
Molecular Characterization of Donacia provosti (Coleoptera: Chrysomelidae) Larval Transcriptome by De Novo Assembly to Discover Genes Associated with Underwater Environmental Adaptations

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Insect Materials and RNA Isolation
2.2. cDNA Library Preparation and Illumina Sequencing
2.3. Bioinformatics Analysis
2.4. Protein-Coding Sequence (CDS) Prediction
2.5. Orthologous Cluster Analysis
2.6. Adaptive Evolution Analysis
3. Results
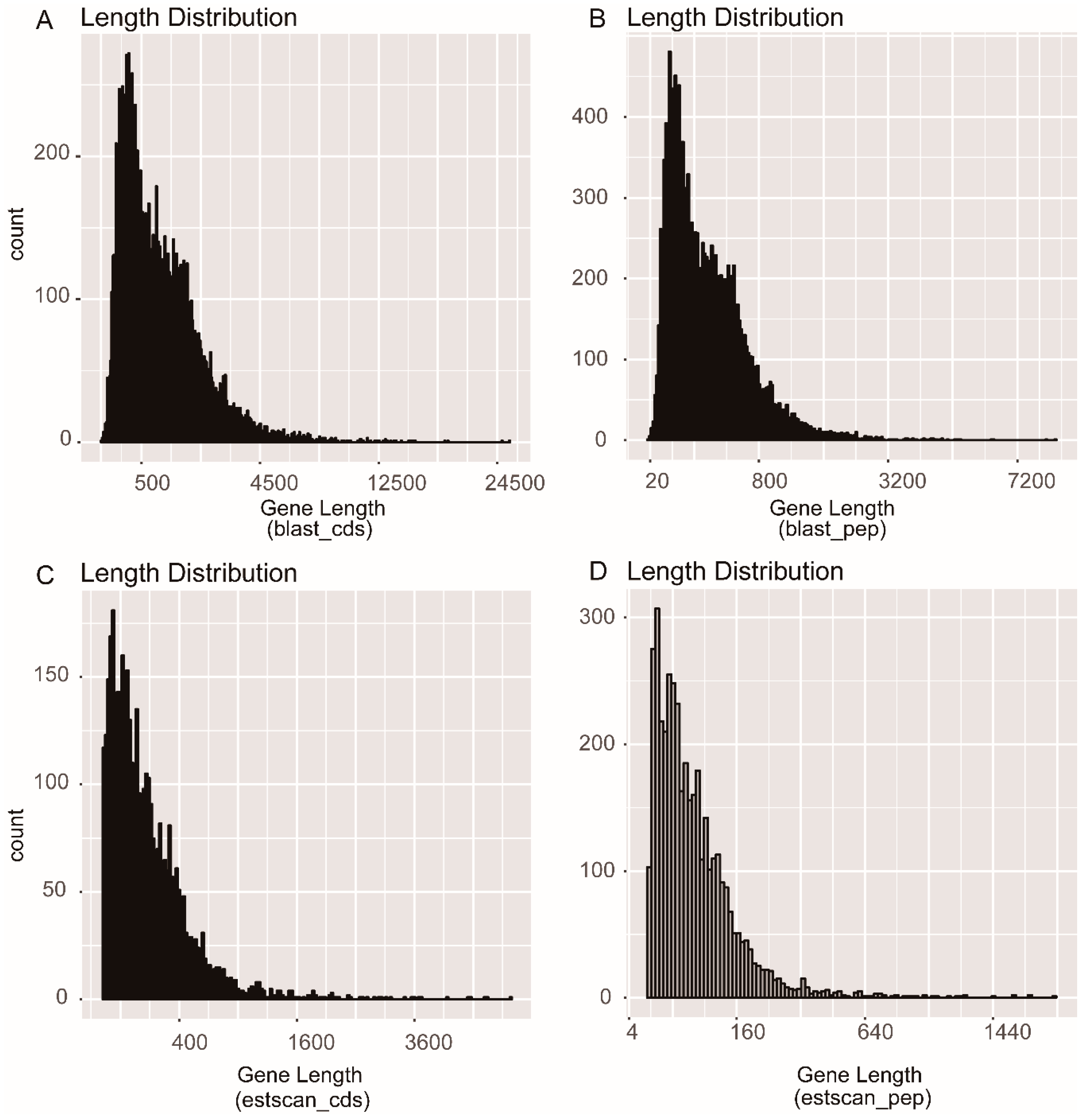
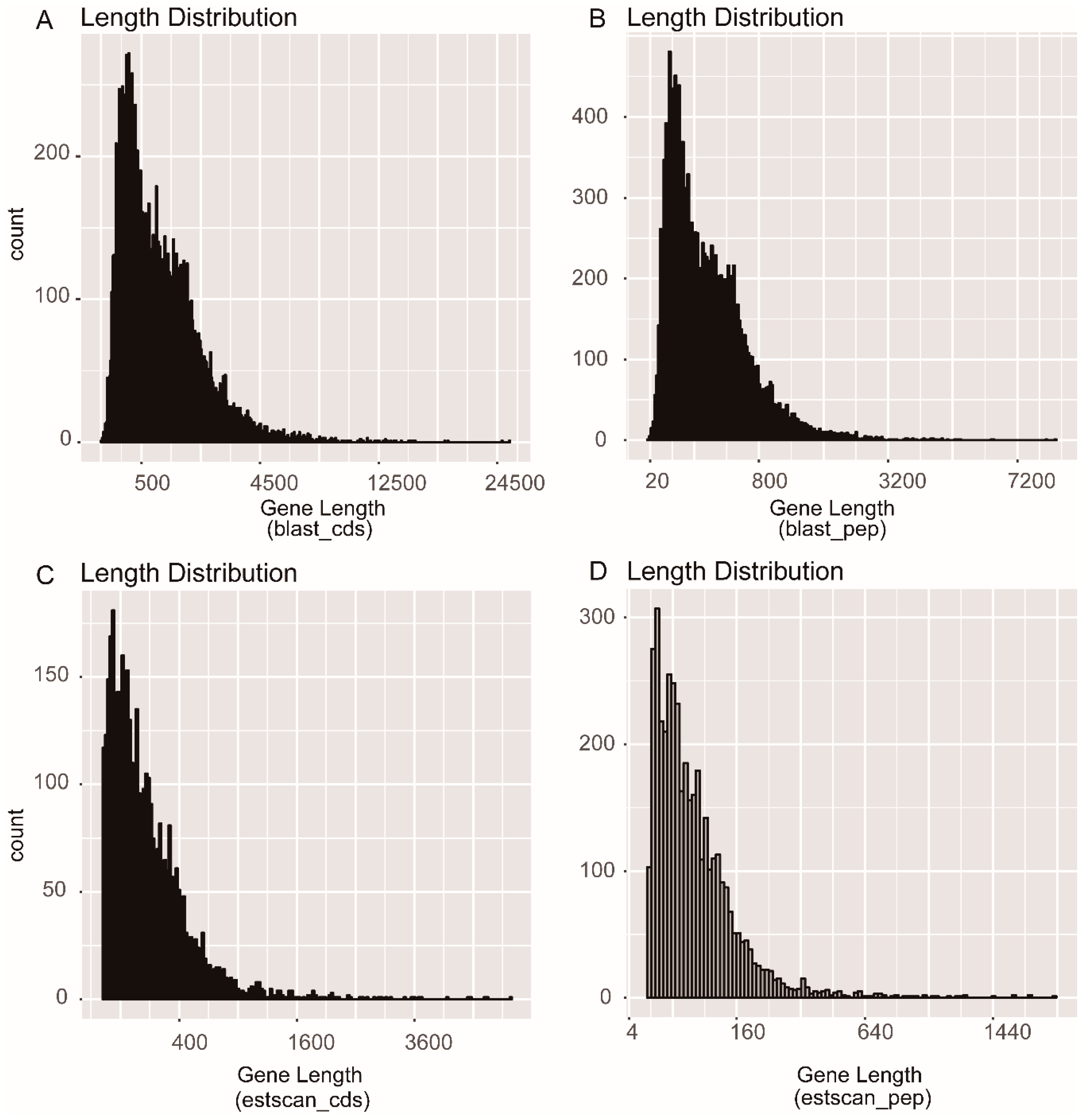
3.1. Transcriptome Sequencing and Assembly
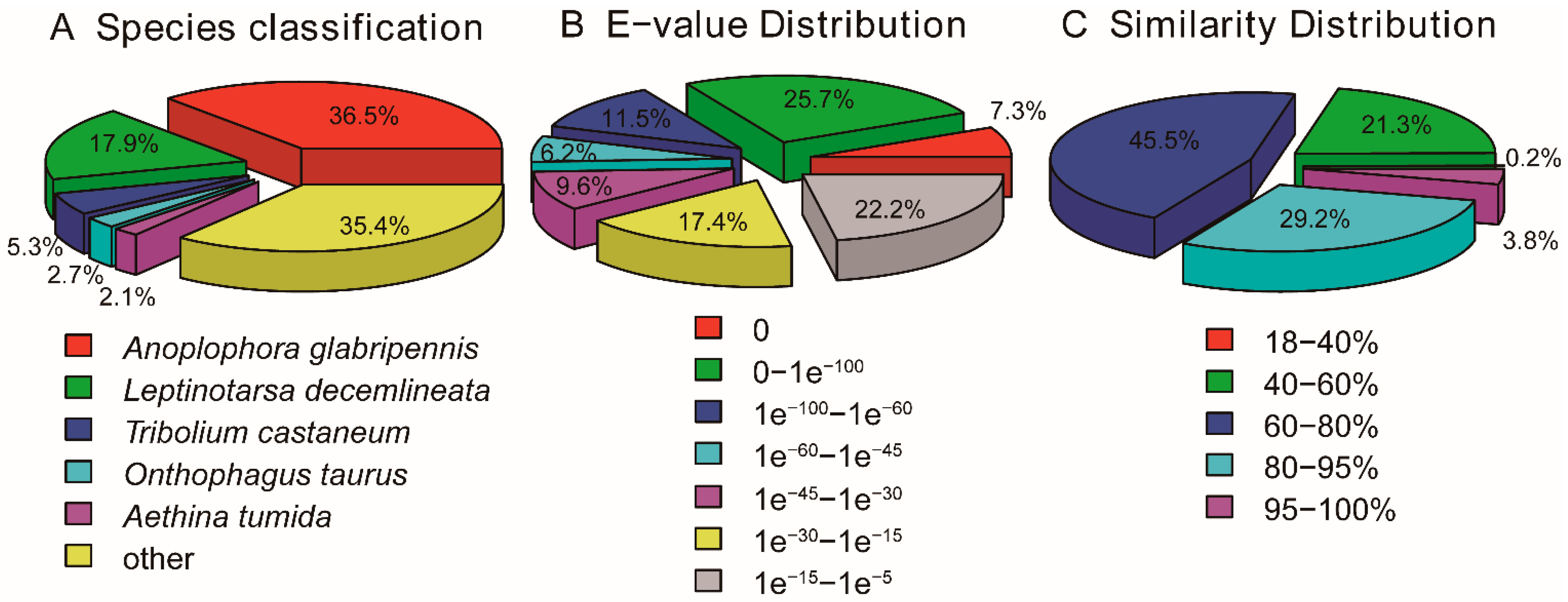
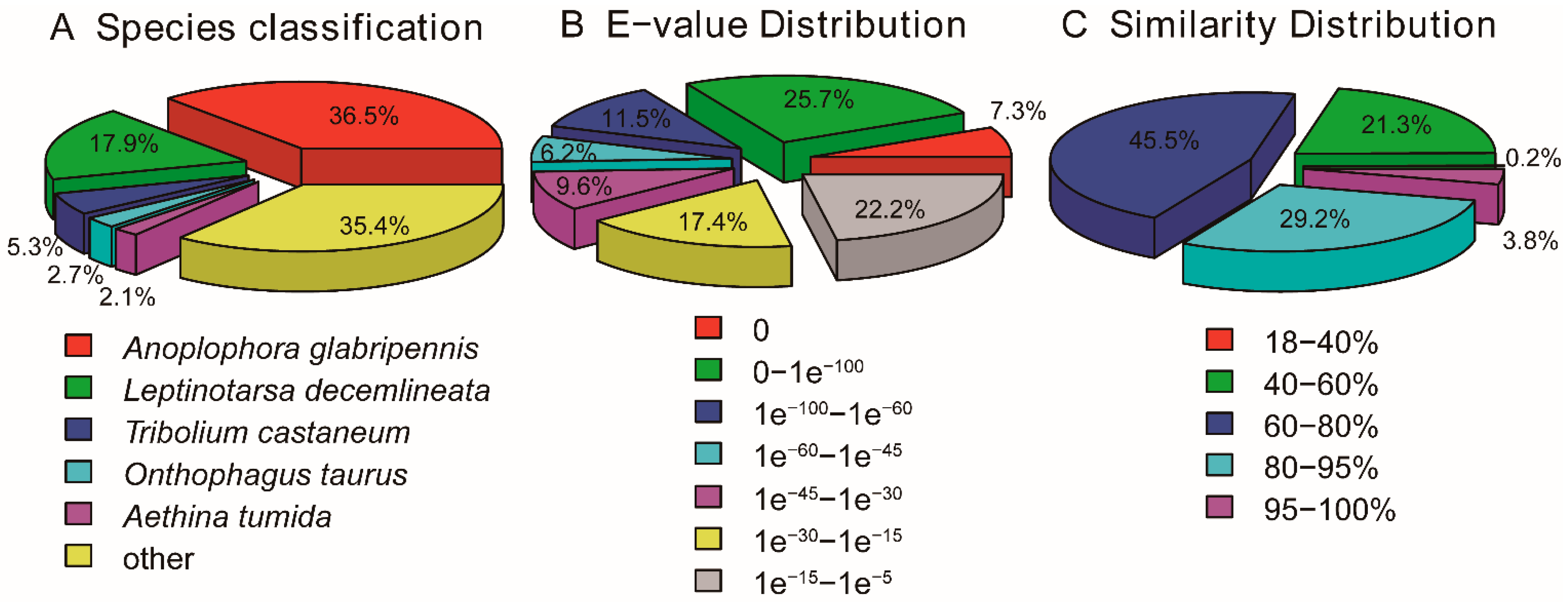
3.2. Function Annotation
3.3. Protein Coding Sequence (CDS) Prediction and Orthologous Analysis
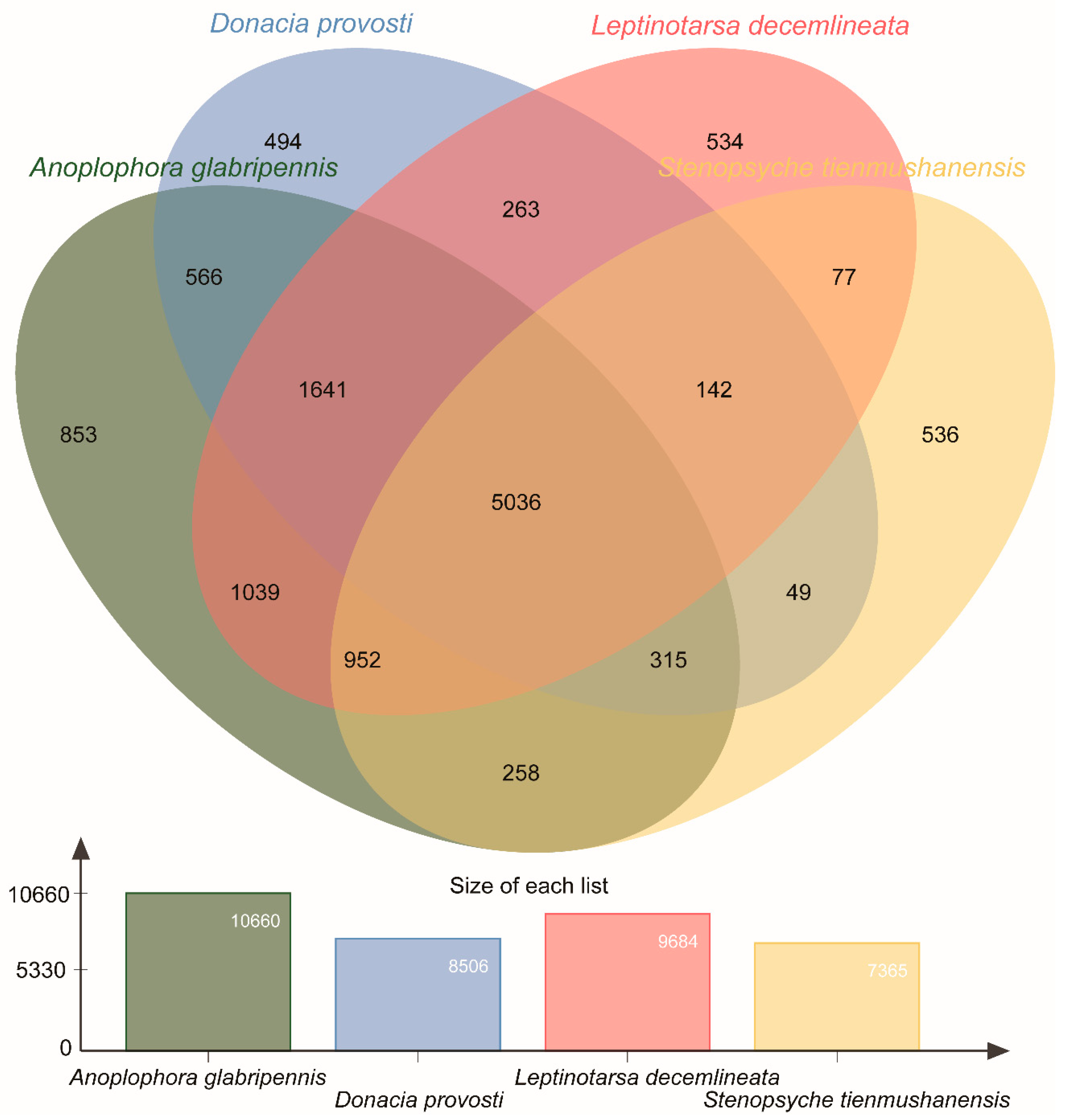
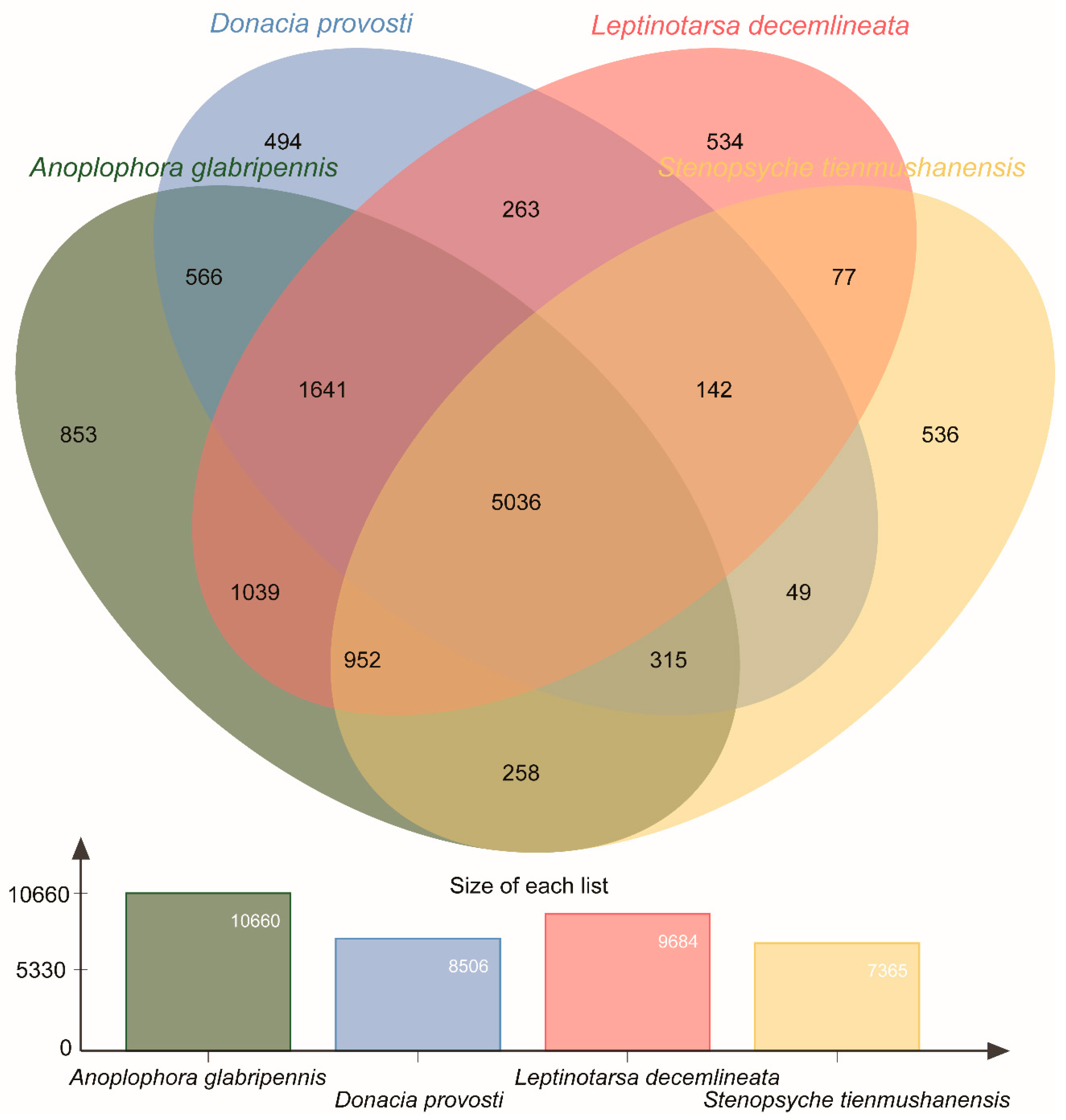
3.4. Orthologous Cluster Analysis
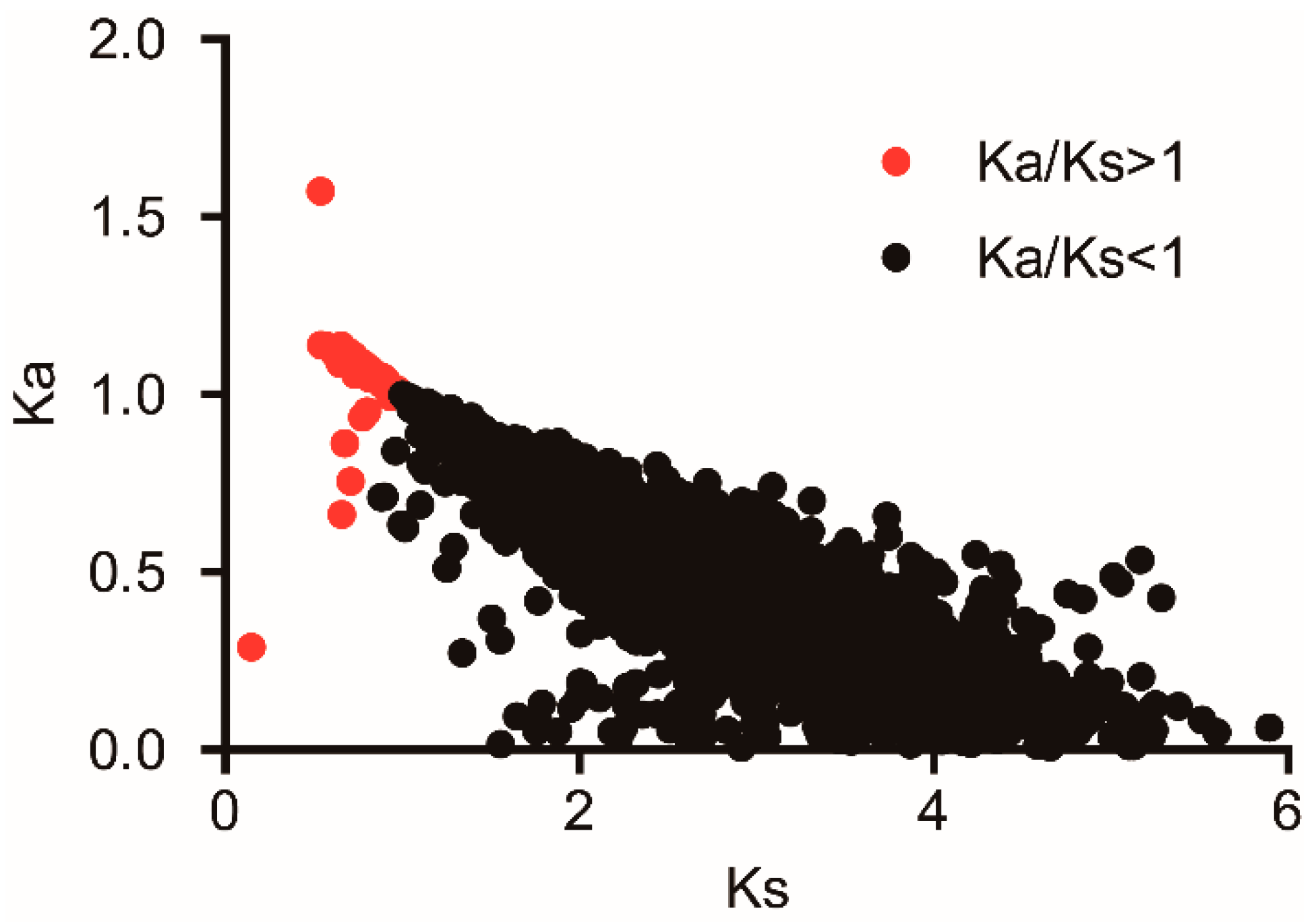
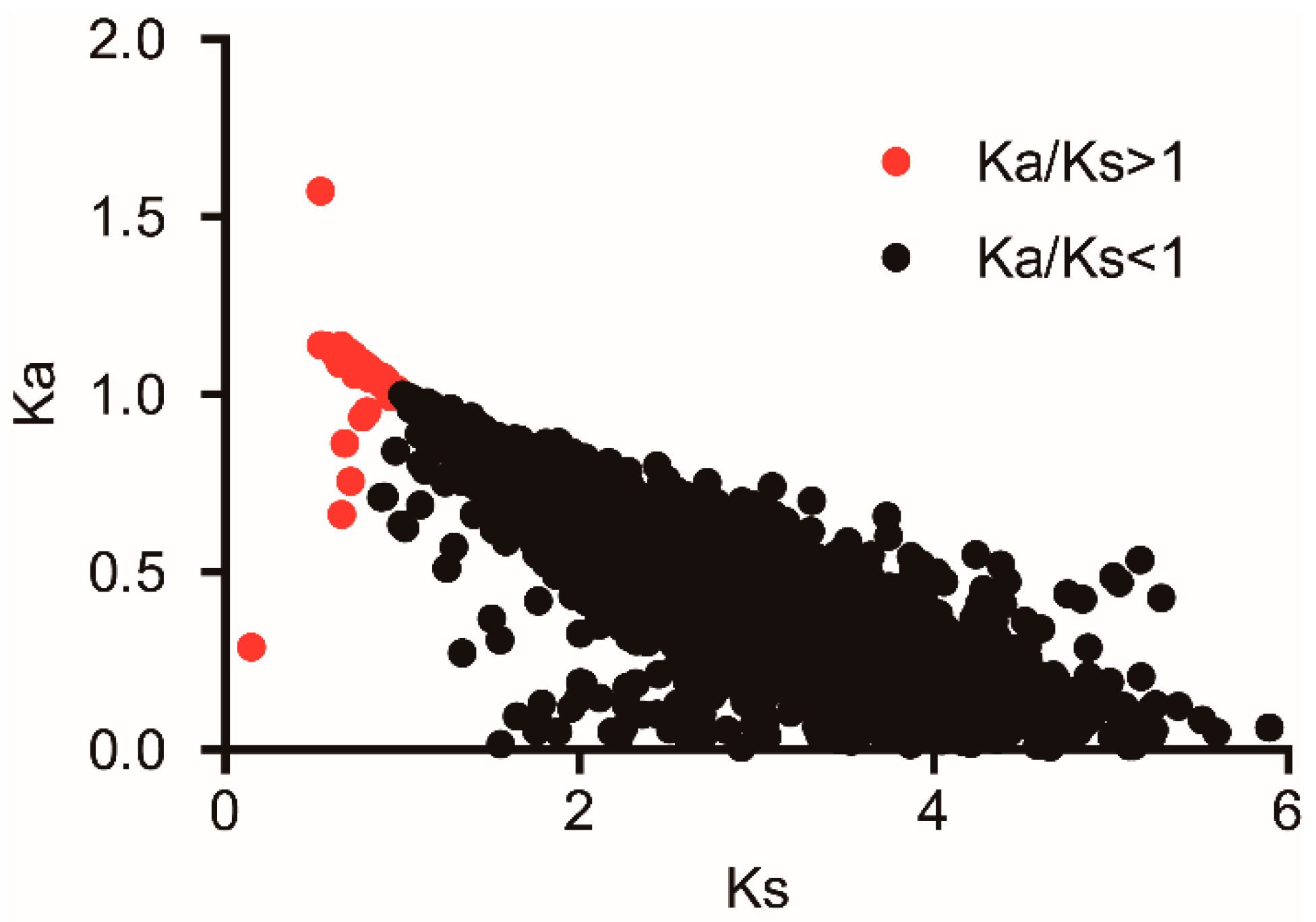
3.5. Adaptive Evolution Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Lobl, L.; Smetana, A. Catalogue of Palaearctic Coleoptera, Chrysomeloidea; Apollo Books: Stenstrup, Denmark, 2010; Volume 6. [Google Scholar]
