Genome-Wide Analysis of Prognostic Alternative Splicing Signature and Splicing Factors in Lung Adenocarcinoma


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
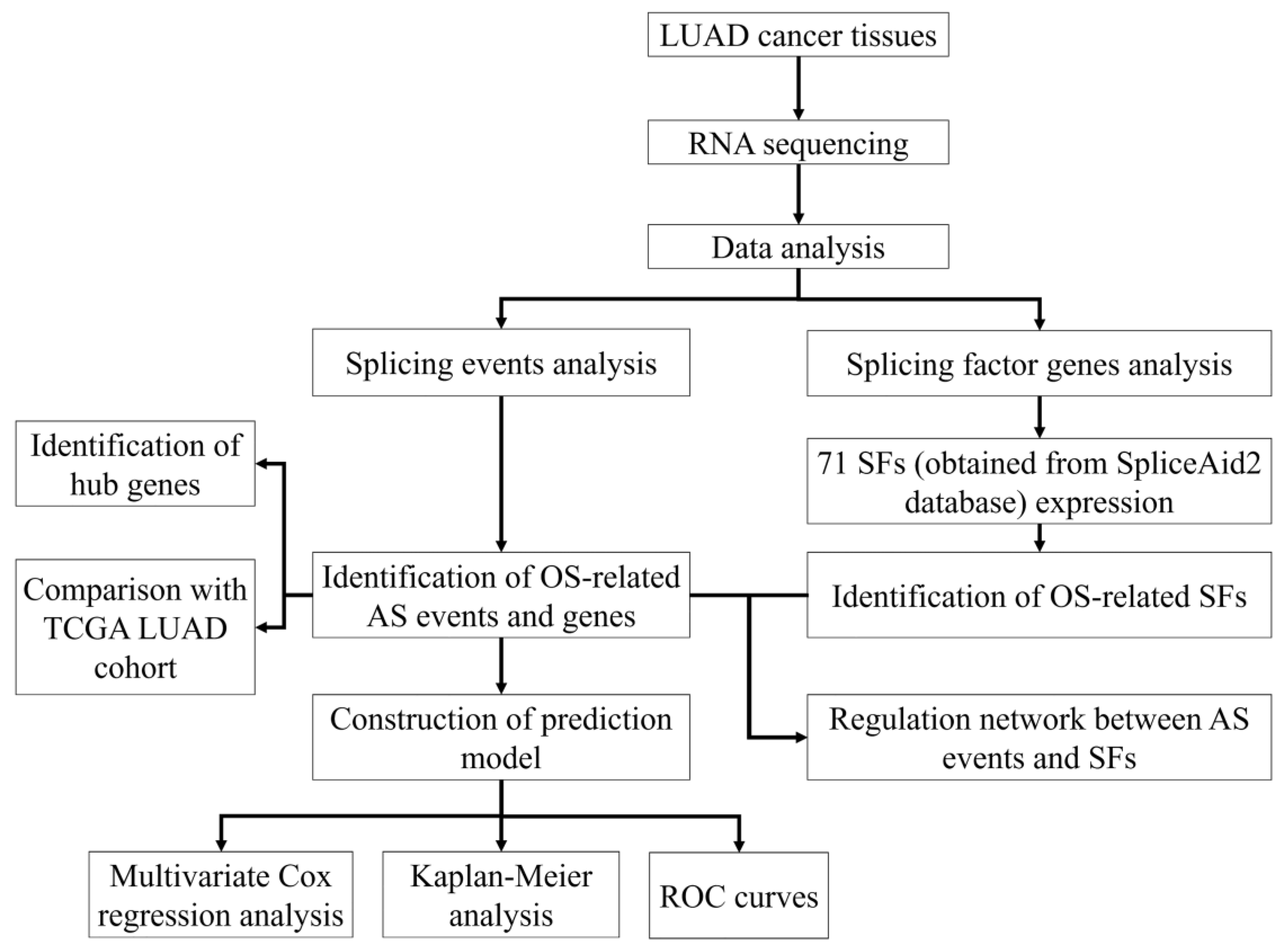
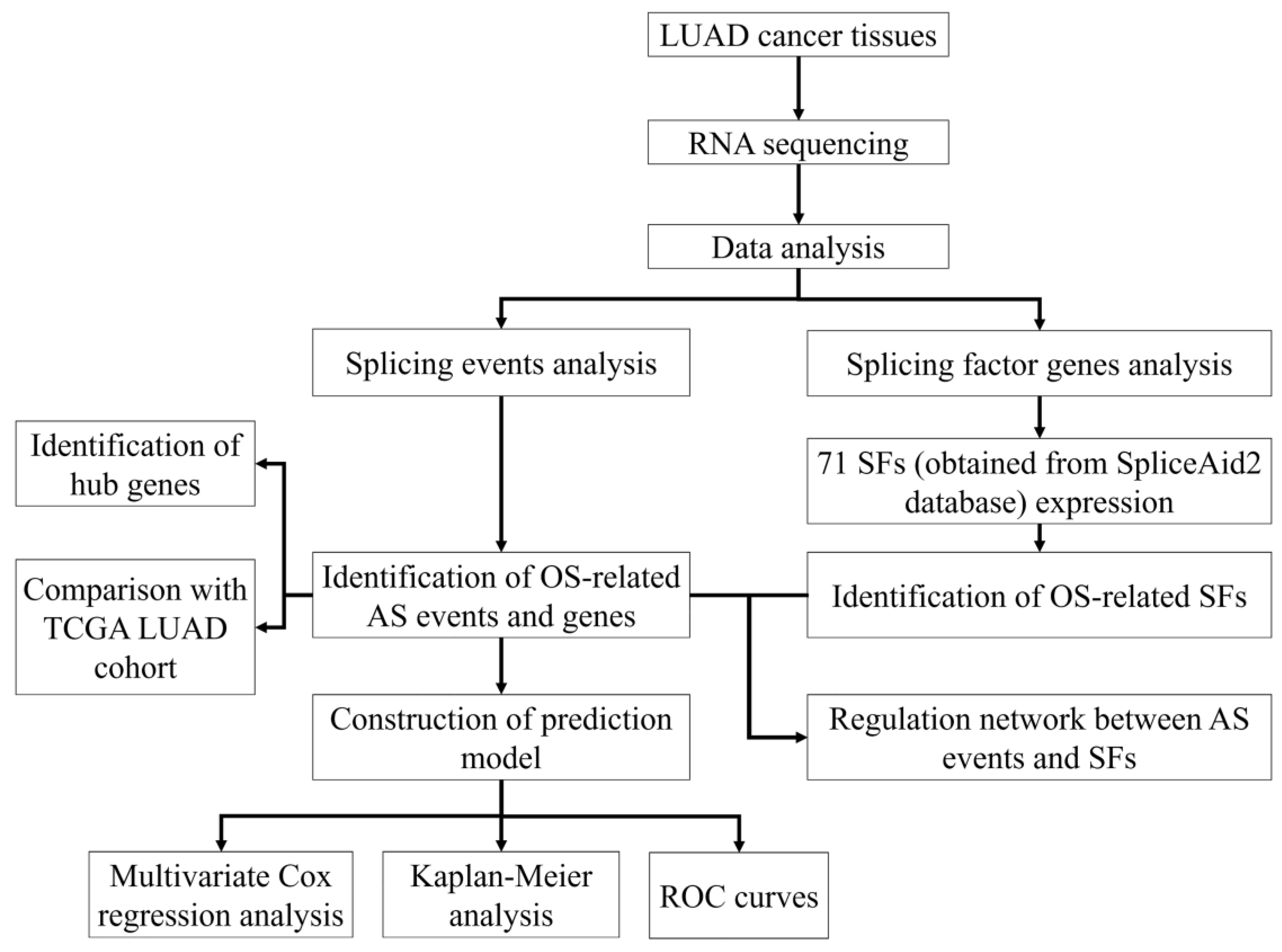
2. Materials and Methods
2.1. Patient Samples
2.2. RNA-Seq
2.3. RNA-Seq Data Analysis
2.4. Bioinformatics Identification of AS Events
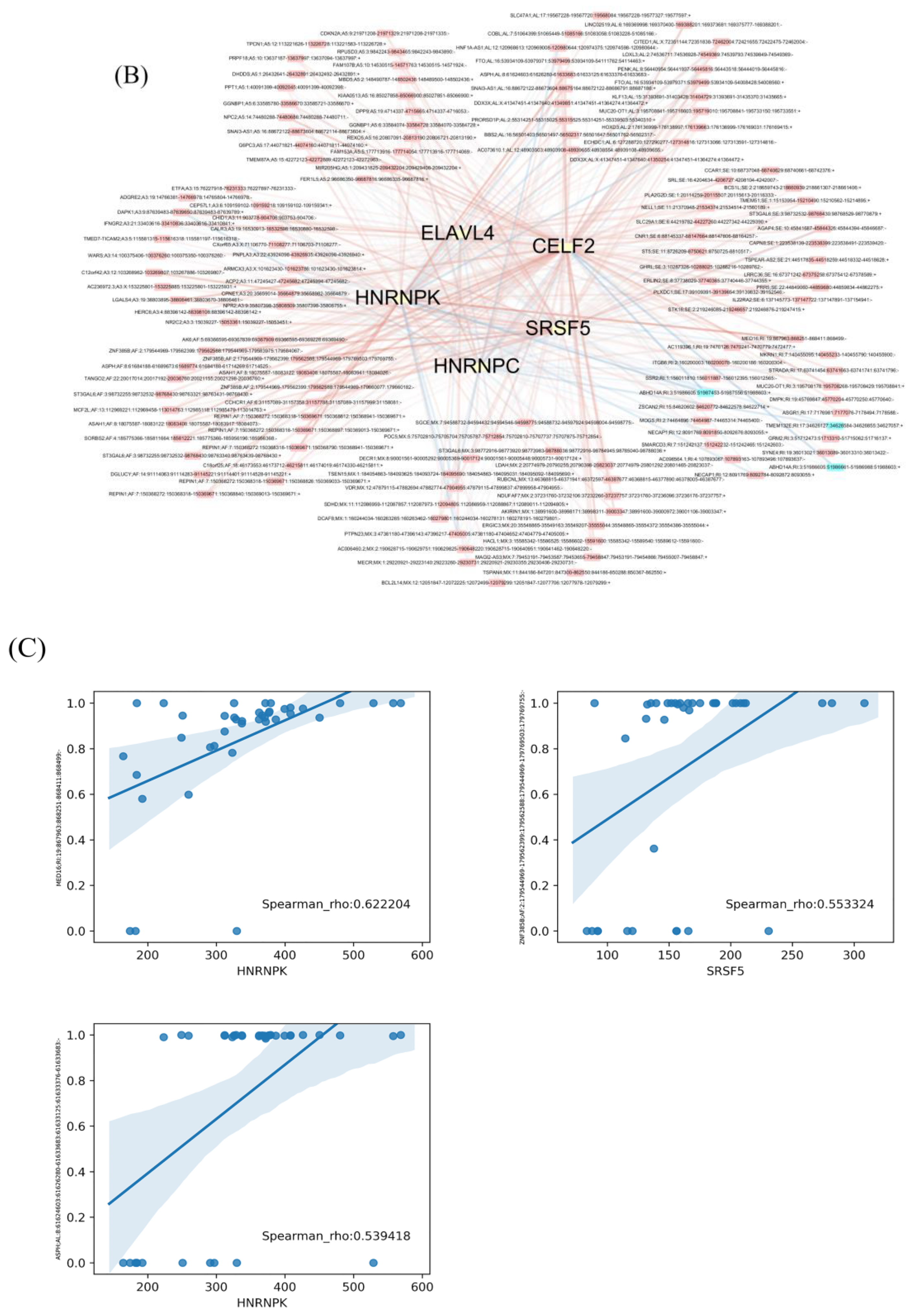
2.5. Construction of Prognostic Models and SF Correlation Network
2.6. Integrative Bioinformatics and Statistical Analysis
3. Results
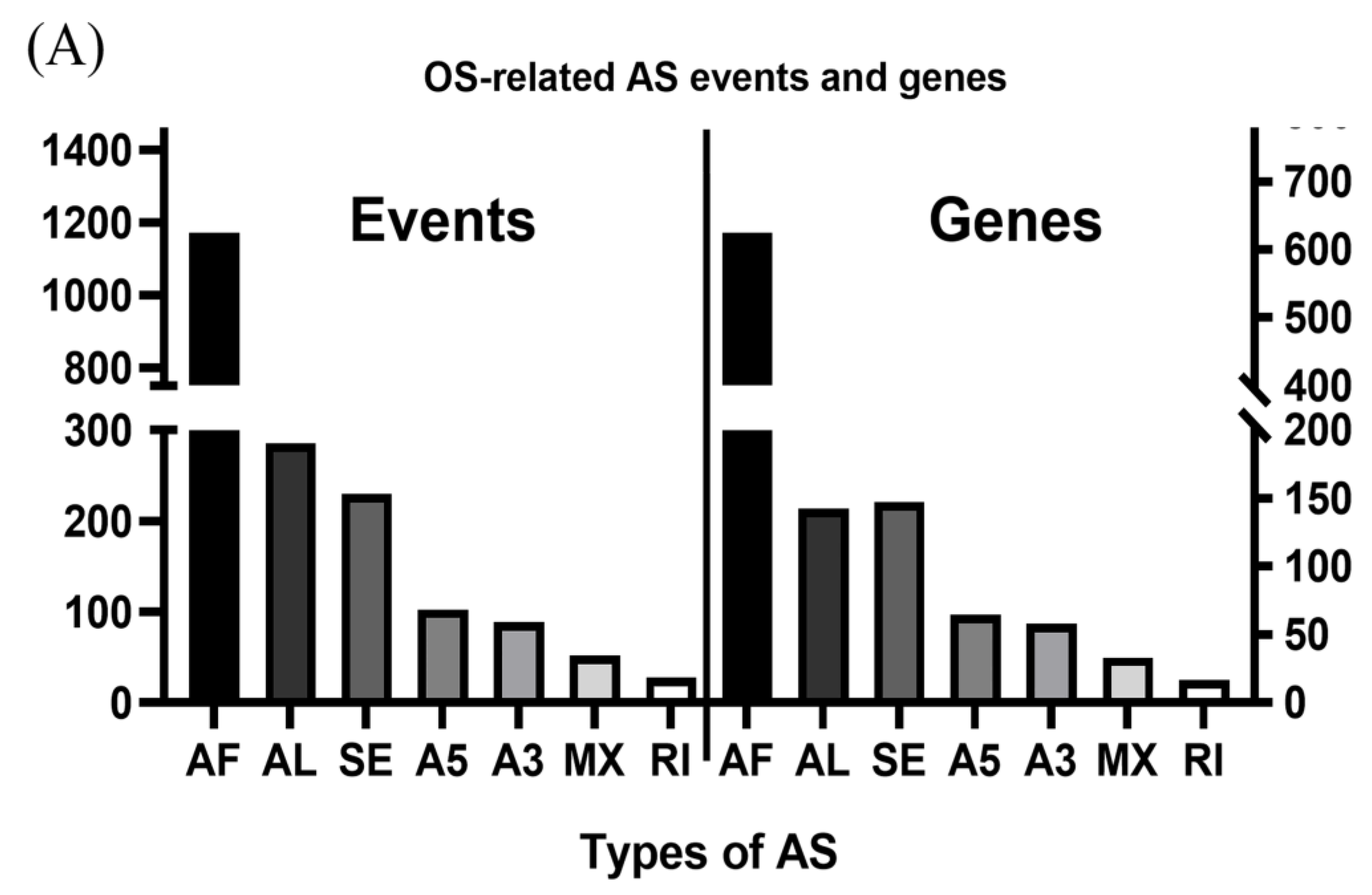
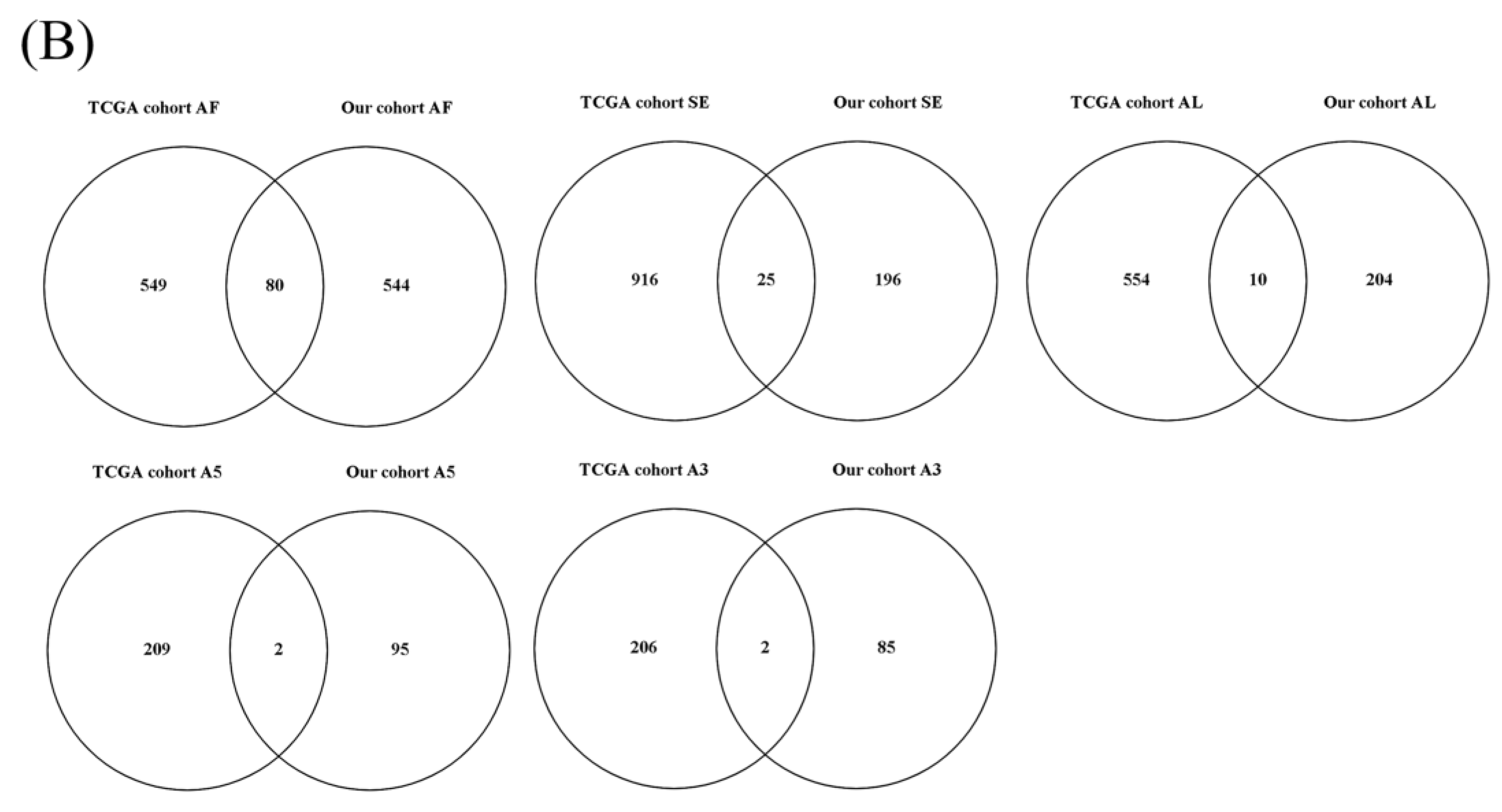
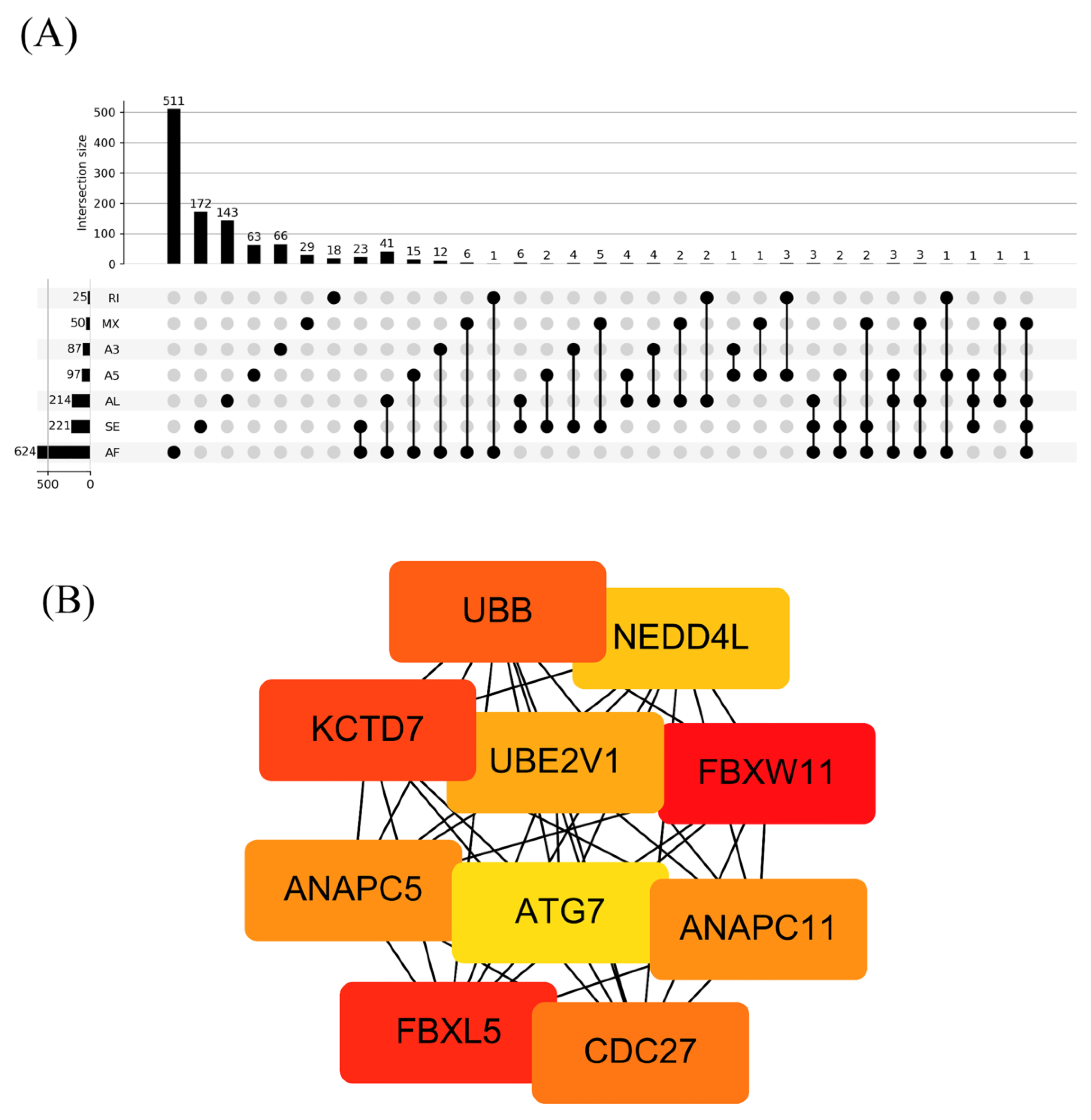
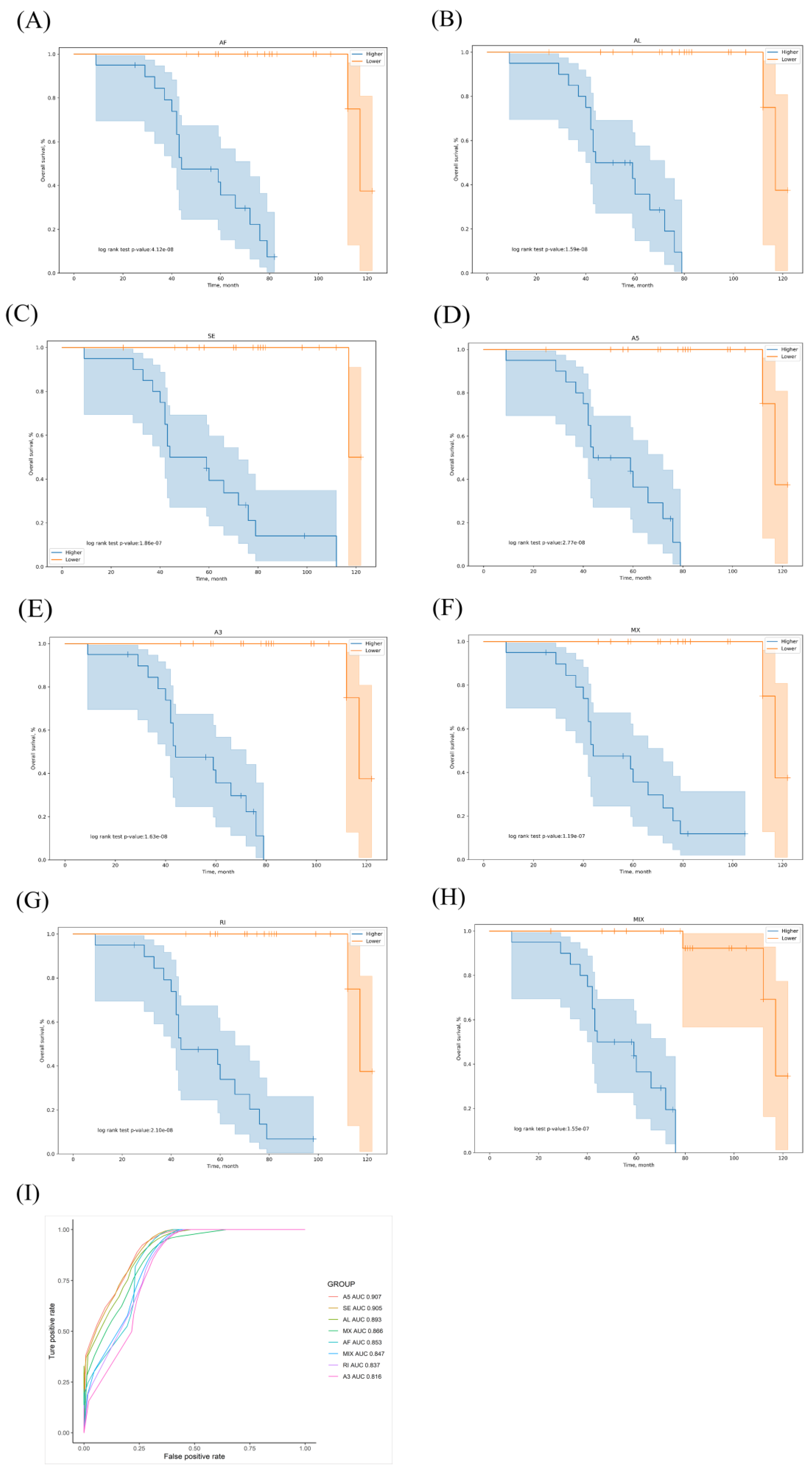
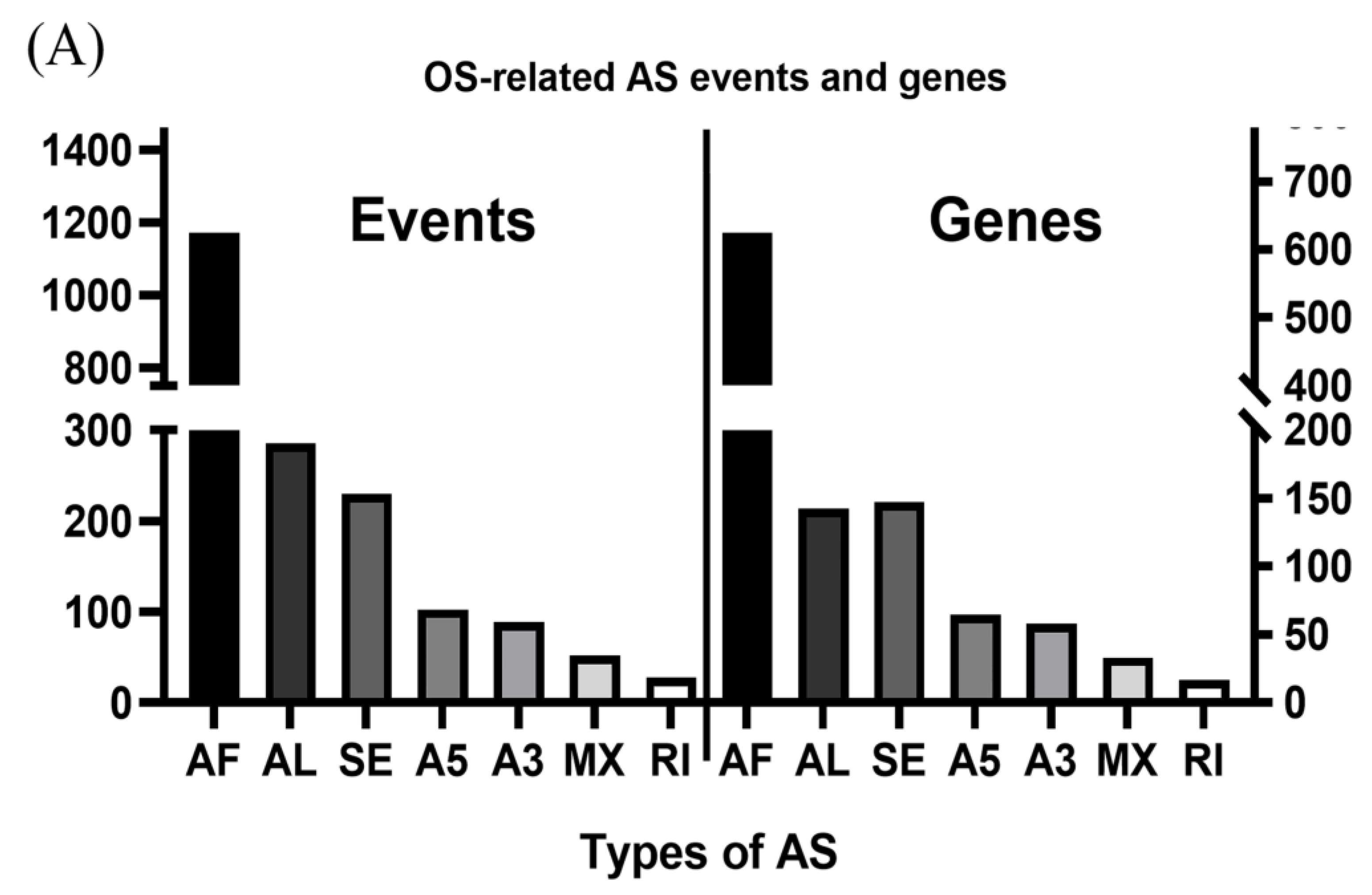
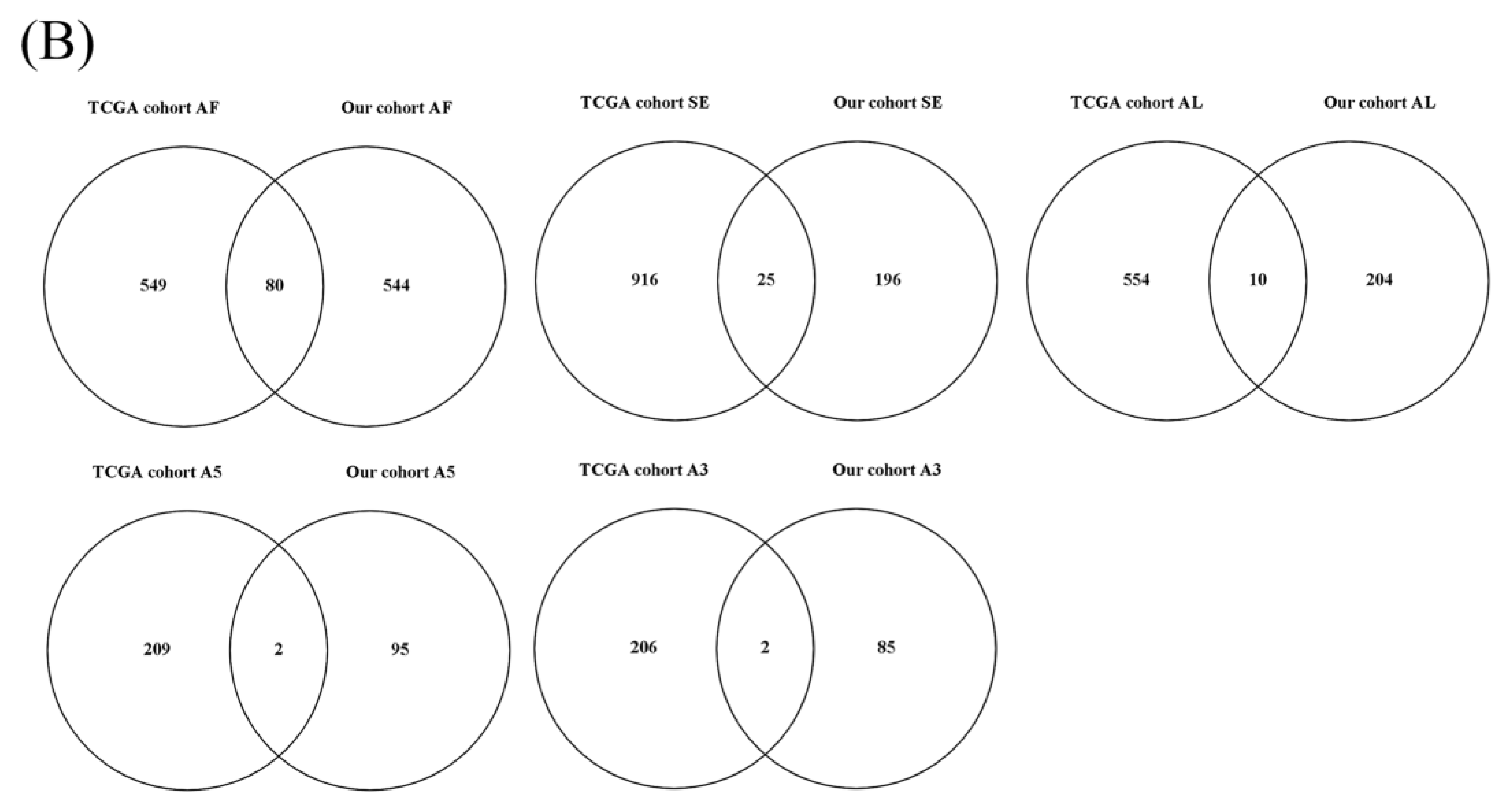
3.1. OS-Related AS Events in LUAD
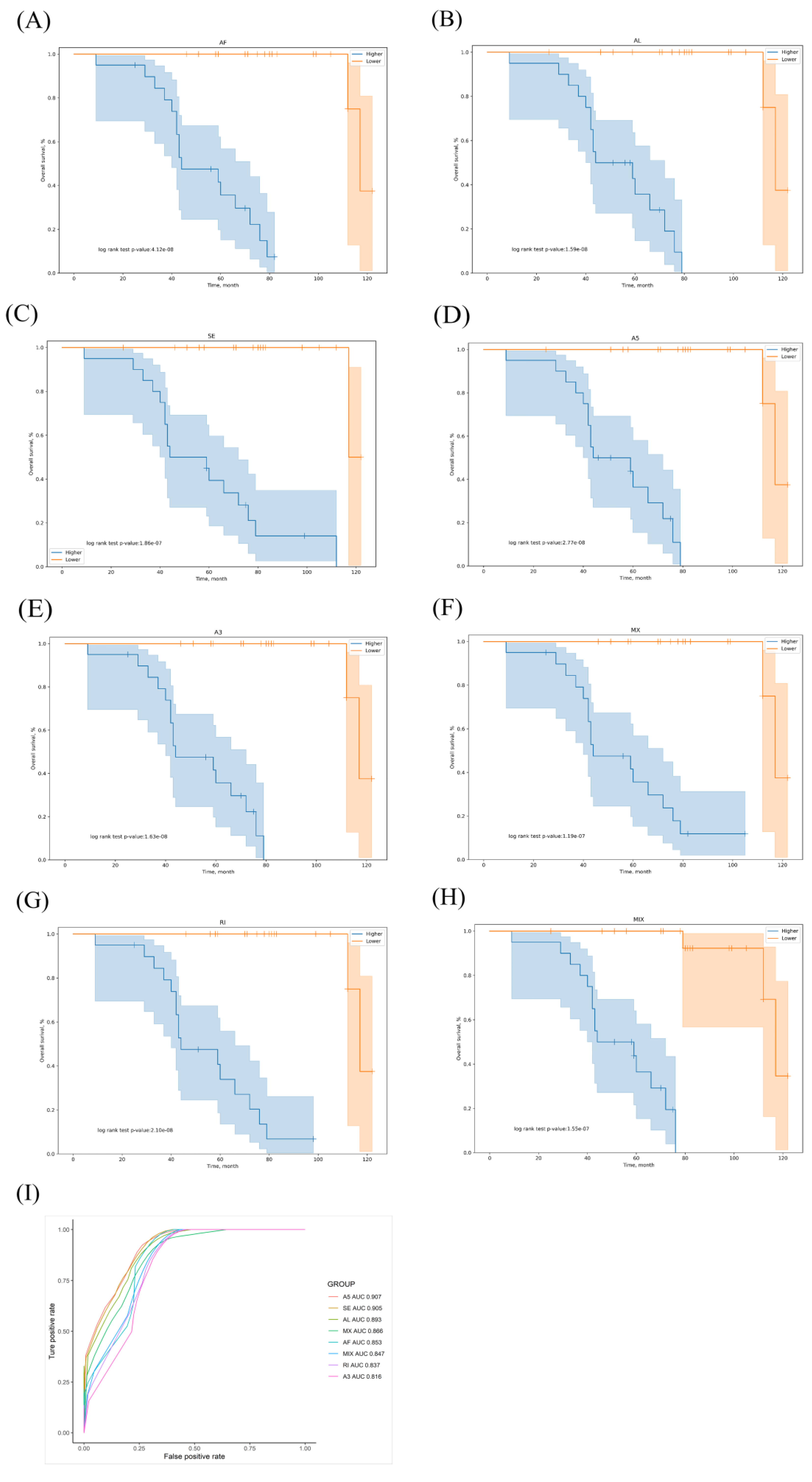
3.2. Prognostic Predictors for the LUAD Cohort
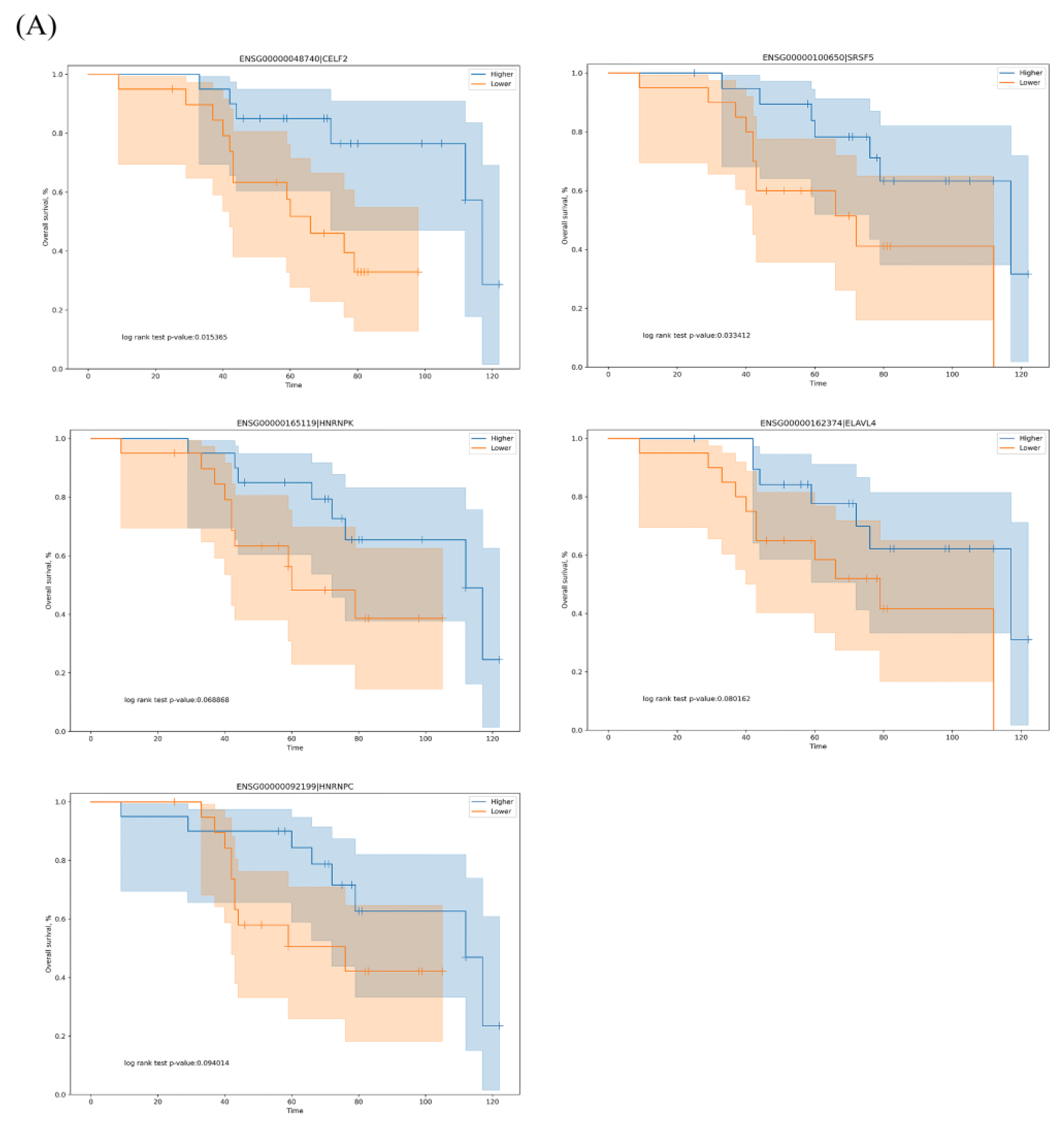
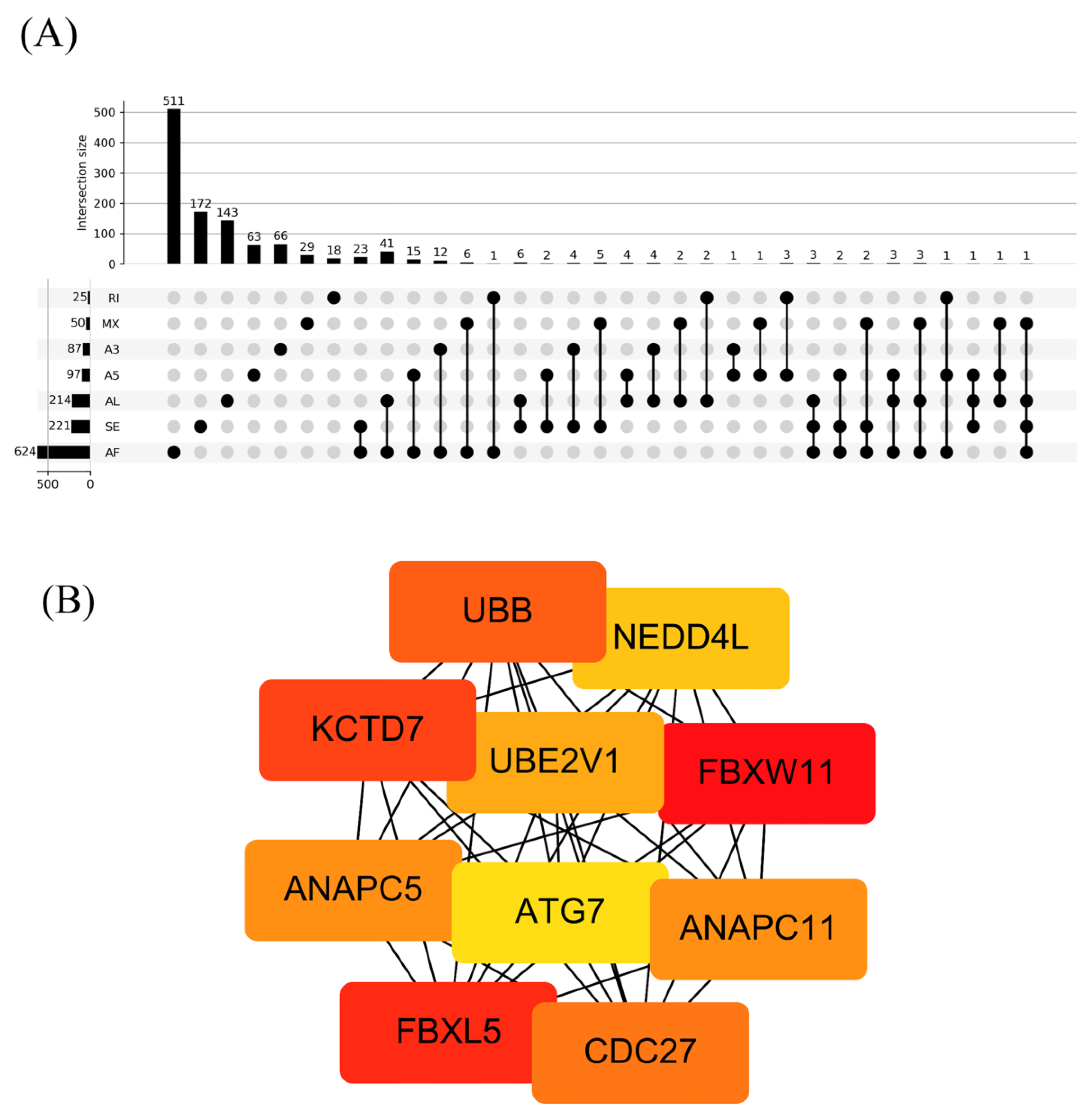
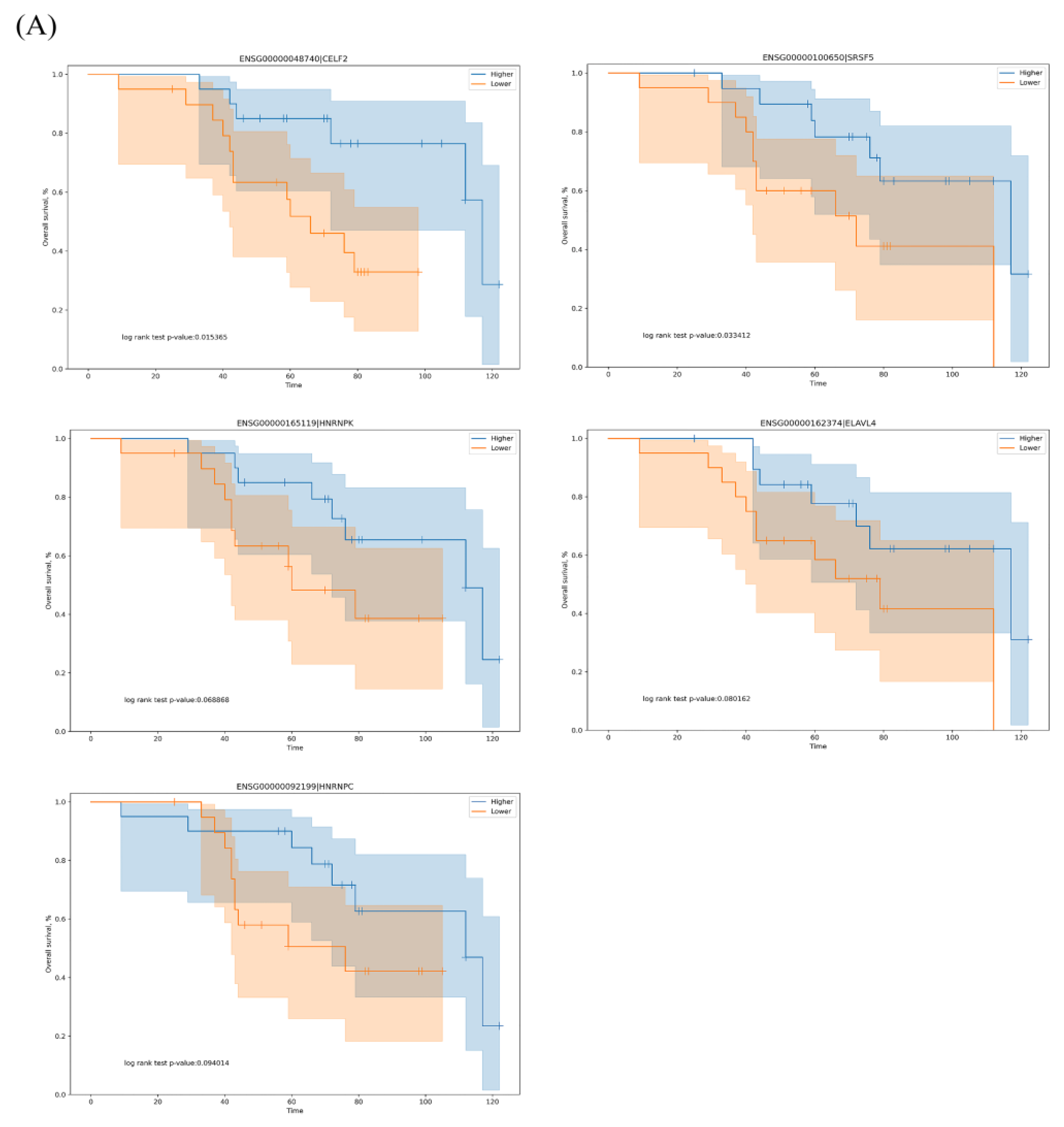
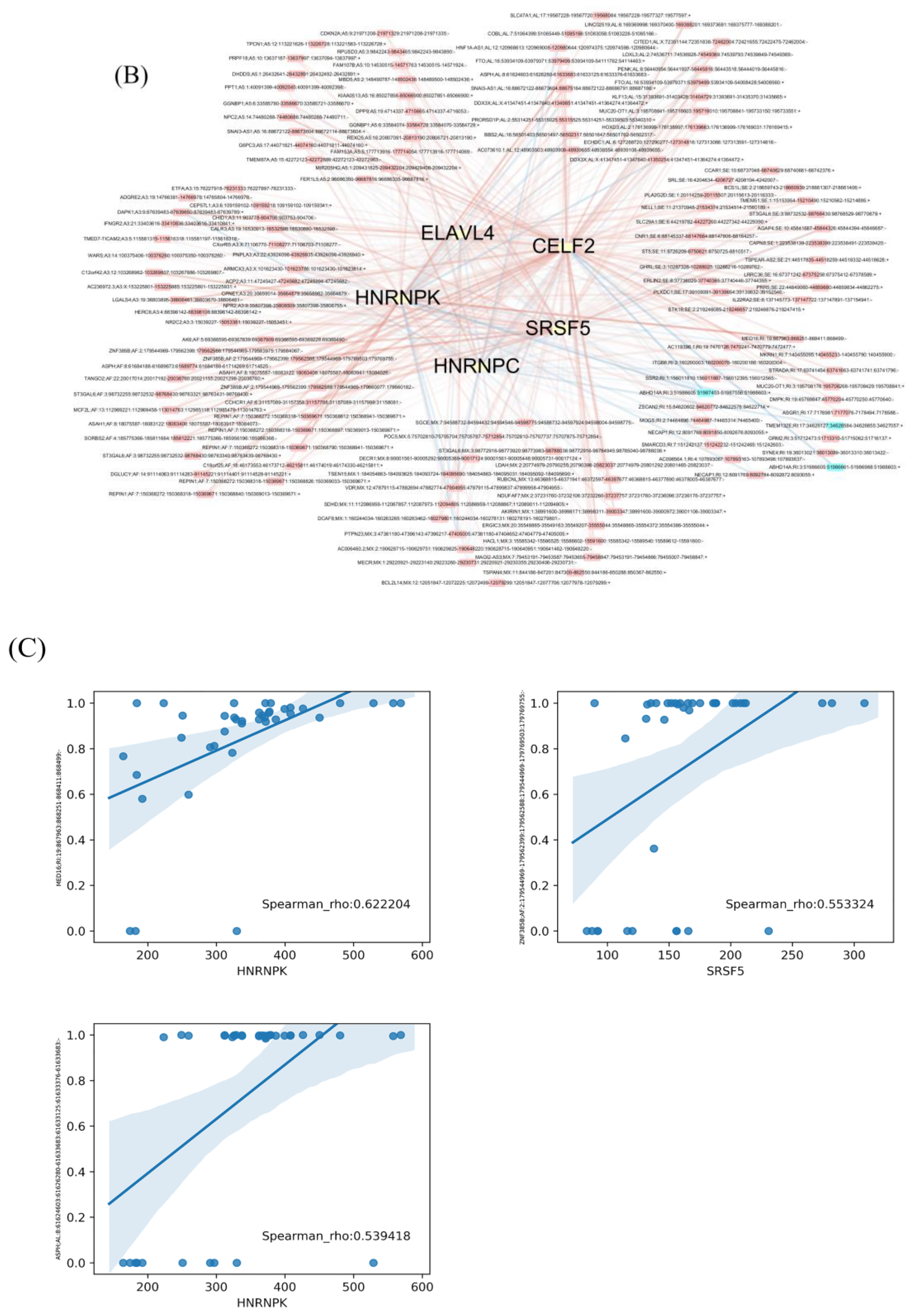
3.3. The Regulation Network of SFs and the Top 20 Significant OS-Related AS Events for the Seven Types of AS
