How to Improve the Antioxidant Defense in Asphyxiated Newborns—Lessons from Animal Models

 , ,
, ,
Abstract
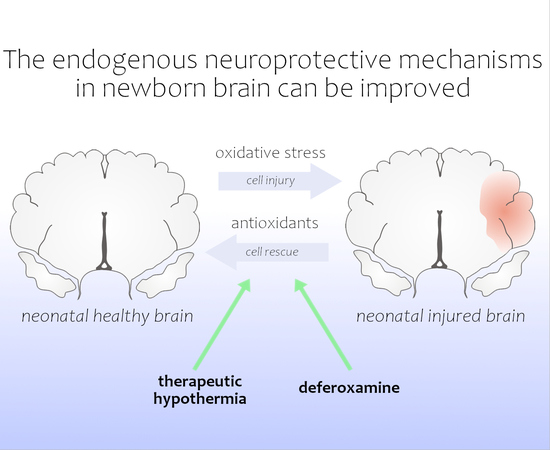
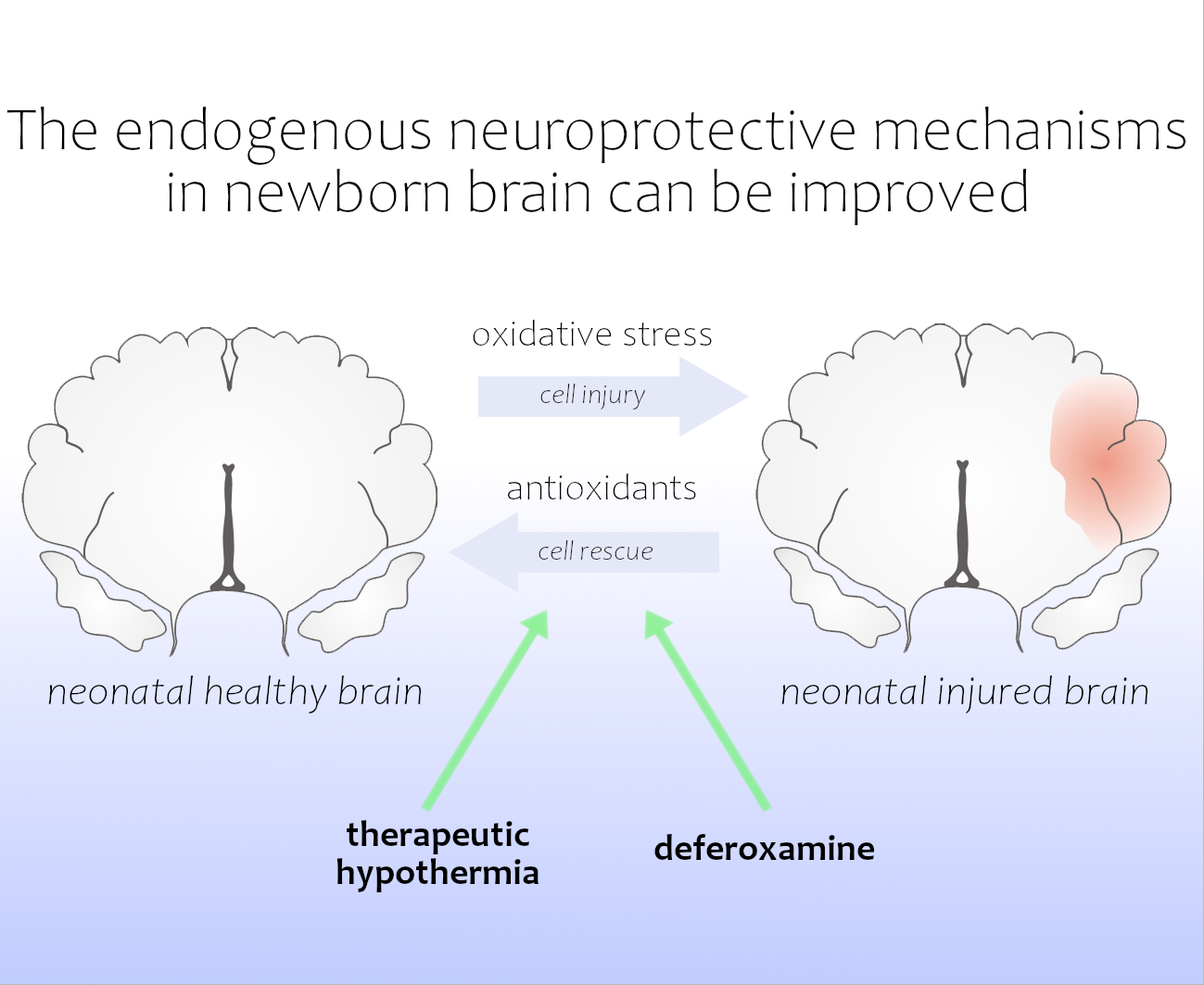
:
1. Introduction
2. Animal Models of Neonatal Hypoxia/Ischemia
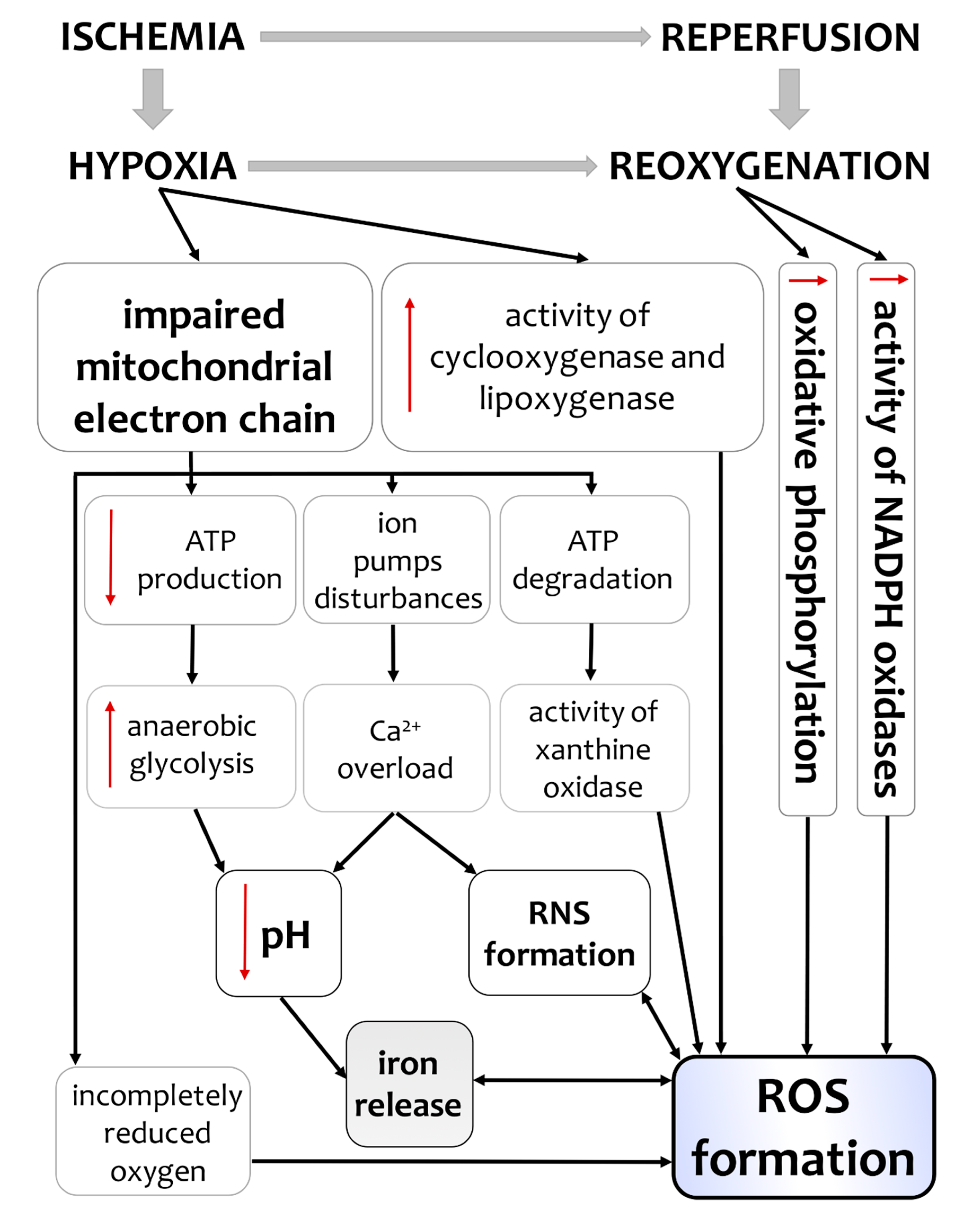
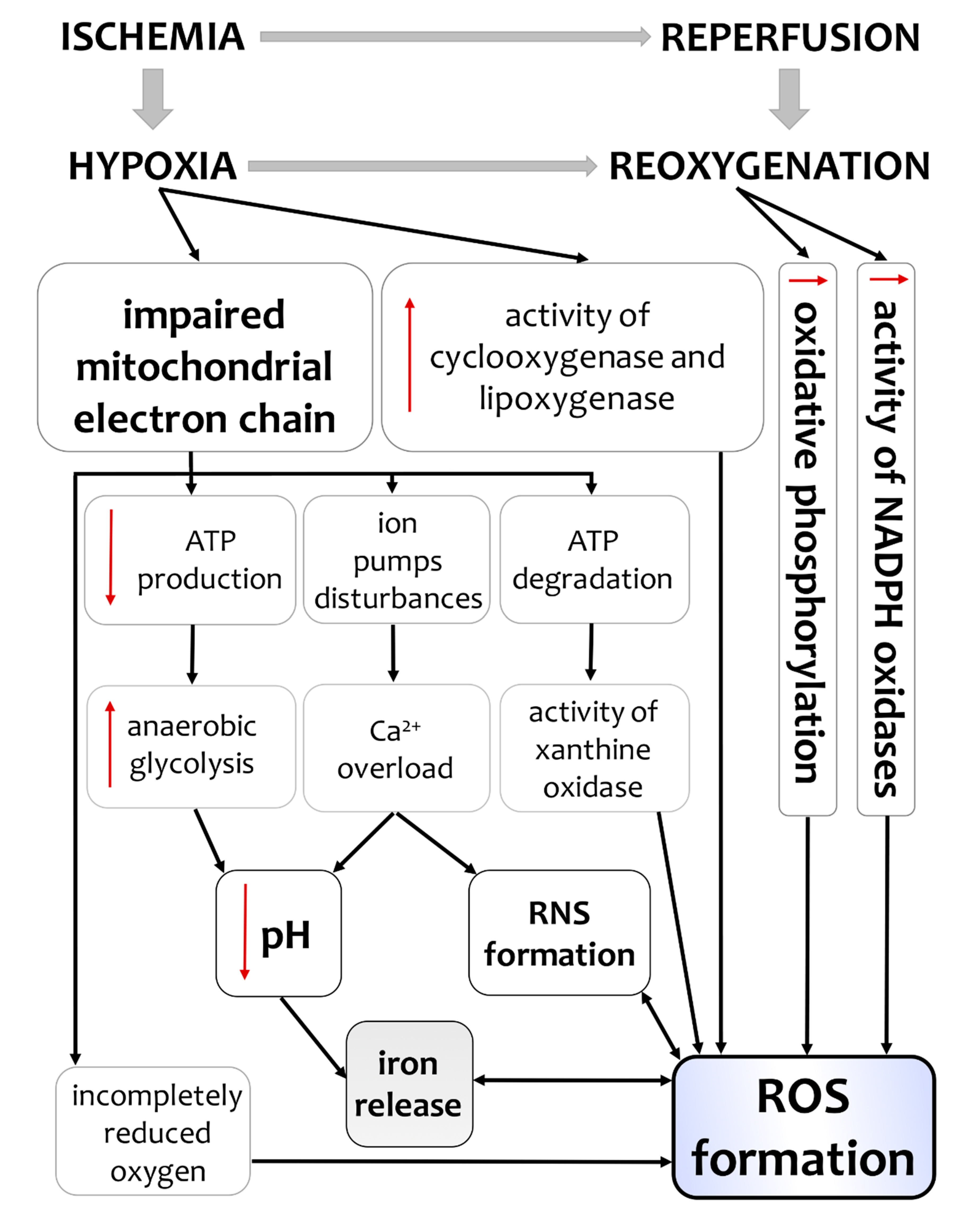
3. Hypoxia/Ischemia-Induced Changes of Oxidative Status in the Brain
4. Therapeutic Hypothermia—Impact on Oxidative Homeostasis under Hypoxic/IschemicConditions
5. DFO—A PromisingAgent in Hypoxic/Ischemic Encephalopathy Therapy?
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hassell, K.J.; Ezzati, M.; Alonso-Alconada, D.; Hausenloy, D.J.; Robertson, N.J. New Horizons for Newborn Brain Protection: Enhancing Endogenous Neuroprotection. Arch. Dis. Child. Fetal Neonatal Ed. 2015, 100, F541–F551. [Google Scholar] [CrossRef] [PubMed]
- Blomgren, K.; Hagberg, H. Free Radicals, Mitochondria, and Hypoxia-Ischemia in the Developing Brain. Free Radic. Biol. Med. 2006, 40, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Johnston, M.V.; Fatemi, A.; Wilson, M.A.; Northington, F. Treatment Advances in Neonatal Neuroprotection and Neurointensive Care. Lancet Neurol. 2011, 10, 372–382. [Google Scholar] [CrossRef] [Green Version]
- Palmer, C.; Vannucci, R.C. Cellular Biology of Perinatal Hypoxia-Ischemia. In Fetal and Neonatal Brain Injury, 3rd ed.; Mechanisms, Management and the Risks of Practise, Stevenson, D.K., Benitz, W.E., Sunshine, P., Eds.; Cambridge University Press: Cambridge, UK, 2003; pp. 58–70. [Google Scholar]
- Flores, K.P.; Blohowiak, S.E.; Winzerling, J.J.; Georgieff, M.K.; Kling, P.J. The Impact of Erythropoietin and Iron Status on Brain Myelination in the Newborn Rat. J. Neurosci. Res. 2018, 96, 1586–1599. [Google Scholar] [CrossRef] [PubMed]
- Buonocore, G.; Zani, S.; Perrone, S.; Caciotti, B.; Bracci, R. Intraerythrocyte Nonprotein-Bound Iron and Plasma Malondialdehyde in the Hypoxic Newborn. Free Radic. Biol. Med. 1998, 25, 766–770. [Google Scholar] [CrossRef]
- Kaur, C.; Ling, E.A. Increased Expression of Transferrin Receptors and Iron in Amoeboid Microglial Cells in Postnatal Rats Following an Exposure to Hypoxia. Neurosci. Lett. 1999, 262, 183–186. [Google Scholar] [CrossRef]
- Bresgen, N.; Eckl, P.M. Oxidative Stress and the Homeodynamics of Iron Metabolism. Biomolecules 2015, 5, 808–847. [Google Scholar] [CrossRef]
- Papanikolaou, G.; Pantopoulos, K. Iron Metabolism and Toxicity. Toxicol. Appl. Pharmacol. 2005, 202, 199–211. [Google Scholar] [CrossRef]
- Palmer, C. Iron and Oxidative Stress in Neonatal Hypoxic-Ischemic Brain Injury Directions for Therapeutic Intervention. Met. Oxidative Damage Neurol. Disord. 1997, 205–236. [Google Scholar] [CrossRef]
- Pabello, N.G.; Tracy, S.J.; Keller, R.W. Protective Effects of Brief Intra- and Delayed Postischemic Hypothermia in a Transient Focal Ischemia Model in the Neonatal Rat. Brain Res. 2004, 995, 29–38. [Google Scholar] [CrossRef]
- Shankaran, S.; Laptook, A.R.; Ehrenkranz, R.A.; Tyson, J.E.; McDonald, S.A.; Donovan, E.F.; Fanaroff, A.A.; Poole, W.K.; Wright, L.L.; Higgins, R.D.; et al. Whole-Body Hypothermia for Neonates with Hypoxic-Ischemic Encephalopathy. N. Engl. J. Med. 2005, 353, 1574–1584. [Google Scholar] [CrossRef]
- Mortola, J.P. Implications of Hypoxic Hypometabolism during Mammalian Ontogenesis. Respir. Physiol. Neurobiol. 2004, 141, 345–356. [Google Scholar] [CrossRef]
- Maier, C.M.; Sun, G.H.; Cheng, D.; Yenari, M.A.; Chan, P.H.; Steinberg, G.K. Effects of Mild Hypothermia on Superoxide Anion Production, Superoxide Dismutase Expression, and Activity Following Transient Focal Cerebral Ischemia. Neurobiol. Dis. 2002, 11, 28–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakita, H.; Hussein, M.H.; Kato, S.; Yamada, Y.; Nagaya, Y.; Asai, H.; Goto, T.; Ito, K.; Sugiura, T.; Daoud, G.A.H.; et al. Hypothermia Attenuates the Severity of Oxidative Stress Development in Asphyxiated Newborns. J. Crit. Care 2012, 27, 469–473. [Google Scholar] [CrossRef] [PubMed]
- Perrone, S.; Szabó, M.; Bellieni, C.V.; Longini, M.; Bangó, M.; Kelen, D.; Treszl, A.; Negro, S.; Tataranno, M.L.; Buonocore, G. Whole Body Hypothermia and Oxidative Stress in Babies with Hypoxic-Ischemic Brain Injury. Pediatr. Neurol. 2010, 43, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Burnard, E.D.; Cross, K.W. Rectal Temperature in the Newborn after Birth Asphyxia. Br. Med. J. 1958, 2, 1197–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westin, B.; Nyberg, R.; Miller, J.A.; Wedenberg, E. Hypothermia and Transfusion with Oxygenated Blood in the Treatment of Asphyxia Neonatorum. Acta Paediatr. Suppl. 1959, 139, 1–80. [Google Scholar] [CrossRef]
- Laptook, A.R.; Watkinson, M. Temperature Management in the Delivery Room. Semin. Fetal Neonatal Med. 2008, 13, 383–391. [Google Scholar] [CrossRef]
- Lemyre, B.; Chau, V. Hypothermia for Newborns with Hypoxic-Ischemic Encephalopathy. Paediatr. Child Health 2018, 23, 285. [Google Scholar] [CrossRef]
- Sarco, D.P.; Becker, J.; Palmer, C.; Sheldon, R.A.; Ferriero, D.M. The Neuroprotective Effect of Deferoxamine in the Hypoxic-Ischemic Immature Mouse Brain. Neurosci. Lett. 2000, 282, 113–116. [Google Scholar] [CrossRef]
- Palmer, C.; Roberts, R.L.; Bero, C. Deferoxamine Posttreatment Reduces Ischemic Brain Injury in Neonatal Rats. Stroke 1994, 25, 1039–1045. [Google Scholar] [CrossRef] [Green Version]
- Rogalska, J.; Danielisova, V.; Caputa, M. Effect of Neonatal Body Temperature on Postanoxic, Potentially Neurotoxic Iron Accumulation in the Rat Brain. Neurosci. Lett. 2006, 393, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Shadid, M.; Buonocore, G.; Groenendaal, F.; Moison, R.; Ferrali, M.; Berger, H.M.; Van Bel, F. Effect of Deferoxamine and Allopurinol on Non-Protein-Bound Iron Concentrations in Plasma and Cortical Brain Tissue of Newborn Lambs Following Hypoxia-Ischemia. Neurosci. Lett. 1998, 248, 5–8. [Google Scholar] [CrossRef]