- Qin, C.H.; Chen, C.; Wan, H.; Li, J.H. Studies on biological characteristics of Donacia provosti. China Veg. 2009, 24, 57–61. [Google Scholar]
- Fairmaire, M.L. Coléoptères de L’intérieur de la Chine: Faitles communications suivantes au sujet de deux Coléoptères. Bull. Société Entomol. Fr. 1885, 6, 64–65. [Google Scholar]
- Hayashi, M.; Shiyake, S. A check-list of the Japanese members of Donaciinae (Coleoptera: Chrysomelidae). Entomol. Rev. Jpn. 2004, 59, 113–125. [Google Scholar]
- Liu, M.G.; Zhang, J.H. A Preliminary Report on the Occurrence of Lotus Root-Eating Golden Flower Insects. Hubei Plant Prot. 2002, 4, 21. [Google Scholar]
- Cronin, G.; Wissing, K.D.; Lodge, D.M. Comparative feeding selectivity of herbivorous insects on water lilies: Aquatic vs. semi-terrestrial insects and submersed vs. floating leaves. Freshw. Biol. 1998, 39, 243–257. [Google Scholar] [CrossRef]
- Liu, X.; Wu, W.W.; He, Z.; Li, L.J. Research progress on the occurrence and control of common lotus root diseases and insect pests. Jiangsu Agr. Sci. 2017, 45, 24–28. [Google Scholar]
