1. Introduction
The coronavirus disease 2019 (COVID-19) outbreak has rapidly involved all countries, resulting in the ongoing pandemic [
1,
2]. The global pandemic still occurs, with continuous issues related to the absence of high-throughput and rapid technologies that are able to deliver rapid diagnostic tests on large-scale populations. Therefore, the reagents and chemicals that are now working on the main hospital routine platforms are generally based on low–medium throughput technologies, with there sometimes being a restriction in the reagent recovery, and market saturation for those working on automated pipelines and higher throughput platforms [
2]. Our lab has been recognized as one of the sixteen accredited centers for molecular diagnostics of COVID-19 infection, and we participated in the Campania region screening as a part of the regional laboratory network, namely ‘Campania Coronet’.
However, since the market saturation did not make available large numbers of kits, reagents, chemicals and disposables [
2,
3,
4], we were forced to use different tools in order not to stop our diagnostic activity, in compliance with the agreement with the regional task force for SARS-CoV-2 screening. However, many practical and technical issues frequently arise in laboratories performing COVID-19 molecular assays, particularly regarding the nucleic acid extraction, nucleic acid amplification reagents, and interpretation of test results [
5,
6].
Therefore, from among the most reliable and effective CE-IVD kits recommended by the Italian health authority (Istituto Superiore di Sanità; ISS) (
https://www.trovanorme.salute.gov.it/norme/renderNormsanPdf?anno=2020&codLeg=73799&parte=1%20&serie=null (accessed on 4 February 2021)), we selected three out of twelve reagents: Allplex 2019-nCov Assay (SEEGENE: kit-A), BOSPHORE-V2 Novel Coronavirus 2019-Ncov (Anatolia: kit-B), and Realquality RQ-2019-NCOV (AB ANALITICA: kit-C), also following the literature reports [
7,
8,
9]. Although the diagnostic market provides lots of solutions, Guglielmi [
10] stated that the current PCR-based assays can test whether someone is infectious, but does not distinguish between individuals who carry the virus and those who are not likely to spread it. In fact, the viral load peaks early in SARS-CoV-2 infections and then gradually declines, with small amounts of virus RNA staying in the nasopharyngeal trait for weeks, or sometimes months [
10]. Thus, it is very difficult to distinguish individuals with active and higher viral loads through the commonly-used routine tests. Nonetheless, in COVID-19 assay-positive individuals, who are screened for the first time and show these high Ct values for the detected targets, it is not possible to determine whether they are initial or previous infections [
7,
8]. In this context, we investigated whether other molecular transcripts of SARS-CoV-2 could provide useful information in terms of potential biomarkers of virus activity in this peculiar situation. Particularly, the transcriptome landscape of SARS-CoV-2 is also characterized by positive-sense sub genomic RNAs (sgRNAs) encoding for both structural and several accessory proteins [
7,
11]. SgRNAs are synthetized by nsp12 harbouring RNA-dependent RNA polymerase (RdRP) activity via a mechanism of discontinuous transcription according to the to the ‘leader–body fusion’ model [
11]. Coronaviruses use these subgenomic RNA fragments, produced by discontinuous transcription, to translate the early proteins necessary for viral replication and encapsidation [
12]: the ratios of the mean normalized sgRNAs per genome are estimated as being about 0.4% [
12]. Kim et al. [
11] reported that the
N RNA is the most abundantly expressed transcript, followed by the
E. Interestingly, as reported by Wölfel et al. [
12], these viral sgRNAs are transcribed only in infected cells, and are not packaged into virions: therefore, they indicate the presence of actively-infected cells. Based on these findings, these authors [
12] argued that, as high viral loads were always shown to be associated with SARS-CoV-2 isolation from early throat swabs, the sgRNAs could be considered as potential virus replication markers in the tissues of the upper respiratory tract [
12]. In order to obtain proof of active virus replication in positive samples, Wölfel et al. [
12] performed RT–qPCR tests to identify viral subgenomic mRNAs directly in clinical nasopharyngeal samples showing a decline of subgenomic RNAs from day ten. However, in throat swabs, all of the samples taken up to day five did not show any subgenomic mRNA detectability. Therefore, Wölfel et al. [
12] stated that their findings indicate that the active replication of SARS-CoV-2 occurs in the throat during the first five days after the onset of symptoms. In support of these findings, we underline that Doddapaneni et al. [
13] were able to obtain complete genomes (genomic and subgenomic regions) only on clinical samples with threshold cycle values <33. Consequently, the different performances of commercial kits could influence our capability both to detect and correlate the genomic viral load with the subgenomic expression.
In this regard, we report herein the data gained on
sgN and
sgE transcripts assayed on oro-nasopharyngeal swab samples in relationship to genomic results obtained by using different kits for SARS-CoV-2 screening (namely, kit-A, kit-B and kit-C). We also evaluated, on the same samples, the performances of two newly-released kits provided by a commercial Korean company (Kit-D and Kit-E), which were not listed among those previously approved by the Italian Ministry. As already reported [
1,
6,
7,
8,
9], most of diagnostic kits are based on the detection of SARS-CoV-2 genes encoding structural (e.g., spike protein [S], envelope protein [E], membrane protein [M], and nucleocapsid protein [N]) or non-structural (Orf1b/RdRp) proteins. We underline that, when the assay results are positive with a high threshold cycle (a Ct from 38 to 40, for example) for one single target (
N,
E,
RdRp,
S,
Orf1b), an ambiguous interpretation is achieved. In this regard, particularly in the presence of lower viral loads [
10], the kit cannot respond to all of the clinical diagnostic questions, such as: (a) does the positivity correspond to an active viral infection? (b) Does the subject have a potential infectious capacity? We aimed to verify the following issues: (a) are all of the kits superimposable in terms of the detection of the specific genomic targets detected? (b) Are the sgRNAs detected in the complete ranges of the threshold cycles? (c) Is there any potential to use subgenomic transcripts, in combination with viral genomic RNA, as ‘higher viral load surrogate markers’? (d) Can the transcription rate of subgenomic RNAs be related to the genomic target detected in the swab samples?
To this purpose, the sgRNAs encoding for Envelope protein (
sgE) and nucleoprotein (
sgN) were assayed through with different molecular approaches [
11,
12,
13,
14] on nasopharyngeal swabs collected from patients admitted to our hospital as being suspected for COVID-19 infection. The RT-qPCR has yet to be used as a method for the detection of SARS-CoV-2 subgenomic transcripts [
12,
15].