- Almli, L.M.; Hamrick, S.E.G.; Koshy, A.A.; Täuber, M.G.; Ferriero, D.M. Multiple Pathways of Neuroprotection against Oxidative Stress and Excitotoxic Injury in Immature Primary Hippocampal Neurons. Dev. Brain Res. 2001, 132, 121–129. [Google Scholar] [CrossRef]
- Kletkiewicz, H.; Nowakowska, A.; Siejka, A.; Mila-Kierzenkowska, C.; Woźniak, A.; Caputa, M.; Rogalska, J. Deferoxamine Prevents Cerebral Glutathione and Vitamin E Depletions in Asphyxiated Neonatal Rats: Role of Body Temperature. Int. J. Hyperth. 2016, 32, 211–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kletkiewicz, H.; Nowakowska, A.; Siejka, A.; Mila-Kierzenkowska, C.; Woźniak, A.; Caputa, M.; Rogalska, J. Deferoxamine Improves Antioxidative Protection in the Brain of Neonatal Rats: The Role of Anoxia and Body Temperature. Neurosci. Lett. 2016, 628, 116–122. [Google Scholar] [CrossRef]
- Barkhuizen, M.; van den Hove, D.L.A.; Vles, J.S.H.; Steinbusch, H.W.M.; Kramer, B.W.; Gavilanes, A.W.D. 25 Years of Research on Global Asphyxia in the Immature Rat Brain. Neurosci. Biobehav. Rev. 2017, 75, 166–182. [Google Scholar] [CrossRef]
- Dickerson, J.W.; Dobbing, J. Prenatal and Postnatal Growth and Development of the Central Nervous System of the Pig. Proc. R. Soc. Lond. B. Biol. Sci. 1967, 166, 384–395. [Google Scholar]
- Buser, J.R.; Segovia, K.N.; Dean, J.M.; Nelson, K.; Beardsley, D.; Gong, X.; Luo, N.L.; Ren, J.; Wan, Y.; Riddle, A.; et al. Timing of Appearance of Late Oligodendrocyte Progenitors Coincides with Enhanced Susceptibility of Preterm Rabbit Cerebral White Matter to Hypoxia-Ischemia. J. Cereb. Blood Flow Metab. 2010, 30, 1053–1065. [Google Scholar] [CrossRef]
- Back, S.A.; Riddle, A.; Hohimer, A.R. Role of Instrumented Fetal Sheep Preparations in Defining the Pathogenesis of Human Periventricular White-Matter Injury. J. Child Neurol. 2006, 21, 582–589. [Google Scholar] [CrossRef]
- Mueller-Burke, D.; Koehler, R.C.; Martin, L.J. Rapid NMDA Receptor Phosphorylation and Oxidative Stress Precede Striatal Neurodegeneration after Hypoxic Ischemia in Newborn Piglets and Are Attenuated with Hypothermia. Int. J. Dev. Neurosci. 2008, 26, 67–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van den Heuij, L.G.; Wassink, G.; Gunn, A.J.; Bennet, L. Using Pregnant Sheep to Model Developmental Brain Damage. Neuromethods 2016, 109, 327–341. [Google Scholar]
- Seehase, M.; Jellema, R.; Jennekens, W.; Zwanenburg, A.; Andriessen, P.; Gavilanes, D.; Kramer, B.W. Fetal Cardiac Arrest Due to Asphyxia in Late Preterm Lambs: Propofol Mediates Neuroprotection for the Fetus When Administered During Emergency Caesarean Section and after Rescusitation. Pediatr. Res. 2011, 70, 61. [Google Scholar] [CrossRef] [Green Version]
- Fahn, S.; Davis, J.; Rowland, L. Cerebral Hypoxia and its Consequences; Raven Press: New York, NY, USA, 1979; Volume 26. [Google Scholar]
- Kyng, K.J.; Skajaa, T.; Kerrn-Jespersen, S.; Andreassen, C.S.; Bennedsgaard, K.; Henriksen, T.B. A Piglet Model of Neonatal Hypoxic-Ischemic Encephalopathy. J. Vis. Exp. 2015, 2015, 52454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerguerian, A.M.; Brambrink, A.M.; Traystman, R.J.; Huganir, R.L.; Martin, L.J. Altered Expression and Phosphorylation of N-Methyl-D-Aspartate Receptors in Piglet Striatum after Hypoxia-Ischemia. Mol. Brain Res. 2002, 104, 66–80. [Google Scholar] [CrossRef]
- Broad, K.D.; Fierens, I.; Fleiss, B.; Rocha-Ferreira, E.; Ezzati, M.; Hassell, J.; Alonso-Alconada, D.; Bainbridge, A.; Kawano, G.; Ma, D.; et al. Inhaled 45–50% Argon Augments Hypothermic Brain Protection in a Piglet Model of Perinatal Asphyxia. Neurobiol. Dis. 2016, 87, 29–38. [Google Scholar] [CrossRef] [Green Version]
- Gunn, A.J.; Parer, J.T.; Mallard, E.C.; Williams, C.E.; Gluckman, P.D. Cerebral Histologie and Electrocorticographic Changes after Asphyxia in Fetal Sheep. Pediatr. Res. 1992, 31, 486–491. [Google Scholar] [CrossRef] [Green Version]
- Keogh, M.J.; Drury, P.P.; Bennet, L.; Davidson, J.O.; Mathai, S.; Gunn, E.R.; Booth, L.C.; Gunn, A.J. Limited Predictive Value of Early Changes in EEG Spectral Power for Neural Injury after Asphyxia in Preterm Fetal Sheep. Pediatr. Res. 2012, 71, 345–353. [Google Scholar] [CrossRef] [Green Version]
- Drury, P.P.; Davidson, J.O.; Bennet, L.; Booth, L.C.; Tan, S.; Fraser, M.; Van Den Heuij, L.G.; Gunn, A.J. Partial Neural Protection with Prophylactic Low-Dose Melatonin after Asphyxia in Preterm Fetal Sheep. J. Cereb. Blood Flow Metab. 2014, 34, 126–135. [Google Scholar] [CrossRef] [Green Version]
- Reddy, K.; Mallard, C.; Guan, J.; Marks, K.; Bennet, L.; Gunning, M.; Gunn, A.; Gluckman, P.; Williams, C.E. Maturational Change in the Cortical Response to Hypoperfusion Injury in the Fetal Sheep. Pediatr. Res. 1998, 43, 674–682. [Google Scholar] [CrossRef] [Green Version]
- Williams, C.E.; Gunn, A.J.; Mallard, C.; Gluckman, P.D. Outcome after Ischemia in the Developing Sheep Brain: An Electroencephalographic and Histological Study. Ann. Neurol. 1992, 31, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.E.; Gunn, A.J.; Synek, B.; Gluckman, P.D. Delayed Seizures Occurring with Hypoxic-Ischemic Encephalopathy in the Fetal Sheep. Pediatr. Res. 1990, 27, 561–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maller, A.I.; Hankins, L.L.; Yeakley, J.W.; Butler, I.J. Rolandic Type Cerebral Palsy in Children as a Pattern of Hypoxic-Ischemic Injury in the Full-Term Neonate. J. Child Neurol. 1998, 13, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Myers, R.E. Atrophic Cortical Sclerosis Associated with Status Marmoratus in a Perinatally Damaged Monkey. Neurology 1969, 19, 1177–1188. [Google Scholar] [CrossRef] [PubMed]
- Myers, R.E. Two Patterns of Perinatal Brain Damage and Their Conditions of Occurrence. Am. J. Obstet. Gynecol. 1972, 112, 246–276. [Google Scholar] [CrossRef]