- Yang, M. Pollution-free control technology of Donacia provosti. China Veg. 2001, 6, 32–33. [Google Scholar]
- Huang, G.H.; Li, J.H. A Map of the Main Pests of Aquatic Vegetables in China; Hubei Science and Technology Press: Wuhan, China, 2013. [Google Scholar]
- Chen, J.H.; Zhao, G.Y.; Zhang, Y.D.; Ma, X.F.; Yang, C.Q.; Li, H.Z.; Wang, C.X. Test on the efficacy of several medicaments to control Donacia provosti. Plant Dr. 2007, 20, 31. [Google Scholar]
- Yu, D. Chlorantraniliprole is effective in controlling Donacia Provosti. Pestic. Mark. Inform. 2007, 14, 35. [Google Scholar]
- Frandsen, P.B.; Bursell, M.G.; Taylor, A.M.; Wilson, S.B.; Steeneck, A.; Stewart, R.J. Exploring the underwater silken architectures of caddisworms: Comparative silkomics across two caddisfly suborders. Philos. Trans. R. Soc. B 2019, 374, 20190206. [Google Scholar] [CrossRef]
- Stewart, R.J.; Wang, C.S. Adaptation of S. tienmushanensis larval silks to aquatic habitats by phosphorylation of H-fibroin serines. Biomacromolecules 2010, 11, 969–974. [Google Scholar] [CrossRef]
