
Emerging Mutations Potentially Related to SARS-CoV-2 Immune Escape: The Case of a Long-Term Patient

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Case Description
3. Materials and Methods
3.1. Specimen Collection and Testing
3.2. cDNA Synthesis and Viral Genome Amplification
3.3. Library Preparation and Whole Genome Sequencing
3.4. Sequence Data Analysis
3.5. Phylogenetic Analysis
3.6. Seroneutralization Assays
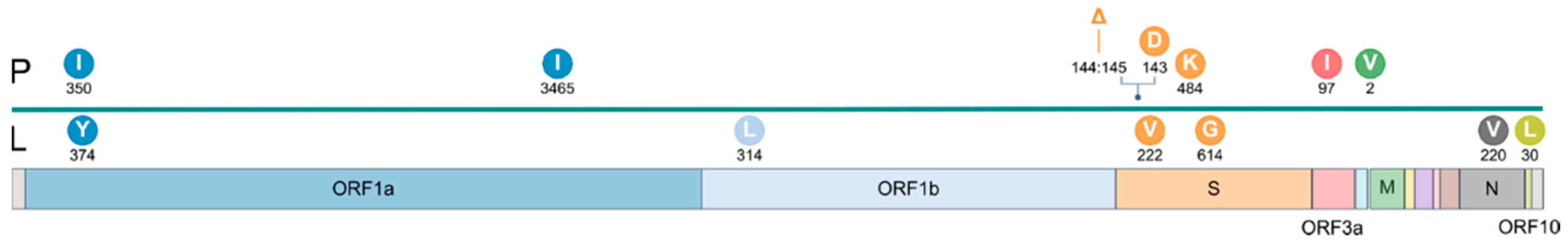
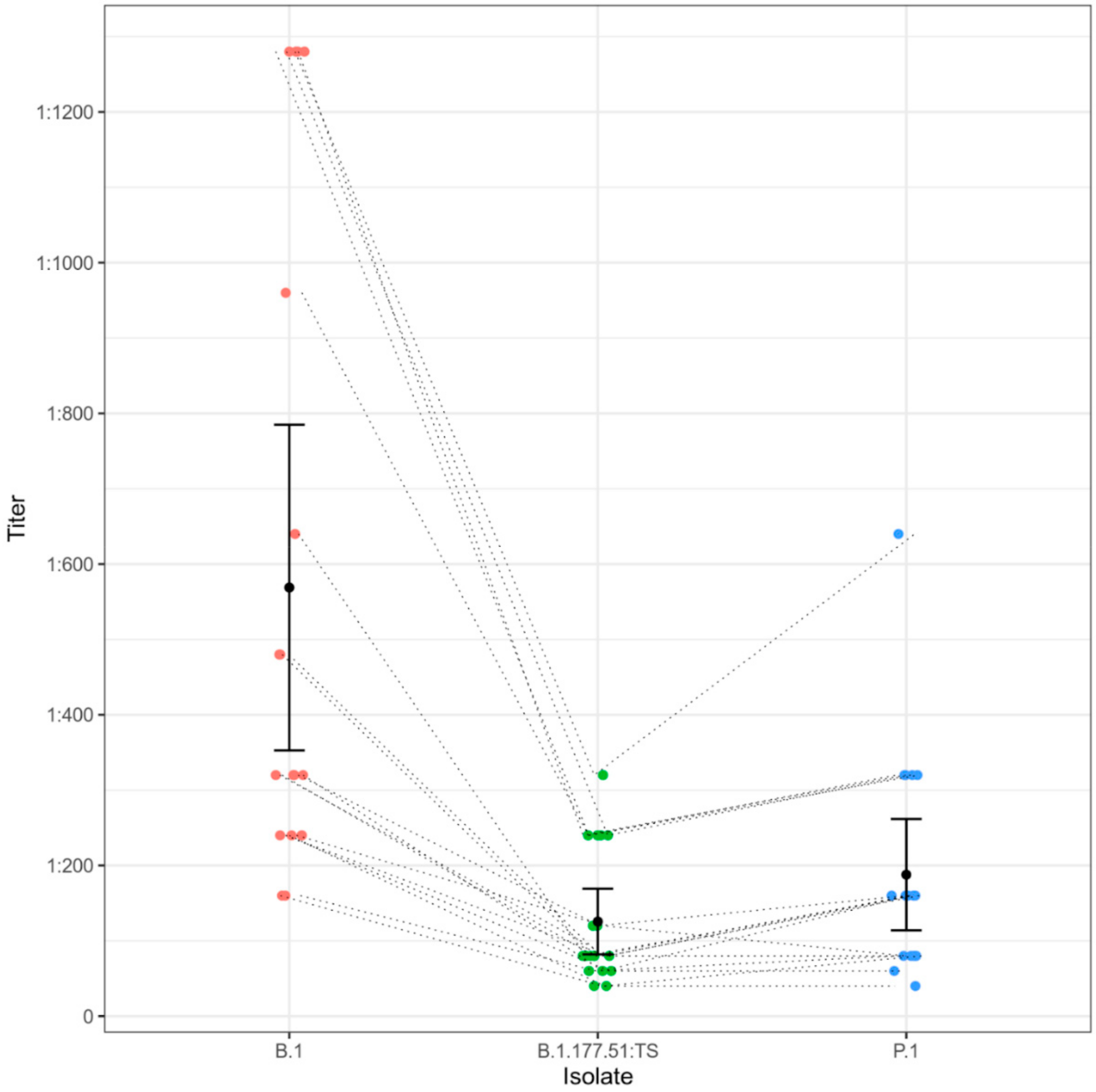
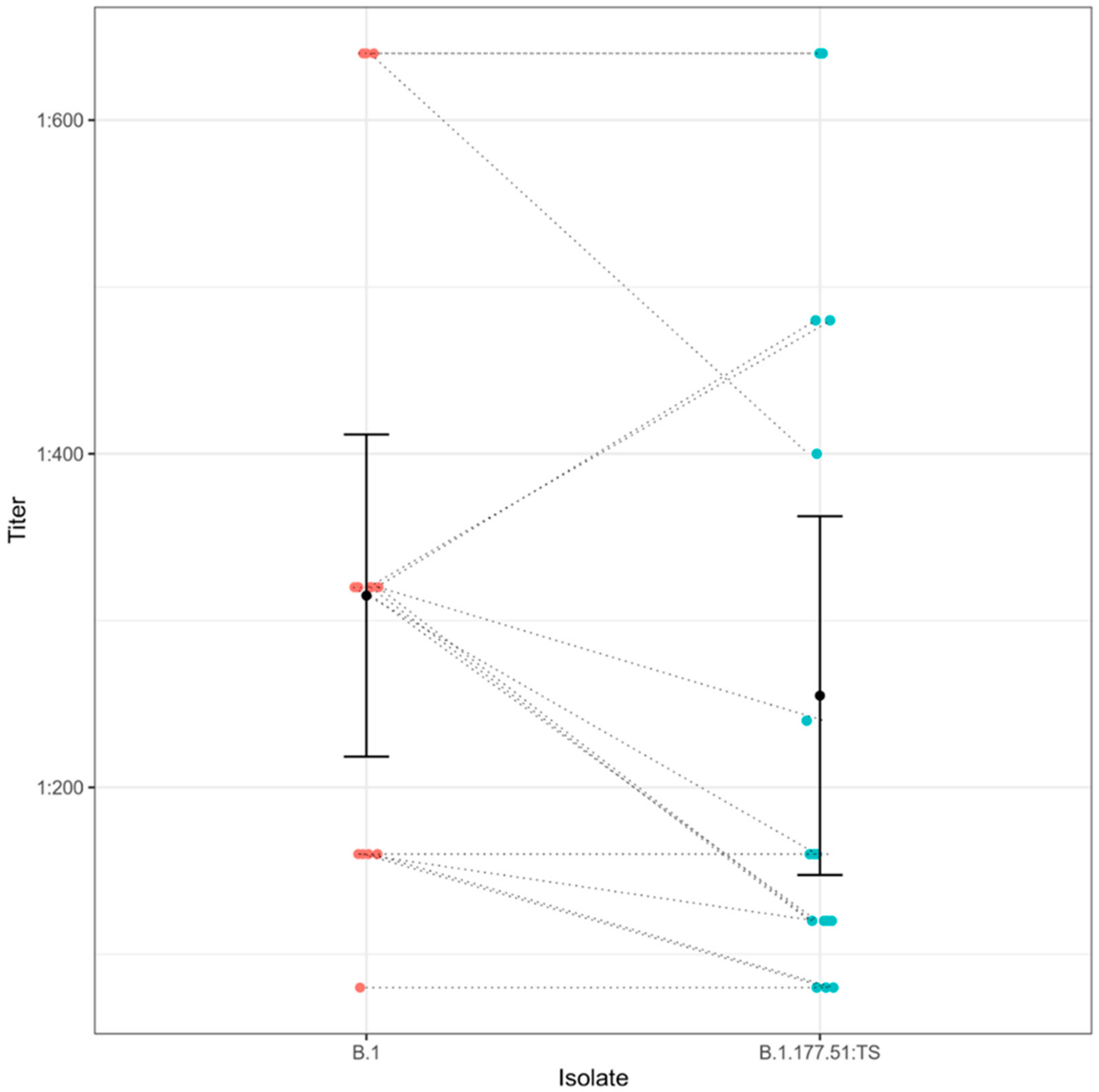
