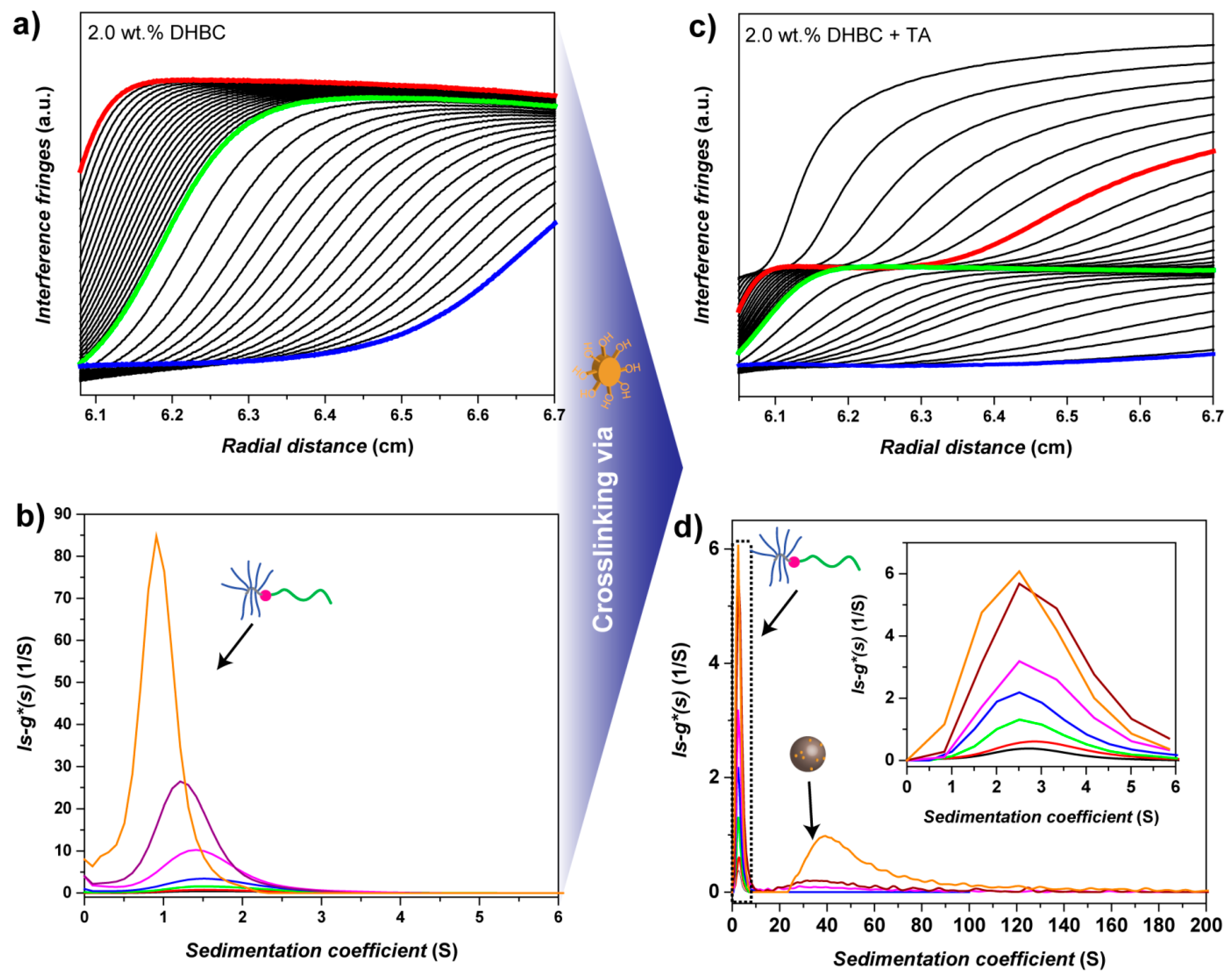
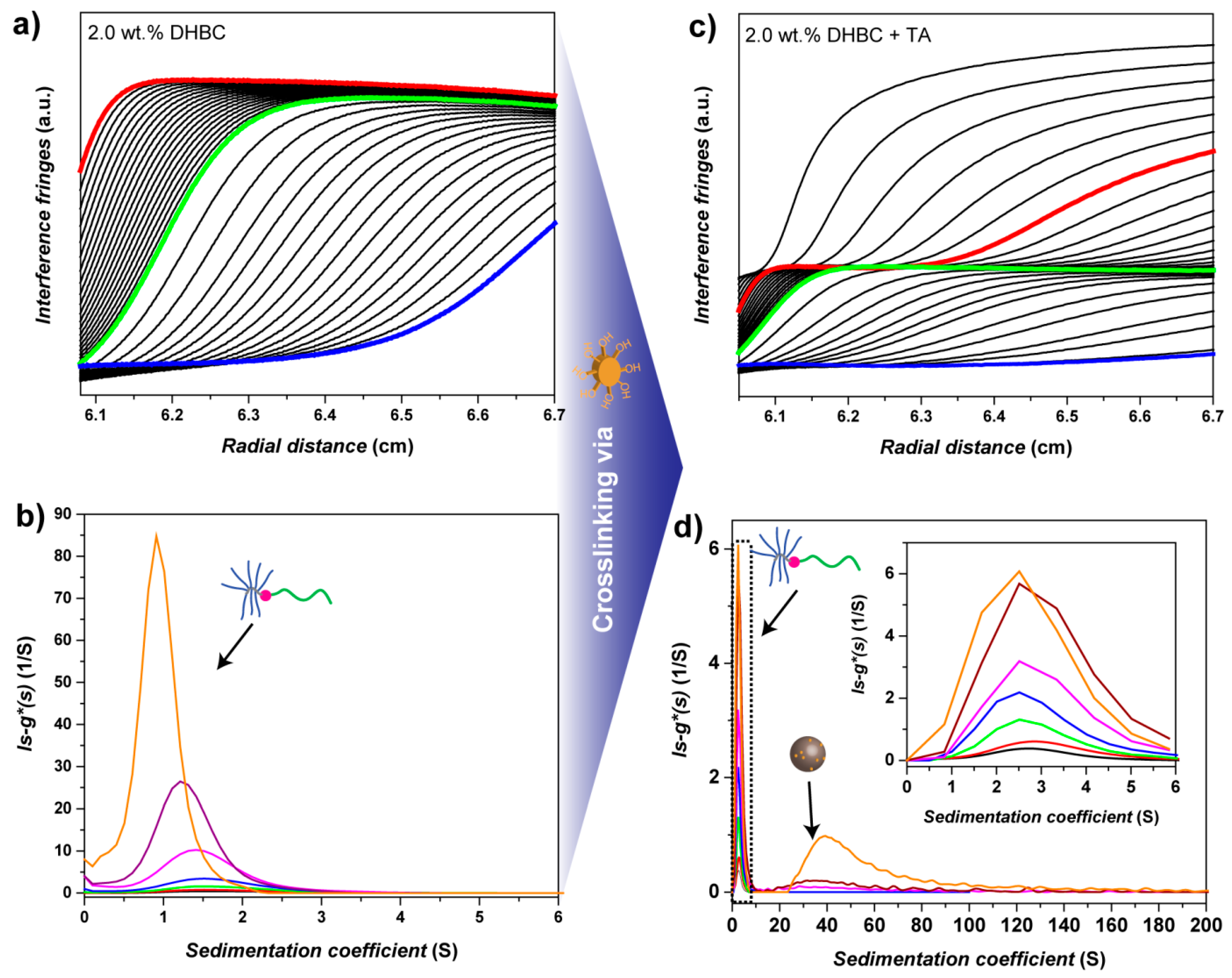
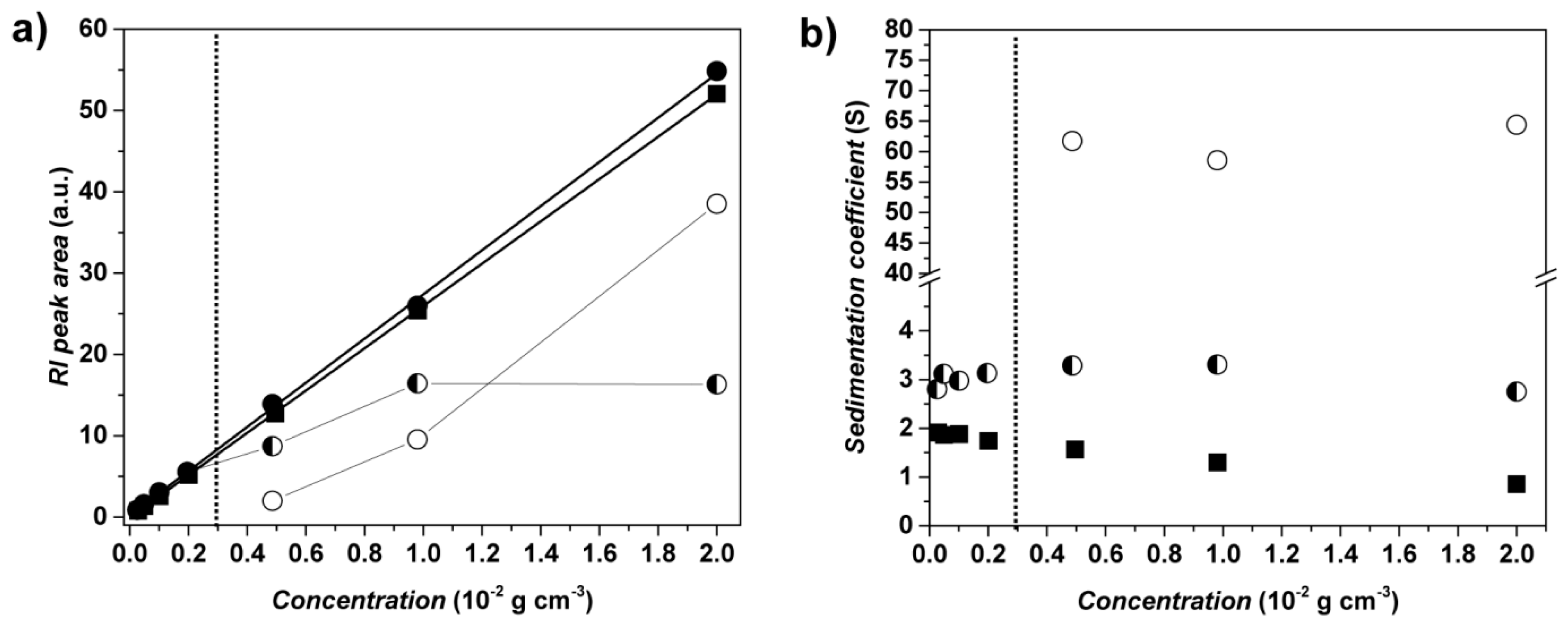
3.5. PVP-b-P(OEGMA) Aggregate Characterization via Analytical Ultracentrifugation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhang, L.; Eisenberg, A. Multiple Morphologies of “Crew-Cut” Aggregates of Polystyrene-b-poly(acrylic acid) Block Copolymers. Science 1995, 268, 1728–1731. [Google Scholar] [CrossRef] [PubMed]
- Derry, M.J.; Fielding, L.A.; Armes, S.P. Polymerization-induced self-assembly of block copolymer nanoparticles via RAFT non-aqueous dispersion polymerization. Prog. Polym. Sci. 2016, 52, 1–18. [Google Scholar] [CrossRef]
- Bang, J.; Jeong, U.; Ryu, D.Y.; Russell, T.P.; Hawker, C.J. Block Copolymer Nanolithography: Translation of Molecular Level Control to Nanoscale Patterns. Adv. Mater. 2009, 21, 4769–4792. [Google Scholar] [CrossRef]
- Jilin, Z.; Xinhong, Y.; Ping, Y.; Juan, P.; Chunxia, L.; Weihuan, H.; Yanchun, H. Microphase Separation of Block Copolymer Thin Films. Macromol. Rapid Commun. 2010, 31, 591–608. [Google Scholar] [CrossRef]
- Gröschel, A.H.; Walther, A.; Löbling, T.I.; Schacher, F.H.; Schmalz, H.; Müller, A.H.E. Guided hierarchical co-assembly of soft patchy nanoparticles. Nature 2013, 503, 247. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, B.V.K.J.; Wang, C.X.; Kraemer, S.; Connal, L.A.; Klinger, D. Highly functional ellipsoidal block copolymer nanoparticles: A generalized approach to nanostructured chemical ordering in phase separated colloidal particles. Polym. Chem. 2018, 9, 1638–1649. [Google Scholar] [CrossRef]
- Ge, Z.; Xie, D.; Chen, D.; Jiang, X.; Zhang, Y.; Liu, H.; Liu, S. Stimuli-Responsive Double Hydrophilic Block Copolymer Micelles with Switchable Catalytic Activity. Macromolecules 2007, 40, 3538–3546. [Google Scholar] [CrossRef]
- Gall, B.; Bortenschlager, M.; Nuyken, O.; Weberskirch, R. Cascade Reactions in Polymeric Nanoreactors: Mono (Rh)- and Bimetallic (Rh/Ir) Micellar Catalysis in the Hydroaminomethylation of 1-Octene. Macromol. Chem. Phys. 2008, 209, 1152–1159. [Google Scholar] [CrossRef]