- Painter, M.J. Animal Models of Perinatal Asphyxia: Contributions, Contradictions, Clinical Relevance. Semin. Pediatr. Neurol. 1995, 2, 37–56. [Google Scholar] [CrossRef]
- Dobbing, J.; Sands, J. Comparative Aspects of the Brain Growth Spurt. Early Hum. Dev. 1979, 3, 79–83. [Google Scholar] [CrossRef]
- Semple, B.D.; Blomgren, K.; Gimlin, K.; Ferriero, D.M.; Noble-Haeusslein, L.J. Brain Development in Rodents and Humans: Identifying Benchmarks of Maturation and Vulnerability to Injury across Species. Prog. Neurobiol. 2013, 106–107, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Rice, J.E.; Vannucci, R.C.; Brierley, J.B. The Influence of Immaturity on Hypoxic-ischemic Brain Damage in the Rat. Ann. Neurol. 1981, 9, 131–141. [Google Scholar] [CrossRef]
- Ditelberg, J.S.; Sheldon, R.A.; Epstein, C.J.; Ferriero, D.M. Brain Injury after Perinatal Hypoxia-Ischemia is Exacerbated in Copper/Zinc Superoxide Dismutase Transgenic Mice. Pediatr. Res. 1996, 39, 204–208. [Google Scholar] [CrossRef] [Green Version]
- Sheldon, R.A.; Sedik, C.; Ferriero, D.M. Strain-Related Brain Injury in Neonatal Mice Subjected to Hypoxia-Ischemia. Brain Res. 1998, 810, 114–122. [Google Scholar] [CrossRef]
- Alexander, M.; Garbus, H.; Smith, A.L.; Rosenkrantz, T.S.; Fitch, R.H. Behavioral and Histological Outcomes Following Neonatal HI Injury in a Preterm (P3) and Term (P7) Rodent Model. Behav. Brain Res. 2014, 259, 85–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Jadhav, V.; Tang, J.; Zhang, J.H. HIF-1α Inhibition Ameliorates Neonatal Brain Injury in a Rat Pup Hypoxic-Ischemic Model. Neurobiol. Dis. 2008, 31, 433–441. [Google Scholar] [CrossRef] [Green Version]
- Vannucci, R.C.; Vannucci, S.J. Perinatal Hypoxic-Ischemic Brain Damage: Evolution of an Animal Model. Dev. Neurosci. 2005, 27, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Hill, A. Current Concepts of Hypoxic-Ischemic Cerebral Injury in the Term Newborn. Pediatr. Neurol. 1991, 7, 317–325. [Google Scholar] [CrossRef]
- Jantzie, L.L.; Robinson, S. Preclinical Models of Encephalopathy of Prematurity. Dev. Neurosci. 2015, 37, 277–288. [Google Scholar] [CrossRef] [Green Version]
- Bjelke, B.; Andersson, K.; Ögren, S.O.; Bolme, P. Asphyctic Lesion: Proliferation of Tyrosine Hydroxylase-Immunoreactive Nerve Cell Bodies in the Rat Substantia Nigra and Functional Changes in Dopamine Neurotransmission. Brain Res. 1991, 543, 1–9. [Google Scholar] [CrossRef]
- Dell’Anna, E.; Chen, Y.; Engidawork, E.; Andersson, K.; Lubec, G.; Luthman, J.; Herrera-Marschitz, M. Delayed Neuronal Death Following Perinatal Asphyxia in Rat. Exp. Brain Res. 1997, 115, 105–115. [Google Scholar] [CrossRef]
- Capani, F.; Loidl, F.; Lopez-Costa, J.J.; Selvin-Testa, A.; Saavedra, J.P. Ultrastructural Changes in Nitric Oxide Synthase Immunoreactivity in the Brain of Rats Subjected to Perinatal Asphyxia: Neuroprotective Effects of Cold Treatment. Brain Res. 1997, 775, 11–23. [Google Scholar] [CrossRef]
- Takada, S.H.; dos Santos Haemmerle, C.A.; Motta-Teixeira, L.C.; Machado-Nils, A.V.; Lee, V.Y.; Takase, L.F.; Cruz-Rizzolo, R.J.; Kihara, A.H.; Xavier, G.F.; Watanabe, I.S.; et al. Neonatal Anoxia in Rats: Hippocampal Cellular and Subcellular Changes Related to Cell Death and Spatial Memory. Neuroscience 2015, 284, 247–259. [Google Scholar] [CrossRef]
- Daval, J.L.; Pourié, G.; Grojean, S.; Lièvre, V.; Strazielle, C.; Blaise, S.; Vert, P. Neonatal Hypoxia Triggers Transient Apoptosis Followed by Neurogenesis in the Rat CA1 Hippocampus. Pediatr. Res. 2004, 55, 561–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Zhan, J.; Wang, X.; Gu, J.; Xie, K.; Zhang, Q.; Liu, D. Sodium Hydrosulfide Prevents Hypoxia-Induced Behavioral Impairment in Neonatal Mice. Brain Res. 2013, 1538, 126–134. [Google Scholar] [CrossRef]
- Caputa, M.; Rogalska, J.; Wentowska, K.; Nowakowska, A. Perinatal Asphyxia, Hyperthermia and Hyperferremia as Factors Inducing Behavioural Disturbances in Adulthood: A Rat Model. Behav. Brain Res. 2005, 163, 246–256. [Google Scholar] [CrossRef] [PubMed]
- Gordon, C.J. Temperature Regulation in Laboratory Rodents; Cambridge University Press: Cambridge, UK, 1993. [Google Scholar]
- Rogalska, J.; Caputa, M. Spontaneously Reduced Body Temperature and Gasping Ability as a Mechanism of Extreme Tolerance to Asphyxia in Neonatal Rats. J. Therm. Biol. 2005, 30, 360–369. [Google Scholar] [CrossRef]
- Rogalska, J.; Caputa, M. Neonatal Asphyxia under Hyperthermic Conditions Alters HPA Axis Function in Juvenile Rats. Neurosci. Lett. 2010, 472, 68–72. [Google Scholar] [CrossRef]
- Millar, L.J.; Shi, L.; Hoerder-Suabedissen, A.; Molnár, Z. Neonatal Hypoxia Ischaemia: Mechanisms, Models, and Therapeutic Challenges. Front. Cell. Neurosci. 2017, 11, 11. [Google Scholar] [CrossRef] [Green Version]
- Rennie, J.M.; Hagmann, C.F.; Robertson, N.J. Outcome after Intrapartum Hypoxic Ischaemia at Term. Semin. Fetal Neonatal Med. 2007, 12, 398–407. [Google Scholar] [CrossRef]
- Guzy, R.D.; Schumacker, P.T. Oxygen Sensing by Mitochondria at Complex III: The Paradox of Increased Reactive Oxygen Species during Hypoxia. Exp. Physiol. 2006, 91, 807–819. [Google Scholar] [CrossRef]
- Arteaga, O.; Álvarez, A.; Revuelta, M.; Santaolalla, F.; Urtasun, A.; Hilario, E. Role of Antioxidants in Neonatal Hypoxic-Ischemic Brain Injury: New Therapeutic Approaches. Int. J. Mol. Sci. 2017, 18, 256. [Google Scholar] [CrossRef] [Green Version]
- Ferriero, D.M. Oxidant Mechanisms in Neonatal Hypoxia-Ischemia. Dev. Neurosci. 2001, 23, 198–202. [Google Scholar] [CrossRef]
- Mccann, S.K.; Roulston, C.L. NADPH Oxidase as a Therapeutic Target for Neuroprotection against Ischaemic Stroke: Future Perspectives. Brain Sci. 2013, 3, 561–598. [Google Scholar] [CrossRef]
- Tarafdar, A.; Pula, G. The Role of NADPH Oxidases and Oxidative Stress in Neurodegenerative Disorders. Int. J. Mol. Sci. 2018, 19, 3824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Cell Biology of Ischemia/Reperfusion Injury. Int. Rev. Cell Mol. Biol. 2012, 298, 229–317. [Google Scholar] [PubMed] [Green Version]