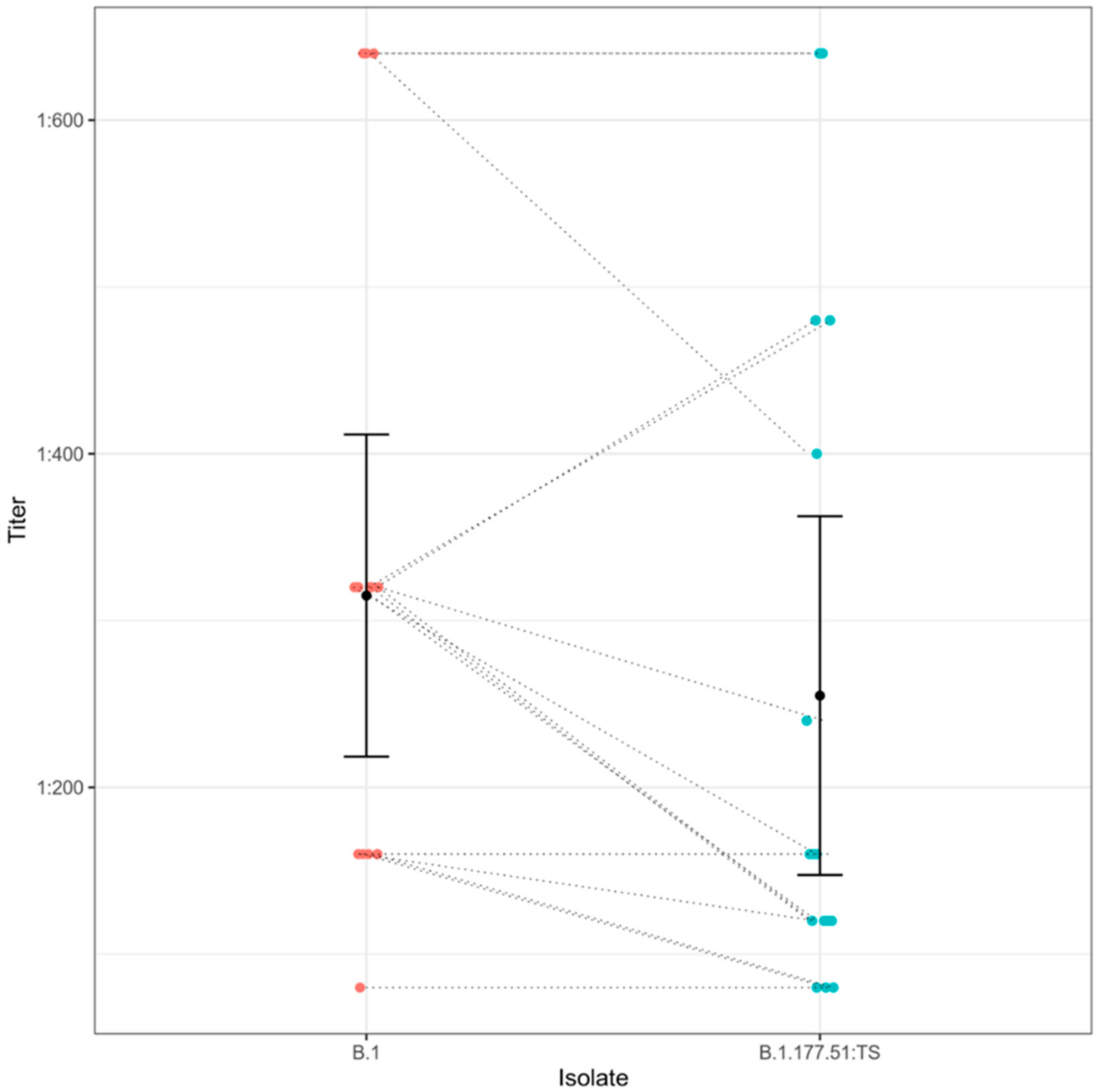
4. Results
5. Discussion
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO Coronavirus Disease (COVID-19) Dashboard. Available online: https://covid19.who.int (accessed on 14 February 2021).
- Pham, V.H.; Gargiulo Isacco, C.; Nguyen, K.; Le, S.H.; Tran, D.K.; Nguyen, Q.V.; Pham, H.T.; Aityan, S.; Pham, S.T.; Cantore, S.; et al. Rapid and sensitive diagnostic procedure for multiple detection of pandemic Coronaviridae family members SARS-CoV-2, SARS-CoV, MERS-CoV and HCoV: A translational research and cooperation