
Occurrence and Distribution of Antibiotic Resistance Genes in Municipal Wastewater Treatment Plants with D-Type Filters


1
School of Environment Science and Engineering, Suzhou University of Science and Technology, Suzhou 215009, China
2
State Key Joint Laboratory of Environment Simulation and Pollution Control, School of Environment, Tsinghua University, Beijing 100084, China
3
School of Energy and Environmental Engineering, University of Science and Technology Beijing, Beijing 100083, China
*
Authors to whom correspondence should be addressed.
Water 2021, 13(23), 3398; https://doi.org/10.3390/w13233398
Submission received: 9 October 2021
/
Revised: 14 November 2021
/
Accepted: 24 November 2021
/
Published: 2 December 2021
(This article belongs to the Section Water and One Health)
Abstract
:Filters are popularly used in municipal wastewater treatment plants (WWTPs) as the final guards against effluent solids; however, their impacts on antibiotic resistance gene (ARG) removal in the WWTPs are still unclear. In this study, metagenomic analysis was used to find out the distribution characteristics of ARGs in two WWTPs equipped with the same D-Type fiber filters. Samples of influent, activated sludge liquor, secondary clarifier effluent, and D-Type filter effluent were found to host 695, 609, 675, and 643 ARG subtypes, respectively. The detected ARGs mainly included macB (4.1–8.9%), sav1866 (1.7–3.4%), and oleC (1.6–3.8%). Co-occurrence network analysis combined with contribution analysis helped to identify the ARG-related risks in the samples. Microbacterium, Acinetobacter, Gordonia, and Streptomyces significantly correlated with more than ten kinds of ARG subtypes, implying that they are potential hosts for these resistance gene subtypes. The number of ARG subtypes in the D-Type filter was less than those in the secondary clarifier effluent, indicating the potential of D-Type filters to effectively reduce the ARGs released into the environment. However, the abundance of two pathogens, Mycobacterium and PmrA, increased after the treatment by the D-Type filter, which may reveal the adverse effects of intercepting ARGs inside the fibers. The results may help the understanding of the complex role of the D-Type fiber filter on ARG distribution in WWTPs.
1. Introduction
As emerging contaminants, antibiotic resistance genes (ARGs) have received wide attention in recent years. Pathogenic bacteria that acquire ARGs will pose a greater threat to human health [1,2]. Many ARG subtypes have been detected in environmental samples, especially in wastewater treatment plants (WWTPs) [3,4,5,6]. As a centralized treatment place for wastewater from residential, medical, and aquaculture, WWTPs are considered the main reservoirs of ARGs. ARGs belonging to types of Quinolones, β-lactam, sulfonamide, tetracycline, and macrolide antibiotics were widely detected in the influent, activated sludge and the supernatant, biofilm, effluent, and aerosol of WWTPs [7,8,9,10]. Moreover, ARGs can be transferred between bacteria through self-proliferation and horizontal gene transfer (HGT), and the rich nutrients and a huge number of microorganisms in activated sludge are beneficial for ARG transfer [11,12]. Therefore, understanding the distribution characteristics of ARGs in WWTPs is important for risk control.
The abundance of ARGs in wastewater depends on the abundance of microorganisms [13]. Therefore, any measure that can greatly reduce the bacterial biomass in wastewater can also effectively reduce the abundance of ARGs in wastewater [13,14]. As an advanced treatment unit, the filter can effectively remove biological flocs in the wastewater. In the past, WWTPs usually used sand filters, fiber ball filters, cloth media filters, and other processes for advanced wastewater treatment. With the technology development and the higher discharge standard for wastewater treatment, new and reconstructive WWTPs began to use more cost-effective filters such as the D-Type filter. The D-Type filter is a kind of fiber filter which belongs to the micron-level filtration technology, with the filtrate diameter of the fiber cloth reaching to tens of microns or even a few microns [15]. The D-Type filter has the characteristics of a larger specific surface area and a smaller filtration resistance. The material used in the D-Type filter can increase the chance of contact between impurity particles in the water and greatly improve the filtration efficiency and interception capacity [16]. As the dense filter media in the filter may enrich drug-resistant bacteria and ARGs, it may promote the horizontal transfer of ARGs between bacteria inside. Nowadays, this kind of filter material is widely used in China, but its effects on the distribution of ARGs have not been carefully considered. In fact, only a few studies have reported and discussed the effects of sand filters and cloth filters on the distribution of typical ARGs [17,18]. Therefore, it is necessary to estimate the potential of D-Type filters to disseminate the ARGs in WWTPs.
With the emergence of a series of culture-free molecular biology techniques, metagenomic analysis has become one of the most effective technologies for studying ARGs in sewage treatment systems [19]. Until now, lots of studies have applied metagenomic technology to obtain ARG distribution in WWTPs [20,21,22]. Metagenomic network analysis is widely used to explore the internal connections between different objects. Studies have shown that a similar abundance trend existed between the ARGs and microbial taxa for certain specific microbial taxa carrying specific ARGs [14]. Previous studies have shown that studying the non-random co-occurrence pattern of microbial communities and ARGs through network analysis can help us explore potential carriers of ARGs [23,24].
In this study, metagenomic technology was used to investigate the ARGs and bacterial community in the influent, activated sludge, the secondary settling tank effluent, and the D-Type filter effluent in two advanced WWTPs. The D-Type filter changed the structure of the microorganisms and ARGs in the water, and the relative abundance of Mycobacterium and PmrA increased. The number of ARG subtypes in the wastewater decreased after the D-Type filter treatment, which reduced the environmental impact of the effluent from the sewage treatment plant.
2. Materials and Methods
2.1. Sample Collection and Pretreatment
Two WWTPs in Sichuan Province in China, named as CD and CN, treated municipal wastewater by similar Anaerobic-Anoxic-Oxic (AAO) processes coupled with the D-Type filter, and were selected as the target of investigation. The process flow chart and sampling point locations are shown in Figure S1. In July 2020, a sampling campaign was conducted in each WWTP. During the sampling, 1L of influent (INF), 500 ml of activated sludge liquor (ASL), 5L of secondary sedimentation tank effluent (SC), and 5L of D-Type filter effluent (DF) were collected from each of the two WWTPs. The samples from WWTP CD were abbreviated as INF1, ASL1, SC1, and DF1, and those from WWTP CN were abbreviated as INF2, ASL2, SC2, and DF2, respectively. All the samples were pretreated in the field labs of the WWTPs. The wastewater samples were filtered through 0.22 μm microfiltration membrane filters (Millipore, Billerica, MA, USA) on a Büchner Funnel equipped by a vacuum pump (Jinlong Company, China). The membrane filters with solid trapped inside were collected for additional DNA extraction. The sludge samples were centrifuged at 5000 r/min for 20 min to remove the supernatant. About 0.1 g of the condensed sludge was taken for additional DNA analysis. Before DNA extraction, all the pretreated samples, including the used membranes and sludge pellets, were stored in a refrigerator at a lower temperature, at −20 °C.
2.2. DNA Extraction and Library Construction
The DNA of the microorganisms in the samples was extracted by the EZNA® DNA Kit (Omega Bio-Tek, Norcross, GA, U.S.). Filtered membranes were cut into pieces and mixed. About 1 g of membrane pieces and 0.5 g of sludge pellets were used for DNA extraction according to the manufacturer’s instructions. The DNA quality was examined with the gel electrophoresis (1% agarose), and the concentration and purity were measured by a NanoDrop2000 spectrophotometer.
After fragmentation, the NEXTFLEX Rapid DNA-SEQ Kit was used to construct a PE library. Magnetic bead screening was used to remove the self-connecting segments of the joint, and then, PCR amplification was used to enrich the library templates. Finally, the PCR products were recovered by magnetic beads to obtain the final library, and the average size of the library was about 300 bp.
2.3. Metagenomic Sequencing and Taxonomy Analysis
Metagenomic sequencing was performed after the library construction and the Adaptor-appended fragments were sequenced using the Illumina HiSeq4000 platform. The original metagenomic sequencing sequences used in this study have been uploaded to the Sequence Read Archive (SRA) database of the National Center for Biotechnology Information (NCBI). The related information can be found in Table S1.
The original sequences were split, mass clipped, and decontaminated by SeqPrep software. Low-quality reads (length < 50 bp or with a quality value < 20 or having N bases) were removed by Sickle and retained were high-quality pair-end reads and single-end reads. The optimized sequence was assembled using MEGAHIT. Contigs > 300 bp were selected as the final assembly result.
Open reading frames (ORFs) from each metagenomic sample were predicted using MetaGene [25]. The predicted ORFs with the length of or over 100 bp were retrieved and translated to amino acid sequences using the NCBI translation table. All sequences from the gene sets with a 95% sequence identity (90% coverage) were clustered as the non-redundant gene catalog by the CD-HIT. Reads after quality control were mapped to the representative genes with 95% identity using SOAPaligner [26], and the gene abundance in each sample was evaluated.
BLAST (Version 2.2.28+) was employed for taxonomic annotations by comparing the non-redundant genes with the NR database (e-value ≤ 1 × 10−5). BLASTP was used to compare non-redundant genes with the Comprehensive Antibiotic Resistance Database (CARD, Version 1.1.3) [26] to obtain annotated information of the antibiotic resistance function (e-value ≤ 1 × 10−5). According to the function and keywords of the CARD database, the sequences can be divided into four categories: AR (Antibiotic Resistance) refers to the antibiotic resistance gene, which can directly resist antibiotics; AS (Antibiotic Sensitive) means that the gene is endowed with antibiotic resistance through mutation, knockout, etc.; AT (Antibiotic Target) refers to the antibiotic target binding gene, which can achieve antibiotic resistance through binding with the target of antibiotics; ABS (Antibiotic Biosynthesis) means that the genes are related to antibiotic biosynthesis.
2.4. Data Analysis Method
In this study, the data were analyzed through the Majorbio cloud platform. The RPKM (Reads Per Kilobases per Million reads) was used to represent the relative abundance of the genes. The RPKM normalized the sequencing depth and gene length, which could eliminate the influence of sequencing depth and gene-length differences on the relative abundance calculation results [27].