- Torchilin, V.P. PEG-based micelles as carriers of contrast agents for different imaging modalities. Adv. Drug Deliv. Rev. 2002, 54, 235–252. [Google Scholar] [CrossRef]
- Appold, M.; Mari, C.; Lederle, C.; Elbert, J.; Schmidt, C.; Ott, I.; Stühn, B.; Gasser, G.; Gallei, M. Multi-stimuli responsive block copolymers as a smart release platform for a polypyridyl ruthenium complex. Polym. Chem. 2017, 8, 890–900. [Google Scholar] [CrossRef]
- Zou, J.; Chen, H.; Chunder, A.; Yu, Y.; Huo, Q.; Zhai, L. Preparation of a Superhydrophobic and Conductive Nanocomposite Coating from a Carbon-Nanotube-Conjugated Block Copolymer Dispersion. Adv. Mater. 2008, 20, 3337–3341. [Google Scholar] [CrossRef]
- Depalo, N.; Mallardi, A.; Comparelli, R.; Striccoli, M.; Agostiano, A.; Curri, M.L. Luminescent nanocrystals in phospholipid micelles for bioconjugation: An optical and structural investigation. J. Colloid Interface Sci. 2008, 325, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Schacher, F.H.; Rupar, P.A.; Manners, I. Functional Block Copolymers: Nanostructured Materials with Emerging Applications. Angew. Chem. Int. Ed. 2012, 51, 7898–7921. [Google Scholar] [CrossRef]
- Ge, Z.; Liu, S. Functional block copolymer assemblies responsive to tumor and intracellular microenvironments for site-specific drug delivery and enhanced imaging performance. Chem. Soc. Rev. 2013, 42, 7289–7325. [Google Scholar] [PubMed]
- Blanazs, A.; Armes, S.P.; Ryan, A.J. Self-Assembled Block Copolymer Aggregates: From Micelles to Vesicles and their Biological Applications. Macromol. Rapid Commun. 2009, 30, 267–277. [Google Scholar] [CrossRef]
- Antonietti, M.; Förster, S. Vesicles and Liposomes: A Self-Assembly Principle Beyond Lipids. Adv. Mater. 2003, 15, 1323–1333. [Google Scholar] [CrossRef]
- Discher, D.E.; Eisenberg, A. Polymer Vesicles. Science 2002, 297, 967–973. [Google Scholar] [CrossRef]
