
Evaluating the Effect of Cell Culture on Gene Expression in Primary Tissue Samples Using Microfluidic-Based Single Cell Transcriptional Analysis

Abstract
:
{kind=link}
{kind=link}
{kind=link}
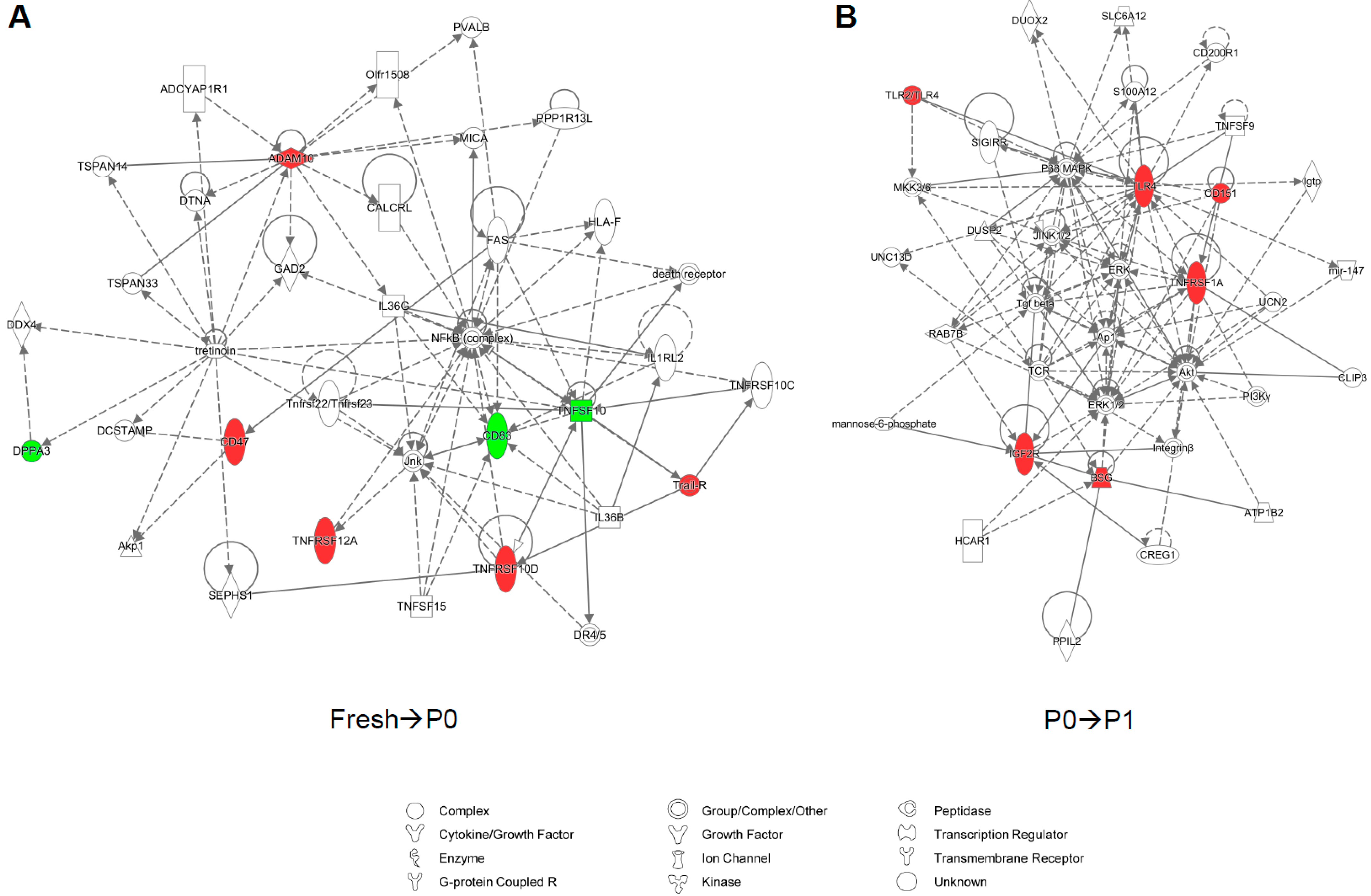{kind=link}
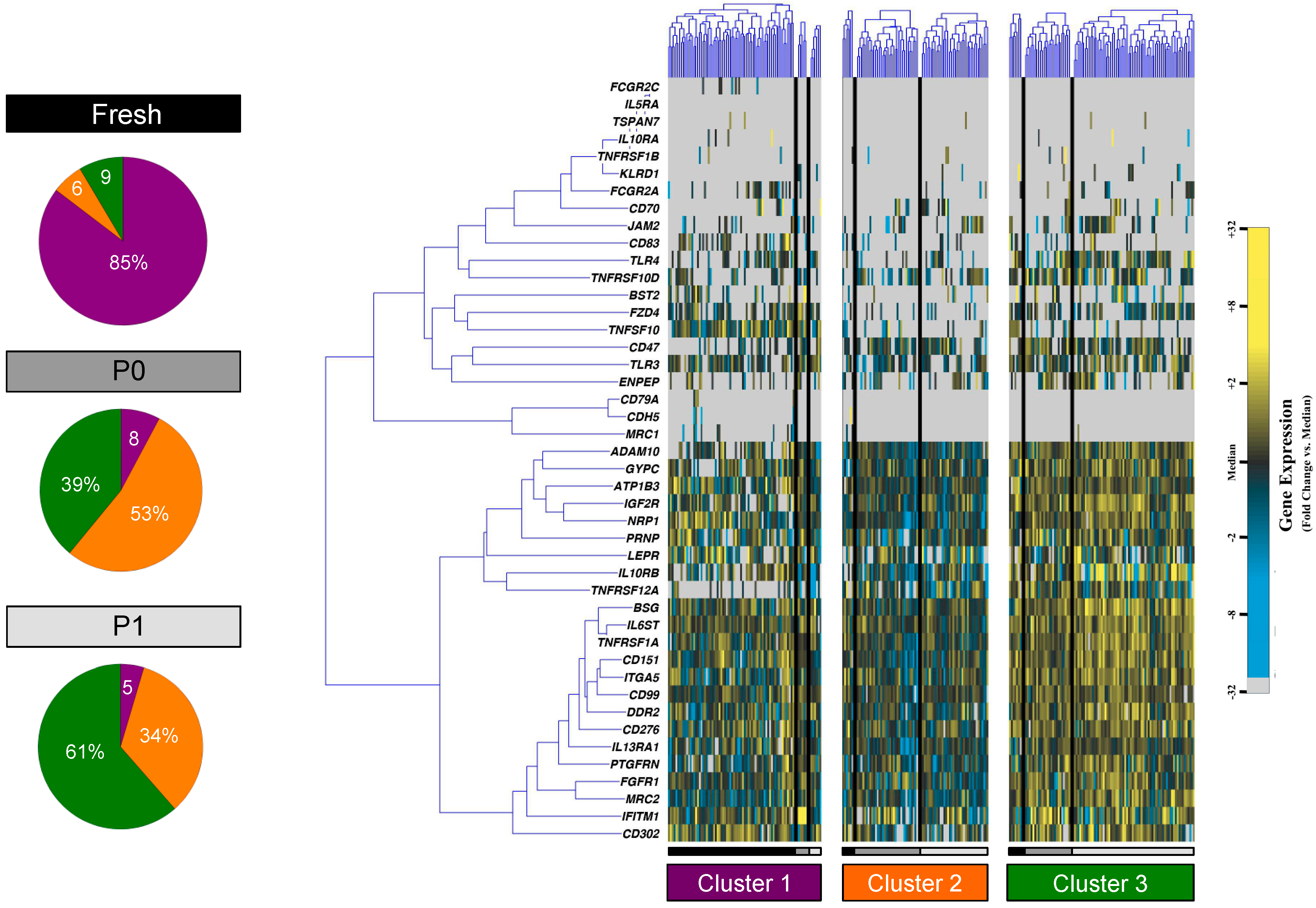{kind=link}
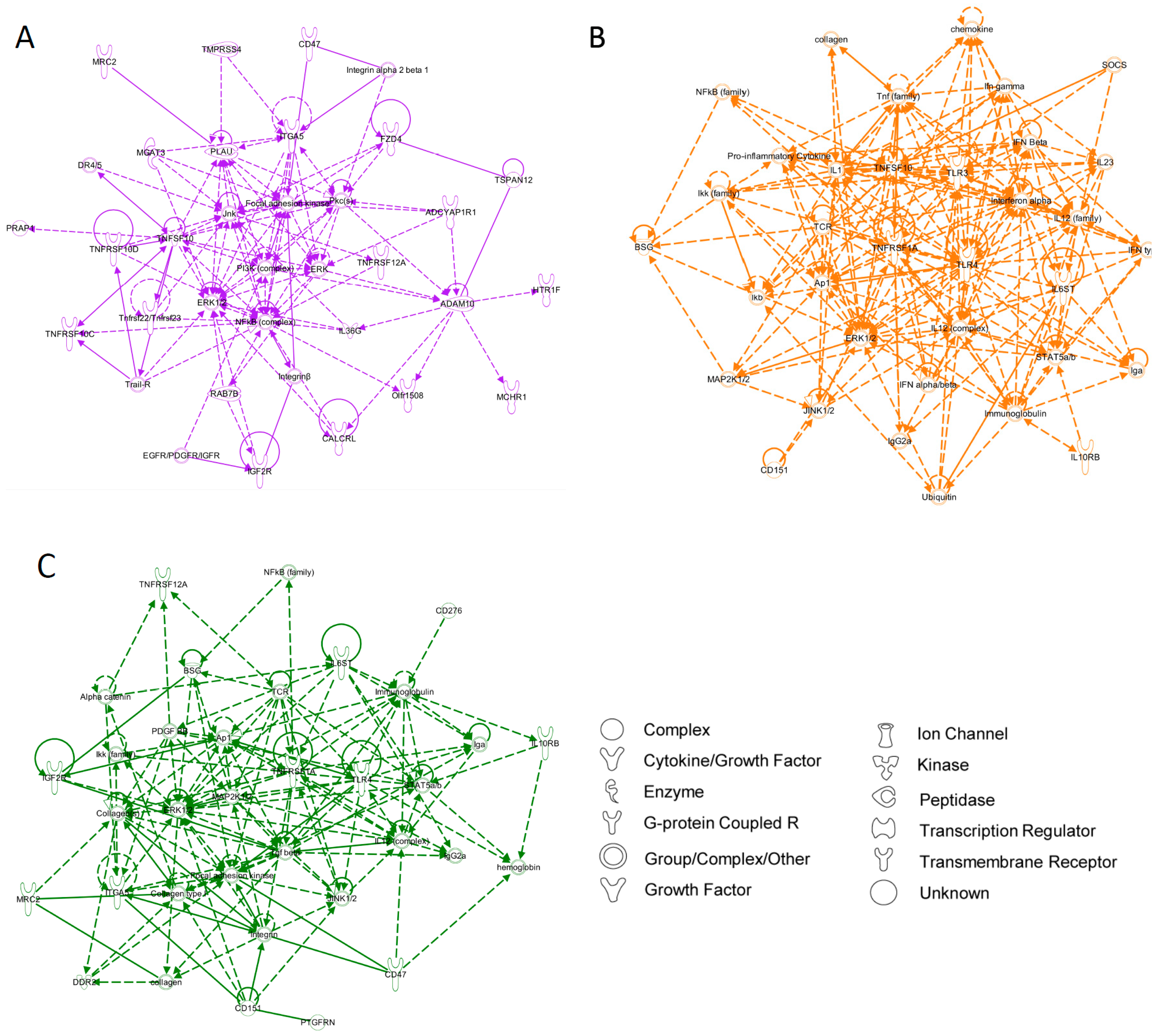{kind=link}
1. Introduction
2. Experimental Section
3. Results and Discussion
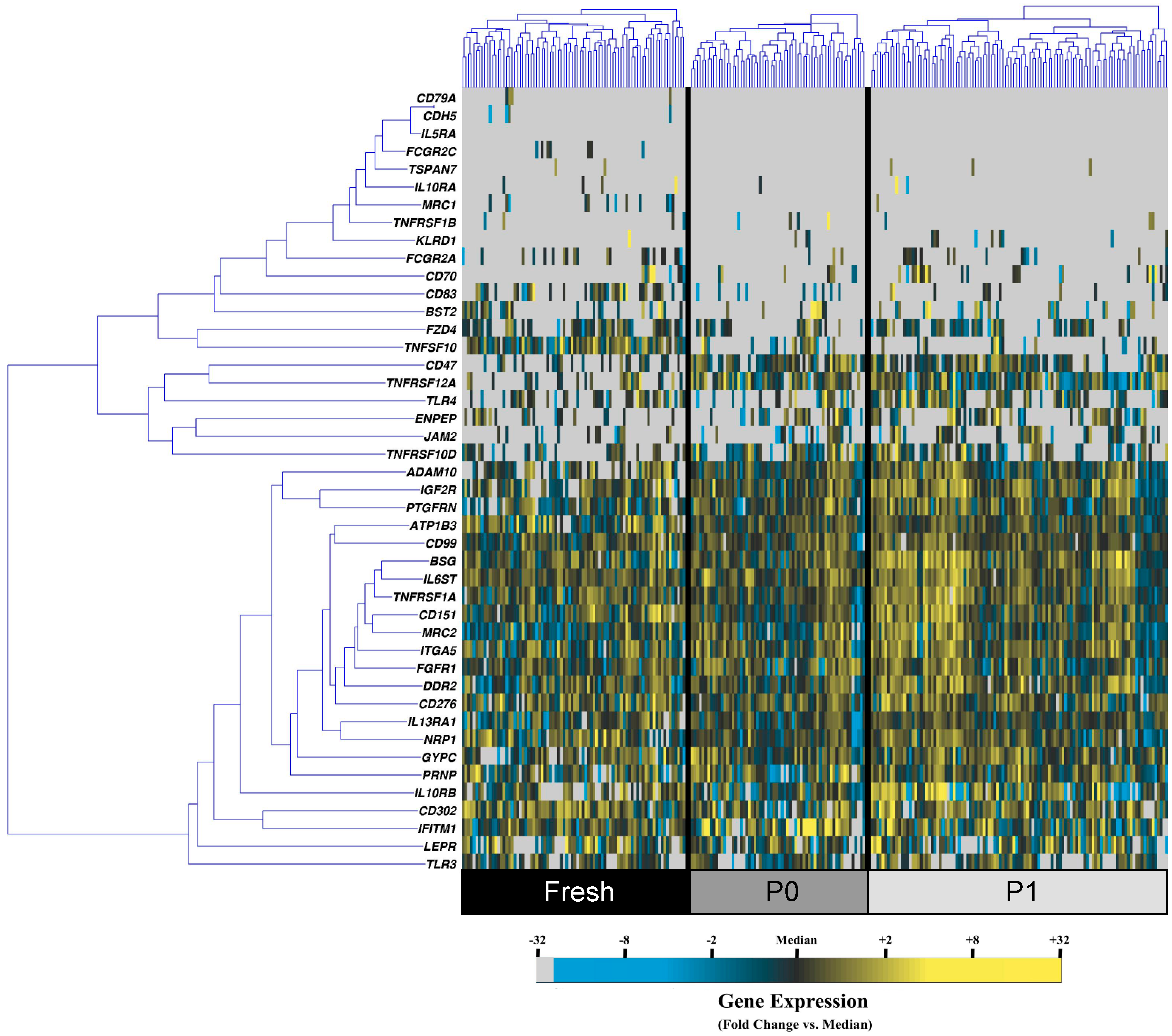
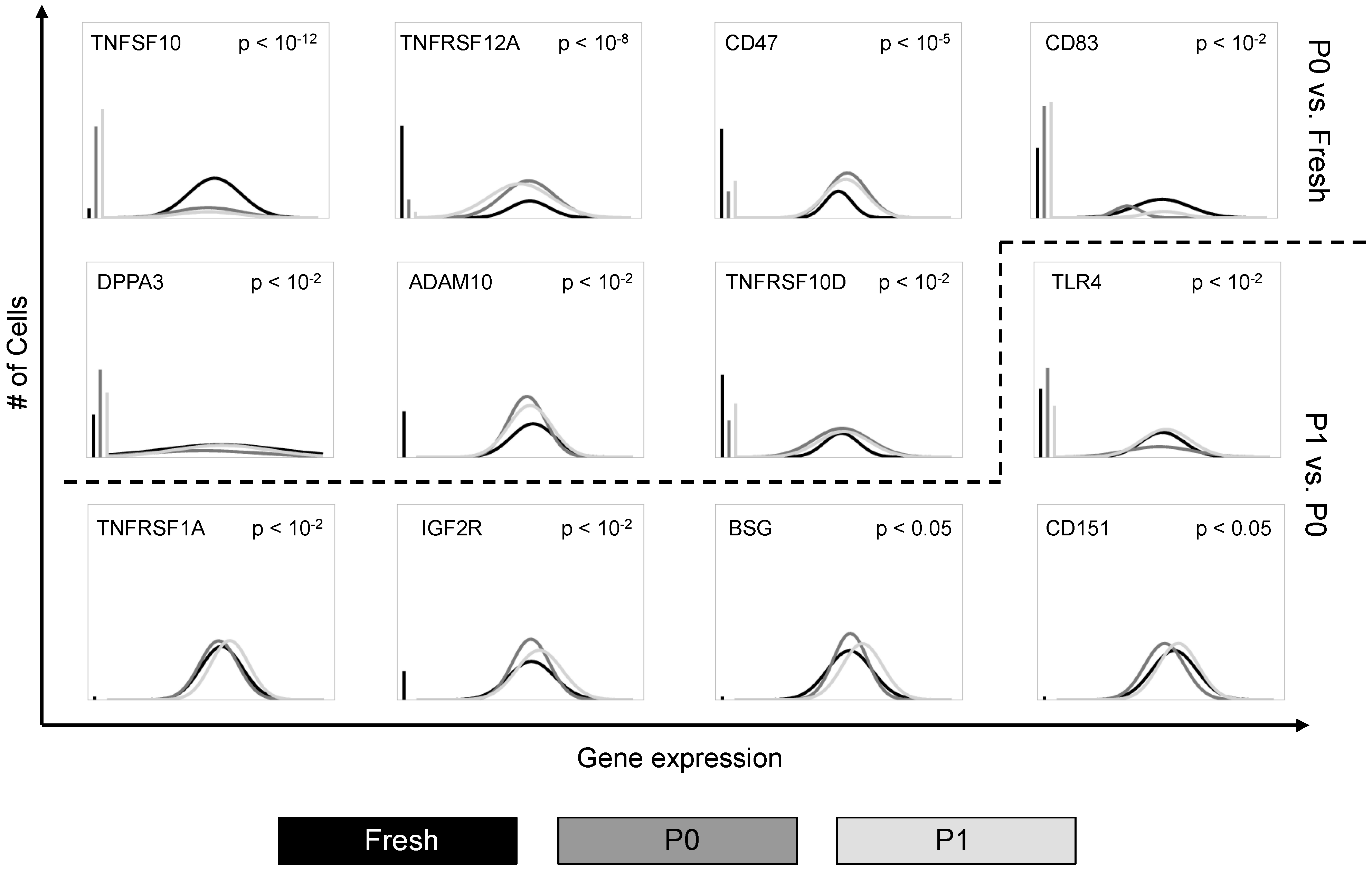
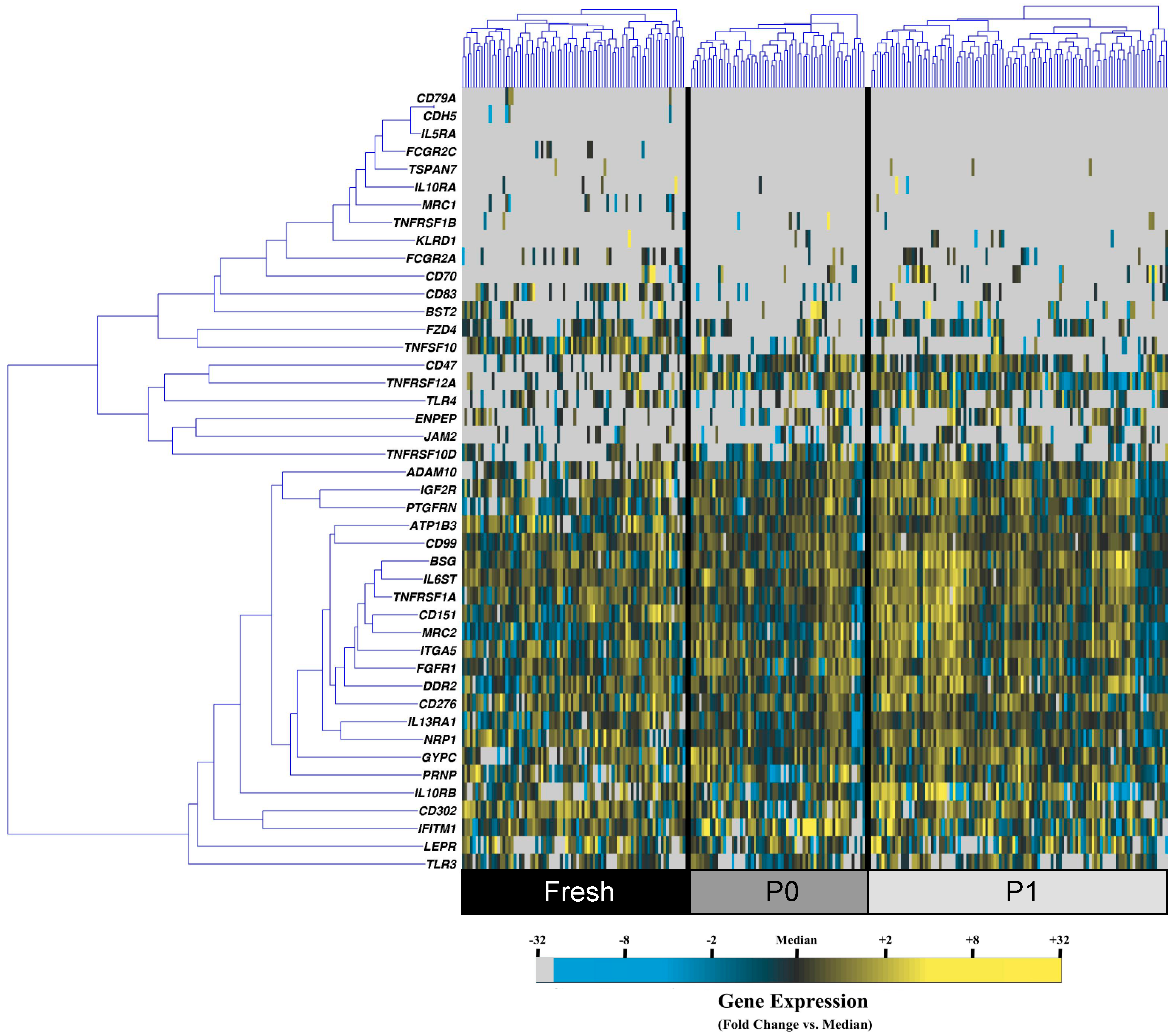
3.1. Cell Culture Alters the Transcriptional Profiles of Human Adipose-Derived Stem Cells

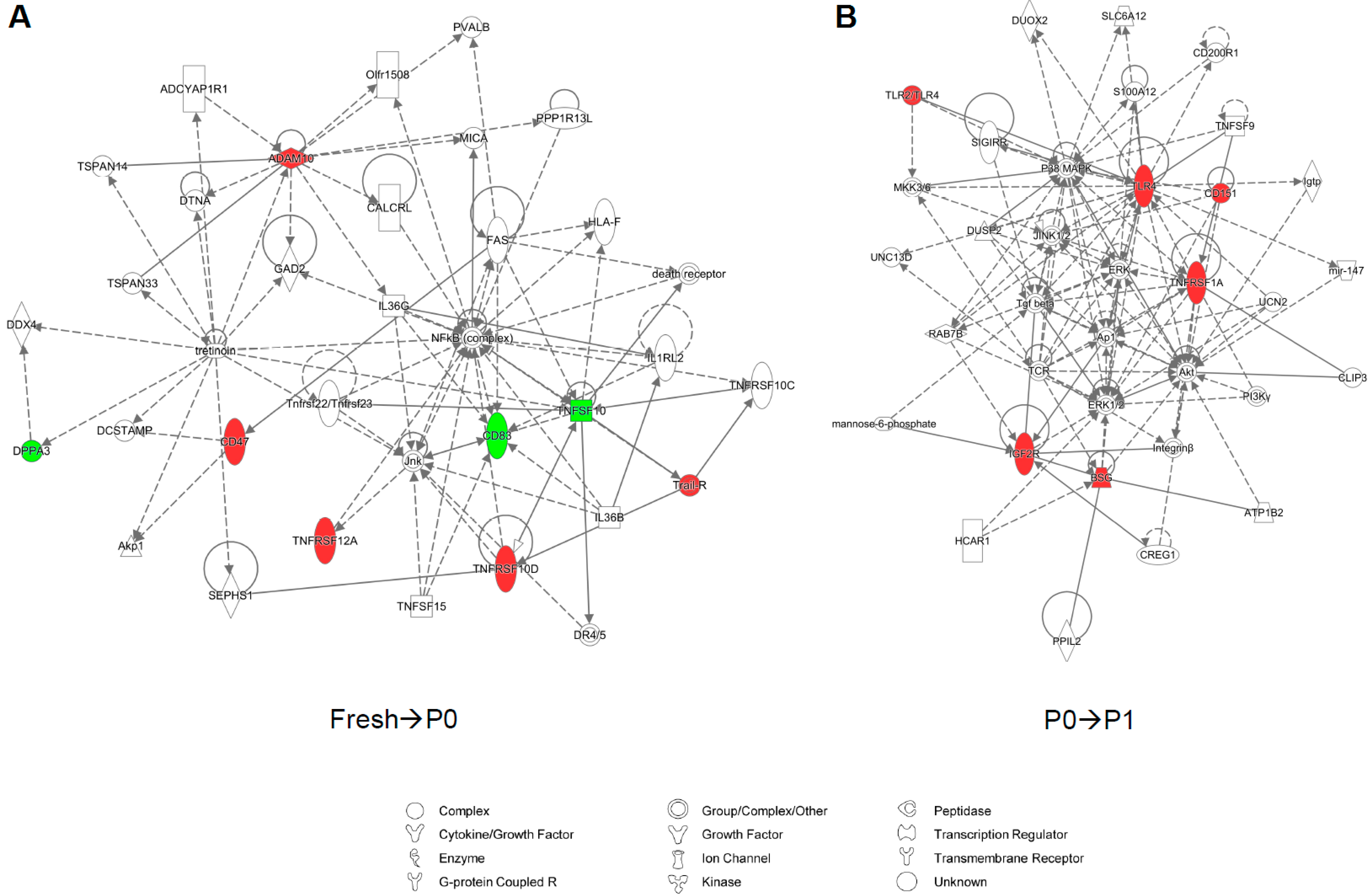
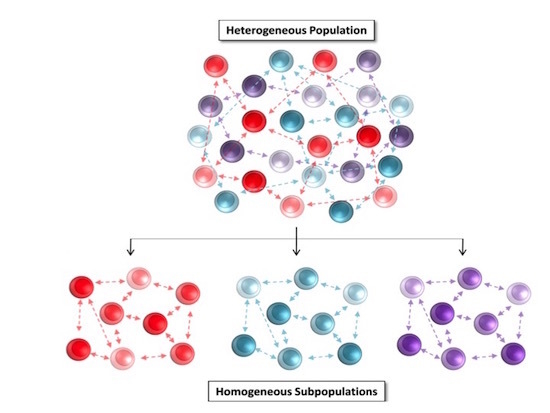
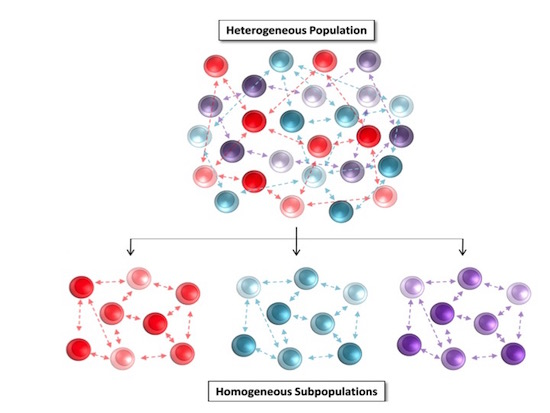
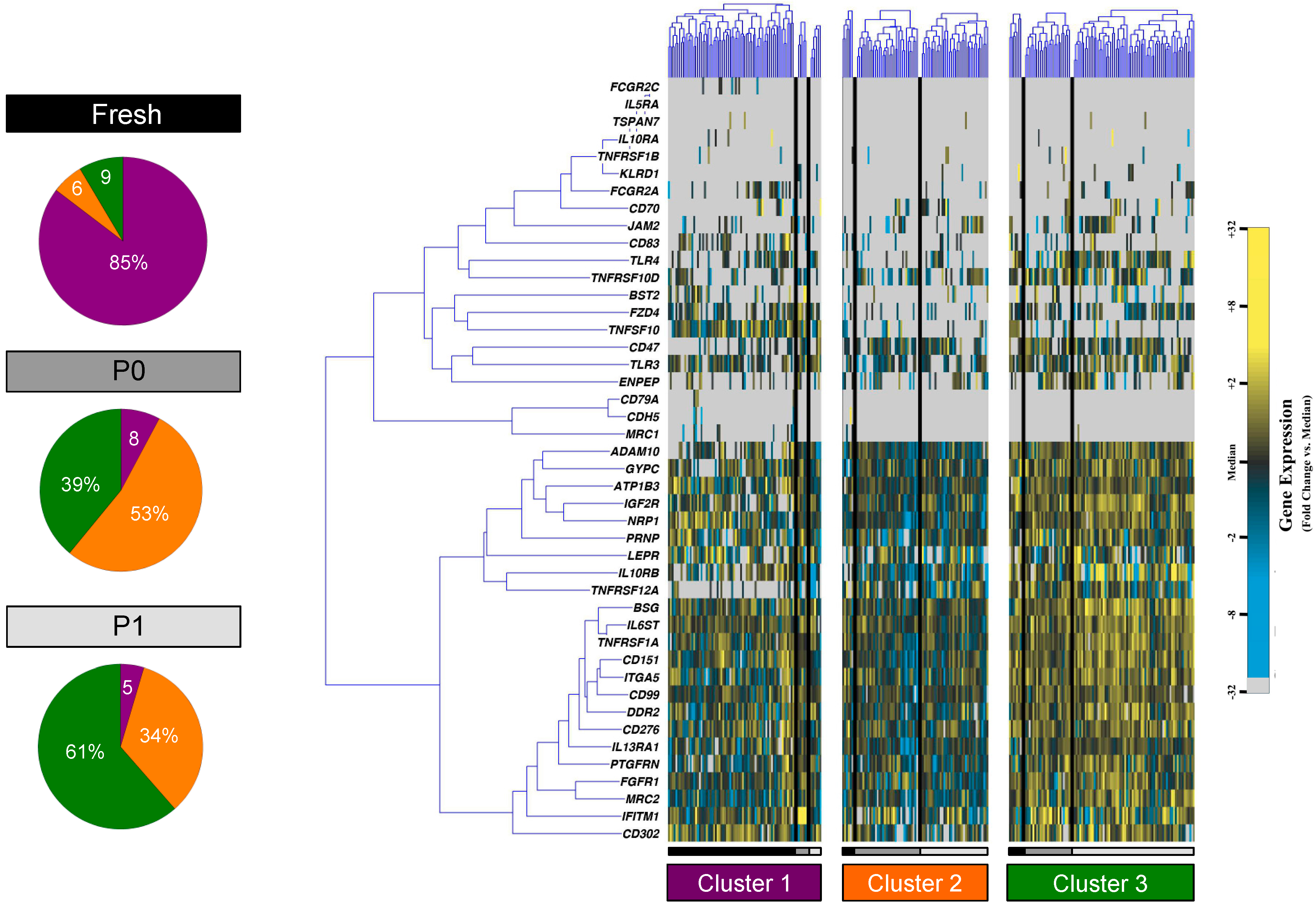
3.2. Partitional Cluster Analysis Reveals Subpopulations of Adipose-Derived Stem Cells that Evolve Throughout Cell Culture Passage
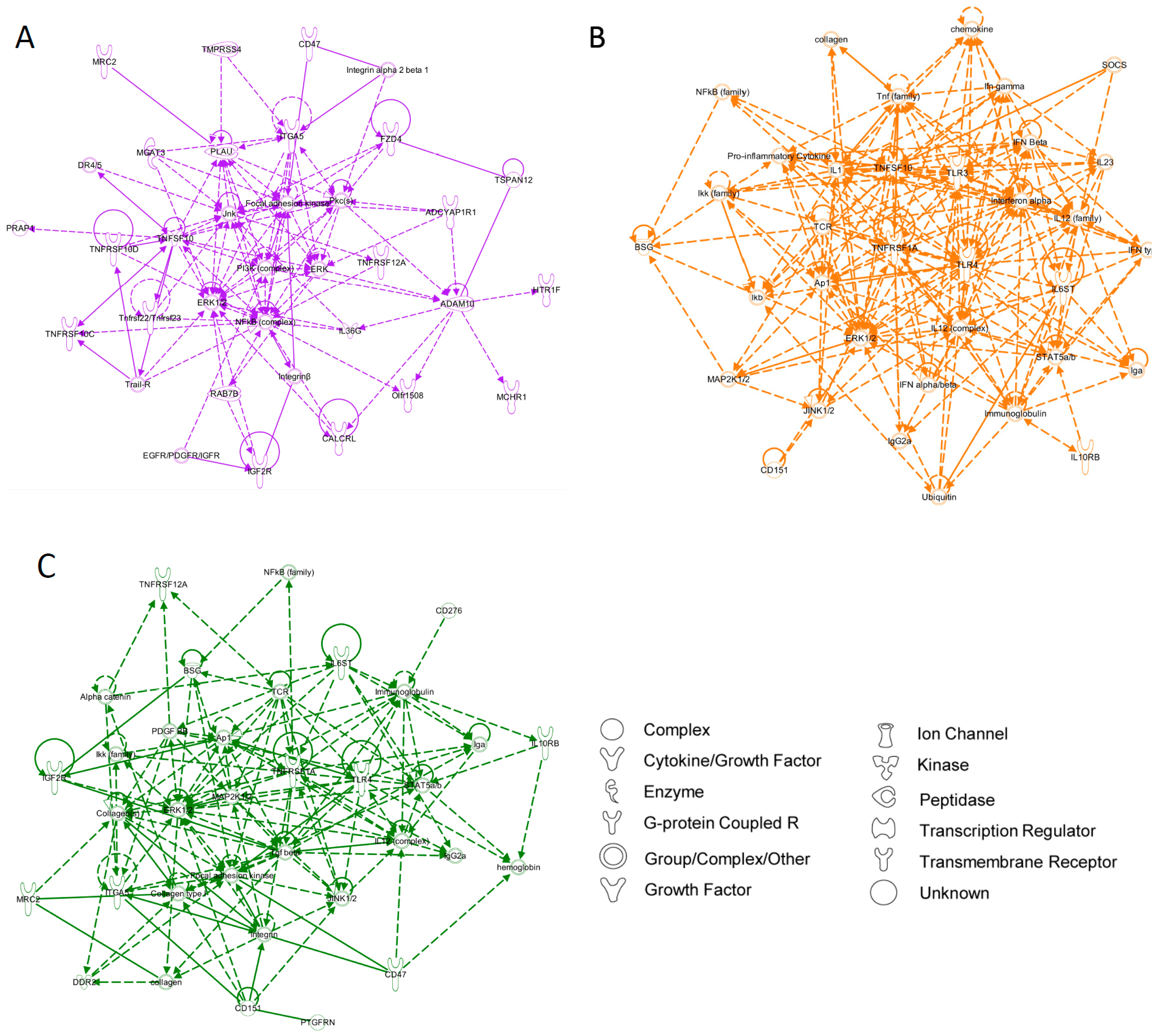