The bacterial communities and ARG subtypes in the annotated sequences were visualized by Heatmap and Circos. The difference in bacterial numbers among the samples was analyzed by Venn diagram analysis [28,29]. The ARG profile similarities among the samples were computed as Bray–Curtis distances and visualized by NMDS [30]. The significance test between the two samples was conducted by the Diff Between Prop Asymptotic CC method to investigate the influence of the D-Type filter on the distribution of the ARGs (Chi-square test, p ≤ 0.05).
2.5. Network Analysis
The Spearman correlation coefficient (ρ > 0.7, p < 0.05) was calculated between the bacterial community and the ARG subtypes. All positive correlation points were selected to construct a correlation matrix. Gephi (Version 0.9.2), as an open graph software platform, was used to visualize the complex network. This method can identify the major bacteria and ARG subtypes, measure the degree of correlation between the bacteria and ARG subtypes, and reduce artificial correlation bias [31].
3. Results and Discussion
3.1. Diversity and Structure of Bacterial Communities in Wastewater and Sludge
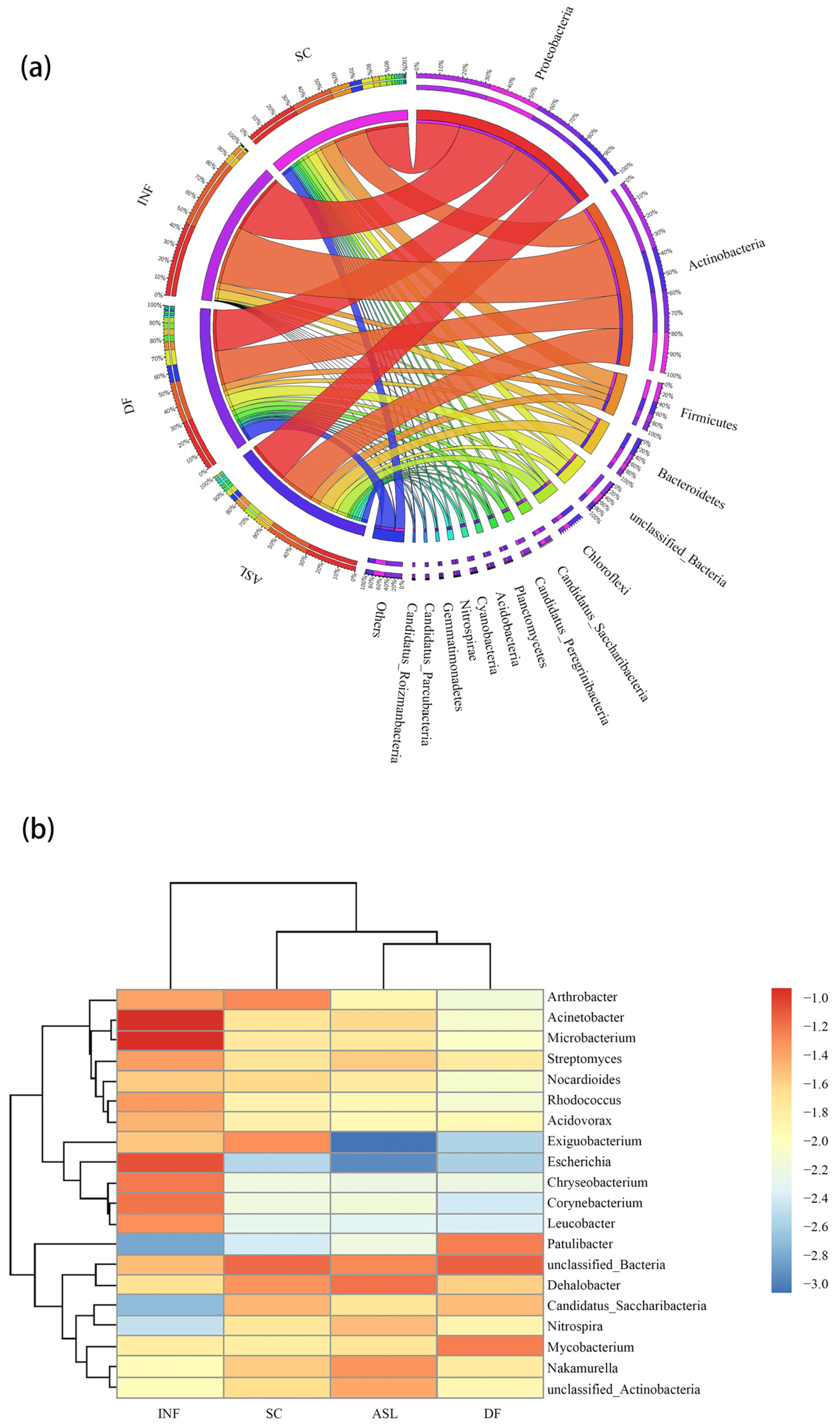
In total, 82 phyla and 1919 genera were detected in all the samples. At the phylum level, the dominant bacteria were Proteobacteria, Actinobacteria, Firmicutes, and Bacteroidete, which are often detected in WWTPs [32,33] (Figure 1a).
Bacterial communities in WWTPs contain abundant pathogens that may cause harm to human health and the environment. The top 20 bacteria at the genus level in relative abundance in the wastewater and activated sludge of the WWTPs are shown in Figure 1b. Among them, Acinetobacter and Mycobacterium are common drug-resistant pathogens in the human body and were listed in urgent new therapy by the World Health Organization (WHO) in 2014 [34,35]. Hospitals around the world have reported many infections due to Acinetobacter outbreaks in the past thirty years. Acinetobacter is resistant to almost all known antibiotics, including aminoglycosides, quinolones, β-lactams, cephalosporins, penicillin, etc. [36,37]. Mycobacterium is widely present in the effluent of WWTPs [35,38] and is one of the typical pathogenic bacteria. The most famous pathogenic bacterium in Mycobacterium is called Mycobacterium tuberculosis, which is the main cause of tuberculosis. Meanwhile, this bacterium is prone to drug resistance.
Without considering unclassified bacteria, the dominant genera were Acinetobacter, Microbacterium, Dehalobacter, Mycobacterium, Arthrobacter, and Escherichia, which have also been found in other studies of WWTPs [32,33]. In the INF, the dominant bacterial genera include Microbacterium (10.0%) and Acinetobacter (10.0%). In the ASL, the dominant bacterial genera include Dehalobacter (5.1%) and Nakamurella (3.2%). In the SC, the dominant bacterial genera include Arthrobacter (4.4%) and Exiguobacterium (4.1%). In the DF, the dominant bacterial genera include Mycobacterium (6.1%) and Patulibacter (6.0%), but their relative abundances were only 0.75% and 0.095% in the SC (Figure S2). The results showed that the D-Type filter can change the composition of the microbial community and lead to an increased proportion of some bacteria in the water. These bacteria, especially pathogens such as Mycobacterium, can be harmful to human health and the environment.
3.2. ARG Distribution along the Treatment Processes
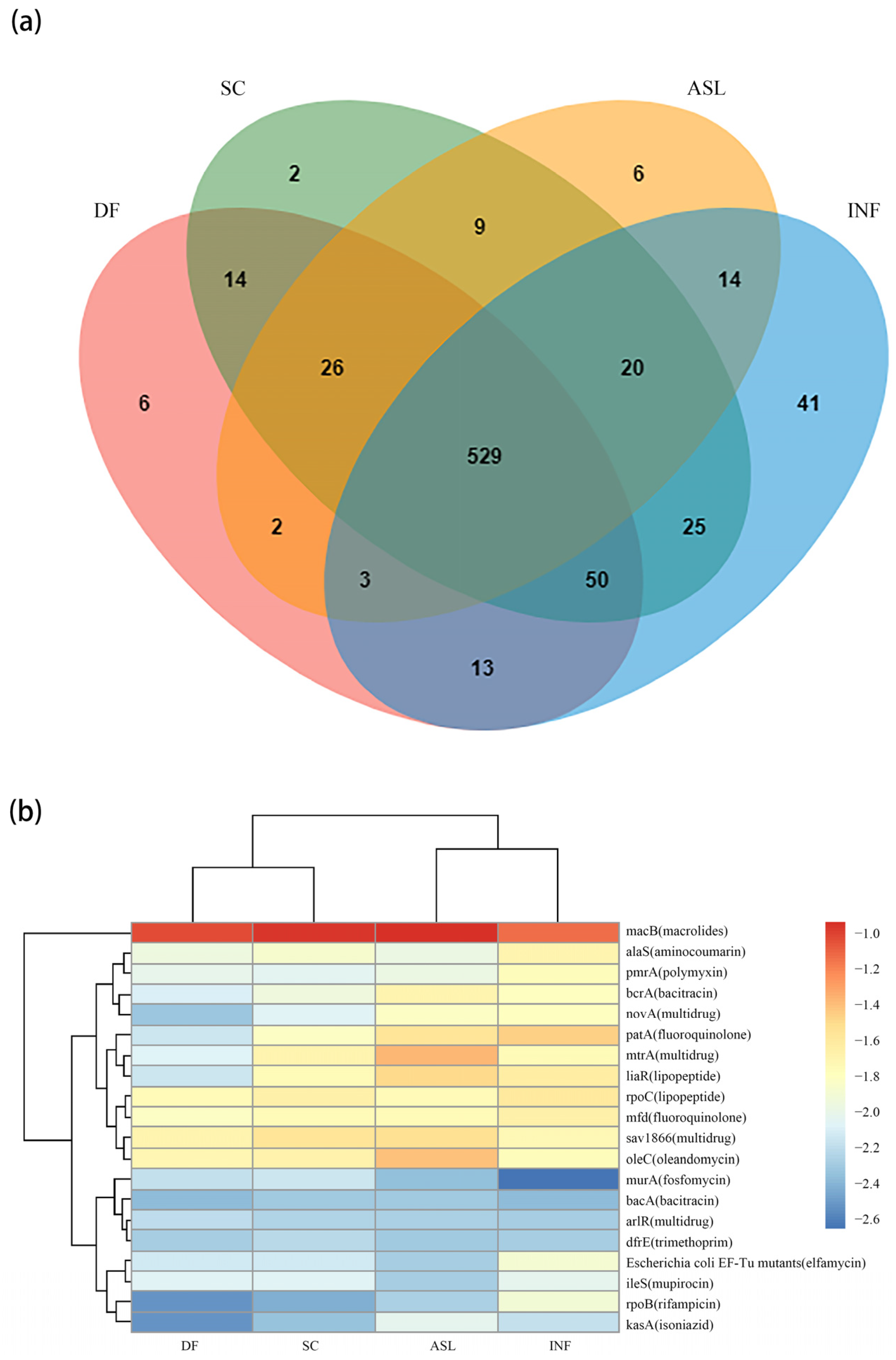
In total, 760 different ARG subtypes were detected in all the samples (Figure 2a), and 695 ARG subtypes were detected in the influent samples (INF) of the two WWTPs. Most of the ARGs in the influent can also be detected in the subsequent process section [39,40,41,42]. In this study, 76% (529 out of 695) of the ARG subtypes in the INF could be detected in other samples. In total, there were 643 ARG subtypes identified in the DF, which was less than those in the SC (675 subtypes). To investigate the influence of the D-Type filter on the distribution of the ARG subtypes, the diversity of ARG subtypes was compared in the secondary sedimentation tank effluent and the D-Type filter sample effluent from the two WWTPs. In plant CD, the number of ARG subtypes in the DF (602) was 2.99% higher than those in the SC (584). Meanwhile, in plant CN, the ARG subtype number in DF was 11.28% higher than those in SC (from 647 in DF to 574 in SC), as shown in Figure S3. Moreover, there were 15 kinds of ARG subtypes removed in both CN and CD, as shown in Table S2. The eliminated ARG subtypes mainly belong to macrolides, β-lactams, aminoglycosides, and sulfonamides. The result indicated that D-Type filters are capable of reducing a certain amount of ARGs in the wastewater treatment process, supporting the control of the risks of their transmission in tailwater.