- Granger, D.N.; Kvietys, P.R. Reperfusion Injury and Reactive Oxygen Species: The Evolution of a Concept. Redox Biol. 2015, 6, 524–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auten, R.L.; Davis, J.M. Oxygen Toxicity and Reactive Oxygen Species: The Devil is in the Details. Pediatr. Res. 2009, 66, 121–127. [Google Scholar] [CrossRef] [Green Version]
- Ciccoli, L.; Rossi, V.; Leoncini, S.; Signorini, C.; Blanco-Garcia, J.; Aldinucci, C.; Buonocore, G.; Comporti, M. Iron Release, Superoxide Production and Binding of Autologous IgG to Band 3 Dimers in Newborn and Adult Erythrocytes Exposed to Hypoxia and Hypoxia-Reoxygenation. Biochim. Biophys. Acta Gen. Subj. 2004, 1672, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wu, Y.; Li, T.; Wang, X.; Zhu, C. Iron Metabolism and Brain Development in Premature Infants. Front. Physiol. 2019, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Qing Kui, L.; Ming, Q.Z. Effect of Asphyxia on Non-Protein-Bound Iron and Lipid Peroxidation in Newborn Infants. Dev. Med. Child Neurol. 2003, 45, 24–27. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M.C. Biologically Relevant Metal Ion-Dependent Hydroxyl Radical Generation An Update. FEBS Lett. 1992, 307, 108–112. [Google Scholar] [CrossRef] [Green Version]
- Kalogeris, T.; Bao, Y.; Korthuis, R.J. Mitochondrial Reactive Oxygen Species: A Double Edged Sword in Ischemia/Reperfusion vs Preconditioning. Redox Biol. 2014, 2, 702–714. [Google Scholar] [CrossRef] [Green Version]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric Oxide and Peroxynitrite in Health and Disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solevåg, A.L.; Schmölzer, G.M.; Cheung, P.Y. Novel Interventions to Reduce Oxidative-Stress Related Brain Injury in Neonatal Asphyxia. Free Radic. Biol. Med. 2019, 142, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Ott, M.; Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Mitochondria, Oxidative Stress and Cell Death. Apoptosis 2007, 12, 913–922. [Google Scholar] [CrossRef]
- Orrenius, S. Mitochondrial Regulation of Apoptotic Cell Death. Toxicol. Lett. 2004, 149, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Arteaga, O.; Revuelta, M.; Montalvo, H.; Cañavate, M.L. Neuroprotective Effect of Antioxidants in Neonatal Rat Brain after Hypoxia- Ischemia. Microsc. Adv. Sci. Res. Educ. 2014, 1, 335–343. [Google Scholar]
- Okado-Matsumoto, A.; Fridovich, I. Subcellular Distribution of Superoxide Dismutases (SOD) in Rat Liver. Cu,Zn-SOD in Mitochondria. J. Biol. Chem. 2001, 276, 38388–38393. [Google Scholar] [CrossRef] [Green Version]
- Zaghloul, N.; Patel, H.; Codipilly, C.; Marambaud, P.; Dewey, S.; Frattini, S.; Huerta, P.T.; Nasim, M.; Miller, E.J.; Ahmed, M. Overexpression of Extracellular Superoxide Dismutase Protects against Brain Injury Induced by Chronic Hypoxia. PLoS ONE 2014, 9, e108168. [Google Scholar] [CrossRef] [Green Version]
- Ullah, R.; Khan, M.; Shah, S.A.; Saeed, K.; Kim, M.O. Natural Antioxidant Anthocyanins—A Hidden Therapeutic Candidate in Metabolic Disorders with Major Focus in Neurodegeneration. Nutrients 2019, 11, 1195. [Google Scholar] [CrossRef] [Green Version]
- Tokarz, P.; Kaarniranta, K.; Blasiak, J. Role of Antioxidant Enzymes and Small Molecular Weight Antioxidants in the Pathogenesis of Age-Related Macular Degeneration (AMD). Biogerontology 2013, 14, 461–482. [Google Scholar] [CrossRef] [Green Version]
- Perrone, S.; Negro, S.; Tataranno, M.L.; Buonocore, G. Oxidative Stress and Antioxidant Strategies in Newborns. J. Matern. Neonatal Med. 2010, 23, 63–65. [Google Scholar] [CrossRef]
- Shouman, B.O.; Mesbah, A.; Aly, H. Iron Metabolism and Lipid Peroxidation Products in Infants with Hypoxic Ischemic Encephalopathy. J. Perinatol. 2008, 28, 487–491. [Google Scholar] [CrossRef] [Green Version]
- Ikonomidou, C.; Kaindl, A.M. Neuronal Death and Oxidative Stress in the Developing Brain. Antioxid. Redox Signal. 2011, 14, 1535–1550. [Google Scholar] [CrossRef] [PubMed]
- Tataranno, M.L.; Perrone, S.; Longini, M.; Buonocore, G. New Antioxidant Drugs for Neonatal Brain Injury. Oxidative Med. Cell. Longev. 2015, 2015, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Cai, Z.; Yan, L.J. Protein Oxidative Modifications: Beneficial Roles in Disease and Health. J. Biochem. Pharmacol. Res. 2013, 1, 15–26. [Google Scholar]
- Yan, L.-J. Protein Redox Modification as a Cellular Defense Mechanism against Tissue Ischemic Injury. Oxidative Med. Cell. Longev. 2014, 2014, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azzopardi, D.; Strohm, B.; Marlow, N.; Brocklehurst, P.; Deierl, A.; Eddama, O.; Goodwin, J.; Halliday, H.L.; Juszczak, E.; Kapellou, O.; et al. Effects of Hypothermia for Perinatal Asphyxia on Childhood Outcomes. N. Engl. J. Med. 2014, 371, 140–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horiguchi, T.; Shimizu, K.; Ogino, M.; Suga, S.; Inamasu, J.; Kawase, T. Postischemic Hypothermia Inhibits the Generation of Hydroxyl Radical Following Transient Forebrain Ischemia in Rats. J. Neurotrauma 2003, 20, 511–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, A.; Jia, L. Crocin Enhances Hypothermia Therapy in Hypoxic Ischemia-Induced Brain Injury in Mice. Acta Neurol. Belg. 2019. [Google Scholar] [CrossRef]
- Nie, X.; Lowe, D.W.; Rollins, L.G.; Bentzley, J.; Fraser, J.L.; Martin, R.; Singh, I.; Jenkins, D. Sex-Specific Effects of N-Acetylcysteine in Neonatal Rats Treated with Hypothermia after Severe Hypoxia-Ischemia. Neurosci. Res. 2016, 108, 24–33. [Google Scholar] [CrossRef] [Green Version]
- Karnatovskaia, L.V.; Wartenberg, K.E.; Freeman, W.D. Therapeutic Hypothermia for Neuroprotection: History, Mechanisms, Risks, and Clinical Applications. Neurohospitalist 2014, 4, 153–163. [Google Scholar] [CrossRef] [Green Version]