between the Phan Chau Trinh University in Vietnam and University of Bari “Aldo Moro” in Italy. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 7173–7191. [Google Scholar] [CrossRef] [PubMed]
- Santacroce, L.; Charitos, I.A.; Ballini, A.; Inchingolo, F.; Luperto, P.; De Nitto, E.; Topi, S. The Human Respiratory System and its Microbiome at a Glimpse. Biology 2020, 9, 318. [Google Scholar] [CrossRef]
- Charitos, I.A.; Ballini, A.; Bottalico, L.; Cantore, S.; Passarelli, P.C.; Inchingolo, F.; D’Addona, A.; Santacroce, L. Special features of SARS-CoV-2 in daily practice. World J. Clin. Cases 2020, 8, 3920–3933. [Google Scholar] [CrossRef]
- Capozzi, L.; Bianco, A.; Del Sambro, L.; Simone, D.; Lippolis, A.; Notarnicola, M.; Pesole, G.; Pace, L.; Galante, D.; Parisi, A. Genomic Surveillance of Circulating SARS-CoV-2 in South East Italy: A One-Year Retrospective Genetic Study. Viruses 2021, 13, 731. [Google Scholar] [CrossRef] [PubMed]
- CDC. Coronavirus Disease 2019 (COVID-19). Available online: https://www.cdc.gov/coronavirus/2019-ncov/variants/variant-info.html (accessed on 3 June 2021).