Figure 2b shows the top 20 ARG subtypes in relative abundance, belonging to macrolides, lipopeptide, fluoroquinolone, aminocoumarin, bacitracin, and other resistance genes. The relative abundance of the dominant ARG subtypes, such as macB (4.1%), sav1866 (1.7%), and oleC (1.6%), in the INF increased in the DF and were 8.9%, 3.4% and 3.3%, respectively. These resistance genes are different types of ABC transporters, which play an important role in bacterial material metabolism, nutrient uptake, and multidrug resistance [43]. Sav1866 is reported as an ABC transporter of Staphylococcus aureus that can transport the lantibiotics to the outside of the bacterial cells, resulting in the inhibition of the growth of other bacteria and achieving self-protection to obtain growth advantages [44].
3.3. ARG Removal Specifically in D-Type Filters
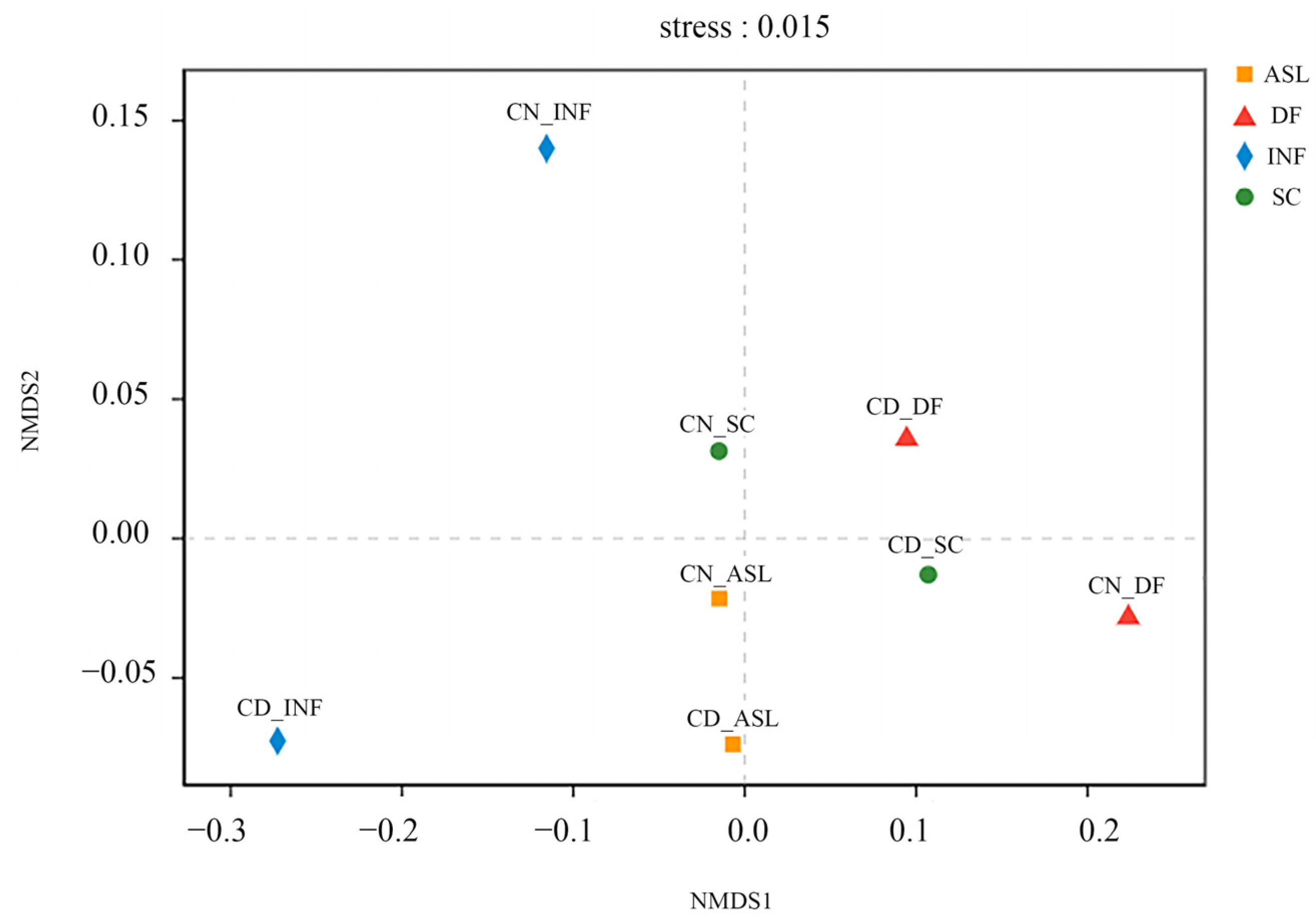
The ARG profile similarities among the samples were visualized by non-metric multidimensional scale (NMDS) analysis (Figure 3). A stress value is used to test the merits and demerits of the NMDS analysis results. When the stress value is less than 0.05, it means that the analysis results are representative. The difference between the CN and the CD influent was significant and was caused by the different sources of the two WWTPs. The distance between the SC and DF samples indicated that filtration could also change the ARG diversity in wastewater.
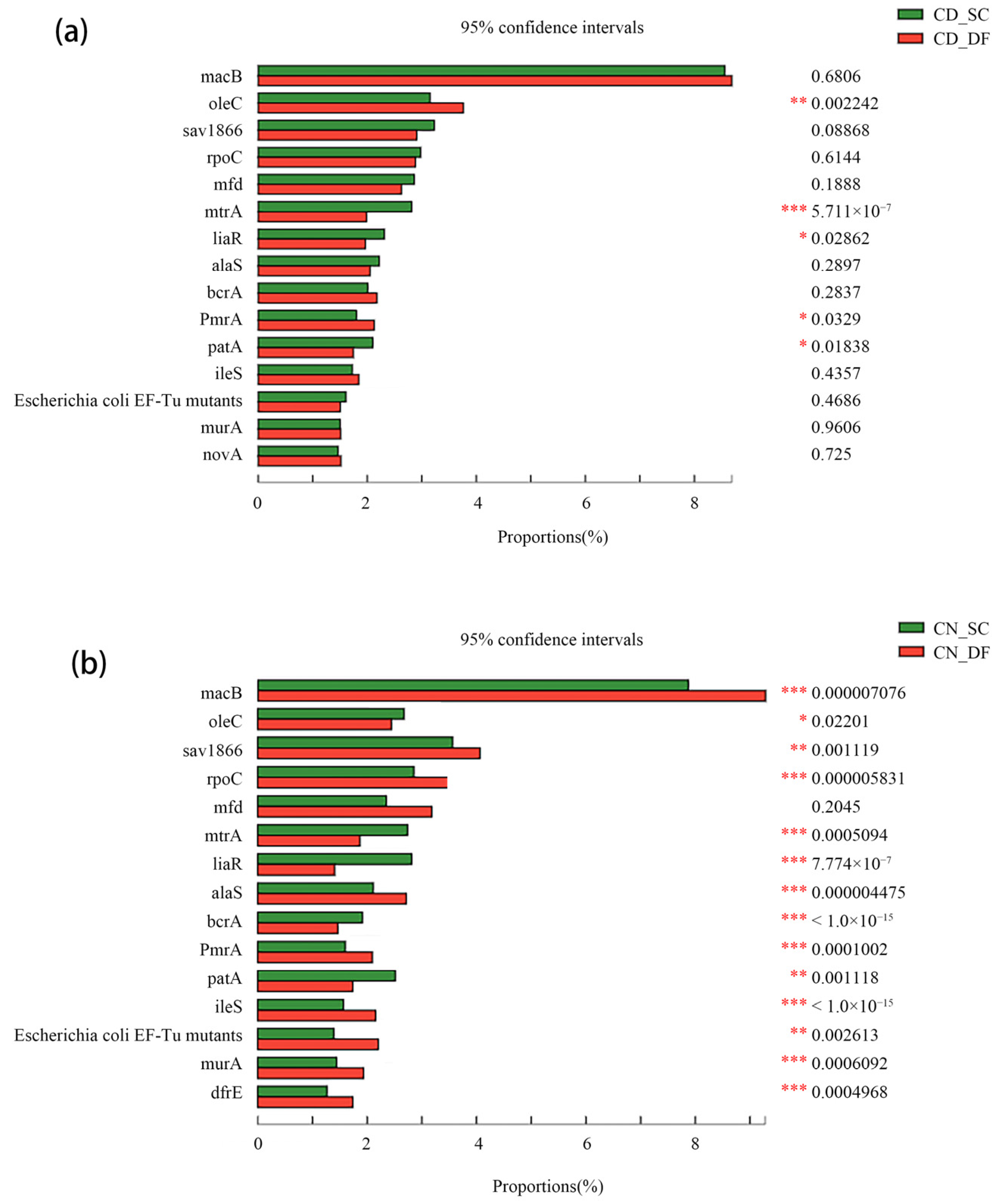
The secondary sedimentation tank is the most efficient unit for removing ARGs among traditional WWTPs [45,46,47], but there are still many suspended solids in its effluent, which carry different ARG subtypes. The effluent of the secondary sedimentation tank is deeply processed by the filter tank to further remove suspended particles. The D-Type filters equipped in CN and CD use comet-type fiber filter material with a diameter between several microns and tens of microns [16]. The differences between the main ARG subtypes in the SC and DF samples are shown in Figure 4.