In summary, we show that sgRNAs (sgN and sgE) detection can provide useful information, to such an extent that we propose to use it in combination with routine assays to assess the early phase of infection in oro-nasopharyngeal swabs, it being the sgRNA N that is able to discriminate between higher and lower viral loads.
2. Materials and Methods
2.1. Sample Collection
Oro-nasopharyngeal sampling was performed by means of commercial-flocked swabs collected in about 1 mL universal transport media (UTM (Copán, Brescia, Italy)) and sent to our Lab in boxes at a controlled temperature within four hours of the collection. The regional health department decided to centralize the swab drawing through a unique unit for samples collection, which consisting of skilled health professionals (coordinated by the ‘Istituto Zooprofilattico Sperimentale del Mezzogiorno’—IZSM) who were trained in performing the oro-nasopharyngeal swabs, which guaranteed homogeneity in the procedures surrounding the sample drawing. In this way, the standardization of the modality of the sample collection was obtained. Our study was approved by the Ethical Committee of Federico II University (n. of protocol 000576 of 10 April 2020), and was performed in compliance with the Declaration of Helsinki.
2.2. RNA Extraction
All of the samples compared in the present paper were extracted using an automated procedure on a MagPurix machine. In detail, a 200 µL volume was used to extract RNA by using a fully-automated system based on a MAGPURIX VIRAL/PATHOGEN NUCLEIC ACIDS kit (Zinexts, commercialized by Resnova, Genzano di Roma, Italy) running on a MAGPURIX 24 INSTRUMENT. MagPurix® CE-IVD Reagent Kits are designed to provide the highest extraction quality through optimized protocols. All of the reagent kits are available in pre-packed environmentally-friendly boxes containing all of the components for 48 extractions: separable reagent cartridges, polygon reaction chambers, tip holders, single-sample tubes, elution tubes, filter tips and piercing pins, as well as additional specific buffer and an optional RNA carrier. No spiking-in with an internal control was performed on the starting swab, in order to obtain viral RNA that was free from RNAs that could potentially interfere with the different methods used. All of the RNA was eluted in 50 µL elution buffer provided by the manufacturers.
2.3. Amplification Kit Selection
Four CE-IVD reagent kits were used, following our Ministry of Health’s indications: the latter, in agreement with the recognized National Covid Reference Laboratories (Istituto Superiore di Sanità and Lazzaro Spallanzani Laboratory), provided a list of eleven kits recognized as being useful in SARS-CoV-2 routine diagnostics, see
Table 1.
The assays selected for the detection of genomic SARS-CoV-2 transcripts are reported in
Table 2, in which all of the characteristics are listed in terms of the target gene, company producing or selling kit, instrumentations validated for the amplificon procedure, input of RNA, number of cycles, range of cycles indicative of positive results, limit of detection, and precision of the methods.
The performances of the three CE-IVD tools were also evaluated against the following new releases: the iONEBIO kit (Kit-D) and the GenePro COVID-19 ((Kit-E); Gencurix, SouthKorea), which has received CE-IVD and FDA clearance. The first was the only one working with a different chemistry, namely the loop-mediated isothermal amplification (LAMP) method, with positive results when the threshold cycle (Ct) values were below or equal to 25.
In the first phase, we used artificial transcripts (see
Section 2.4) to verify the performance of these kits. In a second phase, we switched the evaluation on twenty swabs previously analysed and confirmed as positive in two independent runs.
Following the manufacturers’ instructions, all of the samples were run in single. Nevertheless, in order to verify the performance of each kit, some samples were randomly run in duplicate, blind, pipetting the sample duplicate in different wells on each real-time plate. In this way, the instrument performance was also better evaluated in terms of the homogeneity of the running conditions of the thermal-block during the real-time PCR assay. The personnel should be maintained the templates and performed the run was chosen to be the same. This choice was taken in order to reduce biases related to the sample handling.
2.4. Spike Oligonucleotides Synthesis
We reported, in
Table 3, on RNA oligonucleotides (i.e., SARS-CoV-2_N_F1, 117mer; SARS-CoV-2_E, 113mer; Horizon;
https://horizondiscovery.com (accessed on 4 February 2021)) which were synthetized by using the standard phosphoramidite solid-phase synthesis technology and 5’-hydroxyl (5’-SIL), in combination with an acid-labile orthoester on the 2’-hydroxyl (2’-ACE) as the protecting groups. In addition, the SARS-CoV-2_E oligonucleotide was O2’-methylated. Finally, the oligonucleotides were deprotected by removing the 2’-ACE protecting groups, desalted by ethanol precipitation, and purified with ion exchange high performance liquid chromatography (HPLC).
After this setting, we proceeded to spike-in the detection of both constructs by means of RNA serial dilutions, starting from 1 ng of the gene N and E constructs, obtaining the following dilutions: 1:10 (100 pg); 1:100 (10 pg); 1:1000 (1 pg); 1:10,000 (0.1 pg), as refereed to 1 ng (starting amount). The dilutions were performed by spiking in a TMB medium belonging to a negative sample that was previously tested in triplicate. The same setting was performed by using PBS as the dilution medium, in order to verify a possible matrix effect on the extraction procedure. All of the dilutions were run in triplicate.
2.5. Amplification of the Subgenomic Orf1 Region SgRNAs for N and E Regions Amplification
The oligonucleotide sequence of the primers is described in
Table 4.
The total RNA was extracted from all of the positive samples. The sgRNA RT-PCR assay used the SuperScript IV VILO Master Mix (11756500, Invitrogen, Carlsbad, CA, USA), following the manufacturer’s instructions. The testing for sgRNAs used a leader-specific primer, as well as primers and probes targeting sequences downstream of the start codons of the
E and
N genes [
11,
13]. In addition, the SYBR-green technology was also used to detect the above subgenomic transcripits by a different couple of primers. In detail, the reverse transcription products (cDNA) were amplified by quantitative real-time PCR using a real-time PCR system (Quantstudio 5, Life Technologies, Monza MB, Italy). The target genes were detected using a Brightgreen 2× qPCR Mastermix low-rox (#Mastermix-lr; ABM.). These analyses were run using a PCR machine (Quantstudio 5) under the following conditions: hold stage, 50 °C for 2 min, 95 °C for 10 min; PCR stage, 95 °C for 15 s, 60 °C for 1 min (×60 cycles); melt curve stage, 95 °C for 15 s, 60 °C for 1 min; 95 °C for 15 s. The SYBR Green Primers are reported in
Table 3.