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. IJC 2018, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malhotra, J.; Malvezzi, M.; Negri, E.; La Vecchia, C.; Boffetta, P. Risk factors for lung cancer worldwide. ERS 2016, 48, 889–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gould, M.K.; A Silvestri, G.; Detterbeck, F. Multidisciplinary management of lung cancer. N. Engl. J. Med. 2004, 350, 2008. [Google Scholar] [PubMed]
- Ettinger, D.S.; Akerley, W.; Bepler, G.; Blum, M.G.; Chang, A.; Cheney, R.T.; Chirieac, L.R.; D’Amico, T.A.; Demmy, T.L.; Ganti, A.K.P.; et al. Non–Small Cell Lung Cancer. J. Natl. Compr. Cancer Netw. 2010, 8, 740–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.-C.; Wang, J.; Shao, G.-G.; Wang, Q.; Qu, X.; Wang, B.; Moy, C.; Fan, Y.; Albertyn, Z.; Huang, X.; et al. Comprehensive genomic and immunological characterization of Chinese non-small cell lung cancer patients. Nat. Commun. 2019, 10, 1772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Liu, J.; Shao, D.; Deng, Q.; Tang, H.; Liu, Z.; Chen, X.; Guo, F.; Lin, Y.; Mao, M.; et al. Comprehensive genomic profiling of lung cancer using a validated panel to explore therapeutic targets in East Asian patients. Cancer Sci. 2017, 108, 2487–2494. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef] [PubMed]
- Germann, S.; Gratadou, L.; Dutertre, M.; Auboeuf, D. Splicing Programs and Cancer. J. Nucleic Acids 2011, 2012, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.-D.; Lee, N.H. Aberrant RNA Splicing in Cancer and Drug Resistance. Cancers 2018, 10, 458. [Google Scholar] [CrossRef] [Green Version]
- Venables, J.P. Unbalanced alternative splicing and its significance in cancer. BIOEEJ 2006, 28, 378–386. [Google Scholar] [CrossRef]
- Avery-Kiejda, K.A.; Zhang, X.D.; Adams, L.J.; Scott, R.J.; Vojtesek, B.; Lane, D.P.; Hersey, P. Small Molecular Weight Variants of p53 Are Expressed in Human Melanoma Cells and Are Induced by the DNA-Damaging Agent Cisplatin. Clin. Cancer Res. 2008, 14, 1659–1668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poulikakos, P.I.; Persaud, Y.; Janakiraman, M.; Kong, X.; Ng, C.; Moriceau, G.; Shi, H.; Atefi, M.; Titz, B.; Gabay, M.T.; et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nat. Cell Biol. 2011, 480, 387–390. [Google Scholar] [CrossRef] [Green Version]
- Coomer, A.O.; Black, F.; Greystoke, A.; Munkley, J.; Elliott, D.J. Alternative splicing in lung cancer. Biochim. Biophys. Acta (BBA) Bioenerg. 2019, 1862, 194388. [Google Scholar] [CrossRef] [PubMed]
- Dvinge, H.; Kim, E.; Abdel-Wahab, E.K.O.; Bradley, H.D.R.K. RNA splicing factors as oncoproteins and tumour suppressors. Nat. Rev. Cancer 2016, 16, 413–430. [Google Scholar] [CrossRef]
- Zong, Z.; Li, H.; Yi, C.; Ying, H.; Zhu, Z.; Wang, H. Genome-Wide Profiling of Prognostic Alternative Splicing Signature in Colorectal Cancer. Front. Oncol. 2018, 8, 537. [Google Scholar] [CrossRef]
- Yang, L.; He, Y.; Zhang, Z.; Wang, W. Systematic analysis and prediction model construction of alternative splicing events in hepatocellular carcinoma: A study on the basis of large-scale spliceseq data from The Cancer Genome Atlas. PeerJ 2019, 7, e8245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, L.; Zhang, X.; Tong, D. Systematic Profile Analysis of Prognostic Alternative Messenger RNA Splicing Signatures and Splicing Factors in Head and Neck Squamous Cell Carcinoma. DNA Cell Biol. 2019, 38, 627–638. [Google Scholar] [CrossRef]