- Charleux, B.; Delaittre, G.; Rieger, J.; D’Agosto, F. Polymerization-Induced Self-Assembly: From Soluble Macromolecules to Block Copolymer Nano-Objects in One Step. Macromolecules 2012, 45, 6753–6765. [Google Scholar] [CrossRef]
- Ladmiral, V.; Semsarilar, M.; Canton, I.; Armes, S.P. Polymerization-Induced Self-Assembly of Galactose-Functionalized Biocompatible Diblock Copolymers for Intracellular Delivery. J. Am. Chem. Soc. 2013, 135, 13574–13581. [Google Scholar] [CrossRef]
- Gaitzsch, J.; Huang, X.; Voit, B. Engineering functional polymer capsules toward smart nanoreactors. Chem. Rev. 2015, 116, 1053–1093. [Google Scholar] [CrossRef]
- Nardin, C.; Widmer, J.; Winterhalter, M.; Meier, W. Amphiphilic block copolymer nanocontainers as bioreactors. Eur. Phys. J. E 2001, 4, 403–410. [Google Scholar] [CrossRef]
- Rösler, A.; Vandermeulen, G.W.M.; Klok, H.-A. Advanced drug delivery devices via self-assembly of amphiphilic block copolymers. Adv. Drug Deliv. Rev. 2012, 64, 270–279. [Google Scholar] [CrossRef]
- Blackman, L.D.; Varlas, S.; Arno, M.C.; Houston, Z.H.; Fletcher, N.L.; Thurecht, K.J.; Hasan, M.; Gibson, M.I.; O’Reilly, R.K. Confinement of Therapeutic Enzymes in Selectively Permeable Polymer Vesicles by Polymerization-Induced Self-Assembly (PISA) Reduces Antibody Binding and Proteolytic Susceptibility. ACS Central Sci. 2018, 4, 718–723. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Ge, Z.; Xu, J.; Liu, H.; Liu, S. Fabrication of Multiresponsive Shell Cross-Linked Micelles Possessing pH-Controllable Core Swellability and Thermo-Tunable Corona Permeability. Biomacromolecules 2007, 8, 3184–3192. [Google Scholar] [CrossRef]
- Schmidt, B.V.K.J. Double Hydrophilic Block Copolymer Self-Assembly in Aqueous Solution. Macromol. Chem. Phys. 2018, 219, 1700494. [Google Scholar] [CrossRef]
- Albertsson, P.-Å. Partition of Cell Particles and Macromolecules in Polymer Two-Phase Systems. Adv. Protein Chem. 1970, 24, 309–341. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Hwang, J.; Antonietti, M.; Schmidt, B.V.K.J. Water-in-Water Pickering Emulsion Stabilized by Polydopamine Particles and Cross-Linking. Biomacromolecules 2018, 20, 204–211. [Google Scholar] [CrossRef]