The cDNA was obtained by random primer RT-PCR using the SensiFASTcDNA Synthesis Kit (Bioline, provided by Life Technologies Italia, Monza MB, Italy), using 5ul RNA extracted from nasopharyngeal swabs. We used the following primer setting (
sgN-For AAACCAACCAACTTTCGATCTCTTGTA and
sgN-Rev TCTGGTTACTGCCAGTTGAATC) to amplify the
sgN region, and to perform the Sanger sequencing. All of the primer settings are reported in
Figure S1 (supplementary files). The conditions for the target amplification were the following: cDNA was used as a template to amplify the
sgN sequence by means of the Wonder Taq DNA Polymerase (EuroClone, Via Figino, 20/22 20016 Pero (MI)) according to the manufacturer’s instructions. The amplified products were extracted from agarose gel and purified with the Wizard SV Gel and PCR Clean-Up System kit (Promega). In brief, an equal volume of Membrane Binding Solution was added to the isolated fragment in order to allow the solubilization of the agarose; then, it was aliquoted in a silica column and centrifuged at 14,000 rpm for 1 min. The column was washed with 700 µL washing buffer and centrifuged again at 14,000 rpm for 1 min. In the final step, the DNA was eluted with Nuclease-Free Water, centrifuged at 14,000 rpm for 1 min, and controlled on agarose gel. Finally, the purified PCR product was sequenced by the Sanger methodology.
2.6. Digital Droplet PCR (ddPCR)
The absolute quantification of SARS-CoV-2-RNA was carried out by ddPCR using a two-step reaction; the cDNA synthesized using the SensiFAST cDNA Synthesis Kit (Bioline) was amplified using the 2× ddPCR Supermix (no dUTP) (Bio-Rad, Hercules, CA, USA). The QX200 droplet generator was used to generate the droplets by mixing the cDNA samples, 9 µM of both forward and reverse primers (reported in
Table 4), and 2.5 µM of probe with 70 µL droplet-formation oil. The amplification step was performed on a T100 thermal cycler (Bio-Rad) with the following conditions: heat to 95 °C for 10 min, followed by 95 °C for 30 s, and 55 °C for 1 min, for a total of 45 cycles (at a heating rate of 2 °C/s), followed by 98 °C for 10 min. After the PCR, the positive/negative droplets were analysed in the QX200 droplet reader (Bio-rad), and the QuantaSoft analysis software (Bio-Rad) was used to calculate the number of targets analysed.
2.7. Confirmation of Subgenomic sgN and sgE Transcription in Vero E6 Cells
In order to confirm the levels of
sgE and
