
Reactive, Sparingly Soluble Calcined Magnesia, Tailor-Made as the Reactive Material for Heavy Metal Removal from Contaminated Groundwater Using Permeable Reactive Barrier


Abstract
:1. Introduction
- Mg(OH)2 is much less soluble than Ca(OH)2 in aqueous solutions (KS,Mg(OH)2 = 1 × 10−11; KS,Ca(OH)2 = 4 × 10−6); it is, therefore, an attractive alternative reagent for passive remediation systems [18].
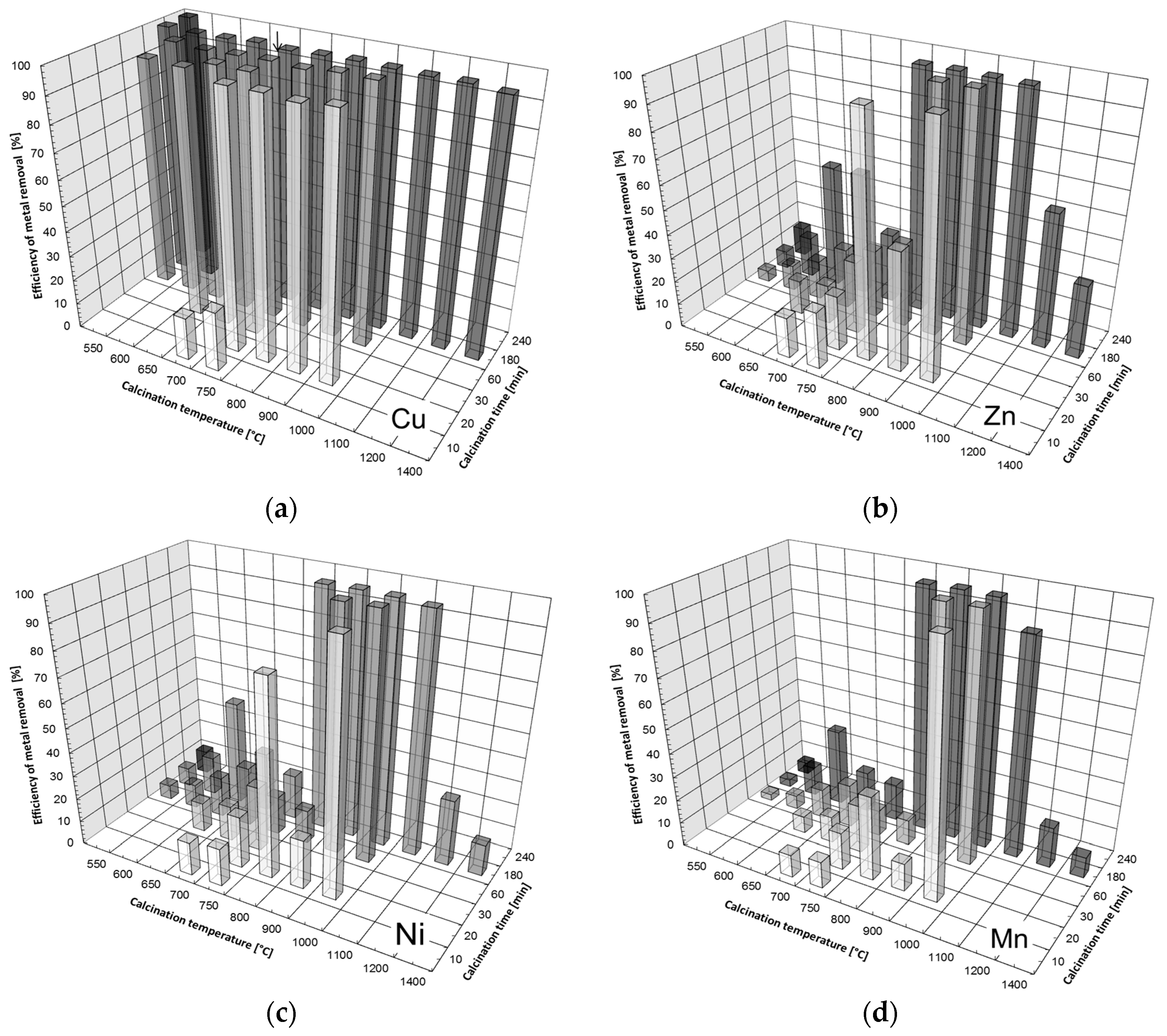
- Upon contact with water, MgO hydrates to form Mg(OH)2, which buffers aqueous