The relative abundance of some ARG subtypes showed a significant difference between the SC and DF samples, especially in the CN plant. After filtering by the D-Type filter, the proportion of PmrA (1.8–2.1% and 1.6–2.1%) increased, and the proportion of mtrA (2.8–2.0% and 2.7–1.9%), liaR (2.3–2.0% and 2.8–1.4%), and patA (2.1–1.7% and 2.5–1.7%) decreased in both CD and CN (the details are shown in Table 1). PmrA is found in many pathogens and is often reported to play an important role in mediating the resistance mechanism of colistin [48,49]. This phenomenon indicates that the D-Type filter may have the risk of the enrichment of some ARGs. Taking into account the working mechanism of the filter, the suspended solids in the wastewater are adsorbed on the surface of the filter material and continuously accumulate in the filter layer. The filter is likely to be a potential ARG repository. Moreover, the compact structure of the filter material of the D-Type filter may also make the bacterial cells close to each other and promote the horizontal transfer of the ARGs between bacteria. Backwashing should be carried out regularly during the use of the filter. The use of water and air washing during the backwashing process could wash away the ARGs adsorbed in the filter material; however, ARGs may enter into the air in the form of bioaerosol. This is similar to the fact that the concentration of ARGs in the aerosol above the aeration tank of WWTPs is higher than that in other functional areas [50], which poses a threat to the health of people around and in the environment [51]. Therefore, the diffusion effect of the filter on ARGs is also worthy of attention.
3.4. Relationship and Co-Occurrence Patterns between ARG and Bacterial Taxa
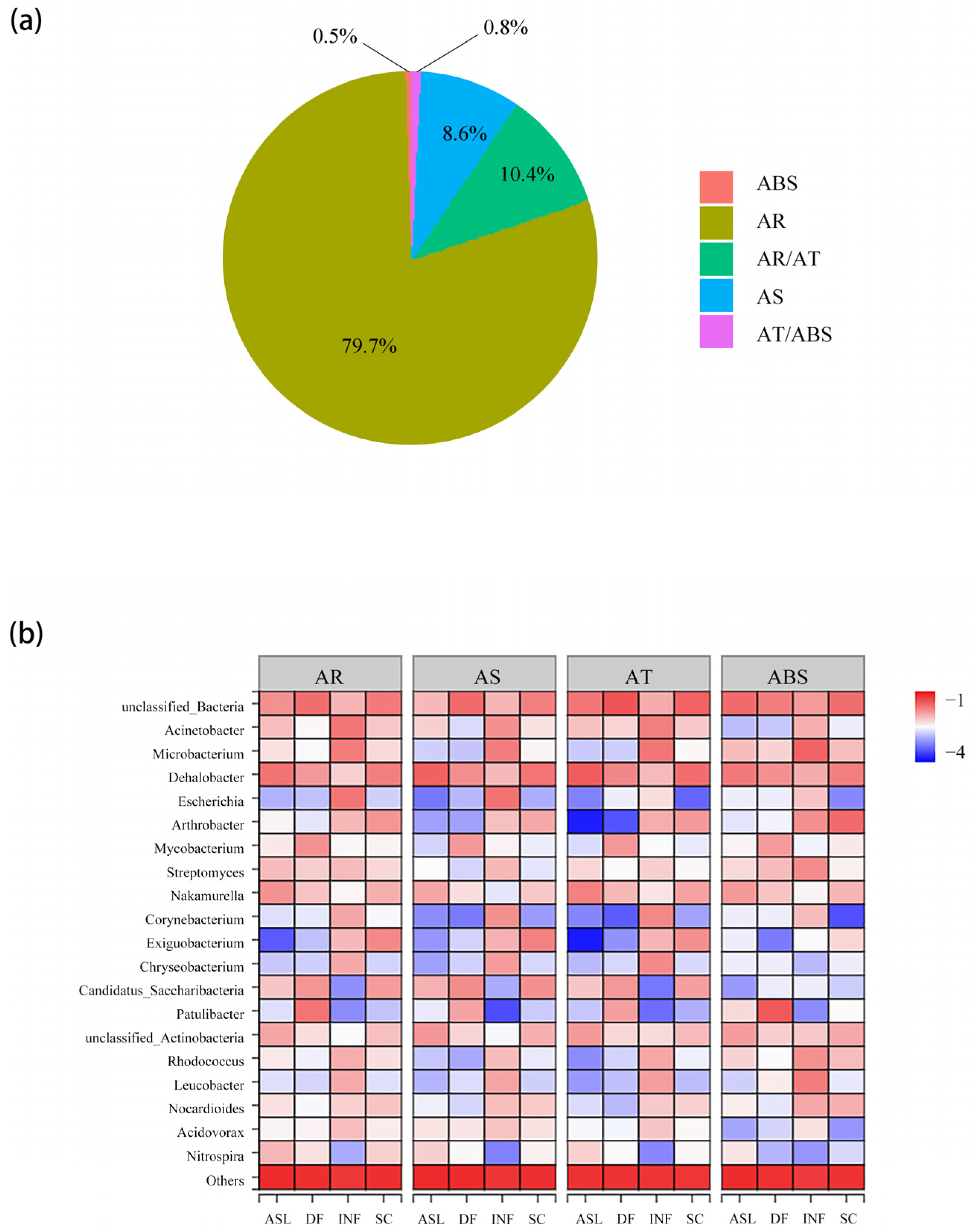
Based on the Comprehensive Antibiotic Research Database (CARD), the ARGs were divided into four functional groups: antibiotic target (AT), antibiotic resistance (AR), antibiotic sensitive (AS), and antibiotic biosynthesis (ABS). Some ARGs have multiple functions; for example, dfrE has both AR and AT functions. As Figure 5a shows, 79.7% (606 out of 760) ARG subtypes belong to the AR group.
Contribution analysis was conducted by correlation analysis of the relative abundance of bacteria at genus level and ARG subtypes in each sample. It can reveal the dominant species that carry specific functions. Figure 5b shows the contribution of the top 20 bacteria at genus level to the four types of functions. Except for the unclassified bacteria, Microbacterium, Acinetobacter, and Escherichia contributed the most to the four functions of the INF on average, with the contributions of 10.5%, 6.1%, and 5.6%. Dehalobacter (5.0%–13.3%) has maintained a high contribution in other samples. In general, the abundance of resistance genes is positively correlated with the abundance of bacteria. Dehalobacter was not the dominant bacteria in the SC and DF but had a high degree of contribution to the ARGs, indicating that Dehalobacter may carry a higher abundance of ARGs than other bacteria. Other bacteria with relatively high contributions are Actinobacteria, Microbacterium, and Escherichia.