sgN in relation to those of the genomic regions amplified by the five kits, Vero E6 cells (8 × 10
5) were infected with SARS-CoV-2 viral particles obtained from an Italian patient affected by COVID-19 (gene bank: MT682732.1), as previously reported [
15]. After 72 h of infection, the total RNA was extracted from SARS-CoV-2–infected Vero E6 cells and reverse transcribed in cDNA [
15]. The qPCR for
sgN,
sgE and
b-Actin amplification was performed using SYBR Green chemistry. The quantity of the total viral RNA is expressed as Multiplicity of Infection (MOI): the latter was calculated by performing a standard curve with RNA related to an N1 fragment derived from standard positive controls (pcs) with a known viral titre (500000-50).
2.8. Running Conditions for Assay Comparisons
The iONEBIO and Gencurix plates were run on BioRad CFX instrumentation, following the manufacturers’ instructions. The remaining ones were all run on ABI7500 Dx fast and LC480-II Roche. On the latter, a specific updated software was used to decipher the results coming from each run. The threshold line values was set by default on the machine, and adjusted—when necessary—following the manufacturer’s indication. The specific threshold cycle (Ct) was used to distinguish the positive and negative samples, respectively.
2.9. Statistical Analysis
The statistical analyses were performed using the IBM SPSS® Statistics (IBM Company, New York, NY, USA) software package (IBM SPSS Statistics for Mac, version 26). Correlation matrixes (Spearman rho coefficient) were used to show the linear relationship between the diagnostic tests. p ≤ 0.05 was regarded as being statistically significant.
3. Results
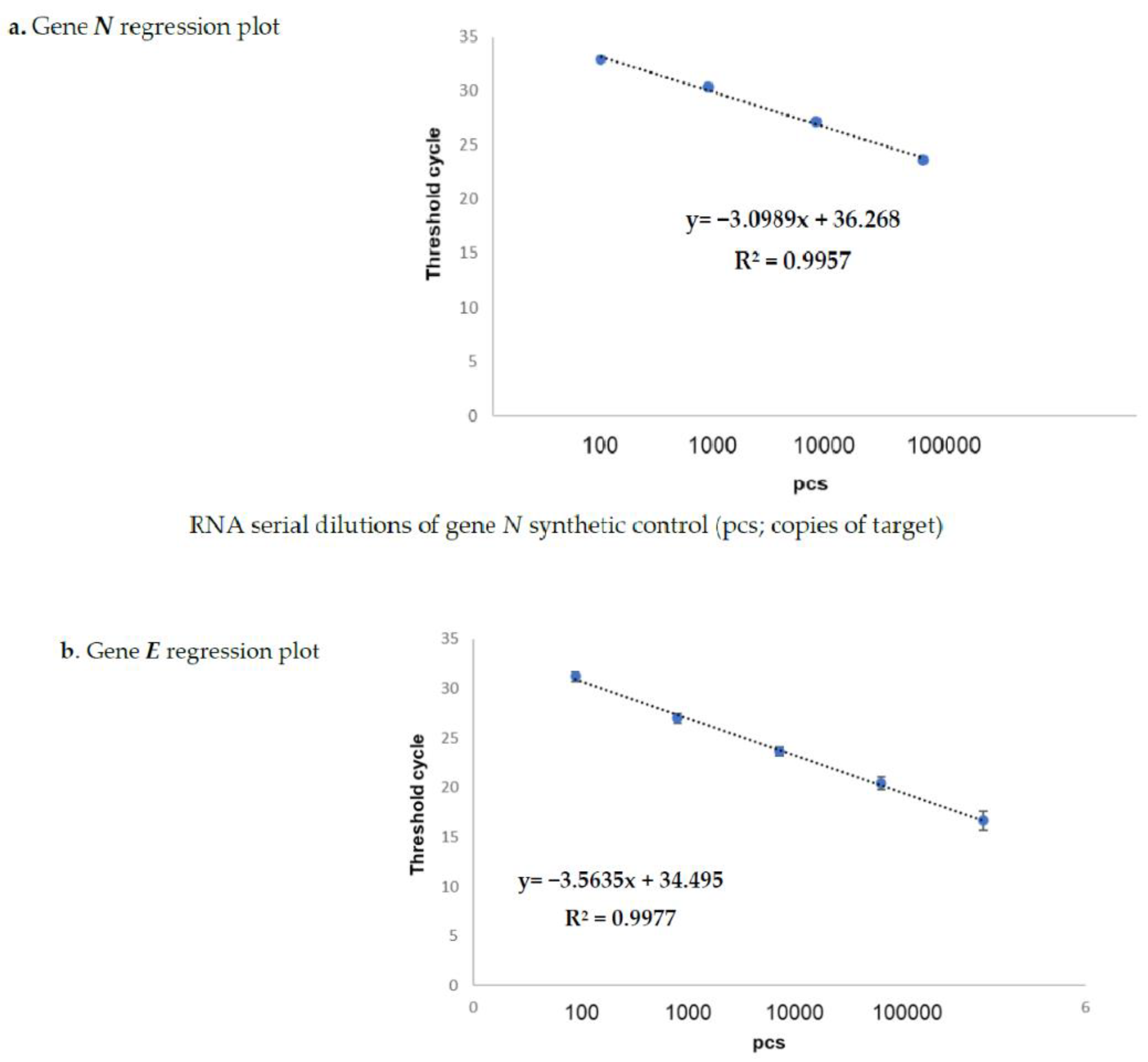
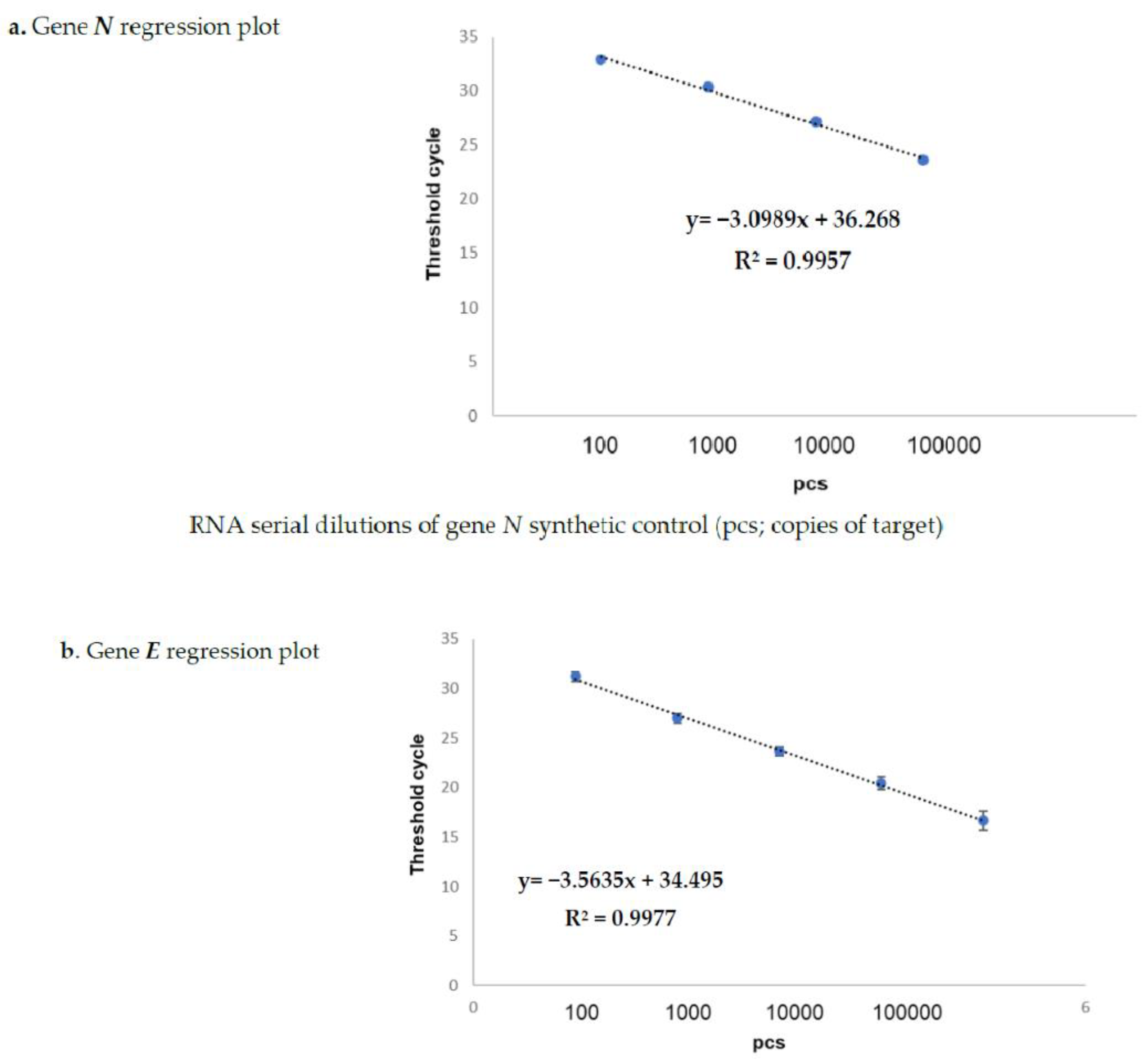
The results obtained on the sample dilutions of the spiked-in constructs are shown in
Figure 1a,b, in which the amplification plots show an excellent evaluation of the regression linear coefficients (R2: 0.9957 and 0.9977) for
N and for gene
E, respectively, in triplicates assayed by means of the Allplex 2019-nCoV assay Seegene kit (KitA; Genova GE, Italy). The other kits reported in
Table 2 also showed superimposable results. In fact, none of these failed in detecting the specific SARS-CoV-2 gene target at any dilution point (data not shown). The same results were also obtained on sampled spiked-in PBS buffer, therefore excluding the matrix effect during the extraction procedure (data not shown).