solutions’ pH between 8.0–10.5 [18,32,33,35]. In this pH range, the solubility of most metal hydroxides is very low, so divalent metals cations, such as Zn2+, Cd2+, Ni2+, or Co2+, can be removed by precipitation [18,33,35,36].
2. Materials and Methods
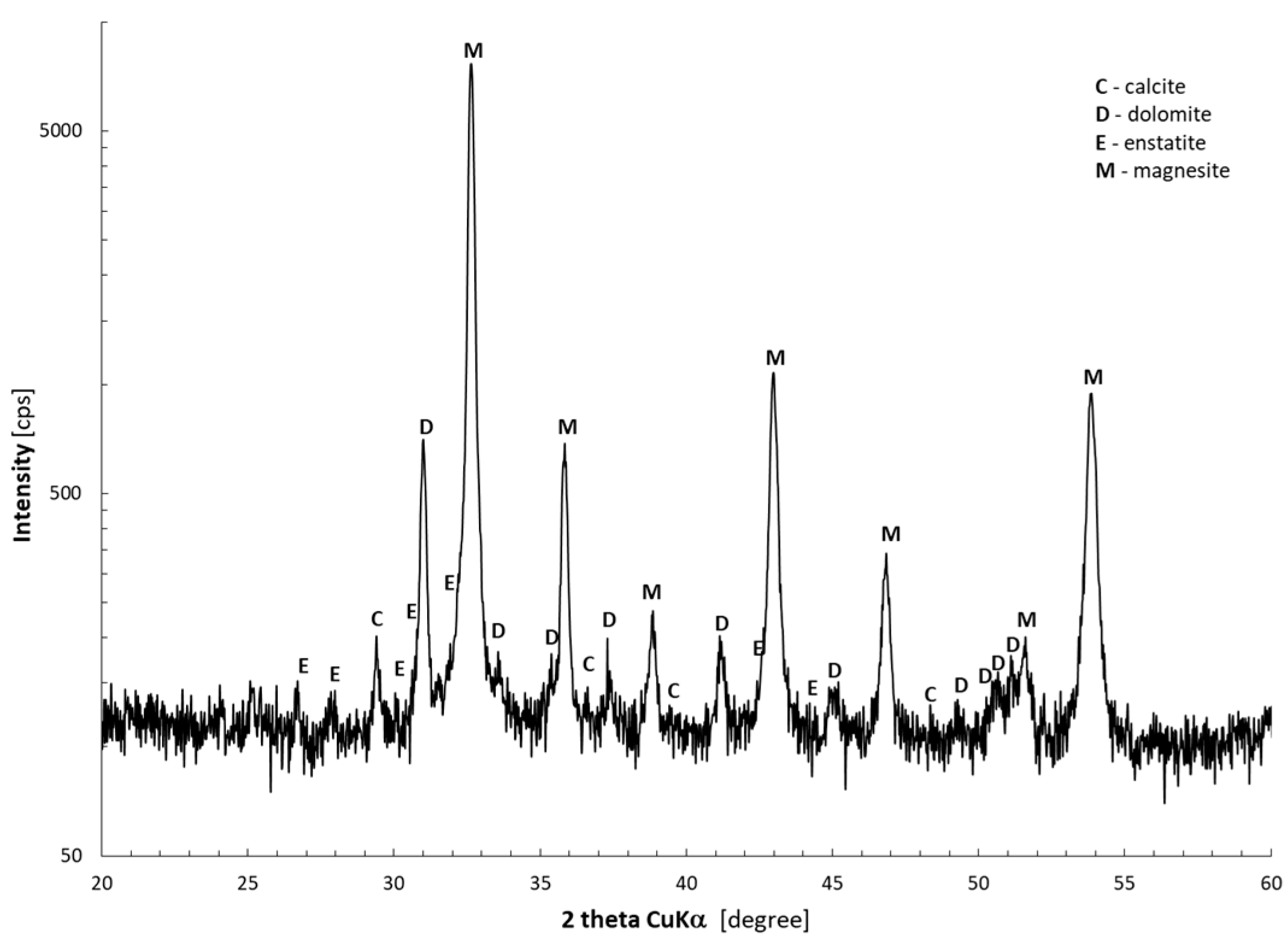
2.1. Materials
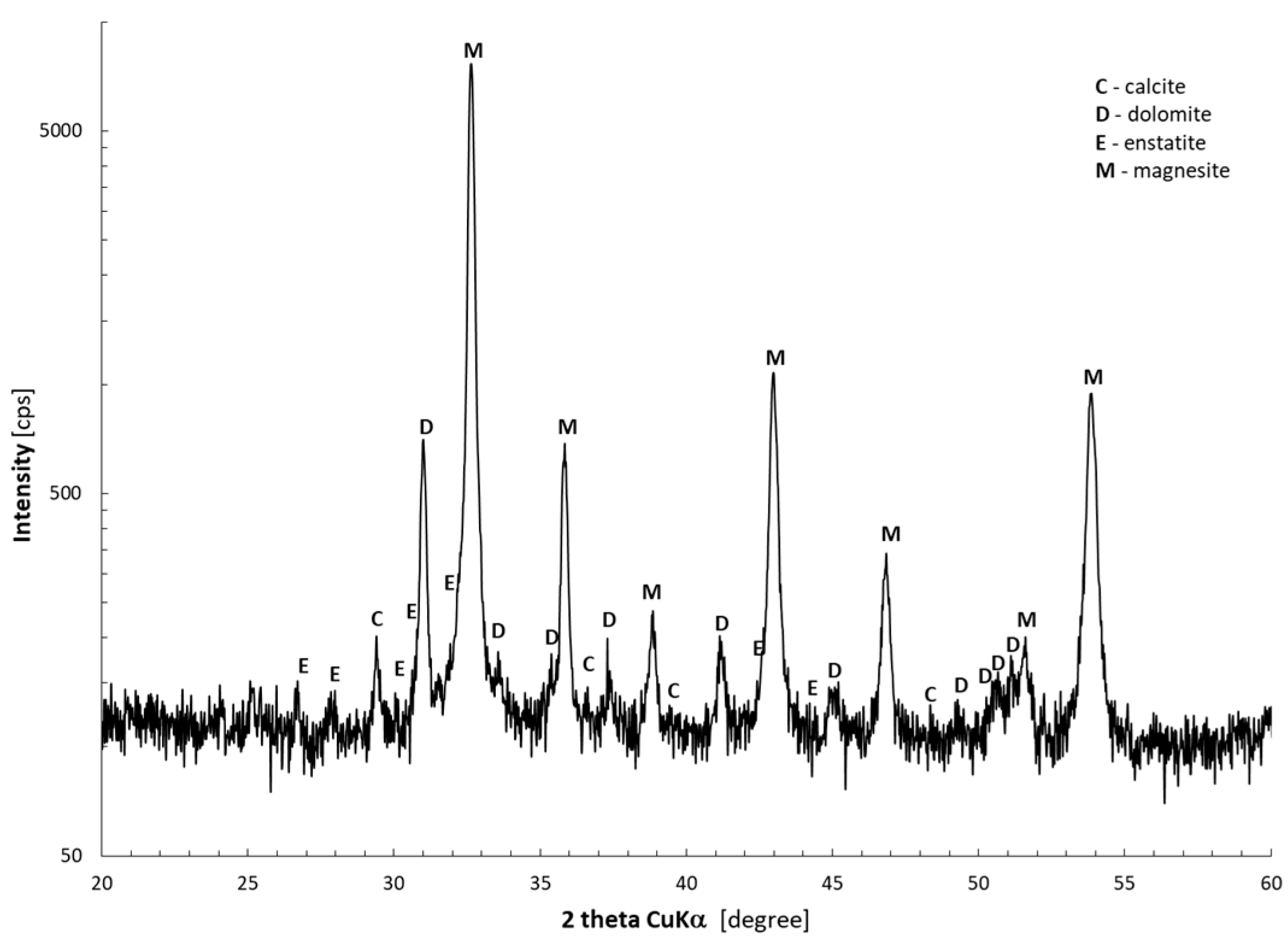
2.2. Calcination of Magnesite
2.3. Batch Precipitation Tests
3. Results and Discussion
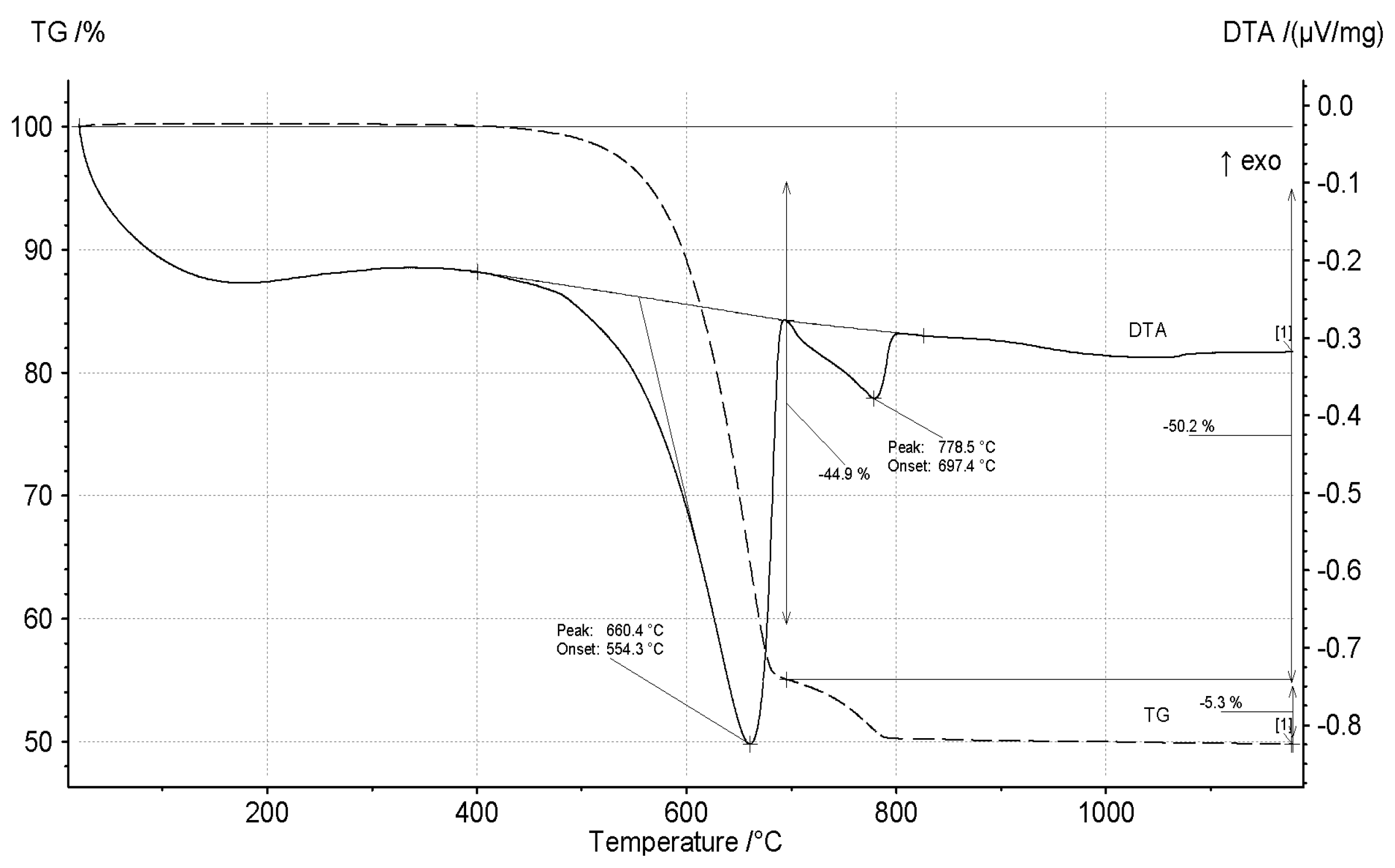
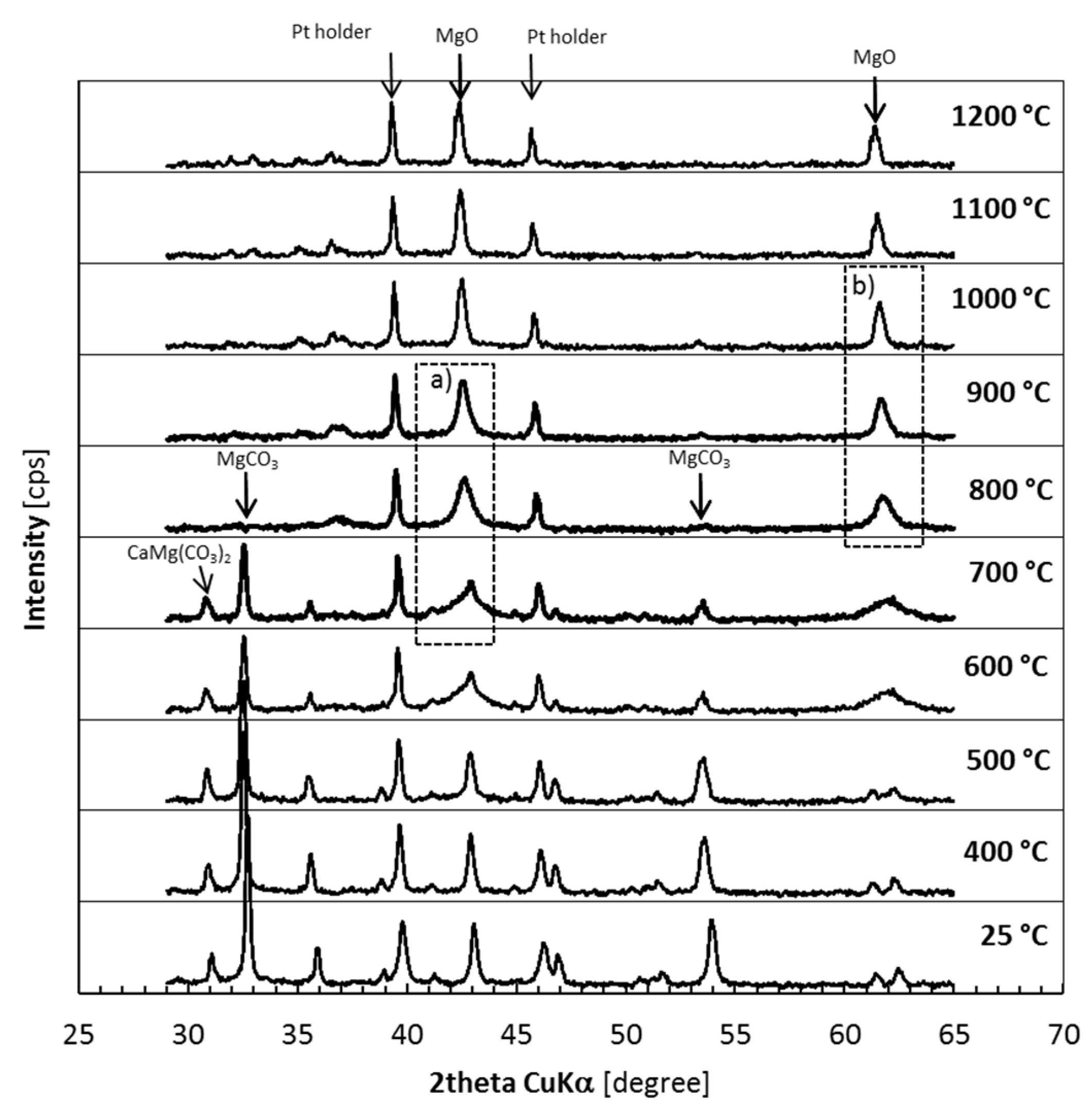