4. Conclusions
Supplementary Materials
Author Contributions
Conflicts of Interest
References
- Raaijmakers, M.H.; Scadden, D.T. Divided within: Heterogeneity within adult stem cell pools. Cell 2008, 135, 1006–1008. [Google Scholar] [CrossRef] [PubMed]
- Januszyk, M.; Gurtner, G.C. High-throughput single-cell analysis for wound healing applications. Adv. Wound Care 2013, 2, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Bar-Even, A.; Paulsson, J.; Maheshri, N.; Carmi, M.; O’Shea, E.; Pilpel, Y.; Barkai, N. Noise in protein expression scales with natural protein abundance. Nat. Genet. 2006, 38, 636–643. [Google Scholar] [CrossRef] [PubMed]
- Elowitz, M.B.; Levine, A.J.; Siggia, E.D.; Swain, P.S. Stochastic gene expression in a single cell. Science 2002, 297, 1183–1186. [Google Scholar] [CrossRef] [PubMed]
- Maheshri, N.; O’Shea, E.K. Living with noisy genes: How cells function reliably with inherent variability in gene expression. Ann. Rev. Biophys. Biomol. Struct. 2007, 36, 413–434. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.; Marcotte, E.M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 2012, 13, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Dalerba, P.; Kalisky, T.; Sahoo, D.; Rajendran, P.S.; Rothenberg, M.E.; Leyrat, A.A.; Sim, S.; Okamoto, J.; Johnston, D.M.; Qian, D.; et al. Single-cell dissection of transcriptional heterogeneity in human colon tumors. Nat. Biotechnol. 2011, 29, 1120–1127. [Google Scholar] [CrossRef] [PubMed]
- Levi, B.; Wan, D.C.; Glotzbach, J.P.; Hyun, J.; Januszyk, M.; Montoro, D.; Sorkin, M.; James, A.W.; Nelson, E.R.; Li, S.; et al. CD105 protein depletion enhances human adipose-derived stromal cell osteogenesis through reduction of transforming growth factor beta1 (TGF-β1) signaling. J. Biol. Chem. 2011, 286, 39497–39509. [Google Scholar] [CrossRef] [PubMed]
- Eisen, M.B.; Spellman, P.T.; Brown, P.O.; Botstein, D. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. USA 1998, 95, 14863–14868. [Google Scholar] [CrossRef] [PubMed]