Therefore, following phase one, we analysed the RNA samples obtained from the collected from n.20 positive COVID-19 symptomatic patients. The positivity on these samples had been previously assigned by the use of kit-B and Kit-C. Here, in order to assess the reliability of RT-PCR, we tested the other three kits on the same RNA samples. As reported in
Table 5, the kits showed different performances in terms of target detection (expressed as Ct values).
The viral E gene was included as a target in four of the kits, and the concordance of the detection rate was 100% (14/20 swabs), 75% (3/20), and 50% (3/20), respectively. For the RdRp gene, the concordance in the rate of detection was 100% in 15/20 swabs, and 66% in 5/20 samples. In 33% of cases, two out of the four kits missed the viral target. This behavior was also observed for the N gene, which was not detected in 5 out of 20 samples. The comparison of the gene ORF1ab performance was not evaluable due to absence of this target in the other commercial kits. Nevertheless, this target was not detectable in 6 out of 20 samples.
Then, in order to verify the superimposability of the results when they referred to the detection of the same viral genomic target (i.e.,
N,
E,
RdRp), correlation matrices were used. As reported in
Table 6 and
Table 7, by comparing three kits targeting gene E, the Genecurix method outperformed because of its significant correlation with the other two kits. The same behavior was observed for the ‘
RdRp’ gene (
Table 6).
Furthermore, we amplified the sgN and sgE targets, with the following results: only the four samples with a higher viral load (presence of mean Ct ≤ 22.5) and corresponding to patients with the most severe symptoms showed sgN positivity, while sgE showed positive results in all of the samples tested. These results, obtained on this explorative group of samples, were confirmed in two different runs, in duplicate, with a mean standard deviation of Ct values close to 0.1 Ct. Following the hypothesis that sgN is associated with higher virus load in the swabs, while sgE is not, we performed an in vitro experiment using Vero E6 cells [
15] infected with the virus (Genebank MT682732.1) at different Multiplicities of Infection (MOI) (with standard positive controls of N1 gene, see
Figure S2 and Table S1—ST1) following and verifying the expression of both sgN and sgE using SYBR Green real-time PCR. Our results unequivocally indicate that, while sgE is expressed at low levels (between 30.3 to 36.81 Ct) at the different MOI used (from 0.1 to 0.05), sgN is mainly expressed (between 25.36 to 33.72 Ct) on higher virus infected cells (MOI 0.15 to 0.02). The use of further dilutions of the virus (MOI < 0.02) corresponded to the undetectability of the sgN transcript (as reported in the
Supplementary Table S1—ST1). Finally, in order to confirm these results in vivo in the swab, we retrospectively recovered n. 48 RNA samples (used as the discovery cohort) extracted from COVID-19 positive patients showing Ct values ranging from 13.5 to 22.5 (
n = 26) to >22.5–40 (
n = 22). The sgN target was detectable only in those samples with Ct values close to or below 22.5. In contrast, sgE was always detected in all of the Ct ranges. These results were obtained by both TaqMan and SYBR Green real-time PCR chemistries. Unfortunately, for these 48 retrospective samples, the contact tracing assessment of COVID-19 transmission was difficult to recover, particularly because we had received swabs from lots of different areas, and the centralized unit performing the swab sampling only reported these as symptomatic or asymptomatic. Consequently, it is difficult to correlate our confirmatory molecular results with the specific period of observation either for asymptomatic contacts or COVID-19 symptomatic individuals, even considering how hectic this swab collection activity has been. However, we can only report them as COVID-19–positive samples, and about 15% of these asymptomatic subjects showed low Ct values (<25.0).