- Peddireddy, K.R.; Nicolai, T.; Benyahia, L.; Capron, I. Stabilization of Water-in-Water Emulsions by Nanorods. ACS Macro Lett. 2016, 5, 283–286. [Google Scholar] [CrossRef]
- Brosnan, S.M.; Schlaad, H.; Antonietti, M. Aqueous Self-Assembly of Purely Hydrophilic Block Copolymers into Giant Vesicles. Angew. Chem. Int. Ed. 2015, 54, 9715–9718. [Google Scholar] [CrossRef]
- Willersinn, J.; Bogomolova, A.; Cabre, M.B.; Schmidt, B.V.K.J. Vesicles of double hydrophilic pullulan and poly(acrylamide) block copolymers: A combination of synthetic- and bio-derived blocks. Polym. Chem. 2017, 8, 1244–1254. [Google Scholar] [CrossRef]
- Willersinn, J.; Schmidt, B.V.K.J. Self-Assembly of Double Hydrophilic Poly(2-ethyl-2-oxazoline)-b-poly(N-vinylpyrrolidone) Block Copolymers in Aqueous Solution. Polymers 2017, 9, 293. [Google Scholar] [CrossRef]
- Oh, T.; Nagao, M.; Hoshino, Y.; Miura, Y. Self-Assembly of a Double Hydrophilic Block Glycopolymer and the Investigation of Its Mechanism. Langmuir 2018, 34, 8591–8598. [Google Scholar] [CrossRef] [PubMed]
- Quan, J.; Shen, F.-W.; Cai, H.; Zhang, Y.-N.; Wu, H. Galactose-Functionalized Double-Hydrophilic Block Glycopolymers and Their Thermoresponsive Self-Assembly Dynamics. Langmuir 2018, 34, 10721–10731. [Google Scholar] [CrossRef]
- Pasparakis, G.; Alexander, C. Sweet Talking Double Hydrophilic Block Copolymer Vesicles. Angew. Chem. Int. Ed. 2008, 47, 4847–4850. [Google Scholar] [CrossRef]
- Casse, O.; Shkilnyy, A.; Linders, J.; Mayer, C.; Häussinger, D.; Völkel, A.; Thünemann, A.F.; Dimova, R.; Cölfen, H.; Meier, W.; et al. Solution Behavior of Double-Hydrophilic Block Copolymers in Dilute Aqueous Solution. Macromolecules 2012, 45, 4772–4777. [Google Scholar] [CrossRef]
- Al Nakeeb, N.; Willersinn, J.; Schmidt, B.V.K.J. Self-Assembly Behavior and Biocompatible Cross-Linking of Double Hydrophilic Linear-Brush Block Copolymers. Biomacromolecules 2017, 18, 3695–3705. [Google Scholar] [CrossRef]