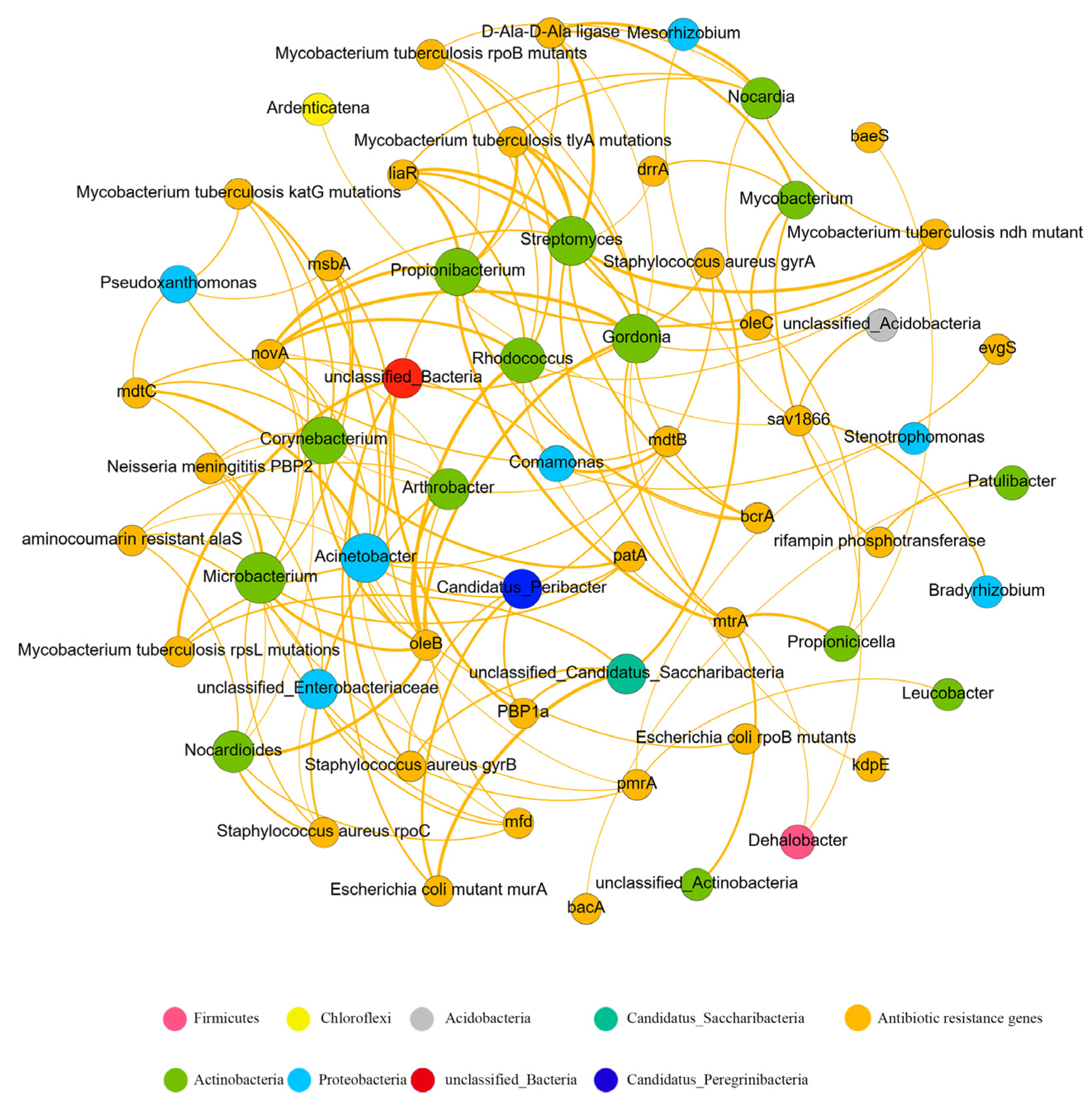
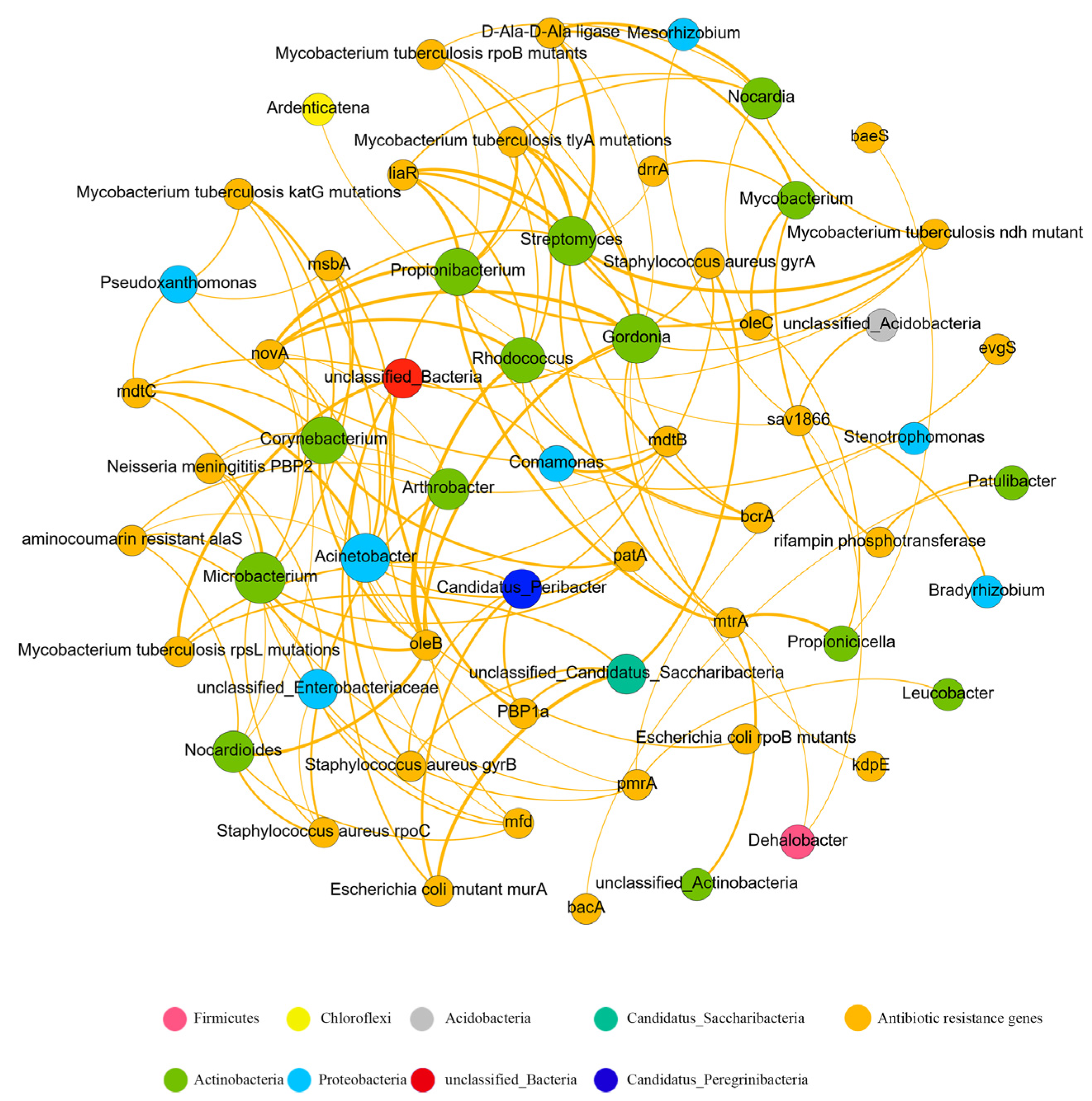
Network analysis provides us with new insights into the ARG subtypes and their possible hosts. Contribution analysis combined with network analysis can help us identify the main risk factors in complex environmental samples. The nonrandom connections between ARG subtypes and bacteria could indicate the possible host information of ARG subtypes [8]. According to Spearman’s correlation analysis (ρ > 0.7, P-value < 0.05), a co-occurrence network of bacterial taxa and ARG subtypes is constructed (Figure 6). The co-occurrence network of bacteria and ARG subtypes consists of 60 nodes (27 bacterial genera and 33 ARG subtypes) and 130 edges. Microbacterium, Acinetobacter, Gordonia, and Streptomyces were positively correlated with more ARG subtypes (11, 10, 10, and 10, respectively) than the other bacterial genera, indicating that these bacterial genera were the main possible hosts of ARG subtypes.
To know more about the risk of wastewater treated by the D-Type filter, bacteria with a higher relative abundance of the DF were investigated. Patulibacter was found to have positively nonrandom connections with the resistance genes against bacitracin (bacA) and rifampin (rifampin phosphotransferase). Mycobacterium is a pathogen and has been found to have positively nonrandom connections with resistance genes against rifampin (rifampin phosphotransferase), oleandomycin (oleC), doxorubicin (ddrA), and glycopeptide (D-Ala-D-Ala-ligase). The related bacterial genera and ARG subtypes were identified in the same contigs, indicating that these three kinds of bacteria were the possible hosts of these ARG subtypes. Some ARG hosts have been verified in previous studies; for example, Mycobacterium has been known to carry D-Ala-D-Ala-ligase [52,53,54].
4. Conclusions
The occurrence and distribution of ARGs in two municipal WWTPs with similar AAO process has been studied through metagenomic and network analysis in this paper. In total, 760 bacterial host ARG subtypes were detected from the samples, and 79.7% of them belonged to the AR category, in which macB was the most dominant ARG subtype.
The proportion of Mycobacterium and PmrA in the water was found to have significantly increased after the D-Type filter as the tertiary treatment, among which Mycobacterium was the potential host of the resistance genes against rifampin phosphotransferase, oleC, ddrA, and D-Ala-D-Ala-ligase. Compared to the secondary clarifier, the D-Type filter did reduce the number of ARG subtypes in the wastewater; however, these trapped ARG subtypes were held and enriched in the filter, increasing the risk of ARGs spreading inside the filter. Microbacterium, Acinetobacter, Gordonia, and Streptomyces significantly correlated with more than ten kinds of ARG subtypes, and more attention should be paid to the risk of spreading ARGs by these bacteria in the filter.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/w13233398/s1, Figure S1: schematic flow diagram of the WWTPs, with the sampling points for two wastewater treatment plants, Figure S2: circular representation of bacterial community in INF, ASL, SC, and DF at genus level. The outermost circles are the names of the sample and microbial composition. The lines inside the circle link the genus to the sample and the lines stand for the relative abundance of different genera in the samples, Figure S3: distribution of ARG subtypes in the SC and DF at two WWTPs, Table S1: information of accession ID, Table S2: ARGs removed from the CN and CD plants through D-type filters.
Author Contributions
Conceptualization, B.L., Y.Q. and Y.T.; methodology, J.Z. and B.L.; investigation, H.W.; data curation, B.L.; writing, H.W.; writing—review and editing, H.W., B.L. and Y.Q.; visualization, H.W. and J.Z.; funding acquisition, Y.Q. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (51778325) and the National Key Research and Development Program of China (2020YFC19092-05).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article and supplementary material.