Nonetheless, due to the recrudescence of COVID-19 in the last months, we were able to collect, in the first weeks of December, an additional 20 independent highly-positive samples (used as the validation group of samples), corresponding to: (a) four symptomatic individuals with severe forms of COVID-19 (mean Ct value ≤22.5); (b) six less severe patients with moderately-positive swabs (mean Ct ranging >22.5 and ≤29.5), and (c) 10 asymptomatic subjects corresponding to the close contacts of infected patients who were screened for the first time, and submitted to a swab drawing one week after the onset of symptoms in the positive individuals (n. 4, with a mean Ct value ≤22.5; n. 4 with mean Ct > 22.5–29.5, and n. 2 with mean Ct > 29.5), where we confirmed the above reported data. Furthermore, in order to exclude false positive results given by any possible off-target signal, we used high resolution melting analysis (HRMA) and Sanger sequencing.
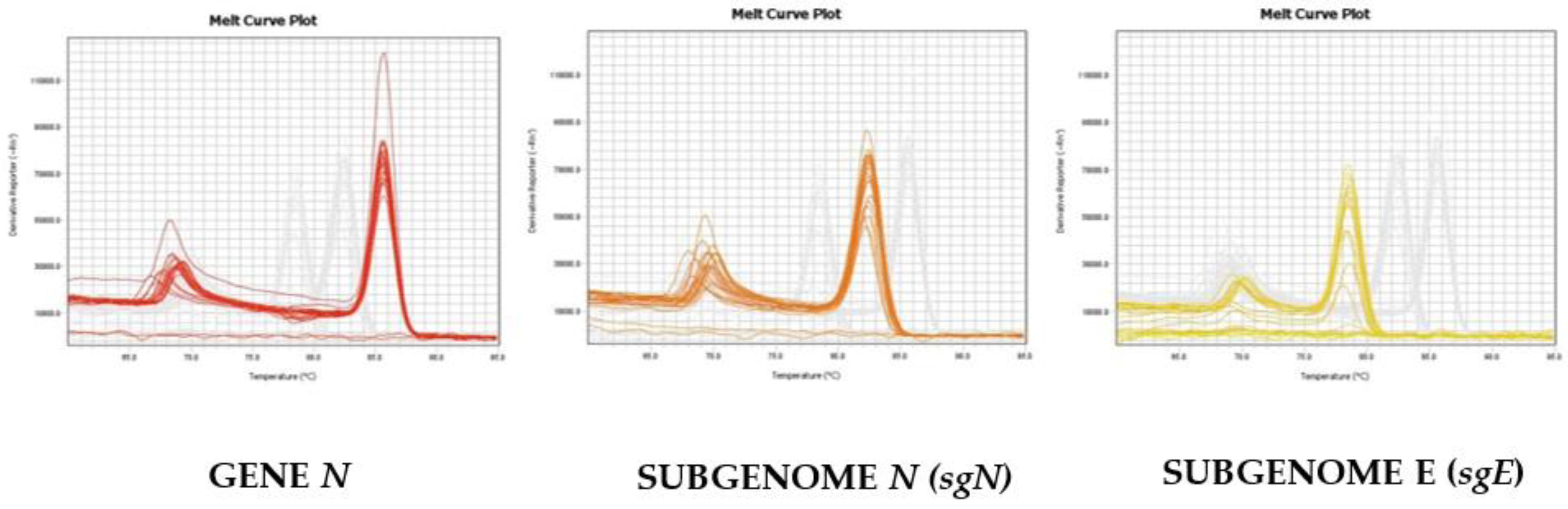
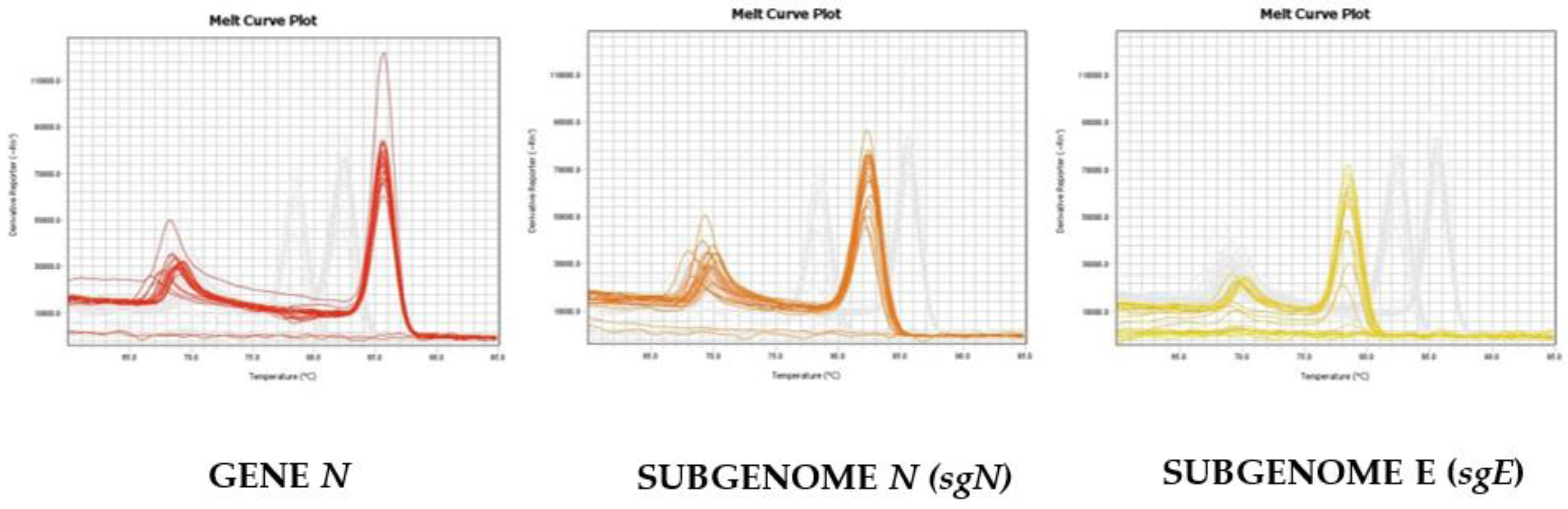
As reported in
Figure 2, we were able to distinguish and confirm the different targets amplified by HRMA. In order to better validate our results, we sequenced the sgN region, obtaining the following amplicon of 141, as reported in
Figure S3. Data regarding subgenomic sequencing by Nanopore technology of the subgenomic RNAs of SARS-CoV-2 have been published by our group [
15]. The virus that was isolated, fully sequenced, and used in our studies has a GeneBank number of MT682732.1 (Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/human/ITA/Naples/2020, complete genome). The GISAID annotations for all of the genomic and subgenomic regions are reported in the Ref [
15].
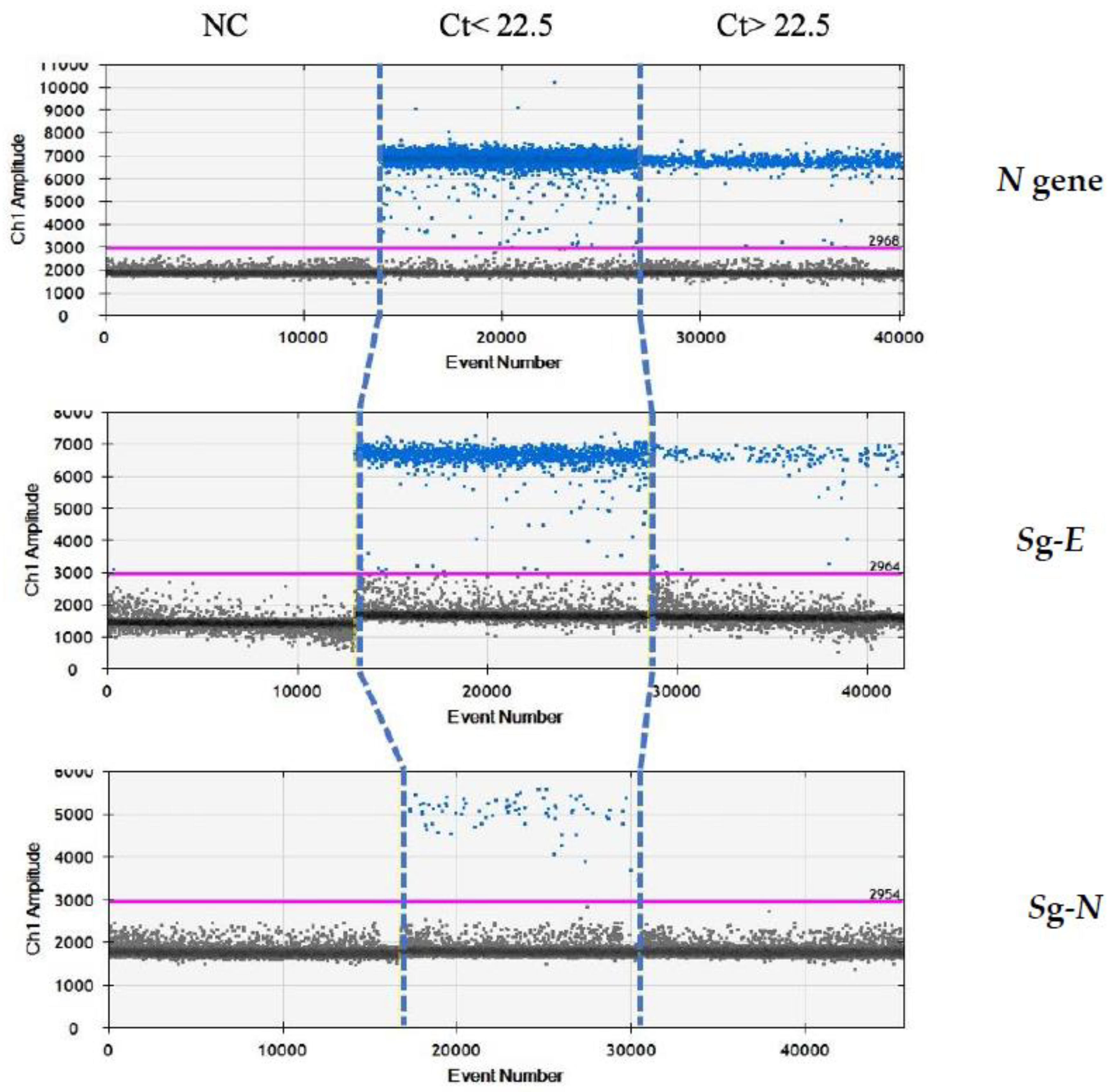
Finally, we re-analysed these samples by droplet-digital-real-time PCR (ddPCR) in order to evaluate whether the absence of
sgN in the samples above Ct 22.5 was due to the limit of detection of the real-time methods, rather than being due to the characteristics of samples containing a lower viral load. Our ddPCR results (reported in
Figure 3) clearly show that
sgE is detected in samples with Ct > or ≤22.5 (from 13.5 to 40), while the
sgN is detected only when Ct was ≤22.5. It is of interest that the
sgN expression does not seem to be influenced by genomic
N gene transcription, with the
N gene being detectable in all of the Ct ranges (from 13.5 to 40.0).
4. Discussion
In the present report, we provide extensive data regarding an evaluation detection of subgenomic transcripts in relationship to the positivity rate (evaluated as Ct values) of three CE-IVD commercial kits used during the first outbreak of COVID-19 pandemic, plus two new kits recently released onto the market. The use of these different reagents—alternatively or as a complementary, especially in the case of uncertain results—was necessary to verify not only the relative performances, but also to correlate the amplification rate of
N,
E, and
RdRp targets with the subgenomic expression of the
sgN and
sgE transcripts. In fact, it is reported that the amplification of the complete SARS-CoV-2 genome is possible only when the threshold cycle is below 33.0. Due to both the preanalytical and analytical variables [
5,
16] affecting the virus detection in oro-nasopharyngeal swabs, we used different methods in order to better evaluate the expression of the subgenomic transcripts in relationship to the genomic transcripts. The high-sensitivity PCR-based tests are nearly 100% accurate in identifying infected people, if they are administered appropriately [
1,
17]. In order to avoid biases related to the amplification process of RNA from unknown swabs, we evaluated the analytical performance of the five kits reported herein using both negative sample swabs and phosphate buffer medium, into which genomic constructs of gene
N and
E were spiked-in at different dilutions. In these theoretically-ideal conditions (the certainty that at least a SARS-CoV-2 RNA target was present), at the different dilutions, we obtained the result that all of the kits were able to detect the target within the limit of detection range declared. This means that both our automated extraction method and the medium used for the dilutions (TMB or PBS) did not influence either the yield or the recovery of the
N and