- Cölfen, H. Double-Hydrophilic Block Copolymers: Synthesis and Application as Novel Surfactants and Crystal Growth Modifiers. Macromol. Rapid Commun. 2001, 22, 219–252. [Google Scholar] [CrossRef]
- Hwang, J.; Heil, T.; Antonietti, M.; Schmidt, B.V.K.J. Morphogenesis of Metal–Organic Mesocrystals Mediated by Double Hydrophilic Block Copolymers. J. Am. Chem. Soc. 2018, 140, 2947–2956. [Google Scholar] [CrossRef]
- Knop, K.; Hoogenboom, R.; Fischer, D.; Schubert, U.S. Poly (ethylene glycol) in drug delivery: pros and cons as well as potential alternatives. Angew. Chem. Int. Ed. 2010, 49, 6288–6308. [Google Scholar] [CrossRef]
- D’souza, A.J.M.; Schowen, R.L.; Topp, E.M. Polyvinylpyrrolidone–drug conjugate: synthesis and release mechanism. J. Controlled Release 2004, 94, 91–100. [Google Scholar] [CrossRef]
- Willersinn, J.; Drechsler, M.; Antonietti, M.; Schmidt, B.V.K.J. Organized Polymeric Submicron Particles via Self-Assembly and Cross-Linking of Double Hydrophilic Poly(ethylene oxide)-b-poly(N-vinylpyrrolidone) in Aqueous Solution. Macromolecules 2016, 49, 5331–5341. [Google Scholar] [CrossRef]
- Al Nakeeb, N.; Kochovski, Z.; Li, T.; Zhang, Y.; Lu, Y.; Schmidt, B.V.K.J. Poly(ethylene glycol) brush-b-poly(N-vinylpyrrolidone)-based double hydrophilic block copolymer particles crosslinked via crystalline a-cyclodextrin domains. RSC Adv. 2019, 9, 4993–5001. [Google Scholar] [CrossRef]
- Willersinn, J.; Schmidt, B.V.K.J. Pure hydrophilic block copolymer vesicles with redox- and pH cleavable crosslinks. Polym. Chem. 2018, 9, 1626–1837. [Google Scholar] [CrossRef]
- Lehn, J.-M. Supramolecular chemistry: Where from? Where to? Chem. Soc. Rev. 2017, 46, 2378–2379. [Google Scholar] [CrossRef] [PubMed]
- Lehn, J.-M. Supramolecular Chemistry—Scope and Perspectives Molecules, Supermolecules, and Molecular Devices (Nobel Lecture). Angew. Chem. Int. Ed. 1988, 27, 89–112. [Google Scholar] [CrossRef]
- Wilson, A.J. Non-covalent polymer assembly using arrays of hydrogen-bonds. Soft Matter 2007, 3, 409–425. [Google Scholar] [CrossRef]