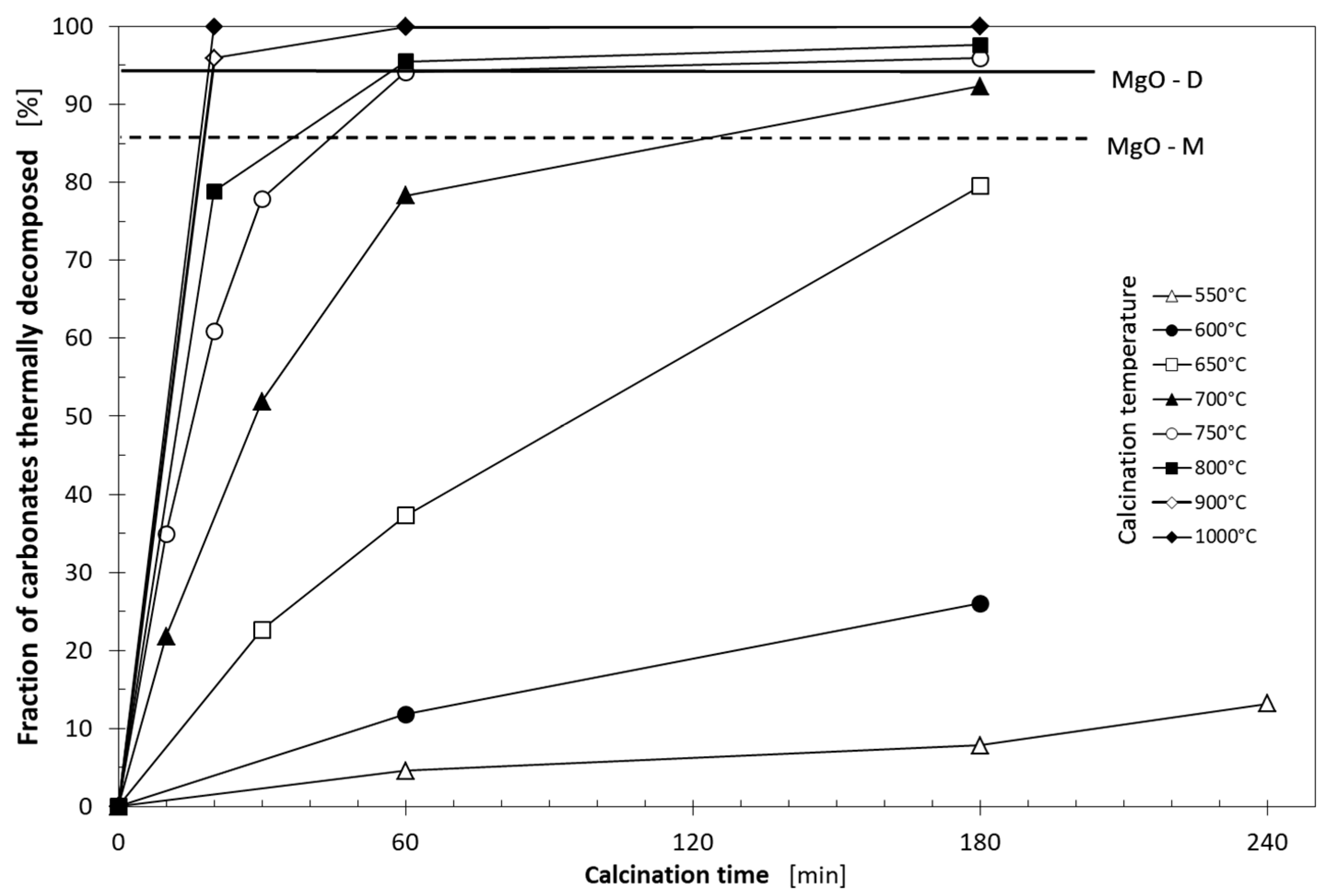
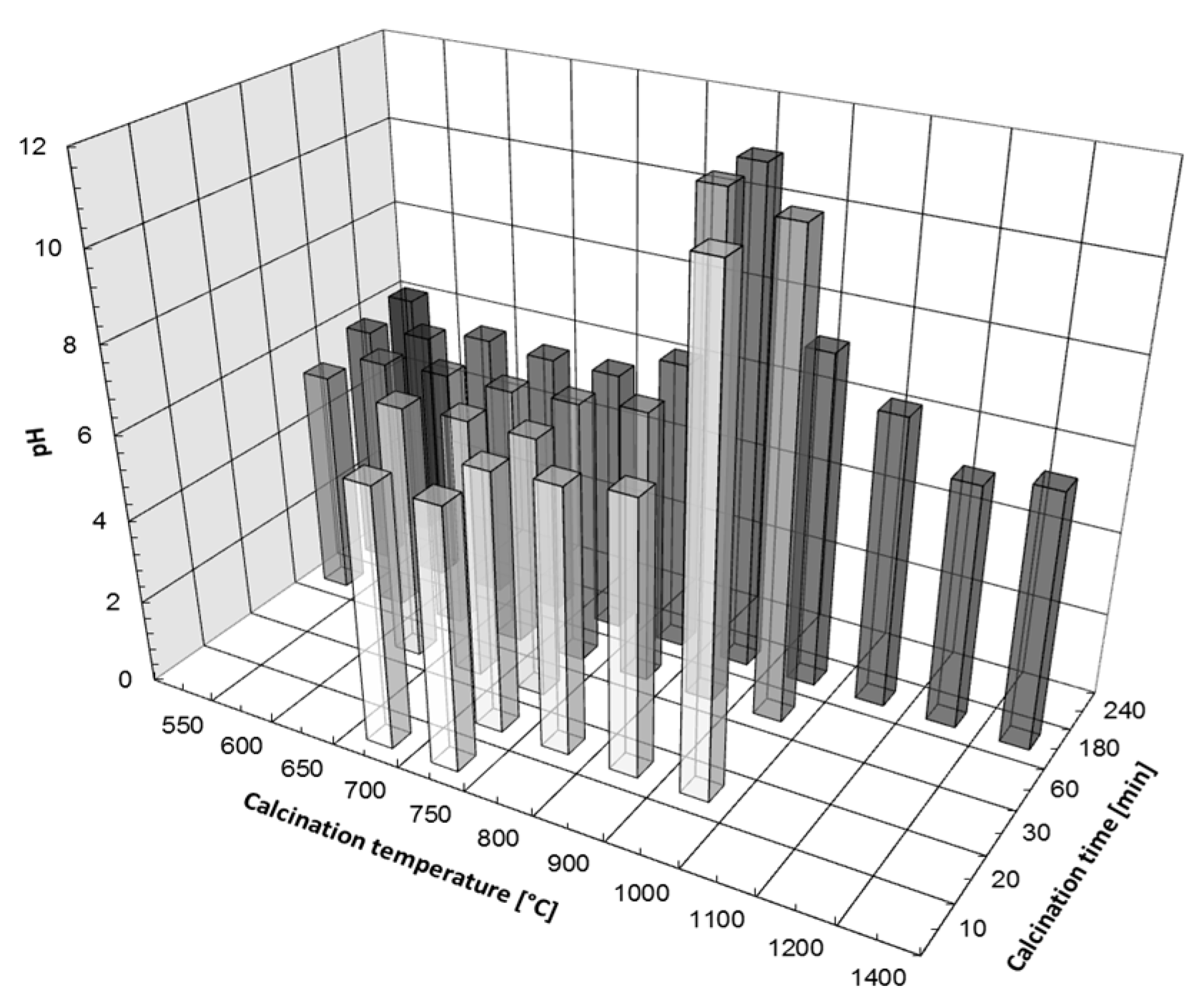
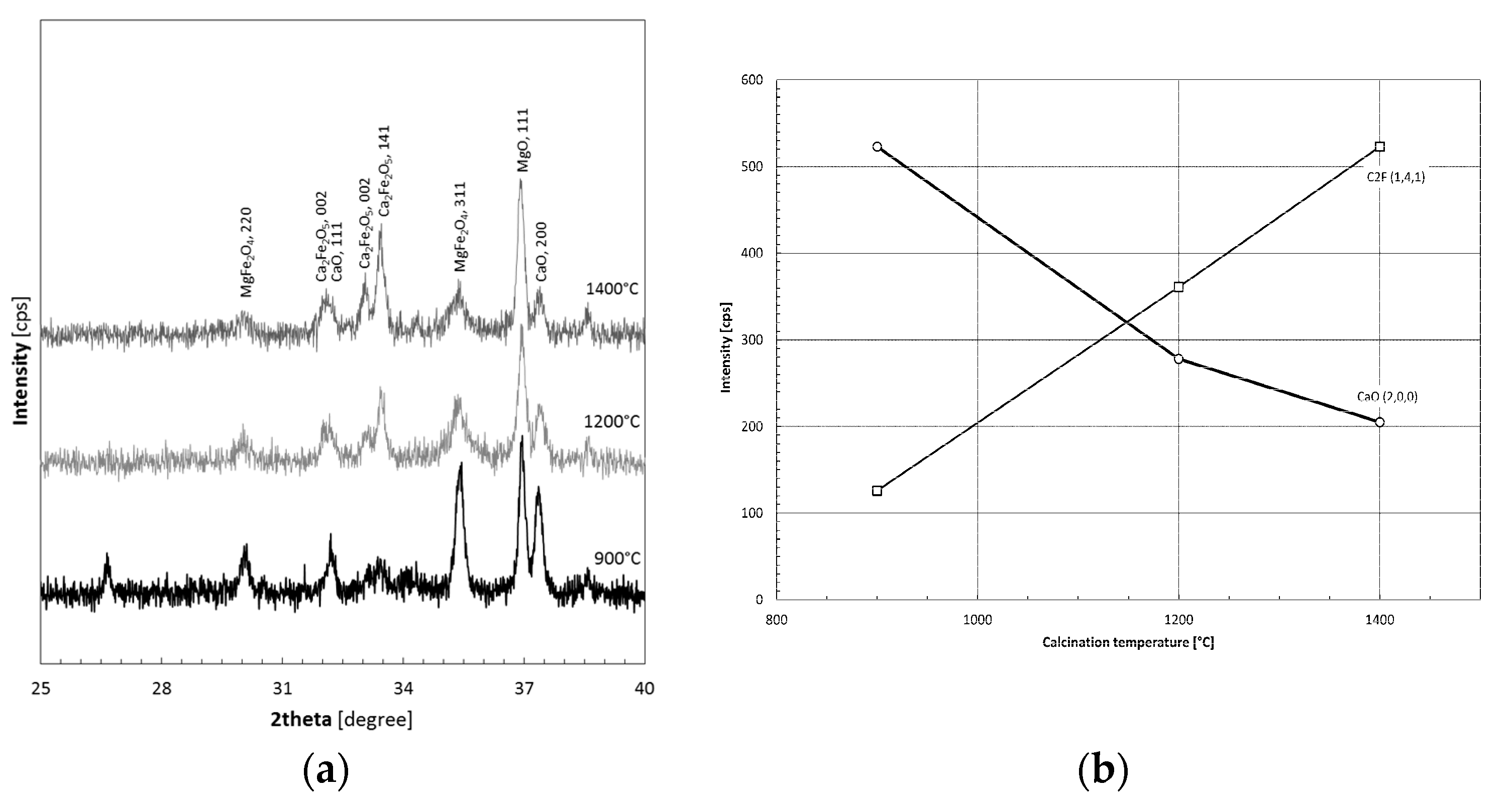
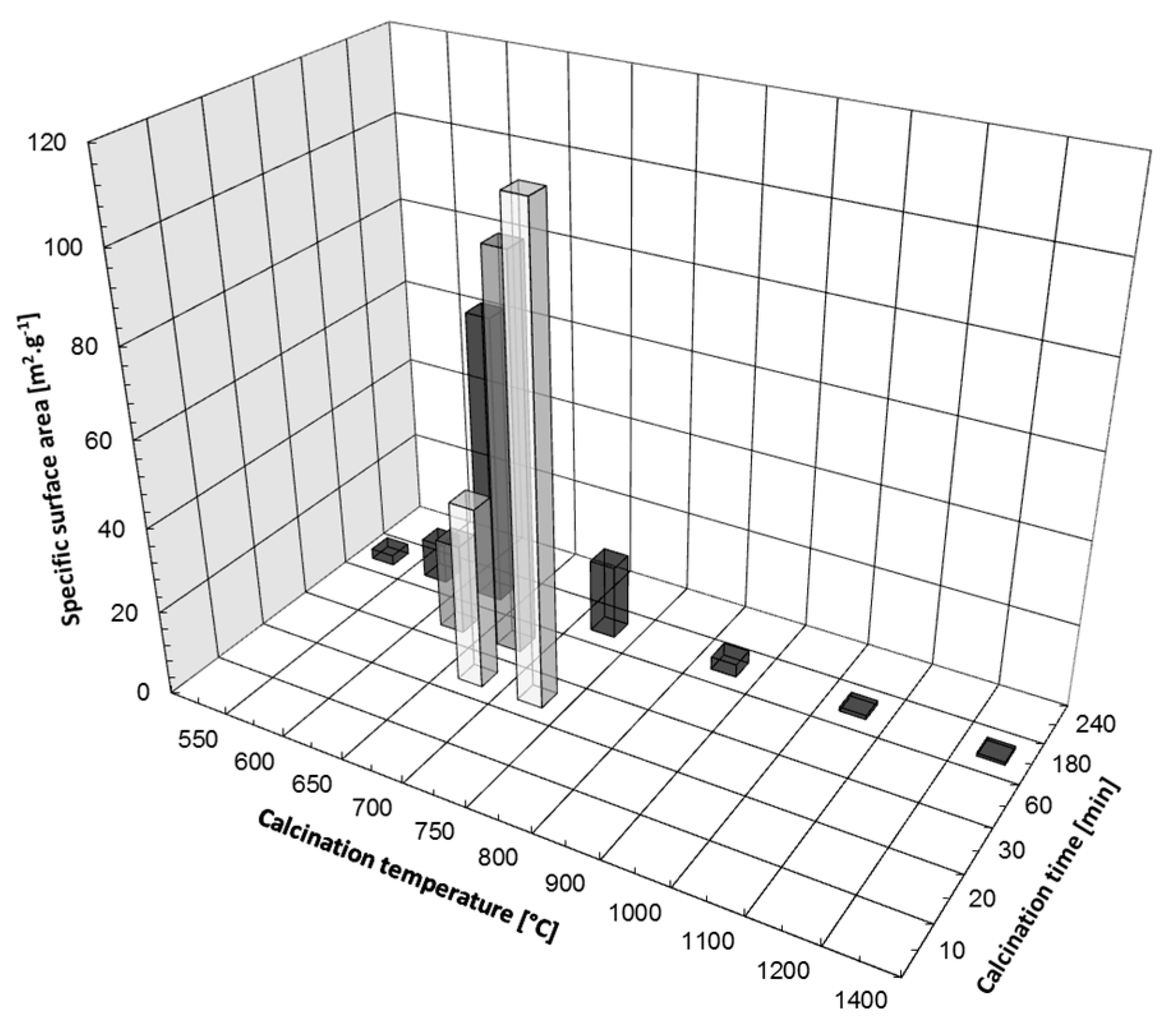
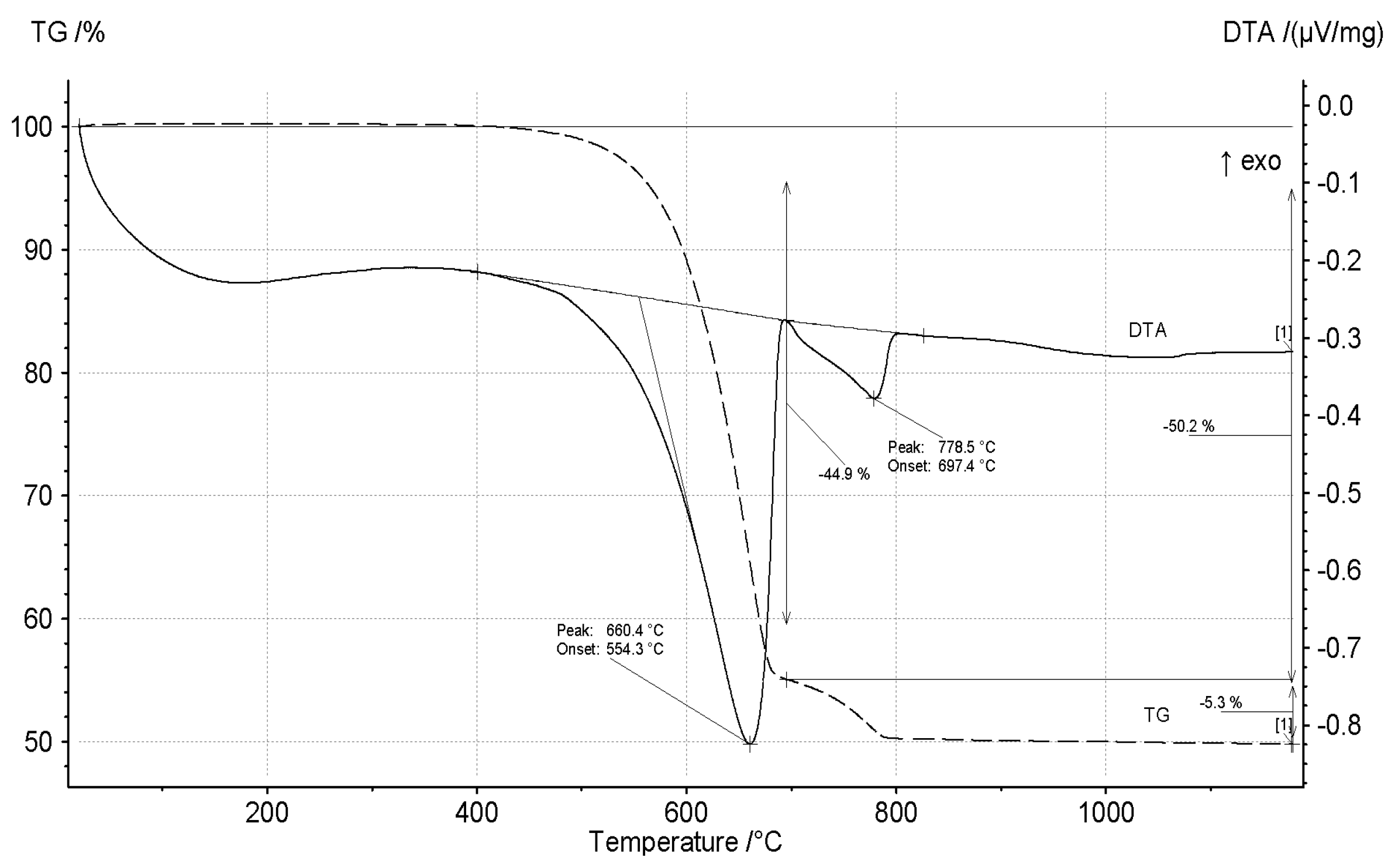
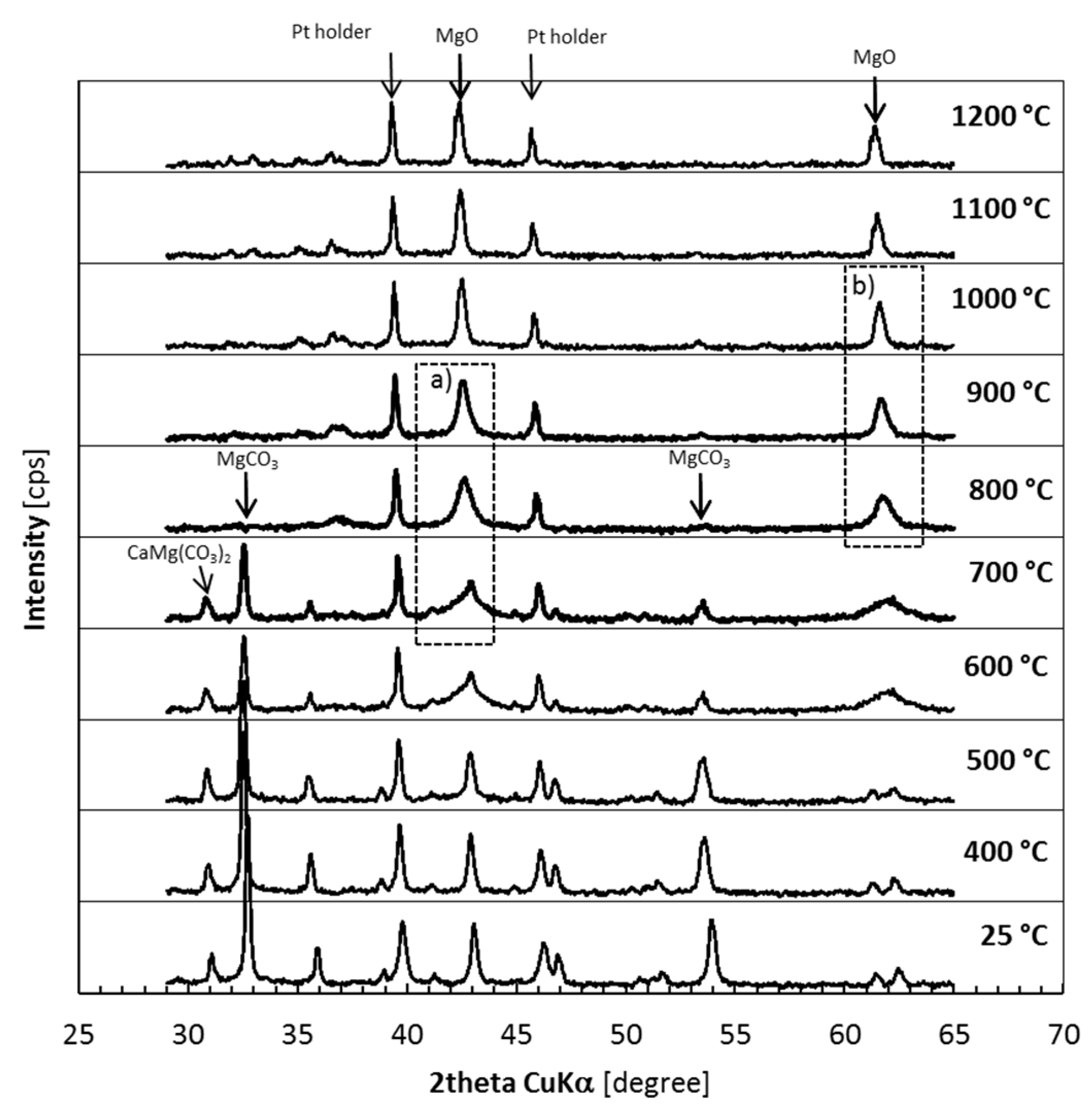
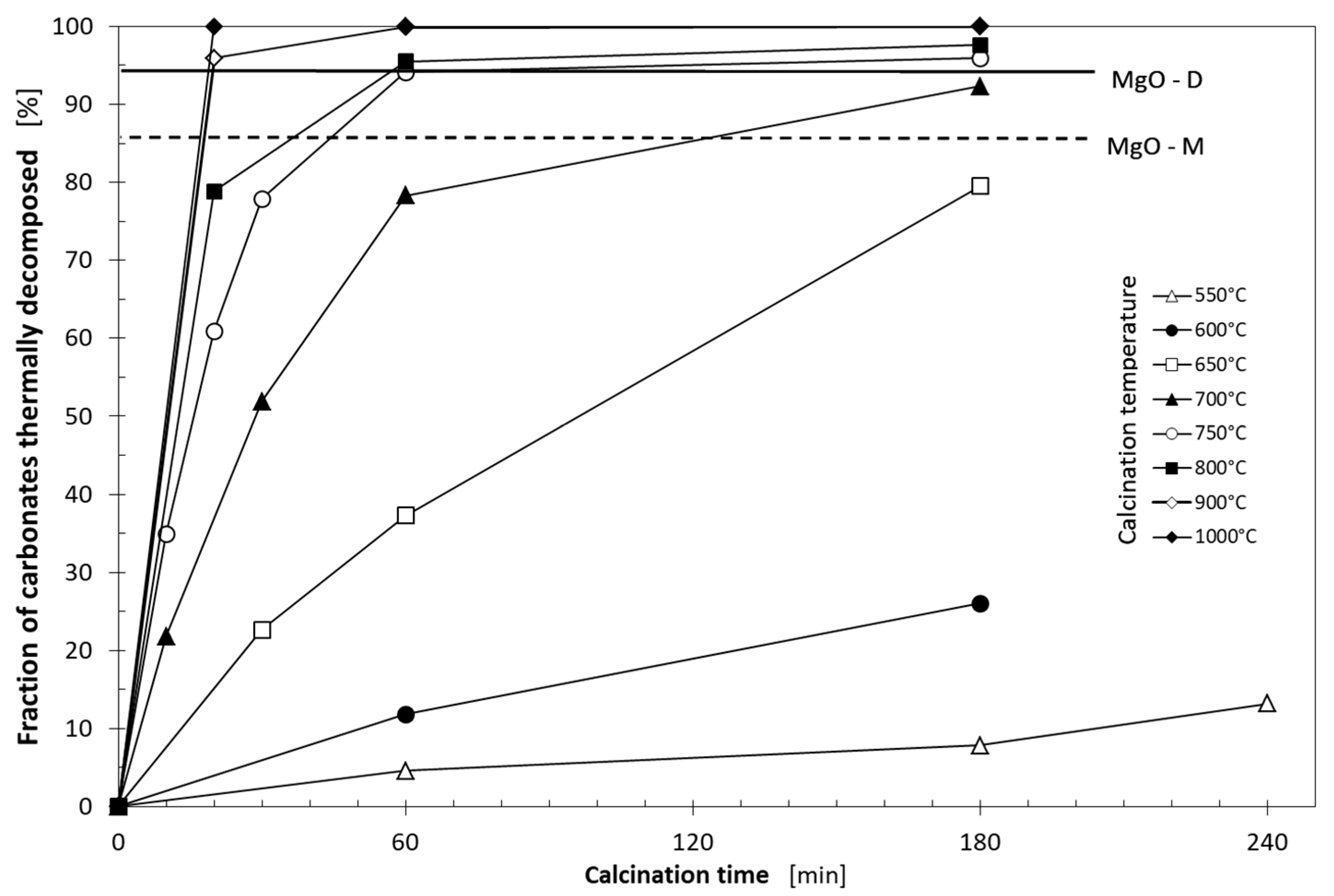
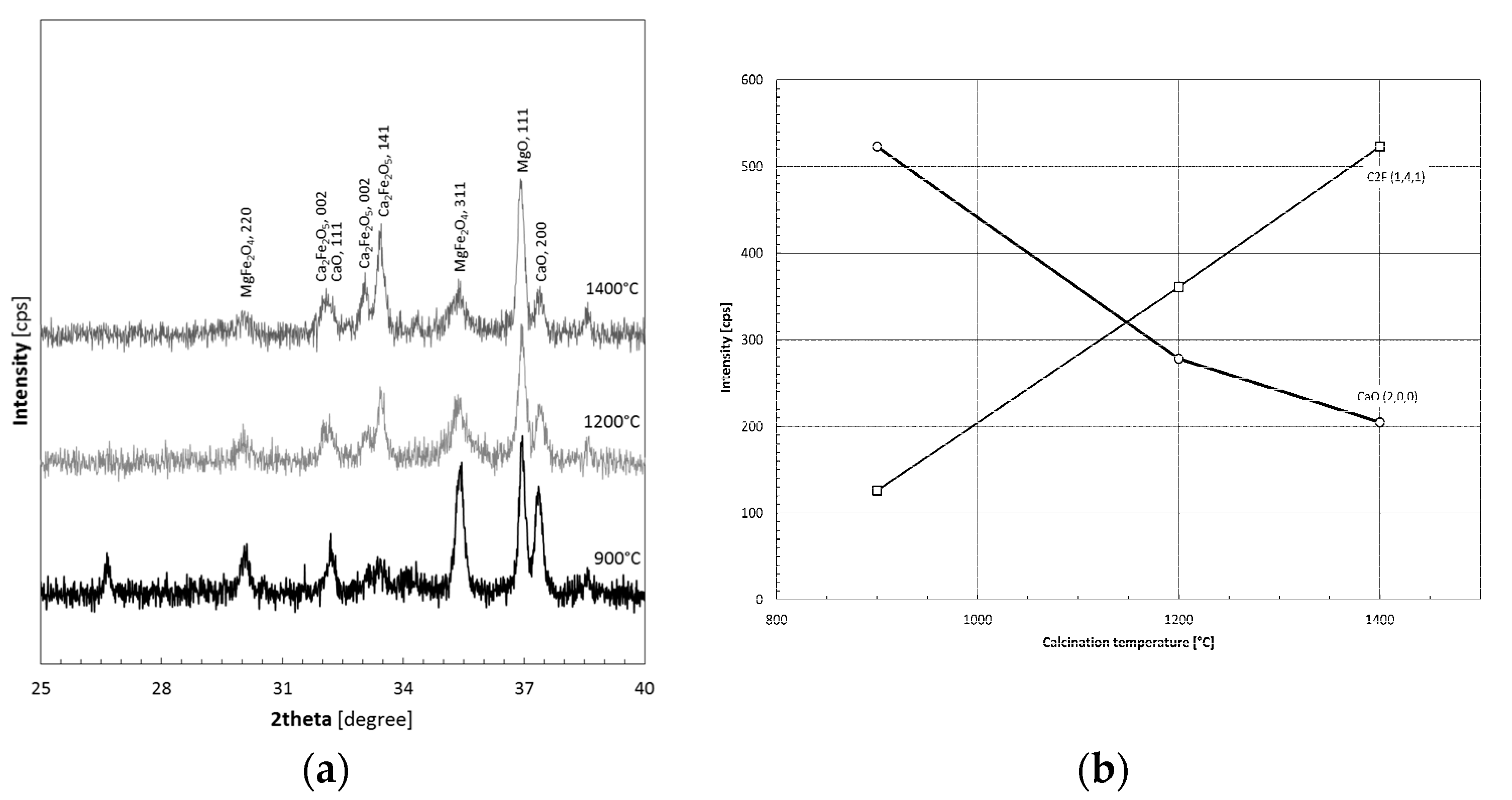
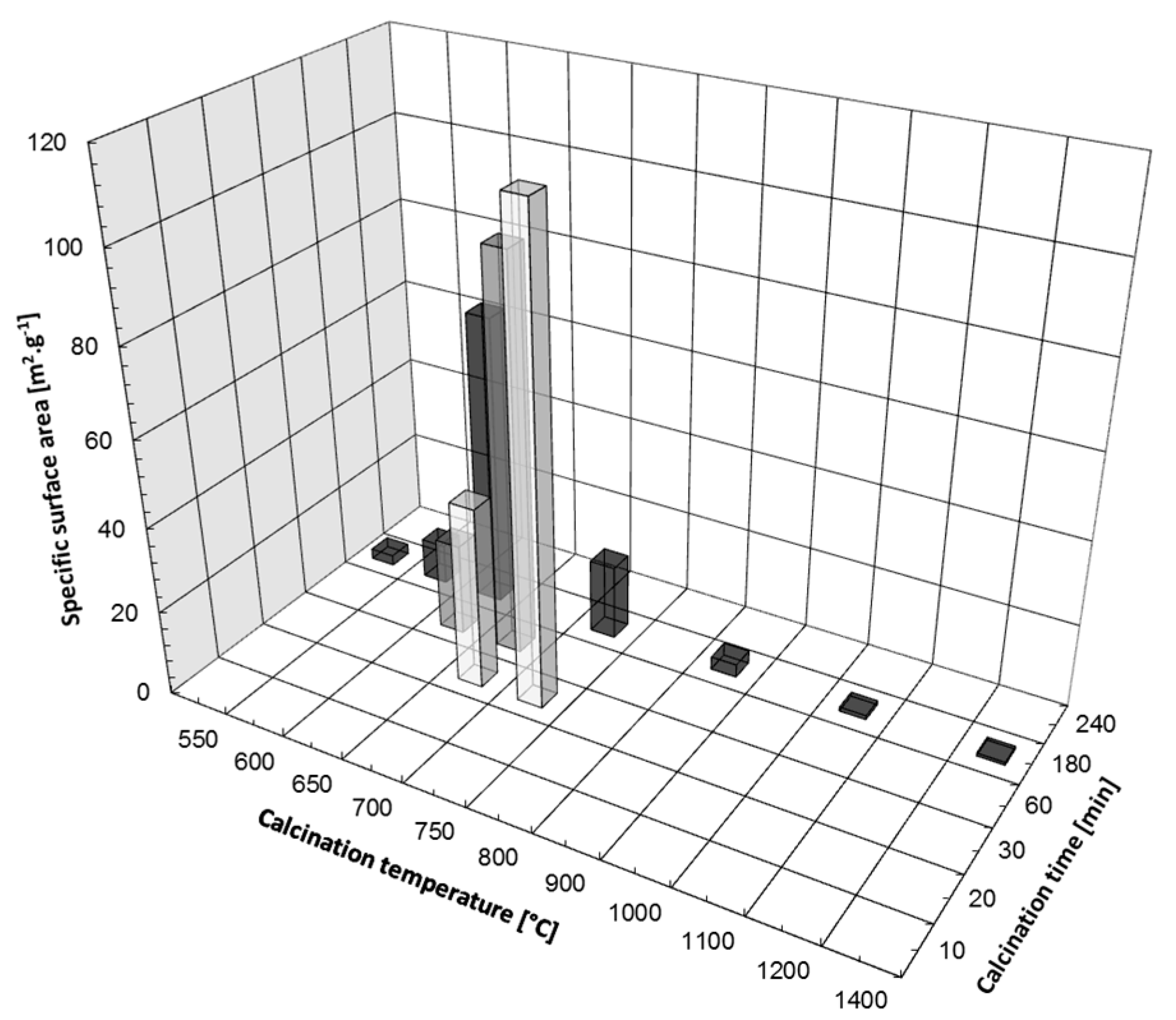
3.1. Thermal Decomposition of Magnesite
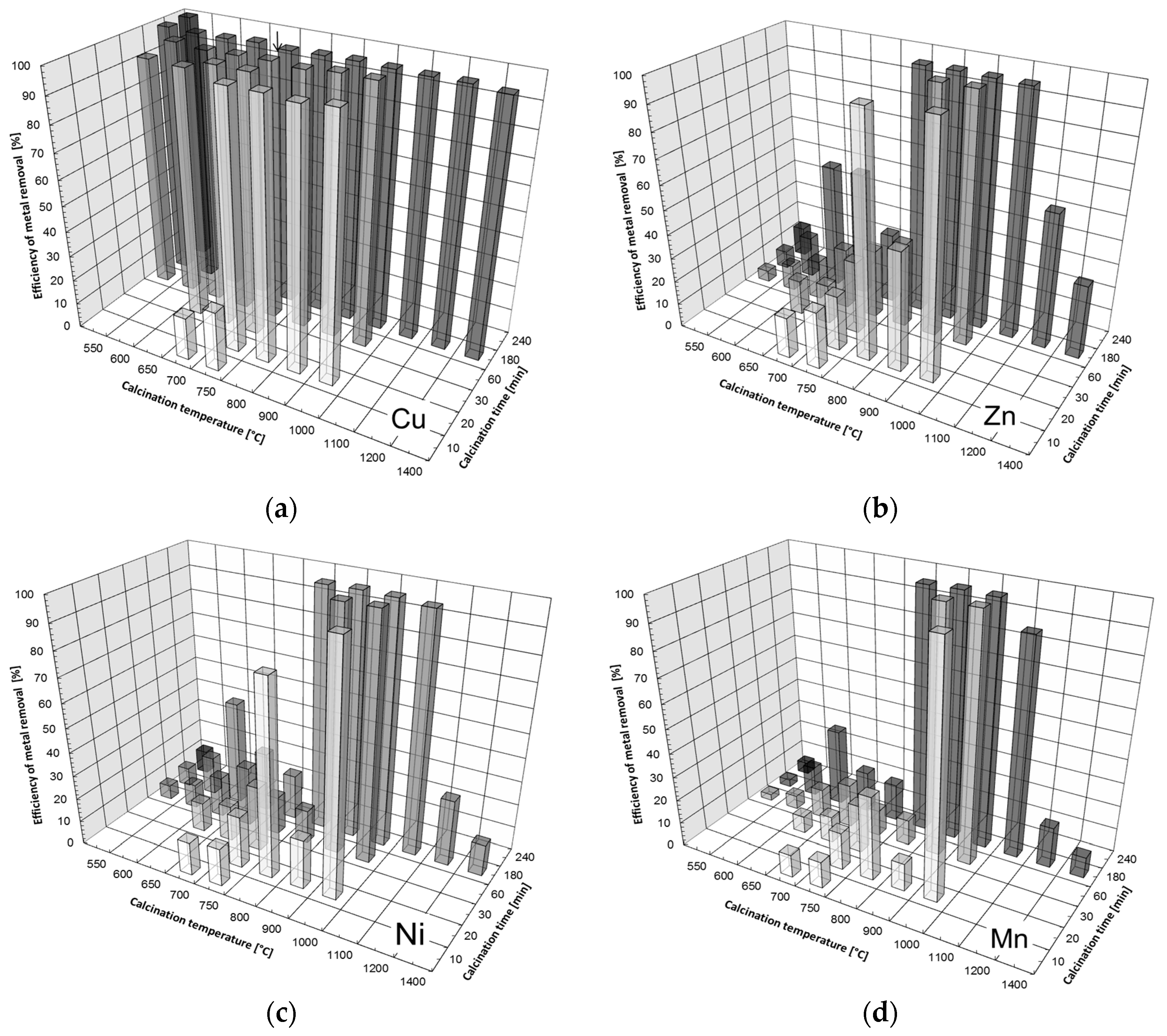
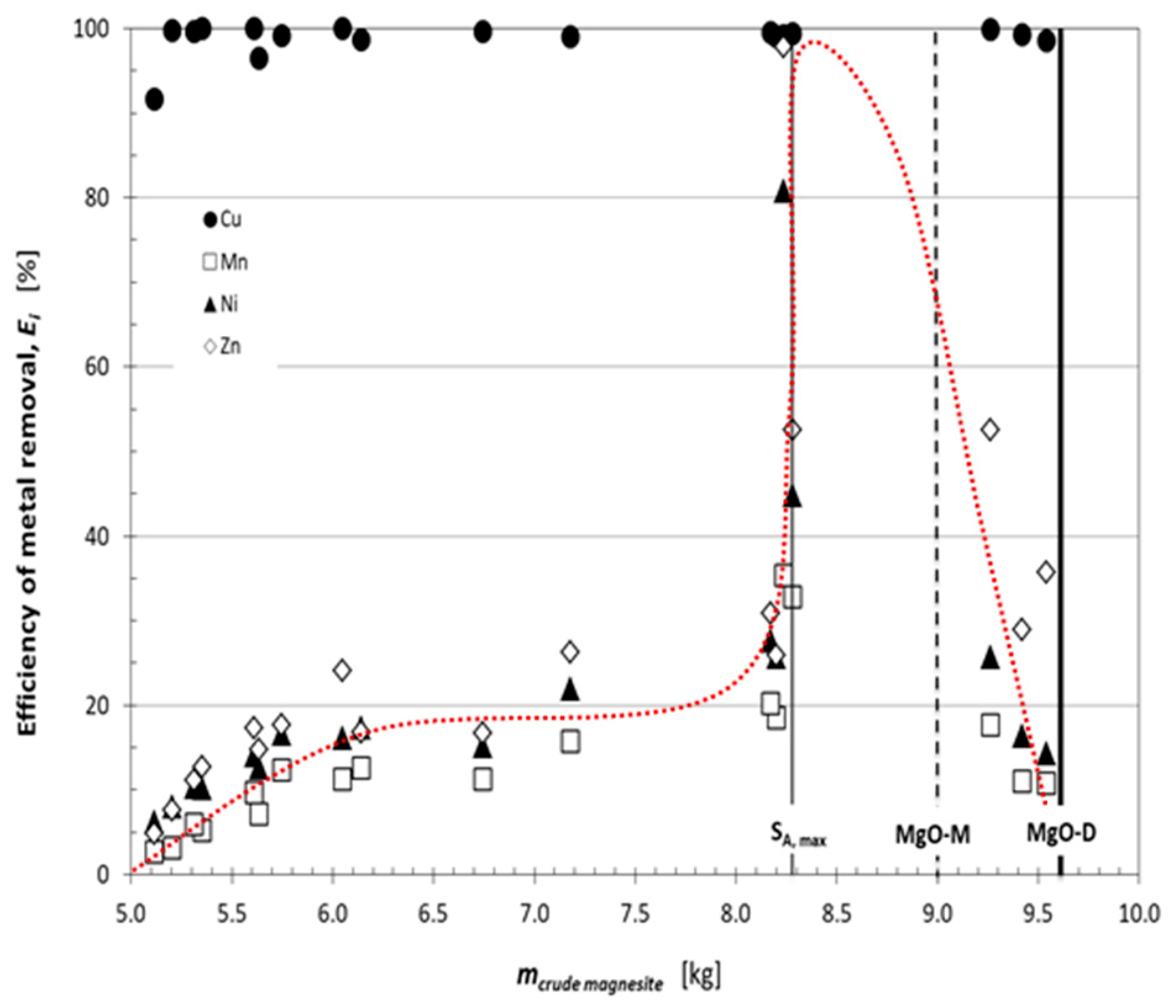
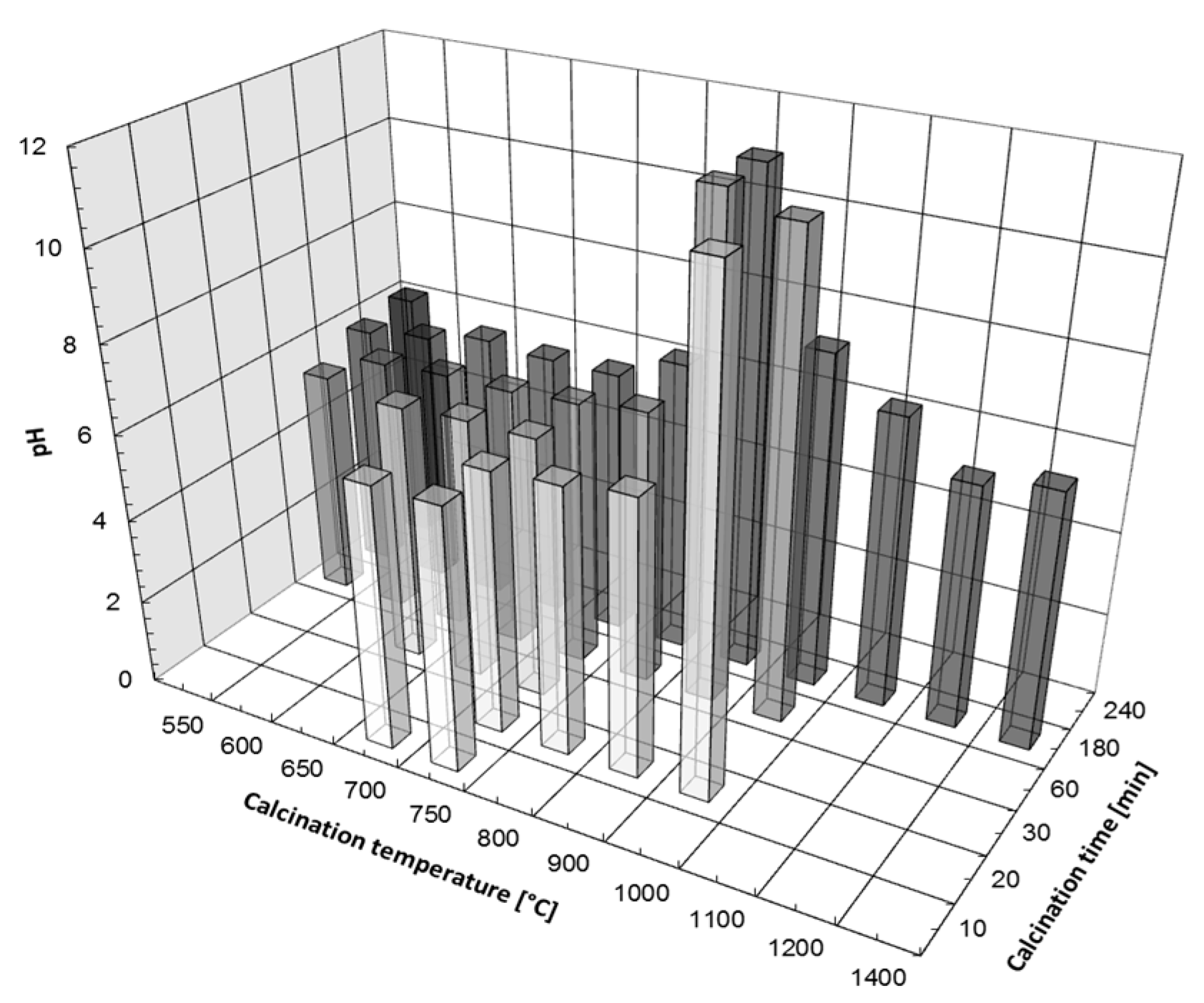