- Luo, S.; Tang, M.; Frandsen, P.B.; Stewart, R.J.; Zhou, X. The genome of an underwater architect, the caddisfly Stenopsyche tienmushanensis Hwang (Insecta: Trichoptera). GigaScience 2018, 7, giy143. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.D.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Davidson, N.M.; Oshlack, A. Corset: Enabling differential gene expression analysis for de novo assembled transcriptomes. Genome Biol. 2014, 15, 1–14. [Google Scholar]
- Bateman, A.; Coin, L.; Durbin, R.; Finn, R.D.; Hollich, V.; Griffiths-Jones, S.; Khanna, A.; Marshall, M.; Moxon, S.; Sonnhammer, E.L.L.; et al. The Pfam protein families database. Nucleic Acids Res. 2004, 32, D138–D141. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed]
- McKenna, D.D.; Scully, E.D.; Pauchet, Y.; Hoover, K.; Kirsch, R.; Geib, S.M.; Richards, S. Genome of the Asian longhorned beetle (Anoplophora glabripennis), a globally significant invasive species, reveals key functional and evolutionary innovations at the beetle plant-interface. Genome Biol. 2016, 17, 1–18. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools-an integrative toolkit developed for interactive analyses of big biological data. BioRxiv 2020, 13, 289660. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Bielawski, J.P. Statistical methods for detecting molecular adaptation. Trends Ecol. Evol. 2000, 15, 496–503. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, J.; Zhao, X.Q.; Wang, J.; Wong, G.K.; Yu, J. KaKs_Calculator: Calculating Ka and Ks through model selection and model averaging. Genom. Proteom. Bioinf. 2006, 4, 259–263. [Google Scholar] [CrossRef]
- Yang, Z.; Nielsen, R. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol. Biol. Evol. 2000, 17, 32–43. [Google Scholar] [CrossRef]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.M.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L.P. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef]
- Backström, N.; Zhang, Q.; Edwards, S.V. Evidence from a house finch (Haemorhous mexicanus) spleen transcriptome for adaptive evolution and biased gene conversion in passerine birds. Mol. Biol. Evol. 2003, 30, 1046–1050. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Wang, R.; Qu, C.; Fu, N.; Luo, C.; Xu, Y. Sequencing and characterization of the invasive sycamore lace bug corythucha ciliata (Hemiptera: Tingidae) transcriptome. PLoS ONE 2016, 11, e0160609. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.W.; Zhao, Q.Y.; Luan, J.B.; Wang, Y.J.; Yan, G.H.; Liu, S.S. Analysis of a native whitefly transcriptome and its sequence divergence with two invasive whitefly species. BMC Genom. 2012, 13, 529. [Google Scholar] [CrossRef]
- Yang, H.B.; Hu, Z.J.; Li, D.X.; Zhu, P.H.; Dong, J.F. Analysis of the antennal transcriptome and olfactory-related genes of the sycamore lace bug (Corythucha ciliata). J. Agricult. Biotechnol. 2018, 26, 2109–2120. [Google Scholar]
- Ferguson, K.B.; Kursch-Metz, T.; Verhulst, E.C.; Pannebakker, B.A. Hybrid genome assembly and evidence-based annotation of the egg parasitoid and biological control agent Trichogramma brassicae. BioRxiv 2020. [Google Scholar] [CrossRef]
- Huang, K.; Fingar, D.C. Growing knowledge of the mTOR signaling network. In Seminars in Cell & Developmental Biology; Academic Press: Cambridge, MA, USA, 2014; Volume 36, pp. 79–90. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Replicates | Total Raw Reads | Total Clean Reads | Clean Bases (G) | Q20% | GC% |
|---|---|---|---|---|---|
| Larvae_1 | 50,681,752 | 49,714,298 | 7.46 | 97.53 | 39.95 |
| Larvae_2 | 50,342,128 | 49,459,666 | 7.42 | 97.58 | 39.98 |
| Larvae_3 | 60,752,838 | 59,043,054 | 8.86 | 97.54 | 40.25 |
| All | 161,776,718 | 158,217,018 | 23.74 |
| De Novo Assembly | Total Number | Total Length (bp) | Mean Length (bp) | N50 |
|---|---|---|---|---|
| Transcripts | 75,658 | 117,120,238 | 1548 | 2722 |
| Unigenes | 34,118 | 44,794,61 | 1304 | 2194 |
| Databases Annotation | Number of Unigenes | Percentage (%) |
|---|---|---|
| Annotated in NR | 18,081 | 52.99 |
| Annotated in NT | 5685 | 16.66 |
| Annotated in KO | 6340 | 18.58 |
| Annotated in SwissProt | 10,040 | 29.42 |
| Annotated in PFAM | 12,036 | 35.27 |
| Annotated in GO | 12,036 | 35.27 |
| Annotated in KOG | 5999 | 17.58 |
| Annotated in all Databases | 2239 | 6.56 |