- Caputa, M.; Rogalska, J.; Nowakowska, A. Effect of Temperature on Postanoxic, Potentially Neurotoxic Changes of Plasma PH and Free Iron Level in Newborn Rats. Brain Res. Bull. 2001, 55, 281–286. [Google Scholar] [CrossRef]
- Yoshida, Y.; Umeno, A.; Shichiri, M. Lipid Peroxidation Biomarkers for Evaluating Oxidative Stress and Assessing Antioxidant Capacity in Vivo. J. Clin. Biochem. Nutr. 2013, 52, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huun, M.U.; Garberg, H.T.; Escobar, J.; Chafer, C.; Vento, M.; Holme, I.M.; Saugstad, O. Di.; Solberg, R. DHA Reduces Oxidative Stress Following Hypoxia-Ischemia in Newborn Piglets: A Study of Lipid Peroxidation Products in Urine and Plasma. J. Perinat. Med. 2018, 46, 209–217. [Google Scholar] [CrossRef]
- Huun, M.U.; Garberg, H.T.; Buonocore, G.; Longini, M.; Belvisi, E.; Bazzini, F.; Proietti, F.; Saugstad, O.D.; Solberg, R. Regional Differences of Hypothermia on Oxidative Stress Following Hypoxia-Ischemia: A Study of DHA and Hypothermia on Brain Lipid Peroxidation in Newborn Piglets. J. Perinat. Med. 2019, 47, 82–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, E.; Karimi Galougahi, K.; Liu, C.C.; Bhindi, R.; Figtree, G.A. Biological Markers of Oxidative Stress: Applications to Cardiovascular Research and Practice. Redox Biol. 2013, 1, 483–491. [Google Scholar] [CrossRef] [Green Version]
- Toader, A.M.; Filip, A.; Decea, N.; Muresan, A. Neuroprotective Strategy in an Experimental Newborn Rat Model of Brain Ischemia and Hypoxia: Effects of Resveratrol and Hypothermia. Clujul Med. 2013, 86, 203–207. [Google Scholar]
- Suzuki, Y.J.; Carini, M.; Butterfield, D.A. Protein Carbonylation. Antioxid. Redox Signal. 2010, 12, 323–325. [Google Scholar] [CrossRef]
- Lafuente, H.; Pazos, M.R.; Alvarez, A.; Mohammed, N.; Santos, M.; Arizti, M.; Alvarez, F.J.; Martinez-Orgado, J.A. Effects of Cannabidiol and Hypothermia on Short-Term Brain Damage in New-Born Piglets after Acute Hypoxia-Ischemia. Front. Neurosci. 2016, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Barata, L.; Arruza, L.; Rodríguez, M.J.; Aleo, E.; Vierge, E.; Criado, E.; Sobrino, E.; Vargas, C.; Ceprián, M.; Gutiérrez-Rodríguez, A.; et al. Neuroprotection by Cannabidiol and Hypothermia in a Piglet Model of Newborn Hypoxic-Ischemic Brain Damage. Neuropharmacology 2019, 146, 1–11. [Google Scholar] [CrossRef]
- Santos, P.T.; O’Brien, C.E.; Chen, M.W.; Hopkins, C.D.; Adams, S.; Kulikowicz, E.; Singh, R.; Koehler, R.C.; Martin, L.J.; Lee, J.K. Proteasome Biology is Compromised in White Matter after Asphyxic Cardiac Arrest in Neonatal Piglets. J. Am. Heart Assoc. 2018, 7. [Google Scholar] [CrossRef] [Green Version]
- Dalen, M.L.; Alme, T.N.; Bjørås, M.; Munkeby, B.H.; Rootwelt, T.; Saugstad, O.D. Reduced Expression of DNA Glycosylases in Post-Hypoxic Newborn Pigs Undergoing Therapeutic Hypothermia. Brain Res. 2010, 1363, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Wang, B.; Lee, J.H.; Armstrong, J.S.; Kulikowicz, E.; Bhalala, U.S.; Martin, L.J.; Koehler, R.C.; Yang, Z.J. Additive Neuroprotection of a 20-HETE Inhibitor with Delayed Therapeutic Hypothermia after Hypoxia-Ischemia in Neonatal Piglets. Dev. Neurosci. 2015, 37, 376–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldon, R.A.; Christen, S.; Ferriero, D.M. Genetic and Pharmacologic Manipulation of Oxidative Stress after Neonatal Hypoxia-Ischemia. Int. J. Dev. Neurosci. 2008, 26, 87–92. [Google Scholar] [CrossRef] [Green Version]
- Bratek, E.; Ziembowicz, A.; Salinska, E. Pretreatment with Group II Metabotropic Glutamate Receptor Agonist LY379268 Protects Neonatal Rat Brains from Oxidative Stress in an Experimental Model of Birth Asphyxia. Brain Sci. 2018, 8, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niki, E. Role of Vitamin E as a Lipid-Soluble Peroxyl Radical Scavenger: In Vitro and in Vivo Evidence. Free Radic. Biol. Med. 2014, 66, 3–12. [Google Scholar] [CrossRef]
- Satoh, T.; Yoshioka, Y. Contribution of Reduced and Oxidized Glutathione to Signals Detected by Magnetic Resonance Spectroscopy as Indicators of Local Brain Redox State. Neurosci. Res. 2006, 55, 34–39. [Google Scholar] [CrossRef]
- Devi, P.U.; Manocha, A.; Vohora, D. Seizures, Antiepileptics, Antioxidants and Oxidative Stress: An Insight for Researchers. Expert Opin. Pharmacother. 2008, 9, 3169–3177. [Google Scholar] [CrossRef]
- Gurer, H.; Ercal, N. Can Antioxidants be Beneficial in the Treatment of Lead Poisoning? Free Radic. Biol. Med. 2000, 29, 927–945. [Google Scholar] [CrossRef]
- Schafer, F.Q.; Buettner, G.R. Redox Environment of the Cell as Viewed through the Redox State of the Glutathione Disulfide/Glutathione Couple. Free Radic. Biol. Med. 2001, 30, 1191–1212. [Google Scholar] [CrossRef]
- Alva, N.; Carbonell, T.; Palomeque, J. Hypothermic Protection in an Acute Hypoxia Model in Rats: Acid-Base and Oxidant/Antioxidant Profiles. Resuscitation 2010, 81, 609–616. [Google Scholar] [CrossRef]
- Flora, G.; Gupta, D.; Tiwari, A. Toxicity of Lead: A Review with Recent Updates. Interdiscip. Toxicol. 2012, 5, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Wisnowski, J.L.; Wu, T.W.; Reitman, A.J.; McLean, C.; Friedlich, P.; Vanderbilt, D.; Ho, E.; Nelson, M.D.; Panigrahy, A.; Blüml, S. The Effects of Therapeutic Hypothermia on Cerebral Metabolism in Neonates with Hypoxic-Ischemic Encephalopathy: An in Vivo 1 H-MR Spectroscopy Study. J. Cereb. Blood Flow Metab. 2016, 36, 1075–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kletkiewicz, H.; Rogalska, J.; Nowakowska, A.; Wozniak, A.; Mila-Kierzenkowska, C.; Caputa, M. Effects of Body Temperature on Post-Anoxic Oxidative Stress from the Perspective of Postnatal Physiological Adaptive Processes in Rats. J. Physiol. Pharmacol. 2016, 67, 287–299. [Google Scholar] [PubMed]
- Zhang, H.; Zhang, J.J.; Mei, Y.W.; Sun, S.G.; Tong, E.T. Effects of Immediate and Delayed Mild Hypothermia on Endogenous Antioxidant Enzymes and Energy Metabolites Following Global Cerebral Ischemia. Chin. Med. J. 2011, 124, 2764–2766. [Google Scholar]