- Schmidt, B.V.K.J.; Barner-Kowollik, C. Dynamic Macromolecular Material Design—The Versatility of Cyclodextrin-Based Host–Guest Chemistry. Angew. Chem. Int. Ed. 2017, 56, 8350–8369. [Google Scholar] [CrossRef]
- Lohmeijer, B.G.G.; Schubert, U.S. Playing LEGO with macromolecules: Design, synthesis, and self-organization with metal complexes. J. Polym. Sci. Part A Polym. Chem. 2003, 41, 1413–1427. [Google Scholar] [CrossRef]
- Rudolph, T.; Schacher, F.H. Selective crosslinking or addressing of individual domains within block copolymer nanostructures. Eur. Polym. J. 2016, 80, 317–331. [Google Scholar] [CrossRef]
- Appel, E.A.; del Barrio, J.; Loh, X.J.; Scherman, O.A. Supramolecular polymeric hydrogels. Chem. Soc. Rev. 2012, 41, 6195–6214. [Google Scholar] [CrossRef]
- Van Buren, J.P.; Robinson, W.B. Formation of complexes between protein and tannic acid. J. Agric. Food Chem. 1969, 17, 772–777. [Google Scholar] [CrossRef]
- Chen, J.; Kozlovskaya, V.; Goins, A.; Campos-Gomez, J.; Saeed, M.; Kharlampieva, E. Biocompatible Shaped Particles from Dried Multilayer Polymer Capsules. Biomacromolecules 2013, 14, 3830–3841. [Google Scholar] [CrossRef] [PubMed]
- Ejima, H.; Richardson, J.J.; Liang, K.; Best, J.P.; van Koeverden, M.P.; Such, G.K.; Cui, J.; Caruso, F. One-Step Assembly of Coordination Complexes for Versatile Film and Particle Engineering. Science 2013, 341, 154–157. [Google Scholar] [CrossRef]
- Erwin, A.J.; Korolovych, V.F.; Iatridi, Z.; Tsitsilianis, C.; Ankner, J.F.; Tsukruk, V.V. Tunable Compartmentalized Morphologies of Multilayered Dual Responsive Star Block Polyampholytes. Macromolecules 2018, 51, 4800–4812. [Google Scholar] [CrossRef]
- Kozlovskaya, V.; Zavgorodnya, O.; Chen, Y.; Ellis, K.; Tse, H.M.; Cui, W.; Thompson, J.A.; Kharlampieva, E. Ultrathin Polymeric Coatings Based on Hydrogen-Bonded Polyphenol for Protection of Pancreatic Islet Cells. Adv. Funct. Mater. 2012, 22, 3389–3398. [Google Scholar] [CrossRef]