- Li, Y.; Sun, N.; Lu, Z.; Sun, S.; Huang, J.; Chen, Z.; He, J. Prognostic alternative mRNA splicing signature in non-small cell lung cancer. Cancer Lett. 2017, 393, 40–51. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Kovaka, S.; Zimin, A.V.; Pertea, G.M.; Razaghi, R.; Salzberg, S.L.; Pertea, M. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 2019, 20, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Hu, Z.; Zhao, Y.; Huang, S.; He, X. Transcriptome-Wide Analysis Reveals the Landscape of Aberrant Alternative Splicing Events in Liver Cancer. Hepatology 2018, 69, 359–375. [Google Scholar] [CrossRef] [Green Version]
- Trincado, J.L.; Entizne, J.C.; Hysenaj, G.; Singh, B.; Skalic, M.; Elliott, D.J.; Eyras, E. SUPPA2: Fast, accurate, and uncertainty-aware differential splicing analysis across multiple conditions. Genome Biol. 2018, 19, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heagerty, P.J.; Lumley, T.; Pepe, M.S. Time-Dependent ROC Curves for Censored Survival Data and a Diagnostic Marker. Biomedical 2000, 56, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Piva, F.; Giulietti, M.; Burini, A.B.; Principato, G. SpliceAid 2: A database of human splicing factors expression data and RNA target motifs. Hum. Mutat. 