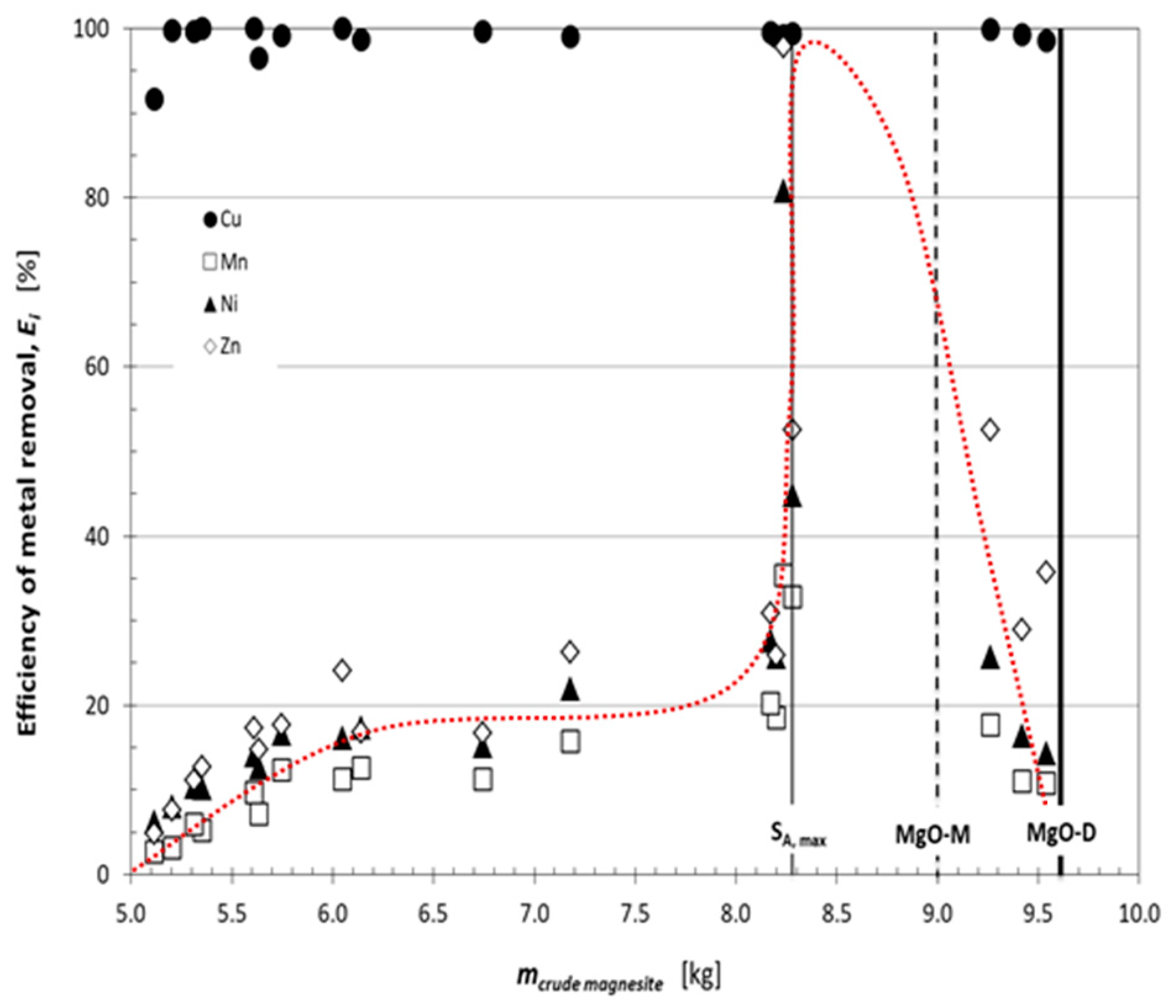
3.2. Removal of Heavy Metals by Precipitation Using the CCM
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CCM | caustic calcined magnesia |
| PRB | permeable reactive barrier |
| HM | heavy metals |
| DAS | dispersed alkaline substrate, i.e., mixture of a fine-grained caustic calcined magnesia and coarse inert matrix (e.g., wood shavings or gravel) |
| Ei | efficiency of individual heavy metals removal from solution (%) |
| Xt | fraction of carbonates thermally decomposed during the raw magnesite heating (%) |