Acknowledgments
The author would like to acknowledge the fruitful comments of the anonymous reviewers who provided beneficial suggestions that improved the quality of this paper.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Arulkumaran, N.; Routledge, M.; Schlebusch, S.; Lipman, J.; Conway Morris, A. Antimicrobial-associated harm in critical care: A narrative review. Intensive Care Med. 2020, 46, 225–235. [Google Scholar] [CrossRef]
- DuPont, H.L.; Steffen, R. Use of antimicrobial agents for treatment and prevention of travellers’ diarrhoea in the face of enhanced risk of transient fecal carriage of multi-drug resistant enterobacteriaceae: Setting the stage for consensus recommendations. J. Travel. Med. 2017, 24, S57–S62. [Google Scholar] [CrossRef]
- Peak, N.; Knapp, C.W.; Yang, R.K.; Hanfelt, M.M.; Smith, M.S.; Aga, D.S.; Graham, D.W. Abundance of six tetracycline resistance genes in wastewater lagoons at cattle feedlots with different antibiotic use strategies. Environ. Microbiol. 2007, 9, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Pruden, A.; Pei, R.; Storteboom, H.; Carlson, K.H. Antibiotic Resistance Genes as Emerging Contaminants: Studies in Northern Colorado. Environ. Sci. Technol. 2006, 40, 7445–7450. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.G.; Johnson, T.A.; Su, J.Q.; Qiao, M.; Guo, G.X.; Stedtfeld, R.D.; Hashsham, S.A.; Tiedje, J.M. Diverse and abundant antibiotic resistance genes in Chinese swine farms. Proc. Natl. Acad. Sci. USA 2013, 110, 3435–3440. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Yuan, K.; Chen, X.; Yang, Y.; Zhang, T.; Wang, Y.; Luan, T.; Zou, S.; Li, X. Metagenomic Analysis Revealing Antibiotic Resistance Genes (ARGs) and Their Genetic Compartments in the Tibetan Environment. Environ. Sci. Technol. 2016, 50, 6670–6679. [Google Scholar] [CrossRef]
- Yu, K.F.; Li, P.; Chen, Y.H.; Zhang, B.; Huang, Y.S.; Huang, F.Y.; He, Y.L. Antibiotic resistome associated with microbial communities in an integrated wastewater reclamation system. Water Res. 2020, 173, 115541. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.J.; Guan, Y.T.; Zhao, R.X.; Feng, J.; Huang, J.; Ma, L.P.; Li, B. Metagenomic and network analyses decipher profiles and co-occurrence patterns of antibiotic resistome and bacterial taxa in the reclaimed wastewater distribution system. J. Hazard. Mater. 2020, 400, 123170. [Google Scholar] [CrossRef]
- Gandolfi, I.; Bertolini, V.; Ambrosini, R.; Bestetti, G.; Franzetti, A. Unravelling the bacterial diversity in the atmosphere. Appl. Microbiol. Biotechnol. 2013, 97, 4727–4736. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.H.; Li, J.; Chen, H.; Bond, P.L.; Yuan, Z.G. Metagenomic analysis reveals wastewater treatment plants as hotspots of antibiotic resistance genes and mobile genetic elements. Water Res. 2017, 123, 468–478. [Google Scholar] [CrossRef]
- Kim, S.; Yun, Z.; Ha, U.H.; Lee, S.; Park, H.; Kwon, E.E.; Cho, Y.; Choung, S.; Oh, J.; Medriano, C.A.; et al. Transfer of antibiotic resistance plasmids in pure and activated sludge cultures in the presence of environmentally representative micro-contaminant concentrations. Sci. Total Environ. 2014, 468–469, 813–820. [Google Scholar] [CrossRef]
- Warnes, S.L.; Highmore, C.J.; Keevil, C.W. Horizontal transfer of antibiotic resistance genes on abiotic touch surfaces: Implications for public health. mBio 2012, 3, e00489-12. [Google Scholar] [CrossRef] [Green Version]
- Ju, F.; Beck, K.; Yin, X.; Maccagnan, A.; McArdell, C.S.; Singer, H.P.; Johnson, D.R.; Zhang, T.; Burgmann, H. Wastewater treatment plant resistomes are shaped by bacterial composition, genetic exchange, and upregulated expression in the effluent microbiomes. ISME J. 2019, 13, 346–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forsberg, K.J.; Patel, S.; Gibson, M.K.; Lauber, C.L.; Knight, R.; Fierer, N.; Dantas, G. Bacterial phylogeny structures soil resistomes across habitats. Nature 2014, 509, 612–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Wang, J. Application of a Fiber Filter in Chengdu Shahe Sewage Treatment Plant. Environ. Pollut. Control 2006, 28, 388–390. [Google Scholar]
- Wang, Z.Z.; Sun, Y.X.; Wu, G.X.; Wu, Q.Y.; Hu, H.Y.; Wu, Y.H.; Guo, F.; Guo, Y.M. Analysis of performance and cost of a micro-flocculation-D type filter process applied in wastewater tertiary treatment process. Chin. J. Environ. Eng. 2014, 8, 3132–3136. [Google Scholar]
- Tong, J.; Tang, A.P.; Wang, H.Y.; Liu, X.X.; Huang, Z.; Wang, Z.H.; Wang, Z.Y.; Zhang, J.Y.; Wei, Y.S.; Su, Y.Y.; et al. Microbial community evolution and fate of antibiotic resistance genes along six different full-scale municipal wastewater treatment processes. Bioresour. Technol. 2019, 272, 489–500. [Google Scholar] [CrossRef]
- Hayward, J.L.; Huang, Y.N.; Yost, C.K.; Hansen, L.T.; Lake, C.; Tong, A.; Jamieson, R.C. Lateral flow sand filters are effective for removal of antibiotic resistance genes from domestic wastewater. Water Res. 2019, 162, 482–491. [Google Scholar] [CrossRef]
- Yuan, L.; Ma, Q.; Zhao, J. Meta-omics and its application in biological wastewater treatment system. Acta Sci. Circumstantiae 2020, 40, 2690–2699. [Google Scholar]
- Yang, Y.; Li, B.; Zou, S.C.; Fang, H.H.P.; Zhang, T. Fate of antibiotic resistance genes in sewage treatment plant revealed by metagenomic approach. Water Res. 2014, 62, 97–106. [Google Scholar] [CrossRef]
- Yoo, K.; Yoo, H.; Lee, J.; Choi, E.J.; Park, J. Exploring the antibiotic resistome in activated sludge and anaerobic digestion sludge in an urban wastewater treatment plant via metagenomic analysis. J. Microbiol. 2020, 58, 123–130. [Google Scholar] [CrossRef]
- Strange, J.E.S.; Leekitcharoenphon, P.; Moller, F.D.; Aarestrup, F.M. Metagenomics analysis of bacteriophages and antimicrobial resistance from global urban sewage. Sci. Rep. 2021, 11, 1600. [Google Scholar] [CrossRef] [PubMed]
- Barberan, A.; Bates, S.T.; Casamayor, E.O.; Fierer, N. Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J. 2012, 6, 343–351. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.M.; Zhao, D.Y.; Huang, R.; Cao, X.Y.; Zeng, J.; Yu, Z.B.; Hooker, K.V.; Hambright, K.D.; Wu, Q.L. Contrasting Network Features between Free-Living and Particle-Attached Bacterial Communities in Taihu Lake. Microb. Ecol. 2018, 76, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Jia, B.F.; Raphenya, A.R.; Alcock, B.; Waglechner, N.; Guo, P.Y.; Tsang, K.K.; Lago, B.A.; Dave, B.M.; Pereira, S.; Sharma, A.N.; et al. CARD 2017: Expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2017, 45, D566–D573. [Google Scholar] [CrossRef] [PubMed]
- Li, R.Q.; Li, Y.R.; Kristiansen, K.; Wang, J. SOAP: Short oligonucleotide alignment program. Bioinformatics 2008, 24, 713–714. [Google Scholar] [CrossRef] [Green Version]
- Lawson, C.E.; Wu, S.; Bhattacharjee, A.S.; Hamilton, J.J.; McMahon, K.D.; Goel, R.; Noguera, D.R. Metabolic network analysis reveals microbial community interactions in anammox granules. Nat. Commun. 2017, 8, 15416. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [Green Version]
- Zhou, G.Y.; Soufan, O.; Ewald, J.; Hancock, R.E.W.; Basu, N.; Xia, J.G. NetworkAnalyst 3.0: A visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res. 2019, 47, W234–W241. [Google Scholar] [CrossRef] [Green Version]