2012, 33, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Lex, A.; Gehlenborg, N.; Strobelt, H.; Vuillemot, R.; Pfister, H. UpSet: Visualization of Intersecting Sets. IEEE Trans. Vis. Comput. Graph. 2014, 20, 1983–1992. [Google Scholar] [CrossRef]
- Davidson-Pilon, C.; Kalderstam, J.; Jacobson, N.; sean-reed; Kuhn, B.; Zivich, P.; Williamson, M.; Abdeali, J.K.; Datta, D.; Fiore-Gartland, A.; et al. CamDavidsonPilon/lifelines: v0.25.2, Zenodo. 2020. Available online: https://zenodo.org/record/3977479#.X50DxVARXIU(accessed on 30 October 2020).
- Savci-Heijink, C.D.; Halfwerk, H.; Koster, J.; Horlings, H.; Van De Vijver, M.J. A specific gene expression signature for visceral organ metastasis in breast cancer. BMC Cancer 2019, 19, 333. [Google Scholar] [CrossRef]
- Zhang, W.; Zhu, J.; Bai, J.; Jiang, H.; Liu, F.; Liu, A.; Liu, P.; Ji, G.; Guan, R.; Sun, D.; et al. Comparison of the inhibitory effects of three transcriptional variants of CDKN2A in human lung cancer cell line A549. J. Exp. Clin. Cancer Res. 2010, 29, 74. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.; Liu, Y.-H.; Wang, L.-L.; Wang, J.; Zhao, Z.-H.; Qu, J.-F.; Wang, S.-F. MiR-182 promotes cell proliferation by suppressing FBXW7 and FBXW11 in non-small cell lung cancer. Am. J. Transl. Res. 2018, 10, 1131–1142. [Google Scholar] [PubMed]
- Wu, W.-D.; Wang, M.; Ding, H.-H.; Qiu, Z.-J. FBXL5 attenuates RhoGDI2-induced cisplatin resistance in gastric cancer cells. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 2551–2557. [Google Scholar] [PubMed]
- Xiong, Y.; Sun, F.; Dong, P.; Watari, H.; Yue, J.; Yu, M.-F.; Lan, C.-Y.; Wang, Y.; Ma, Z.-B. iASPP induces EMT and cisplatin resistance in human cervical cancer through miR-20a-FBXL5/BTG3 signaling. J. Exp. Clin. Cancer Res. 2017, 36, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Su, S.; Xia, D.; Wang, M.; Li, Z.; Chen, W.; Ren, L.; Xu, L. F-box and leucine-rich repeat protein 5 promotes colon cancer progression by modulating PTEN/PI3K/AKT signaling pathway. Biomed. Pharmacother. 2018, 107, 1712–1719. [Google Scholar] [CrossRef]