- Nelson, G.; Buzko, O.; Spilman, P.; Niazi, K.; Rabizadeh, S.; Soon-Shiong, P. Molecular Dynamic Simulation Reveals E484K Mutation Enhances Spike RBD-ACE2 Affinity and the Combination of E484K, K417N and N501Y Mutations (501Y.V2 Variant) Induces Conformational Change Greater than N501Y Mutant Alone, Potentially Resulting in an Escape Mutant. bioRxiv 2021. [Google Scholar] [CrossRef]
- Ferrareze, P.A.G.; Franceschi, V.B.; de Mayer, A.M.; Caldana, G.D.; Zimerman, R.A.; Thompson, C.E. E484K as an Innovative Phylogenetic Event for Viral Evolution: Genomic Analysis of the E484K Spike Mutation in SARS-CoV-2 Lineages from Brazil. bioRxiv 2021. [Google Scholar] [CrossRef] [PubMed]
- Diamond, M.; Chen, R.; Xie, X.; Case, J.; Zhang, X.; VanBlargan, L.; Liu, Y.; Liu, J.; Errico, J.; Winkler, E.; et al. SARS-CoV-2 Variants Show Resistance to Neutralization by Many Monoclonal and Serum-Derived Polyclonal Antibodies. Res. Sq. 2021. [Google Scholar] [CrossRef]
- Artic-Network/Artic-Ncov2019. Available online: https://github.com/artic-network/artic-ncov2019 (accessed on 14 February 2021).
- Bianco, A.; Capozzi, L.; Monno, M.R.; Del Sambro, L.; Manzulli, V.; Pesole, G.; Loconsole, D.; Parisi, A. Characterization of Bacillus Cereus Group Isolates From Human Bacteremia by Whole-Genome Sequencing. Front. Microbiol. 2021, 11, 599524. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and Accurate Long-Read Alignment with Burrows–Wheeler Transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grubaugh, N.D.; Gangavarapu, K.; Quick, J.; Matteson, N.L.; De Jesus, J.G.; Main, B.J.; Tan, A.L.; Paul, L.M.; Brackney, D.E.; Grewal, S.; et al. An Amplicon-Based Sequencing Framework for Accurately Measuring Intrahost Virus Diversity Using PrimalSeq and IVar. Genome Biol. 2019, 20. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A Novel Method for Rapid Multiple Sequence Alignment Based on Fast Fourier Transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [Green Version]
- Darriba, D.; Posada, D.; Kozlov, A.M.; Stamatakis, A.; Morel, B.; Flouri, T. ModelTest-NG: A New and Scalable Tool for the Selection of DNA and Protein Evolutionary Models. Mol. Biol. Evol. 2020, 37, 291–294. [Google Scholar] [CrossRef] [Green Version]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letunic, I.; Bork, P. Interactive Tree Of Life (ITOL) v4: Recent Updates and New Developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rondinone, V.; Pace, L.; Fasanella, A.; Manzulli, V.; Parisi, A.; Capobianchi, M.R.; Ostuni, A.; Chironna, M.; Caprioli, E.; Labonia, M.; et al. VOC 202012/01 Variant Is Effectively Neutralized by Antibodies Produced by Patients Infected before Its Diffusion in Italy. Viruses 2021, 13, 276. [Google Scholar] [CrossRef] [PubMed]
- Hodcroft, E.B.; Zuber, M.; Nadeau, S.; Comas, I.; Candelas, F.G.; Consortium, S.-S.; Stadler, T.; Neher, R.A. Emergence and Spread of a SARS-CoV-2 Variant through Europe in the Summer of 2020. medRxiv 2020. [Google Scholar] [CrossRef]