- Levsky, J.M.; Singer, R.H. Gene expression and the myth of the average cell. Trends Cell Biol. 2003, 13, 4–6. [Google Scholar] [CrossRef]
- Warren, L.; Bryder, D.; Weissman, I.L.; Quake, S.R. Transcription factor profiling in individual hematopoietic progenitors by digital RT-PCR. Proc. Natl. Acad. Sci. USA 2006, 103, 17807–17812. [Google Scholar] [CrossRef] [PubMed]
- Melin, J.; Quake, S.R. Microfluidic large-scale integration: The evolution of design rules for biological automation. Ann. Rev. Biophys. Biomol. Struct. 2007, 36, 213–231. [Google Scholar] [CrossRef] [PubMed]
- Januszyk, M.; Sorkin, M.; Glotzbach, J.P.; Vial, I.N.; Maan, Z.N.; Rennert, R.C.; Duscher, D.; Thangarajah, H.; Longaker, M.T.; Butte, A.J.; et al. Diabetes irreversibly depletes bone marrow-derived mesenchymal progenitor cell subpopulations. Diabetes 2014, 63, 3047–3056. [Google Scholar] [CrossRef] [PubMed]
- Rennert, R.C.; Sorkin, M.; Januszyk, M.; Duscher, D.; Kosaraju, R.; Chung, M.T.; Lennon, J.; Radiya-Dixit, A.; Raghvendra, S.; Maan, Z.N.; et al. Diabetes impairs the angiogenic potential of adipose derived stem cells by selectively depleting cellular subpopulations. Stem Cell Res. Ther. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Suga, H.; Rennert, R.C.; Rodrigues, M.; Sorkin, M.; Glotzbach, J.P.; Januszyk, M.; Fujiwara, T.; Longaker, M.T.; Gurtner, G.C. Tracking the elusive fibrocyte: Identification and characterization of collagen-producing hematopoietic lineage cells during murine wound healing. Stem Cells 2014, 32, 1347–1360. [Google Scholar] [CrossRef] [PubMed]
- Sawada, H.; Saito, T.; Nickel, N.P.; Alastalo, T.-P.; Glotzbach, J.P.; Chan, R.; Haghighat, L.; Fuchs, G.; Januszyk, M.; Cao, A.; et al. Reduced BMPR2 expression induces GM-CSF translation and macrophage recruitment in humans and mice to exacerbate pulmonary hypertension. J. Exp. Med. 2014, 211, 263–280. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.; Laurenti, E.; Oser, G.; van der Wath, R.C.; Blanco-Bose, W.; Jaworski, M.; Offner, S.; Dunant, C.F.; Eshkind, L.; Bockamp, E.; Lió, P.; et al. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell 2008, 135, 1118–1129. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Zhu, P.; Wu, X.L.; Li, X.L.; Wen, L.; Tang, F. Single-cell methylome landscapes of mouse embryonic stem cells and early embryos analyzed using reduced representation bisulfite sequencing. Genome Res. 2013, 23, 2126–2135. [Google Scholar] [CrossRef] [PubMed]
- Zuk, P.A.; Zhu, M.; Ashjian, P.; de Ugarte, D.A.; Huang, J.I.; Mizuno, H.; Alfonso, Z.C.; Fraser, J.K.; Benhaim, P.; Hedrick, M.H. Human adipose tissue is a source of multipotent stem cells. Mol. Biol. Cell 2002, 13, 4279–4295. [Google Scholar] [CrossRef] [PubMed]
- Glotzbach, J.P.; Januszyk, M.; Vial, I.N.; Wong, V.W.; Gelbard, A.; Kalisky, T.; Thangarajah, H.; Longaker, M.T.; Quake, S.R.; Chu, G.; Gurtner, G.C.; et al. An information theoretic, microfluidic-based single cell analysis permits identification of subpopulations among putatively homogeneous stem cells. PLoS ONE 2011, 6, e21211. [Google Scholar] [CrossRef] [PubMed]
- Wong, V.W.; Rustad, K.C.; Akaishi, S.; Sorkin, M.; Glotzbach, J.P.; Januszyk, M.; Nelson, E.R.; Levi, K.; Paterno, J.; Vial, I.N.; et al. Focal adhesion kinase links mechanical force to skin fibrosis via inflammatory signaling. Nat. Med. 2012, 18, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Gaudineau, B.; Fougère, M.; Guaddachi, F.; Lemoine, F.; de la Grange, P.; Jauliac, S. Lipocalin 2, the TNF-like receptor TWEAKR and its ligand TWEAK act downstream of NFAT1 to regulate breast cancer cell invasion. J. Cell Sci. 2012, 125, 4475–4486. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Januszyk, M.; Rennert, R.C.; Sorkin, M.; Maan, Z.N.; Wong, L.K.; Whittam, A.J.; Whitmore, A.; Duscher, D.; Gurtner, G.C. Evaluating the Effect of Cell Culture on Gene Expression in Primary Tissue Samples Using Microfluidic-Based Single Cell Transcriptional Analysis. Microarrays 2015, 4, 540-550. https://doi.org/10.3390/microarrays4040540
Januszyk M, Rennert RC, Sorkin M, Maan ZN, Wong LK, Whittam AJ, Whitmore A, Duscher D, Gurtner GC. Evaluating the Effect of Cell Culture on Gene Expression in Primary Tissue Samples Using Microfluidic-Based Single Cell Transcriptional Analysis. Microarrays. 2015; 4(4):540-550. https://doi.org/10.3390/microarrays4040540
Chicago/Turabian StyleJanuszyk, Michael, Robert C. Rennert, Michael Sorkin, Zeshaan N. Maan, Lisa K. Wong, Alexander J. Whittam, Arnetha Whitmore, Dominik Duscher, and Geoffrey C. Gurtner. 2015. "Evaluating the Effect of Cell Culture on Gene Expression in Primary Tissue Samples Using Microfluidic-Based Single Cell Transcriptional Analysis" Microarrays 4, no. 4: 540-550. https://doi.org/10.3390/microarrays4040540