E artificial targets spiked in at the different concentrations. Thus, we excluded the presence of any possible interference by the matrix effect. Nevertheless, the same findings were not so exciting when they referred to the twenty positive samples contemporarily analyzed with the five alternative methods: in fact, in no case have we obtained a complete superimposition of the results, with a correlation from 50% to 100% among the different commercial kits. We could hypothesize that this lack of correlation depends either on the viral load or on the type of components within each reagent. Moreover, we did not observe any signal related to probe degradation or increases in the background signals within each microplate. Consequently, we can only report the discrepancies among the different methods evaluated as the results of different sensitivities on the true clinical samples as compared to those obtained on the spiked-in materials. The best correlation matrix was found for GenePro COVID-19 (Kit-E), which was shown to be the kit with the best performances in terms of both target detection and concordance with the tools targeting the same regions of SARS-CoV-2. This commercial kit was also reported by Kubina R. et al. [
18] as being superimposable to other commercial CE-IVD assays. We underline that one important feature of the SARS-CoV-2 diagnostic path is to anticipate—as soon as possible—its detection in order to rapidly screen the population and avoid or limit its spread. Unfortunately, in the presence of the tools and reagent not overlapping in terms of target detection, it would be very difficult to distinguish people who are under an active viral infection. In this regard, Guglielmi G. et al. [
10] underlined the fact that it is crucial to identify the infected individuals who are not able to spread the virus as only ‘passive’ carriers. In order to overcome the limits given by the kits targeting the SARS-CoV-2 genomic transcripts, we wanted to test whether other molecular markers of SARS-CoV-2 could be detectable as also being able to describe the kinetics of the virus replication inside each individual. Therefore, within the complex replicative machine of SARS-CoV-2, we designed a specific assay to amplify the
sgN and
sgE, which correspond to the subgenomic regions, within positive swabs. Surprisingly, we found that
sgN mRNA is transcribed only in the samples with the highest viral loads, as extrapolated by the Ct values, regardless of the kit that is routinely used. In fact, the
sgN detection was achievable whenever the mean Ct value of the run is below or equal to 22.5. These findings were further validated in vitro with different virus load on Vero E6 cells, in which the
sgN was highly expressed in samples where the genomic targets
N and
E showed the lower Ct. Then, a further confirmation of our results was also obtained by ddPCR, which did not detect any
sgN copy in any of the samples below the 22.5 value, suggesting that
sgN could be further investigated as a reliable marker of the highest viral load on a larger sample number. Our results, confirmed in all of the eighty-eight swab samples analyzed, therefore do not depend on the limit of detection of our assays being ddPCR more sensitive that real-time PCR. These results appear not to be in contrast with previous experimental findings [
7,
11,
12] showing that the
N protein is required for efficient coronavirus subgenomic mRNA transcription. In order to confirm our hypothesis of a potential use of subgenomic transcripts in molecular diagnostics, we confirmed in vitro
, using Vero E6 cells with different MOI virus infection rates, that
sgN was detectable every time the viral load was highly evaluated. These results were not obtained when
sgE was analyzed. Regarding the behavior of
sgE, we verified that this target is expressed in all of the samples, regardless of the individual Ct value of the specific genomic targets amplified by the different kits. Therefore, we could speculate that
sgN is a surrogate marker of both the highest viral load and potential infectivity, although this result needs to be confirmed on a standardized cohort of patients, with a precise indication of the infection time, severity of symptoms, and clinical and molecular evaluation during the follow-up. We highlight here that the mechanism for the generation of these subgenomic mRNAs is not fully understood, although they are strongly regulated to guarantee the best ratio of virus proteins and their survival in the cells. Furthermore, the expression of the N protein seems to be required for an efficient coronavirus subgenomic mRNA transcription, as confirmed by the detection of high numbers of copies of this gene by ddPCR: the N protein stabilizes, in fact, the virus genome copies once it is replicated into the cell.