- Chevin, M.; Guiraut, C.; Maurice-Gelinas, C.; Deslauriers, J.; Grignon, S.; Sébire, G. Neuroprotective Effects of Hypothermia in Inflammatory-Sensitized Hypoxic-Ischemic Encephalopathy. Int. J. Dev. Neurosci. 2016, 55, 1–8. [Google Scholar] [CrossRef]
- Jaiswal, A.K. Nrf2 Signaling in Coordinated Activation of Antioxidant Gene Expression. Free Radic. Biol. Med. 2004, 36, 1199–1207. [Google Scholar] [CrossRef]
- Gong, P.; Li, C.S.; Hua, R.; Zhao, H.; Tang, Z.R.; Mei, X.; Zhang, M.Y.; Cui, J. Mild Hypothermia Attenuates Mitochondrial Oxidative Stress by Protecting Respiratory Enzymes and Upregulating Mnsod in a Pig Model of Cardiac Arrest. PLoS ONE 2012, 7, 35313. [Google Scholar] [CrossRef] [Green Version]
- Joy, R.; Pournami, F.; Bethou, A.; Bhat, V.B.; Bobby, Z. Effect of Therapeutic Hypothermia on Oxidative Stress and Outcome in Term Neonates with Perinatal Asphyxia: A Randomized Controlled Trial. J. Trop. Pediatr. 2013, 59, 17–22. [Google Scholar] [CrossRef] [Green Version]
- Bayir, H.; Adelson, P.D.; Wisniewski, S.R.; Shore, P.; Lai, Y.; Brown, D.; Janesko-Feldman, K.L.; Kagan, V.E.; Kochanek, P.M. Therapeutic Hypothermia Preserves Antioxidant Defenses after Severe Traumatic Brain Injury in Infants and Children. Crit. Care Med. 2009, 37, 689–695. [Google Scholar] [CrossRef]
- Edwards, A.D.; Brocklehurst, P.; Gunn, A.J.; Halliday, H.; Juszczak, E.; Levene, M.; Strohm, B.; Thoresen, M.; Whitelaw, A.; Azzopardi, D. Neurological Outcomes at 18 Months of Age after Moderate Hypothermia for Perinatal Hypoxic Ischaemic Encephalopathy: Synthesis and Meta-Analysis of Trial Data. BMJ 2010, 340, c363. [Google Scholar] [CrossRef] [Green Version]
- Wood, T.; Osredkar, D.; Puchades, M.; Maes, E.; Falck, M.; Flatebø, T.; Walløe, L.; Sabir, H.; Thoresen, M. Treatment Temperature and Insult Severity Influence the Neuroprotective Effects of Therapeutic Hypothermia. Sci. Rep. 2016, 6, 23430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Videla, L.A.; Villena, M.I.; Salgado, C.; Canales, P.; Lissi, E.A. Antioxidant Capacity of Desferrioxamine in Biological Systems. Biochem. Int. 1987, 15, 205–214. [Google Scholar] [PubMed]
- Laptook, A.R. Birth Asphyxia and Hypoxic-Ischemic Brain Injury in the Preterm Infant. Clin. Perinatol. 2016, 43, 529–545. [Google Scholar] [CrossRef] [PubMed]
- Rathnasamy, G.; Ling, E.A.; Kaur, C. Iron and Iron Regulatory Proteins in Amoeboid Microglial Cells are Linked to Oligodendrocyte Death in Hypoxic Neonatal Rat Periventricular White Matter through Production of Proinflammatory Cytokines and Reactive Oxygen/Nitrogen Species. J. Neurosci. 2011, 31, 17982–17995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emerit, J.; Beaumont, C.; Trivin, F. Iron Metabolism, Free Radicals, and Oxidative Injury. Biomed. Pharmacother. 2001, 55, 333–339. [Google Scholar] [CrossRef]
- Gutteridge, J.M.; Richmond, R.; Halliwell, B. Inhibition of the Iron-Catalysed Formation of Hydroxyl Radicals from Superoxide and of Lipid Peroxidation by Desferrioxamine. Biochem. J. 1979, 184, 469–472. [Google Scholar] [CrossRef] [Green Version]
- Halliwell, B. Protection against Tissue Damage in Vivo by Desferrioxamine: What is its Mechanism of Action? Free Radic. Biol. Med. 1989, 7, 645–651. [Google Scholar] [CrossRef]
- Feng, Y.; LeBlanc, M.H.; LeBlanc, E.B.; Parker, C.C.; Fratkin, J.D.; Qian, X.B.; Patel, D.M.; Huang, M.; Smith, E.E.; Vig, P.J.S. Desmethyl Tirilazad Improves Neurologic Function after Hypoxic Ischemic Brain Injury in Piglets. Crit. Care Med. 2000, 28, 1431–1438. [Google Scholar] [CrossRef]
- Lu, Q.; Harris, V.A.; Rafikov, R.; Sun, X.; Kumar, S.; Black, S.M. Nitric Oxide Induces Hypoxia Ischemic Injury in the Neonatal Brain via the Disruption of Neuronal Iron Metabolism. Redox Biol. 2015, 6, 112–121. [Google Scholar] [CrossRef] [Green Version]
- Hartley, A.; Davies, M.; Rice-Evans, C. Desferrioxamine as a Lipid Chain-Breaking Antioxidant in Sickle Erythrocyte Membranes. FEBS Lett. 1990, 264, 145–148. [Google Scholar] [CrossRef] [Green Version]
- McDonald, J.W.; Johnston, M.V. Physiological and Pathophysiological Roles of Excitatory Amino Acids during Central Nervous System Development. Brain Res. Rev. 1990, 15, 41–70. [Google Scholar] [CrossRef] [Green Version]
- Hagberg, H.; Andersson, P.; Kjellmer, I.; Thiringer, K.; Thordstein, M. Extracellular Overflow of Glutamate, Aspartate, GABA and Taurine in the Cortex and Basal Ganglia of Fetal Lambs during Hypoxia-Ischemia. Neurosci. Lett. 1987, 78, 311–317. [Google Scholar] [CrossRef]
- Choi, D.W.; Rothman, S.M. The Role of Glutamate Neurotoxicity in Hypoxic-Ischemic Neuronal Death. Annu. Rev. Neurosci. 1990, 13, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Coyle, J.T.; Puttfarcken, P. Oxidative Stress, Glutamate, and Neurodegenerative Disorders. Science 1993, 262, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Groenendaal, F.; Shadid, M.; McGowan, J.E.; Mishra, O.P.; Van Bel, F. Effects of Deferoxamine, a Chelator of Free Iron, on Na+, K+-ATPase Activity of Cortical Brain Cell Membrane during Early Reperfusion after Hypoxia-Ischemia in Newborn Lambs. Pediatr. Res. 2000, 48, 560–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peeters-Scholte, C.; Braun, K.; Koster, J.; Kops, N.; Blomgren, K.; Buonocore, G.; van Buul-Offers, S.; Hagberg, H.; Nicolay, K.; van Bel, F.; et al. Effects of Allopurinol and Deferoxamine on Reperfusion Injury of the Brain in Newborn Piglets after Neonatal Hypoxia-Ischemia. Pediatr. Res. 2003, 54, 516–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papazisis, G.; Pourzitaki, C.; Sardeli, C.; Lallas, A.; Amaniti, E.; Kouvelas, D. Deferoxamine Decreases the Excitatory Amino Acid Levels and Improves the Histological Outcome in the Hippocampus of Neonatal Rats after Hypoxia-Ischemia. Pharmacol. Res. 2008, 57, 73–78. [Google Scholar] [CrossRef]