- Guthrie, J.L.; Teatero, S.; Zittermann, S.; Chen, Y.; Sullivan, A.; Rilkoff, H.; Joshi, E.; Sivaraman, K.; de Borja, R.; Sundaravadanam, Y.; et al. Detection of the Novel SARS-CoV-2 European Lineage B.1.177 in Ontario, Canada. medRxiv 2020. [Google Scholar] [CrossRef]
- McCarthy, K.R.; Rennick, L.J.; Nambulli, S.; Robinson-McCarthy, L.R.; Bain, W.G.; Haidar, G.; Duprex, W.P. Recurrent Deletions in the SARS-CoV-2 Spike Glycoprotein Drive Antibody Escape. Science 2021, 371, 1139–1142. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Zou, J.; Fontes-Garfias, C.R.; Xia, H.; Swanson, K.A.; Cutler, M.; Cooper, D.; Menachery, V.D.; Weaver, S.; Dormitzer, P.R.; et al. Neutralization of N501Y Mutant SARS-CoV-2 by BNT162b2 Vaccine-Elicited Sera. bioRxiv Prepr. Serv. Biol. 2021. [Google Scholar] [CrossRef]
- Xie, X.; Liu, Y.; Liu, J.; Zhang, X.; Zou, J.; Fontes-Garfias, C.R.; Xia, H.; Swanson, K.A.; Cutler, M.; Cooper, D.; et al. Neutralization of SARS-CoV-2 Spike 69/70 Deletion, E484K and N501Y Variants by BNT162b2 Vaccine-Elicited Sera. Nat. Med. 2021, 27, 620–621. [Google Scholar] [CrossRef] [PubMed]
- Yadav, P.D.; Sapkal, G.N.; Abraham, P.; Ella, R.; Deshpande, G.R.; Patil, D.Y.; Nyayanit, D.A.; Gupta, N.; Sahay, R.R.; Shete, A.M.; et al. Neutralization of Variant under Investigation B.1.617 with Sera of BBV152 Vaccinees. bioRxiv 2021. [Google Scholar] [CrossRef]
- Rathnasinghe, R.; Jangra, S.; Cupic, A.; Martínez-Romero, C.; Mulder, L.C.F.; Kehrer, T.; Yildiz, S.; Choi, A.; Mena, I.; De Vrieze, J.; et al. The N501Y Mutation in SARS-CoV-2 Spike Leads to Morbidity in Obese and Aged Mice and Is Neutralized by Convalescent and Post-Vaccination Human Sera. medRxiv Prepr. Serv. Health Sci. 2021. [Google Scholar] [CrossRef]
- Zhou, D.; Dejnirattisai, W.; Supasa, P.; Liu, C.; Mentzer, A.J.; Ginn, H.M.; Zhao, Y.; Duyvesteyn, H.M.E.; Tuekprakhon, A.; Nutalai, R.; et al. Evidence of Escape of SARS-CoV-2 Variant B.1.351 from Natural and Vaccine-Induced Sera. Cell 2021, 184, 2348–2361. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Capozzi, L.; Simone, D.; Bianco, A.; Del Sambro, L.; Rondinone, V.; Pace, L.; Manzulli, V.; Iacobellis, M.; Parisi, A. Emerging Mutations Potentially Related to SARS-CoV-2 Immune Escape: The Case of a Long-Term Patient. Life 2021, 11, 1259. https://doi.org/10.3390/life11111259
Capozzi L, Simone D, Bianco A, Del Sambro L, Rondinone V, Pace L, Manzulli V, Iacobellis M, Parisi A. Emerging Mutations Potentially Related to SARS-CoV-2 Immune Escape: The Case of a Long-Term Patient. Life. 2021; 11(11):1259. https://doi.org/10.3390/life11111259
Chicago/Turabian StyleCapozzi, Loredana, Domenico Simone, Angelica Bianco, Laura Del Sambro, Valeria Rondinone, Lorenzo Pace, Viviana Manzulli, Michela Iacobellis, and Antonio Parisi. 2021. "Emerging Mutations Potentially Related to SARS-CoV-2 Immune Escape: The Case of a Long-Term Patient" Life 11, no. 11: 1259. https://doi.org/10.3390/life11111259