- Taguchi, Y.H.; Oono, Y. Relational patterns of gene expression via non-metric multidimensional scaling analysis. Bioinformatics 2005, 21, 730–740. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Yang, Y.; Ma, L.P.; Ju, F.; Guo, F.; Tiedje, J.M.; Zhang, T. Metagenomic and network analysis reveal wide distribution and co-occurrence of environmental antibiotic resistance genes. ISME J. 2015, 9, 2490–2502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, J.; Schmitz, B.W.; Caton, K.; Zhang, B.; Zabaleta, J.; Garai, J.; Taylor, C.M.; Romanchishina, T.; Gerba, C.P.; Pepper, I.L.; et al. Assessing the spatial and temporal variability of bacterial communities in two Bardenpho wastewater treatment systems via Illumina MiSeq sequencing. Sci. Total Environ. 2019, 657, 1543–1552. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.G.; Li, B.X.; Li, N.; Sardar, M.F.; Song, T.T.; Zhu, C.X.; Lv, X.W.; Li, H.N. Effects of UV disinfection on phenotypes and genotypes of antibiotic-resistant bacteria in secondary effluent from a municipal wastewater treatment plant. Water Res. 2019, 157, 546–554. [Google Scholar] [CrossRef]
- Cai, L.; Zhang, T. Detecting human bacterial pathogens in wastewater treatment plants by a high-throughput shotgun sequencing technique. Environ. Sci. Technol. 2013, 47, 5433–5441. [Google Scholar] [CrossRef]
- Kummerer, K. Resistance in the environment. J. Antimicrob. Chemother. 2004, 54, 311–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paton, R.; Miles, R.S.; Hood, J.; Amyes, S.G.B.; Miles, R.S.; Amyes, S.G.B. ARI 1: β-lactamase-mediated imipenem resistance in Acinetobacter baumannii. Int. J. Antimicrob. Agents 1993, 2, 81–87. [Google Scholar] [CrossRef]
- Lyytikäinen, O.; Köljalg, S.; Härmä, M.; Vuopio-Varkila, J. Outbreak caused by two multi-resistant Acinetobacter baumannii clones in a burns unit: Emergence of resistance to imipenem. J. Hosp. Infection 1995, 31, 41–54. [Google Scholar] [CrossRef]
- Ye, L.; Zhang, T. Bacterial communities in different sections of a municipal wastewater treatment plant revealed by 16S rDNA 454 pyrosequencing. Appl. Microbiol. Biotechnol. 2013, 97, 2681–2690. [Google Scholar] [CrossRef] [Green Version]
- Ju, F.; Xia, Y.; Guo, F.; Wang, Z.P.; Zhang, T. Taxonomic relatedness shapes bacterial assembly in activated sludge of globally distributed wastewater treatment plants. Environ. Microbiol. 2014, 16, 2421–2432. [Google Scholar] [CrossRef]
- Ju, F.; Zhang, T. Bacterial assembly and temporal dynamics in activated sludge of a full-scale municipal wastewater treatment plant. ISME J. 2015, 9, 683–695. [Google Scholar] [CrossRef]
- Wu, L.W.; Ning, D.L.; Zhang, B.; Li, Y.; Zhang, P.; Shan, X.Y.; Zhang, Q.T.; Brown, M.R.; Li, Z.X.; Van Nostrand, J.D.Y.; et al. Global diversity and biogeography of bacterial communities in wastewater treatment plants. Nat. Microbiol. 2019, 4, 1183–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munir, M.; Wong, K.; Xagoraraki, I. Release of antibiotic resistant bacteria and genes in the effluent and biosolids of five wastewater utilities in Michigan. Water Res. 2011, 45, 681–693. [Google Scholar] [CrossRef] [PubMed]
- Davidson, A.L.; Dassa, E.; Orelle, C.; Chen, J. Structure, function, and evolution of bacterial ATP-binding cassette systems. Microbiol. Mol. Biol. Rev. 2008, 72, 317–364. [Google Scholar] [CrossRef] [Green Version]
- Otto, M.; Götz, F. ABC transporters of staphylococci. Res. Microbiol. 2001, 152, 351–356. [Google Scholar] [CrossRef]
- Gao, P.; Munir, M.; Xagoraraki, I. Correlation of tetracycline and sulfonamide antibiotics with corresponding resistance genes and resistant bacteria in a conventional municipal wastewater treatment plant. Sci. Total Environ. 2012, 421–422, 173–183. [Google Scholar] [CrossRef]
- Auerbach, E.A.; Seyfried, E.E.; McMahon, K.D. Tetracycline resistance genes in activated sludge wastewater treatment plants. Water Res. 2007, 41, 1143–1151. [Google Scholar] [CrossRef]
- Zhang, X.X.; Zhang, T.; Fang, H.H.P. Antibiotic resistance genes in water environment. Appl. Microbiol. Biotechnol. 2009, 82, 397–414. [Google Scholar] [CrossRef] [PubMed]
- Leila, H.; Shahriar, S.; Mohammad, D.; Reyhaneh, J. Molecular analysis of PmrA and PmrB genes in colistin-resistant Pseudomonas aeruginosa strains via PCR method. Pak. J. Pharm. Sci. 2019, 32, 1175–1177. [Google Scholar]
- Murray, S.R.; Ernst, R.K.; Bermudes, D.; Miller, S.I.; Low, K.B. PmrA(Con) confers pmrHFIJKL-dependent EGTA and polymyxin resistance on msbB Salmonella by decorating lipid A with phosphoethanolamine. J. Bacteriol. 2007, 189, 5161–5169. [Google Scholar] [CrossRef] [Green Version]
- Gotkowska-Płachta, A.; Filipkowska, Z.; Korzeniewska, E.; Janczukowicz, W.; Dixon, B.; Gołaś, I.; Szwalgin, D. Airborne Microorganisms Emitted from Wastewater Treatment Plant Treating Domestic Wastewater and Meat Processing Industry Wastes. CLEAN-Soil Air Water 2013, 41, 429–436. [Google Scholar] [CrossRef]
- Elena, E.; Aurora, B.; Miguel, E.; Ramón, G. Isolation of Salmonella serotypes in wastewater and effluent: Effect of treatment and potential risk. Int. J. Hyg. Environ. Health 2006, 209, 103–107. [Google Scholar]
- Bruning, J.B.; Murillo, A.C.; Chacon, O.; Barletta, R.G.; Sacchettini, J.C. Structure of the Mycobacterium tuberculosis D-alanine:D-alanine ligase, a target of the antituberculosis drug D-cycloserine. Antimicrob. Agents Chemother. 2011, 55, 291–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Z.Y.; Barletta, R.G. Roles of Mycobacterium smegmatis D-alanine:D-alanine ligase and D-alanine racemase in the mechanisms of action of and resistance to the peptidoglycan inhibitor D-cycloserine. Antimicrob. Agents Chemother. 2003, 47, 283–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halouska, S.; Fenton, R.J.; Zinniel, D.K.; Marshall, D.D.; Barletta, R.G.; Powers, R. Metabolomics Analysis Identifies d-Alanine-d-Alanine Ligase as the Primary Lethal Target of d-Cycloserine in Mycobacteria. J. Proteome Res. 2014, 13, 1065–1076. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Distribution of bacterial communities in wastewater. (a) Circular representation of the bacterial community in INF, ASL, SC, and DF at phylum level. The outermost circles are names of sample and microbial. The lines inside the circle connect the genus to the sample and these lines stand for the relative abundance of different genera in the samples; (b) the top 20 abundant bacteria at genus level are based on the RPKM method (the color bar indicates log10 transformed values of relative abundance).
Figure 1.
Distribution of bacterial communities in wastewater. (a) Circular representation of the bacterial community in INF, ASL, SC, and DF at phylum level. The outermost circles are names of sample and microbial. The lines inside the circle connect the genus to the sample and these lines stand for the relative abundance of different genera in the samples; (b) the top 20 abundant bacteria at genus level are based on the RPKM method (the color bar indicates log10 transformed values of relative abundance).

Figure 2.
Distribution of ARG subtypes in different samples. (a) Distribution of ARG subtypes in INF, ASL, SC, and DF; (b) the top 20 abundant ARG subtypes are based on the RPKM method (the color bar indicates log10-transformed values of relative abundance).
Figure 2.
Distribution of ARG subtypes in different samples. (a) Distribution of ARG subtypes in INF, ASL, SC, and DF; (b) the top 20 abundant ARG subtypes are based on the RPKM method (the color bar indicates log10-transformed values of relative abundance).
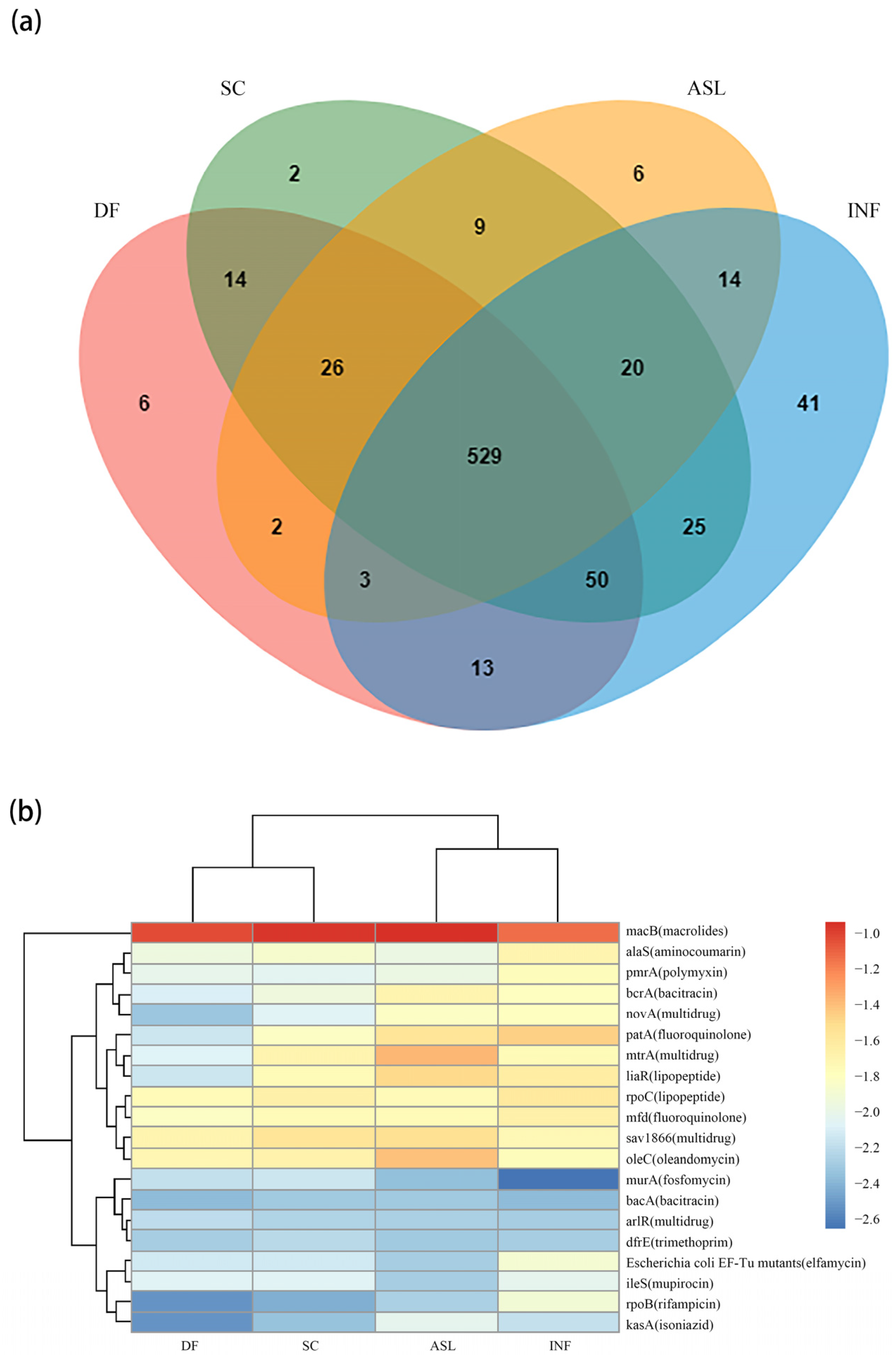
Figure 3.
The similarity analysis of the total ARG relative abundance, using NMDS based on Bray–Curtis metrics. The distance between points represents the degree of difference between samples.
Figure 3.
The similarity analysis of the total ARG relative abundance, using NMDS based on Bray–Curtis metrics. The distance between points represents the degree of difference between samples.