| Annotated in at least one Database | 20,692 | 60.64 |
| Total Unigenes | 34,118 | 100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhan, H.; Dewer, Y.; Qu, C.; Yang, S.; Luo, C.; Li, L.; Li, F. Molecular Characterization of Donacia provosti (Coleoptera: Chrysomelidae) Larval Transcriptome by De Novo Assembly to Discover Genes Associated with Underwater Environmental Adaptations. Insects 2021, 12, 281. https://doi.org/10.3390/insects12040281
Zhan H, Dewer Y, Qu C, Yang S, Luo C, Li L, Li F. Molecular Characterization of Donacia provosti (Coleoptera: Chrysomelidae) Larval Transcriptome by De Novo Assembly to Discover Genes Associated with Underwater Environmental Adaptations. Insects. 2021; 12(4):281. https://doi.org/10.3390/insects12040281
Chicago/Turabian StyleZhan, Haixia, Youssef Dewer, Cheng Qu, Shiyong Yang, Chen Luo, Liangjun Li, and Fengqi Li. 2021. "Molecular Characterization of Donacia provosti (Coleoptera: Chrysomelidae) Larval Transcriptome by De Novo Assembly to Discover Genes Associated with Underwater Environmental Adaptations" Insects 12, no. 4: 281. https://doi.org/10.3390/insects12040281