References
- Permeable Reactive Barriers for Inorganic and Radionuclide Contamination. Kate Bronstein National Network of Environmental Management Studies Fellow for U.S. Environmental Protection Agency Office of Solid Waste and Emergency Response Office of Superfund Remediation and Technology Innovation, Washington, DC, August 2005. Available online: https://clu-in.org/download/studentpapers/bronsteinprbpaper.pdf/ (accessed on 30 November 2016).
- Thiruvenkatachari, R.; Vigneswaran, S.; Naidu, R. Permeable reactive barrier for groundwater remediation. J. Ind. Eng. Chem. 2008, 14, 145–156. [Google Scholar] [CrossRef]
- Fu, F.; Wang, Q. Removal of heavy metal ions from wastewaters: A review. J. Environ. Manag. 2011, 92, 407–418. [Google Scholar] [CrossRef]
- Johnson, D.B.; Hallberg, K.B. Acid mine drainage remediation options: A review. Sci. Total Environ. 2005, 338, 3–14. [Google Scholar] [CrossRef]
- Barbooti, M.M.; Abid, B.A.; Al-Shuwaiki, N.M. Removal of heavy metals using chemicals precipitation. Eng. Tech. J. 2011, 29, 595–612. [Google Scholar]
- Taylor, J.; Pape, S.; Murphy, N. A Summary of Passive and Active Treatment Technologies for Acid and Metalliferous Drainage (AMD), Fifth Australian Workshop on Acid Drainage, 29–31 August 2005 Fremantle, Western Australia. The Australian Centre for Minerals Extension and Research (ACMER), Earth Systems PTY Ltd., Australian Business Number 29 006 227 532. Available online: http://www.earthsystems.com.au/wp-content/uploads/2012/02/AMD_Treatment_Technologies_06.pdf/ (accessed on 30 November 2016).