- Dierendonck, M.; Fierens, K.; De Rycke, R.; Lybaert, L.; Maji, S.; Zhang, Z.; Zhang, Q.; Hoogenboom, R.; Lambrecht, B.N.; Grooten, J.; et al. Nanoporous Hydrogen Bonded Polymeric Microparticles: Facile and Economic Production of Cross Presentation Promoting Vaccine Carriers. Adv. Funct. Mater. 2014, 24, 4634–4644. [Google Scholar] [CrossRef]
- Schuck, P.; Rossmanith, P. Determination of the sedimentation coefficient distribution by least-squares boundary modeling. Biopolymers 2000, 54, 328–341. [Google Scholar] [CrossRef]
- Quemener, D.; Davis, T.P.; Barner-Kowollik, C.; Stenzel, M.H. RAFT and click chemistry: A versatile approach to well-defined block copolymers. Chem. Commun. 2006, 48, 5051–5053. [Google Scholar] [CrossRef]
- Li, T.; Kumru, B.; Al Nakeeb, N.; Willersinn, J.; Schmidt, B.V.K.J. Thermoadaptive Supramolecular α-Cyclodextrin Crystallization-Based Hydrogels via Double Hydrophilic Block Copolymer Templating. Polymers 2018, 10, 576. [Google Scholar] [CrossRef]
- Pan, X.; Guo, X.; Choi, B.; Feng, A.; Wei, X.; Thang, S.H. A facile synthesis of pH stimuli biocompatible block copolymer poly(methacrylic acid)-block-poly(N-vinylpyrrolidone) utilizing switchable RAFT agents. Polym. Chem. 2019. [Google Scholar] [CrossRef]
- Pound, G.; Aguesse, F.; McLeary, J.B.; Lange, R.F.; Klumperman, B. Xanthate-mediated copolymerization of vinyl monomers for amphiphilic and double-hydrophilic block copolymers with poly (ethylene glycol). Macromolecules 2007, 40, 8861–8871. [Google Scholar] [CrossRef]
- Bernard, J.; Save, M.; Arathoon, B.; Charleux, B. Preparation of a xanthate-terminated dextran by click chemistry: Application to the synthesis of polysaccharide-coated nanoparticles via surfactant-free ab initio emulsion polymerization of vinyl acetate. J. Polym. Sci. Part A Polym. Chem. 2008, 46, 2845–2857. [Google Scholar] [CrossRef]
- Liu, F.; Kozlovskaya, V.; Zavgorodnya, O.; Martinez-Lopez, C.; Catledge, S.; Kharlampieva, E. Encapsulation of anticancer drug by hydrogen-bonded multilayers of tannic acid. Soft Matter 2014, 10, 9237–9247. [Google Scholar] [CrossRef] [PubMed]