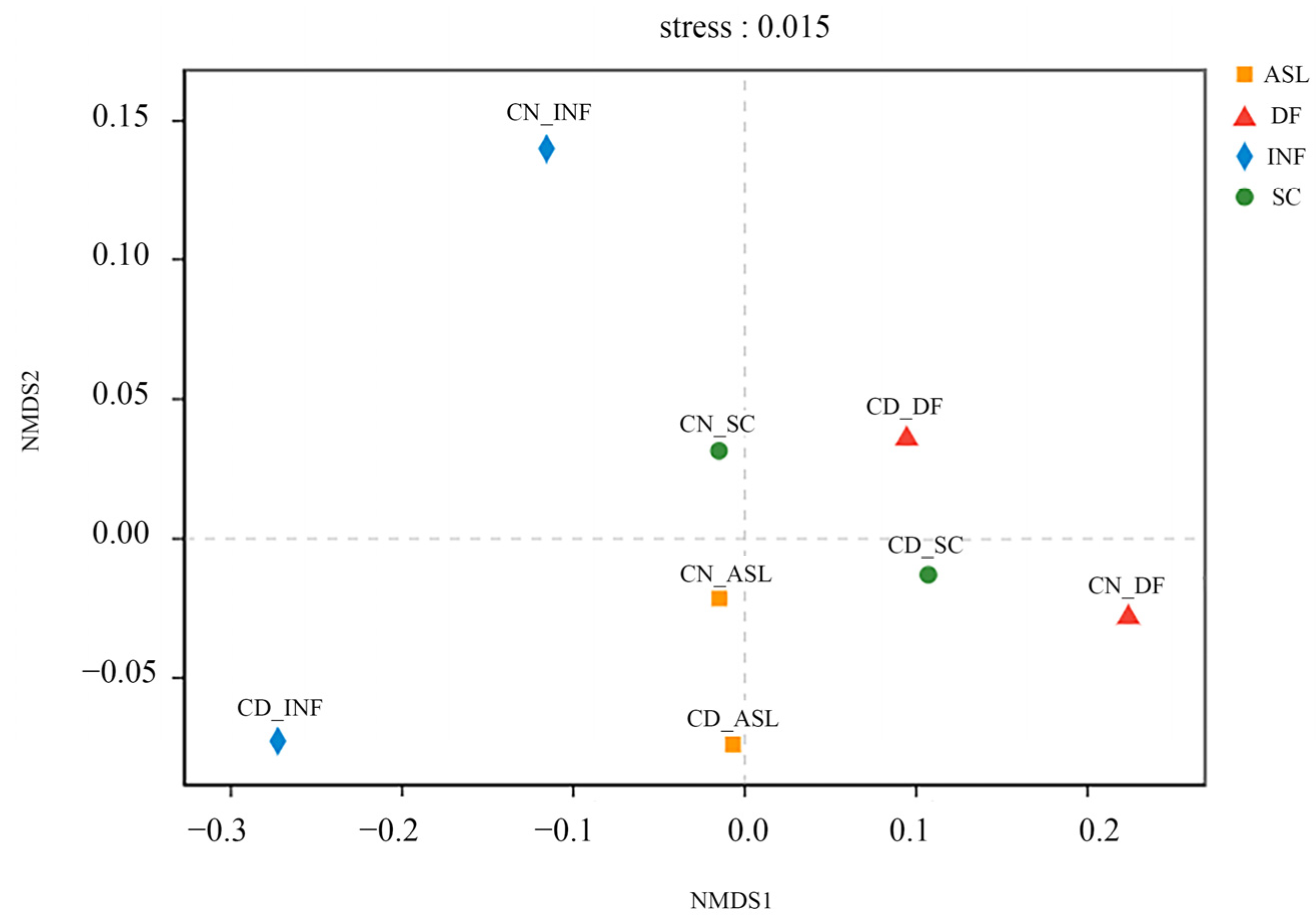
Figure 4.
The differences in relative abundance of ARG subtypes between SC and DF samples based on Chi-square test. (a) CD plant; (b) CN plant (* means 0.01 < p ≤ 0.05, ** means 0.001 < p ≤ 0.01, *** means p ≤ 0.001).
Figure 4.
The differences in relative abundance of ARG subtypes between SC and DF samples based on Chi-square test. (a) CD plant; (b) CN plant (* means 0.01 < p ≤ 0.05, ** means 0.001 < p ≤ 0.01, *** means p ≤ 0.001).
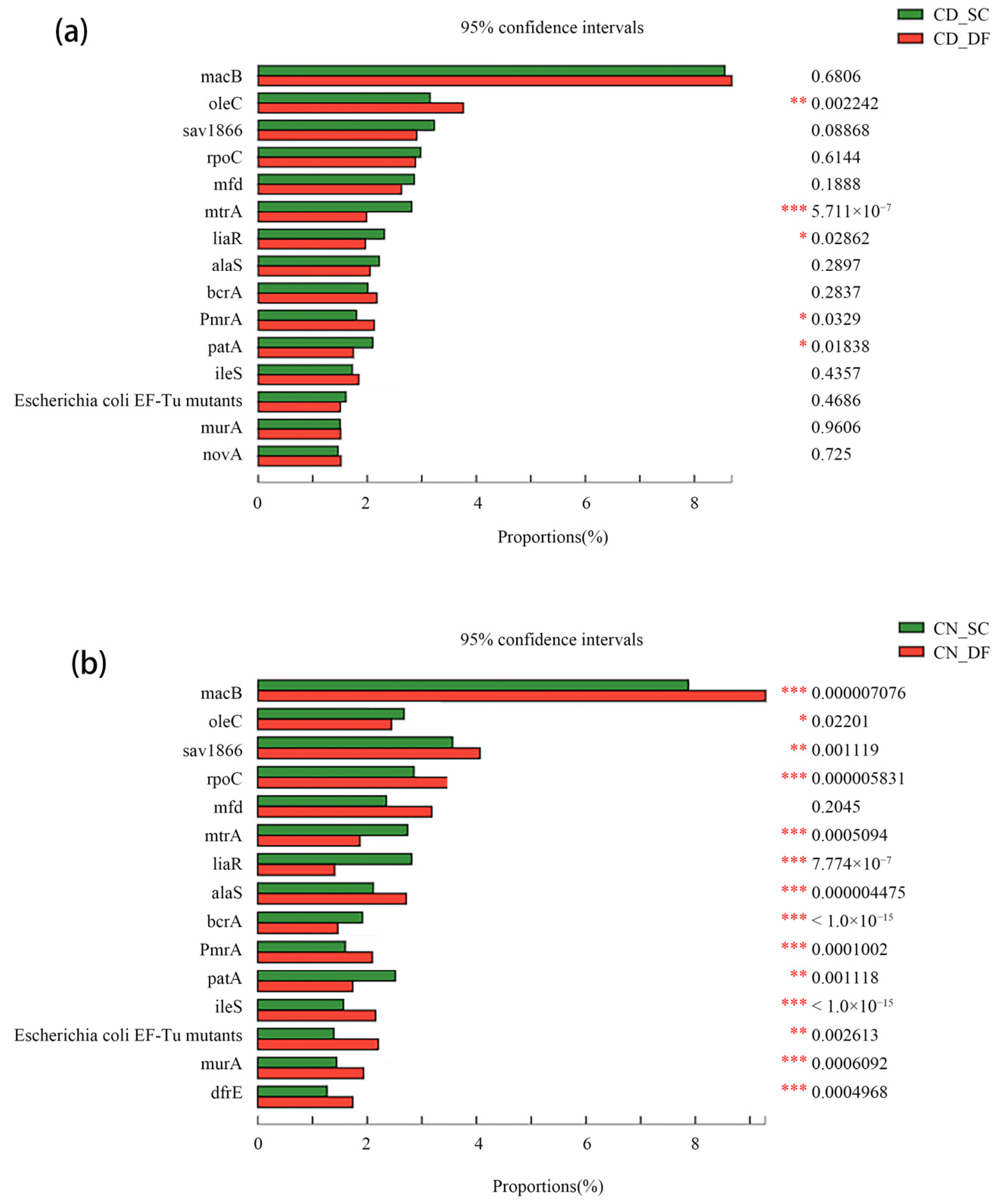
Figure 5.
The number of ARG subtypes of different types and their contribution to bacteria. (a) The proportion of ARG subtypes to the antibiotic resistance types; (b) contribution of bacteria at genus level to the four types of antibiotic resistance.
Figure 5.
The number of ARG subtypes of different types and their contribution to bacteria. (a) The proportion of ARG subtypes to the antibiotic resistance types; (b) contribution of bacteria at genus level to the four types of antibiotic resistance.

Figure 6.
The co-occurrence network of bacterial genera with the ARG subtypes of different categories. The nodes were colored according to phylum. The size of each node is proportional to the number of connections.
Figure 6.
The co-occurrence network of bacterial genera with the ARG subtypes of different categories. The nodes were colored according to phylum. The size of each node is proportional to the number of connections.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
ARGs subtypes with the same trend through D-Type filters.
| ARGs Name | SC1 | DF1 | SC2 | DF2 |
|---|---|---|---|---|
| PmrA | 1.8% | 2.1% | 1.6% | 2.1% |
| mtrA | 2.8% | 2.0% | 2.7% | 1.9% |
| liaR | 2.3% | 2.0% | 2.8% | 1.4% |
| patA | 2.1% | 1.7% | 2.5% | 1.7% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Wang, H.; Li, B.; Zhao, J.; Tian, Y.; Qiu, Y. Occurrence and Distribution of Antibiotic Resistance Genes in Municipal Wastewater Treatment Plants with D-Type Filters. Water 2021, 13, 3398. https://doi.org/10.3390/w13233398
AMA Style
Wang H, Li B, Zhao J, Tian Y, Qiu Y. Occurrence and Distribution of Antibiotic Resistance Genes in Municipal Wastewater Treatment Plants with D-Type Filters. Water. 2021; 13(23):3398. https://doi.org/10.3390/w13233398
Chicago/Turabian StyleWang, Haoze, Bing Li, Jiaheng Zhao, Yongjing Tian, and Yong Qiu. 2021. "Occurrence and Distribution of Antibiotic Resistance Genes in Municipal Wastewater Treatment Plants with D-Type Filters" Water 13, no. 23: 3398. https://doi.org/10.3390/w13233398
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.