We further underline that subgenomic RNAs are considered to be particularly abundant during the early infection (up to 70 times more abundant than virus genomic RNA at the peak of RNA transcription in the cell culture), as it occurs as early as 6-8 h after infection [
7,
12]. The detection of these subgenomic RNAs in our clinical nasopharyngeal swabs confirms that these transcripts are moderately stable, being the
sgN RNA mainly associated to the highest viral load. Moreover, in contrast to the data reported by Alexandersen et al. [
17], who reported that the relative abundance of subgenomic RNAs may be more related to the sample’s quality and storage before the laboratory assay than to the actual stage of infection, we can emphasize that the persistence of
sgN RNAs might be considered as a candidate biomarker of the active viral load, regardless of the pre-analytical conditions and analytical diagnostic kit. It is noteworthy that
sgN was reported as the most expressed transcript in other sample types, like stool [
19]; the authors of this research concluded that the detection of
sgN and s
gE improves the diagnosis of COVID-19, particularly in patients who are suspected of being infected but with negative results in the upper respiratory tract. Although the setting of this study [
19] is different, we can agree with the authors’ conclusion, which emphasized the role of these subgenomic transcripts. Our preliminary findings, although they were confirmed on a group of eighty-eight samples, should be deepened on larger cohorts, particularly to confirm the diagnostic potential of the
sgN target in terms of the prediction of higher and active viral loads. Nevertheless, it is not so common to find clinical swabs with these high viral loads, as confirmed—in our experience—on more than 70,000 swabs processed in the last six months, for which the recovery of samples with very high viral loads (intended as with a Ct ≤22.5) is not very frequent. Overall, we recovered about sixty samples with Ct ≤22.5 in the March–October pandemic infection period. We point out that the main limitation of our study is that we were not able to precisely trace the timing of the infection of the individuals who submitted to oro-nasopharyngeal swabs, particularly for the samples collected outside of our hospital. In this regard, we agree with Cheng et al. [
20], who reported that the dynamics of COVID-19 transmissibility are far from being fully understood. However, it is important to investigate possible new markers for a better understanding of the transmission dynamics for the development and evaluation of effective control policies. Cheng et al. [
20] reported that the short serial interval of COVID-19, and the results from viral shedding studies indicate that most transmission occurred near—or even before—the time of the onset of symptoms. Nevertheless, it is not known when and for how long the individual with COVID-19 should be isolated, or whether close contacts should be quarantined. Therefore, we emphasize that every study can be affected by these pre-analytical biases.
However, Alexandersen et al. [
17] stated that SARS-CoV-2 subgenomic RNAs were detectable in about thirteen diagnostic samples up to 17 days after the initial detection of infection. This evidence seems to be related to their nuclease resistance and protection by cellular membranes. Nevertheless, they performed an untargeted analysis in all of the subgenomic regions, which was able to amplify some subgenomic RNAs by NGS, using various amplification steps to amplify the short targets. This approach, on a limited number of samples, cannot be superimposable to our setting, in which we employed different methodologies in an unbiased bioinformatic method of nucleotide leader sequence selection. Therefore, based on our findings, we do not agree with their conclusions assessing that the detection of subgenomic RNAs in clinical samples may not be a suitable indicator of active coronavirus replication/infection.
Thus, we show here results underlining that
sgN positivity can unequivocally reflect a stage at the highest viral load, discriminating between more active (early) virus infection and middle–long-term carrier status. This hypothesis is not in contrast with findings by Wölfel et al. [
12], who reported that “viral subgenomic mRNA is transcribed only in infected cells indicating the presence of actively infected cells in samples”. Moreover, the capability of the virus to grow, particularly when it is isolated from samples with lower Ct values and
sgN detectability, has been demonstrated by our group in a parallel research work that is under publication elsewhere. These findings seem to be in contrast with those published by Kim et al. [
11], who found that the
N RNA is the most represented during the viral replication; nevertheless, these data were obtained from SARS-CoV-2 viral RNA extracted from Vero cells infected with BetaCoV/Korea/KCDC03/2020, the latter being an experimental setting that was different from ours. In support of our considerations, we can provide evidence by Winnett et al. [
21] stating that low-sensitivity tests are comparable to high-sensitivity tests in detecting early infections when the viral load rises quickly (within hours) after infection and reaches high levels (>10
5–10
6 RNA copies/mL). However, although there are no human data testing these assumptions, they provided evidence that, in at least some human cases of SARS-CoV-2, the viral load rises slowly (over days, not hours) and not to such high levels as to be detectable reliably by any low-sensitivity test. We showed herein that our commercially-available kits fail in detecting some SARS-CoV-2 genomic targets in swab-deriving RNA; therefore, the use of subgenomic transcripts might improve the rate of virus detection.
This evidence is of impact because it could be mainly useful during follow-ups in which the positivity of the molecular test cannot discriminate between higher or lower viral loads, while the clearance of SARS-CoV-2 from the respiratory tract occurs, sometimes asymptomatically.
We encourage other laboratories providing SARS-CoV-2 assays to further investigate in this setting, because conflicting findings around reinfection, as well as discrepancies among diagnostic PCRs detecting targets in different regions of the SARS-CoV-2 genome, are reported in the literature, mostly when they are related to subgenomic transcripts.


 ,
, 

{kind=link}
{kind=link}
{kind=link}