- Blais, J.F.; Djedidi, Z.; Ben Cheikh, R.; Tyagi, R.D.; Mercier, G. Metals precipitation from effluents: Review. Pract. Period. Hazard. Toxic Radioact. Waste Manag. 2008, 12, 135–149. [Google Scholar] [CrossRef]
- Cantor, K.P. Drinking water and cancer. Cancer Causes Control. 1997, 8, 292–308. [Google Scholar] [CrossRef] [PubMed]
- Calderon, R.L. The epidemiology of chemical contaminants of drinking water. Food Chem. Toxicol. 2000, 38, 13–20. [Google Scholar] [CrossRef] [Green Version]
- Kavcar, P.; Sofuoglu, A.; Sofuoglu, S.C. A health risk assessment for exposure to trace metals via drinking water ingestion pathway. Int. J. Hyg. Environ. Health 2009, 212, 216–227. [Google Scholar] [CrossRef] [Green Version]
- Kelly, B.C.; Myo, A.N.; Pi, N.; Bayen, S.; Leakhena, P.C.; Chou, M.; Tan, B.H. Human exposure to trace elements in central Cambodia: Influence of seasonal hydrology and food-chain bioaccumulation behaviour. Ecotox. Environ. Saf. 2018, 162, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Kang, H.; Zhang, X.; Shao, H.; Chu, L.; Ruan, C. A critical review on the bio-removal of hazardous heavy metals from contaminated soils: Issues, progress, eco-environmental concerns and opportunities. J. Hazard. Mater. 2010, 174, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Nthunya, L.N.; Masheane, M.L.; George, M.; Kime, M.B.; Mhlanga, S.D. Removal of Fe and Mn from polluted water sources in Lesotho using modified clays. J. Water Chem. Technol. 2019, 41, 81–86. [Google Scholar] [CrossRef]
- Yang, S.; Li, J.; Shao, D.; Hu, J.; Wang, X. Adsorption of Ni (II) on oxidized multi-walled carbon nanotubes: Effect of contact time, pH, foreign ions and PAA. J. Hazard. Mater. 2009, 166, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Rehman, I.; Ishaq, M.; Ali, L.; Khan, S.; Ahmad, I.; Ud Din, I.; Ullah, H. Enrichment, spatial distribution of potential ecological and human healthrisk assessment via toxic metals in soil and surface water ingestion in the vicinity of Sewakht mines, district Chitral, Northern Pakistan. Ecotox. Environ. Saf. 2018, 154, 127–136. [Google Scholar] [CrossRef]
- Sani, H.A.; Ahmad, M.B.; Hussein, M.Z.; Ibrahim, N.A.; Musa, A.; Saleh, T.A. Nanocomposite of ZnO with montmorillonite for removal of lead and copper ions from aqueous solutions. Process Saf. Environ. Protect. 2017, 109, 97–105. [Google Scholar] [CrossRef]
- Gibert, O.; De Pablo, J.; Cortina, J.L.; Ayora, C. In situ removal of arsenic from groundwater by using permeable reactive barriers of organic mater/limestone/zero-valent iron mixtures. Environ. Geochem. Health 2010, 32, 373–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rötting, T.S.; Ayora, C.; Carrera, J. Improved passive treatment of high Zn and Mn concentrations using caustic magnesia (MgO): Particle size effects. Environ. Sci. Technol. 2008, 42, 9370–9377. [Google Scholar] [CrossRef] [PubMed]
- Simon, F.; Meggyes, T. Removal of organic and inorganic pollutants from groundwater using permeable reactive barriers. Land Contam. Reclamat. 2000, 8, 103–116. [Google Scholar]
- Hashim, M.A.; Mukhopadhyay, S.; Sahu, J.N.; Sengupta, B. Remediation technologies for heavy metal contaminated ground water. J. Environ. Manag. 2011, 92, 2355–2388. [Google Scholar] [CrossRef] [PubMed]
- Permeable Reactive Barriers: Lessons Learned/New Directions. Interstate Technology & Regulatory Council: Washington, DC, USA, 2005; p. 9. Available online: http://www.itrcweb.org/GuidanceDocuments/PRB-4.pdf/ (accessed on 30 November 2016).
- Lee, M.K.; Saunders, J.A. Effects of pH on metals precipitation and sorption: Field bioremediation and geochemical modeling approaches. Vadose Zone J. 2003, 2, 177–185. [Google Scholar]
- Obiri-Nyarko, F.; Grajales-Mesa, S.J.; Malina, G. An overview of permeable reactive barriers for in situ sustainable groundwater remediation. Chemosphere 2014, 111, 243–259. [Google Scholar] [CrossRef]
- Army Corps. Engineering Manuals, Manual No. 1110-1-4012: Engineering and Design Precipitation/Coagulation/Flocculation, Department of the Army U.S. Army Corps of Engineers, 2001, Washington, DC 20314-1000. Available online: http://www.publications.usace.army.mil/Portals/76/Publications/EngineerManuals/EM_1110-1-4012.pdf/ (accessed on 30 November 2016).
- Peters, R.W.; Shem, L. Separation of Heavy Metals: Removal from Industrial Wastewaters and Contaminated Soil. In Proceedings of the Symposium on Emerging Separation Technologies for Metals and Fuels, Palm Coast, FL, USA, 13–18 March 1993; Available online: http://www.osti.gov/scitech/servlets/purl/6504209-mKij3S/ (accessed on 30 November 2016).
- Sasaki, K.; Moriyama, S. Effect of calcination temperature for magnesite on interaction of MgO-rich phases with boric acid. Ceram. Int. 2014, 40, 1651–1660. [Google Scholar]
- Barakat, M.A. New trends in removing heavy metals from industrial wastewater. Arab. J. Chem. 2011, 4, 361–377. [Google Scholar] [CrossRef] [Green Version]
- Skousen, J. Anoxic limestone drains for acid mine drainage treatment. Green Lands 1991, 21, 30–35. [Google Scholar]
- Golab, A.N.; Peterson, M.A.; Indraratna, B. Selection of potential reactive materials for a permeable reactive barrier for remediating acidic groundwater in acid sulphate soil terrains. Q. J. Eng. Geol. Hydrogeol. 2006, 39, 209–223. [Google Scholar] [CrossRef] [Green Version]
- Akcil, A.; Koldas, S. Acid mine drainage (AMD): Causes, treatment and case studies. J. Clean. Prod. 2006, 14, 1139–1145. [Google Scholar] [CrossRef]
- Cortina, J.L.; Lagreca, I.; De Pablo, J. Passive in situ remediation of metal-polluted water with caustic magnesia: Evidence from column experiments. Environ. Sci. Technol. 2003, 37, 1971–1977. [Google Scholar] [CrossRef] [PubMed]
- Rötting, T.; Cama, J.; Ayora, C.; Cortina, J.L.; De Pablo, J. The Use of Caustic Magnesia to Remove Cadmium from Water. In Proceedings of the 9th International Mine Water Association Congress, Oviedo, Spain, 5–7 September 2005; pp. 635–639. Available online: http://www.imwa.info/docs/imwa_2005/IMWA2005_090_Roetting.pdf/ (accessed on 30 November 2016).
- Caraballo, M.A.; Rötting, T.S.; Silva, V. Implementation of an MgO-based metal removal step in the passive treatment system of Shilbottle, UK: Column experiments. J. Hazard. Mater. 2010, 181, 923–930. [Google Scholar] [CrossRef]
- Macías, F.; Caraballo, M.A.; Nieto, J.M.; Rötting, T.S.; Ayora, C. Natural pretreatment and passive remediation of highly polluted acid mine drainage. J. Environ. Manag. 2012, 104, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Caraballo, M.A.; Rötting, T.S.; Macías, F.; Nieto, J.M.; Ayora, C. Field multi-step limestone and MgO passive system to treat acid mine drainage with high metal concentrations. Appl. Geochem. 2009, 24, 2301–2311. [Google Scholar] [CrossRef]
- Lin, X.; Burns, R.C.; Lawrance, G.A. Heavy metals in wastewater: The effect of electrolyte composition on the precipitation of cadmium (II) using lime and magnesia. Water Air Soil Poll. 2005, 165, 131–152. [Google Scholar] [CrossRef]
- Sasaki, K.; Fukumoto, N.; Moriyama, S.; Hirajima, T. Sorption characteristics of fluoride on to magnesium oxide-rich phases calcined at different temperatures. J. Hazard. Mater. 2011, 191, 240–248. [Google Scholar] [CrossRef]
- Moore, R.C.; Anderson, D.R. System for Removal of Arsenic from Water. U.S. Patent 6,821,434B1, 23 November 2004. [Google Scholar]
- Navarro, A.; Chimenos, J.; Muntaner, D.; Fernández, I. Permeable reactive barriers for the removal of heavy metals: Lab-scale experiments with low-grade magnesium oxide. Ground Water Monit. Remediat. 2006, 26, 142–152. [Google Scholar] [CrossRef]
- Rotting, T.S.; Cama, J.; Ayora, C.; Cortina, J.L.; De Pablo, J. Use of caustic magnesia to remove cadmium, nickel, and cobalt from water in passive treatment systems: Column experiments. Environ. Sci. Technol. 2006, 40, 6438–6443. [Google Scholar] [CrossRef] [PubMed]
- Ayora, C.; Caraballo, M.A.; Macías, F.; Rötting, T.S.; Carrera, J.; Nieta, J.M. Acid mine drainage in the Iberian Pyrite Belt: 2. Lessons learned from recent passive remediation experiences. Environ. Sci. Pollut. Res. 2013, 20, 7837–7853. [Google Scholar] [CrossRef]
- Macías, F.; Caraballo, M.A.; Rötting, T.S.; Pérez-López, R.; Nieto, J.M.; Ayora, C. From highly polluted Zn-rich acid mine drainage to non-metallic waters: Implementation of a multi-step alkaline passive treatment system to remediate metal pollution. Sci. Total Environ. 2012, 433, 323–330. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Element | Mg | Fe | Ca | Si | Al | Loss on Ignition (L.O.I.) |
|---|---|---|---|---|---|---|
| Weight % | 25.1 | 3.0 | 2.3 | 0.14 | 0.10 | 50.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fedoročková, A.; Raschman, P.; Sučik, G.; Švandová, M.; Doráková, A. Reactive, Sparingly Soluble Calcined Magnesia, Tailor-Made as the Reactive Material for Heavy Metal Removal from Contaminated Groundwater Using Permeable Reactive Barrier. Minerals 2021, 11, 1153. https://doi.org/10.3390/min11111153
Fedoročková A, Raschman P, Sučik G, Švandová M, Doráková A. Reactive, Sparingly Soluble Calcined Magnesia, Tailor-Made as the Reactive Material for Heavy Metal Removal from Contaminated Groundwater Using Permeable Reactive Barrier. Minerals. 2021; 11(11):1153. https://doi.org/10.3390/min11111153
Chicago/Turabian StyleFedoročková, Alena, Pavel Raschman, Gabriel Sučik, Mária Švandová, and Agnesa Doráková. 2021. "Reactive, Sparingly Soluble Calcined Magnesia, Tailor-Made as the Reactive Material for Heavy Metal Removal from Contaminated Groundwater Using Permeable Reactive Barrier" Minerals 11, no. 11: 1153. https://doi.org/10.3390/min11111153