- Erel-Unal, I.; Sukhishvili, S.A. Hydrogen-Bonded Multilayers of a Neutral Polymer and a Polyphenol. Macromolecules 2008, 41, 3962–3970. [Google Scholar] [CrossRef]
- Kozlovskaya, V.; Kharlampieva, E.; Drachuk, I.; Cheng, D.; Tsukruk, V.V. Responsive microcapsule reactors based on hydrogen-bonded tannic acid layer-by-layer assemblies. Soft Matter 2010, 6, 3596–3608. [Google Scholar] [CrossRef]
- Grube, M.; Leiske, M.N.; Schubert, U.S.; Nischang, I. POx as an Alternative to PEG? A Hydrodynamic and Light Scattering Study. Macromolecules 2018, 51, 1905–1916. [Google Scholar] [CrossRef]
- Nischang, I.; Perevyazko, I.; Majdanski, T.; Vitz, J.; Festag, G.; Schubert, U.S. Hydrodynamic Analysis Resolves the Pharmaceutically-Relevant Absolute Molar Mass and Solution Properties of Synthetic Poly(ethylene glycol)s Created by Varying Initiation Sites. Anal. Chem. 2017, 89, 1185–1193. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al Nakeeb, N.; Nischang, I.; Schmidt, B.V.K.J. Tannic Acid-Mediated Aggregate Stabilization of Poly(N-vinylpyrrolidone)-b-poly(oligo (ethylene glycol) methyl ether methacrylate) Double Hydrophilic Block Copolymers. Nanomaterials 2019, 9, 662. https://doi.org/10.3390/nano9050662
Al Nakeeb N, Nischang I, Schmidt BVKJ. Tannic Acid-Mediated Aggregate Stabilization of Poly(N-vinylpyrrolidone)-b-poly(oligo (ethylene glycol) methyl ether methacrylate) Double Hydrophilic Block Copolymers. Nanomaterials. 2019; 9(5):662. https://doi.org/10.3390/nano9050662
Chicago/Turabian StyleAl Nakeeb, Noah, Ivo Nischang, and Bernhard V.K.J. Schmidt. 2019. "Tannic Acid-Mediated Aggregate Stabilization of Poly(N-vinylpyrrolidone)-b-poly(oligo (ethylene glycol) methyl ether methacrylate) Double Hydrophilic Block Copolymers" Nanomaterials 9, no. 5: 662. https://doi.org/10.3390/nano9050662
APA StyleAl Nakeeb, N., Nischang, I., & Schmidt, B. V. K. J. (2019). Tannic Acid-Mediated Aggregate Stabilization of Poly(N-vinylpyrrolidone)-b-poly(oligo (ethylene glycol) methyl ether methacrylate) Double Hydrophilic Block Copolymers. Nanomaterials, 9(5), 662. https://doi.org/10.3390/nano9050662