- Zhao, D.; Zhang, C.; Jiang, M.; Wang, Y.; Liang, Y.; Wang, L.; Qin, K.; Rehman, F.U.; Zhang, X. Survival-associated alternative splicing signatures in non-small cell lung cancer. Aging 2020, 12, 5878–5893. [Google Scholar] [CrossRef] [PubMed]
- Kędzierska, H.; Piekiełko-Witkowska, A. Splicing factors of SR and hnRNP families as regulators of apoptosis in cancer. Cancer Lett. 2017, 396, 53–65. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, Y.-S.; Tu, S.-J.; Chiang, H.-S.; Yen, J.-C.; Lee, Y.-T.; Fang, H.-Y.; Chang, J.-G. Genome-Wide Analysis of Prognostic Alternative Splicing Signature and Splicing Factors in Lung Adenocarcinoma. Genes 2020, 11, 1300. https://doi.org/10.3390/genes11111300
Chang Y-S, Tu S-J, Chiang H-S, Yen J-C, Lee Y-T, Fang H-Y, Chang J-G. Genome-Wide Analysis of Prognostic Alternative Splicing Signature and Splicing Factors in Lung Adenocarcinoma. Genes. 2020; 11(11):1300. https://doi.org/10.3390/genes11111300
Chicago/Turabian StyleChang, Ya-Sian, Siang-Jyun Tu, Hui-Shan Chiang, Ju-Chen Yen, Ya-Ting Lee, Hsin-Yuan Fang, and Jan-Gowth Chang. 2020. "Genome-Wide Analysis of Prognostic Alternative Splicing Signature and Splicing Factors in Lung Adenocarcinoma" Genes 11, no. 11: 1300. https://doi.org/10.3390/genes11111300