- Semenza, G.L. Regulation of Mammalian O2 Homeostasis by Hypoxia-Inducible Factor 1. Annu. Rev. Cell Dev. Biol. 1999, 15, 551–578. [Google Scholar] [CrossRef]
- Callapina, M.; Zhou, J.; Schmid, T.; Köhl, R.; Brüne, B. NO Restores HIF-1α Hydroxylation during Hypoxia: Role of Reactive Oxygen Species. Free Radic. Biol. Med. 2005, 39, 925–936. [Google Scholar] [CrossRef]
- Huang, L.E.; Arany, Z.; Livingston, D.M.; Bunn, H.F. Activation of Hypoxia-Inducible Transcription Factor Depends Primarily upon Redox-Sensitive Stabilization of its Alpha Subunit. J. Biol. Chem. 1996, 271, 32253–32259. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Huang, Q.; Zou, L.; Cao, X.; Huang, H.; Chu, X. Beneficial Effects of Deferoxamine against Astrocyte Death Induced by Modified Oxygen Glucose Deprivation. Brain Res. 2014, 1583, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Chu, P.W.Y.; Beart, P.M.; Jones, N.M. Preconditioning Protects against Oxidative Injury Involving Hypoxia-Inducible Factor-1 and Vascular Endothelial Growth Factor in Cultured Astrocytes. Eur. J. Pharmacol. 2010, 633, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Semenza, G.L. Desferrioxamine Induces Erythropoietin Gene Expression and Hypoxia-Inducible Factor 1 DNA-Binding Activity: Implications for Models of Hypoxia Signal Transduction. Blood 1993, 82, 3610–3615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemus-Varela, M.L.; Flores-Soto, M.E.; Cervantes-Munguía, R.; Torres-Mendoza, B.M.G.; Gudiño-Cabrera, G.; Chaparro-Huerta, V.; Ortuño-Sahagún, D.; Beas-Zárate, C. Expression of HIF-1α, VEGF and EPO in Peripheral Blood from Patients with Two Cardiac Abnormalities Associated with Hypoxia. Clin. Biochem. 2010, 43, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Van der Kooij, M.A.; Groenendaal, F.; Kavelaars, A.; Heijnen, C.J.; van Bel, F. Neuroprotective Properties and Mechanisms of Erythropoietin in in Vitro and in Vivo Experimental Models for Hypoxia/Ischemia. Brain Res. Rev. 2008, 59, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Heijnen, C.J.; van der Kooij, M.A.; Groenendaal, F.; van Bel, F. The Role and Regulation of Hypoxia-Inducible Factor-1α Expression in Brain Development and Neonatal Hypoxic-Ischemic Brain Injury. Brain Res. Rev. 2009, 62, 99–108. [Google Scholar] [CrossRef]
- Hamrick, S.E.G.; McQuillen, P.S.; Jiang, X.; Mu, D.; Madan, A.; Ferriero, D.M. A Role for Hypoxia-Inducible Factor-1α in Desferoxamine Neuroprotection. Neurosci. Lett. 2005, 379, 96–100. [Google Scholar] [CrossRef]
- Mu, D.; Chang, Y.S.; Vexler, Z.S.; Ferriero, D.M. Hypoxia-Inducible Factor 1alpha and Erythropoietin Upregulation with Deferoxamine Salvage after Neonatal Stroke. Exp. Neurol. 2005, 195, 407–415. [Google Scholar] [CrossRef]
- Li, L.; Yin, X.; Ma, N.; Lin, F.; Kong, X.; Chi, J.; Feng, Z. Desferrioxamine Regulates HIF-1 Alpha Expression in Neonatal Rat Brain after Hypoxia-Ischemia. Am. J. Transl. Res. 2014, 6, 377–383. [Google Scholar]

{kind=link}
{kind=link}
| References | Animal Models | Sample Size | Hypoxic/Anoxic Damage | Temperature | Impact on Oxidative Stress |
|---|---|---|---|---|---|
| Barata et al., 2019 | male piglets; one-day-old | No data | Oxygen decreased to 10% for 25 min. | Normothermia: 37.5–38.5 °C (rectal) Hypothermia: 34–34.5 °C (rectal) | ↑ oxidative stress after H/I; ↓ protein carbonyl levels (markers of oxidative stress) after the combination of cannabidiol and hypothermia |
| Dalen et al., 2010 | Noroc (LYxLD) pigs; newborn (25 ± 4.8 h of age) | 52, weight: 1980 ± 106 g, | Oxygen decreased to 8% for 61 min. | Normothermia: 39 °C (rectal) Hypothermia: 35 °C (rectal) | ↑ oxidative stress after hypoxia; ↓ expression of genes of DNA repair after hypothermia, with no effect on accumulation of oxidative damage in genomic DNA |
| Huang et al., 2019 | C57BL/6J mice; post-natal day 7 | no data | Oxygen decreased to 8% for 30 min | Normothermia: 36 °C (rectal) Hypothermia: 33 °C (rectal) | ↓ ROS and NO as a result of hypothermia ↓ MDA, ROS and NO after hypothermia combined with crocin |
| Huun et al., 2018a | Noroc (LyxLD) pigs; newborn (12–36 h of age) | 55 | Oxygen decreased to 8% for 20 min | Normothermia: 38.5–39.5 °C (rectal) Hypothermia: 34.5 °C (rectal) | ↓ oxidative stress markers: 8-iso-PGF2α (in urine) after hypoxia and hypothermia |
| Huun et al., 2018b | Noroc (LyxLD) pigs; newborn (12–36 h of age) | 81 | Oxygen decreased to 8% for 20 min | Normothermia: 39 °C (rectal) Hypothermia: 34.5 °C (rectal) | ↓ oxidative stress markers: F4-NeuroPs, F2-IsoPs, DH-isoprostanes (in the white matter) after hypoxia and hypothermia |
| Kletkiewicz et al., 2016a | Wistar rats; two-days-old | 108, both sexes, weight: 7–8 g | 100% nitrogen atmosphere for 10 min | Normothermia for newborn rat: 33 °C (rectal) Hyperthermia: 37–39 °C (rectal) | ↑ lipid peroxidation ↓ antioxidant enzymes activity after perinatal anoxia at elevated body temperatures, however there was no decrease in enzymes activity in the group with body temperature of 33°C |
| Kletkiewicz et al., 2016b | Wistar rats; two-days-old | 180, both sexes, weight: 7–8 g | 100% nitrogen atmosphere for 10 min | Normothermia for newborn rat: 33 °C (rectal) Hyperthermia: 39 °C (rectal) | Normothermic (33°C) body temperature prevents post-asphyxic disturbances in cerebral oxidant homeostasis (markers: level of low-molecular antioxidants) |
| Kletkiewicz et al., 2016c | Wistar rats; two-days-old | 192, both sexes, weight: 7–8 g | 100% nitrogen atmosphere for 10 min | Hypothermia: 31 °C (rectal) Normothermia for newborn rat: 33 °C (rectal) Hyperthermia: 37 °C & 39 °C (rectal) | ↑ MDA, ↑ CD and ↓GPx in both hyper-thermic groups ↑ SOD and ↓ CAT in extremely hypothermic and hyperthermic newborns, no changes in the levels of° MDA, CD and in enzymes activity in rats with body temperature of 33 °C |
| Lafuente et al., 2016 | male piglets; 1 to 2-day-old | no data | Oxygen decreased to 10% for 30 min | Normothermia: 38 °C (rectal) Hypothermia: 33–34 °C (rectal) | ↓ protein carbonyls formation in parietal cortex and striatum 6h after H/I and hypothermia, cannabidiol enhance the protective effect of hypothermia |
| Mueller-Burke et al., 2008 | male piglets; 5 to 7-day-old | 26, weighing 3.0–4.5 kg, | Oxygen decreased to 10% for 30 min | Normothermia: 38.5 °C (rectal) Hypothermia: 34 °C (rectal) | ↓ protein oxidation after post-hypoxic mild whole-body hypothermia |
| Nie et al., 2016 | Sprague-Dawley rats; post-natal day 7 | 21 | Oxygen decreased to 8% for 120 min | Normothermia: 36.3 ± 0.5 °C Hypothermia: 30 ± 0.5 °C | ↓expression of nitric oxide synthase (iNOS) after post-hypoxic hypothermia combined with N-acetylcysteine |
| Santos et al., 2018 | male piglets; 2 to 3-day-old | 98, weight: 1.0–2.5 kg | Oxygen decreased to 10% for 45 min | Normothermia: 38.0 to 39.5 °C (rectal), Hypothermia: 34.0 °C | ↑ carbonylated protein levels after H/I and hypothermia |
| Toader et al., 2013 | Wistar rats; Newbornpost-natal day 7 | 80, both genders, weight: 10 g | Oxygen decreased to 8% for 90 min | Normothermia: no data on value Hypothermia: 33–34 °C (intra-rectal), | ↑ MDA ↓ SOD and GPx in hypothermia, ↓ MDA ↑ SOD in H/I and hypothermia |
| Zhu et al., 2014 | male piglets; 3–5 days of age | 50 | Oxygen decreased to 10% for 45 min | Normothermia: 38.5 to 39 °C (rectal), Hypothermia: 34.0 °C | the use of inhibitor of oxidative stress promoter enhances the effect of delayed hypothermia |
| References | In Vitro/In Vivo Models | DFO Dose/Time of Administration | Hypoxic/Ischemic Damage | Suggested Mechanisms of Action | Result of DFO Administration |
|---|---|---|---|---|---|
| Papazisis et al., 2008 | Wistar rats; seven-day-old | 150 mg/kg s.c; subcutaneously, immediately after insult and 24 h later | Oxygen decreased to 8% for 60 min | impact on the neurotransmitters’ release | decreases the excitatory amino acid levels; reduces the number of damaged neurons in the CA1 region |
| Kletkiewicz et al., 2016a | Wistar rats; two-days-old | 100 mg/kg s.c; subcutaneously, immediately after insult and 24 h later | 100% nitrogen atmosphere for 10 min | antioxidant action | prevents SOD, CAT and GPx depletion; decreases MDA level |
| Kletkiewicz et al., 2016b | Wistar rats; two-days-old | 100 mg/kg s.c; subcutaneously, immediately after insult and 24h later | 100% nitrogen atmosphere for 10 min | antioxidant action | prevents cerebral glutathione and vitamin E depletion; decreases MDA level |
| Chu et al., 2010 | Primary astrocyte cultures from PD1–2 Swiss white mice | Preconditioning for 0.5 to 24 h with 0.1–1 mM of DFO | hydrogen peroxide exposure (0.1–1 mM) for a further 24 h | iron chelation - removing the PHD-bound ferrous ion | protects the astrocytes against H2O2-induced injury; changes the expression of HIF-1α and VEGF |
| Hamrick et al., 2005 | Hippocampal neurons from E16 CD1 mice | pretreatment with 10 mmol/L DFO for 1h | 95% N and 5% CO2 for 5 min | iron chelation | reduces cell death; induces HIF-1α |
| Almli et al., 2001 | Primary hippocampal cell culture from fetal (E16) CD-1 mice | Pretreatment for 1 h at various doses ranging 50–20 mM | H2O2 and NMDA exposure for 24 h | antioxidant action and iron chelation | reduces cell death; protects against H2O2 and NMDA-induced toxicity |
| Sarco et al., 2000 | tg mice, carrying the SOD1 gene; seven-day-old | 100 mg/kg s.c; subcutaneously, immediately after insult and 24h later | Oxygen decreased to 8% for 30 min | iron chelation | reduces brain iron content and hypoxic-ischemic brain damage |
| Feng et al., 2000 | Piglets; 0 to 3-days-old | 100 mg/kg s.c; 15 min after recovery | Oxygen decreased to 6% for 15 min | iron chelation | inhibits lipid peroxidation |
| Peeters-Scholte et al., 2003 | Piglets; 1 to 3-days-old | 10 mg/kg upon reperfusion and a repeated dose of 2.5 mg/kg at 12 h, injected intravenously | 1 h of hypoxia-ischemia by occluding both carotid arteries and reducing the fraction of inspired oxygen | iron chelation | maintains cerebral energy status after global hypoxia-ischemia |
| Lu et al., 2015 | P7 hippocampal slice cultures exposed to oxygen–glucose deprivation (OGD) | 100 mM; 2 h before OGD | Slices exposed to 0.1% O2, 5% CO2, 94.4% nitrogen for 90 min | iron chelation | reduces hydroxyl radical levels and neuronal cell death |
| Mu et al., 2005 | Sprague–Dawley rats; ten-days-old | 200 mg/kg s.c.; immediately after reperfusion administered intraperitoneally | middle cerebral artery (MCA) occlusion | Induction of HIF1α expression | increases HIF-1α and EPO level |
| Rogalska et al., 2006 | Wistar rats; two-days-old | 100 mg/kg s.c; subcutaneously, immediately after insult and 24 h later | 100% nitrogen atmosphere for 10 min | iron chelation | protects against the brain hyperferremia |
| Caputa et al., 2005 | Wistar rats; two-days-old | 100 mg/kg s.c; subcutaneously, immediately after insult and 24 h later | 100% nitrogen atmosphere for 10 min | iron chelation | prevents the behavioral disturbances |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kletkiewicz, H.; Klimiuk, M.; Woźniak, A.; Mila-Kierzenkowska, C.; Dokladny, K.; Rogalska, J. How to Improve the Antioxidant Defense in Asphyxiated Newborns—Lessons from Animal Models. Antioxidants 2020, 9, 898. https://doi.org/10.3390/antiox9090898
Kletkiewicz H, Klimiuk M, Woźniak A, Mila-Kierzenkowska C, Dokladny K, Rogalska J. How to Improve the Antioxidant Defense in Asphyxiated Newborns—Lessons from Animal Models. Antioxidants. 2020; 9(9):898. https://doi.org/10.3390/antiox9090898
Chicago/Turabian StyleKletkiewicz, Hanna, Maciej Klimiuk, Alina Woźniak, Celestyna Mila-Kierzenkowska, Karol Dokladny, and Justyna Rogalska. 2020. "How to Improve the Antioxidant Defense in Asphyxiated Newborns—Lessons from Animal Models" Antioxidants 9, no. 9: 898. https://doi.org/10.3390/antiox9090898