Identification and Characterization of a Novel CLCN7 Variant Associated with Osteopetrosis

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Case Report
2.2. DNA Sequencing
2.3. Sequence Acquisition
2.4. Bioinformatics Analysis
3. Results
3.1. Case Representation
3.2. Mutation Analysis
3.3. Comparative Protein Sequence Analysis and Modelling
3.4. Bioinformatics Analysis for Variant Significance Confirmation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stark, Z.; Savarirayan, R. Osteopetrosis. Orphanet. J. Rare. Dis. 2009, 4, 5. [Google Scholar]
- Sobacchi, C.; Schulz, A.; Coxon, F.P.; Villa, A.; Helfrich, M.H. Osteopetrosis: Genetics, treatment and new insights into osteoclast function. Nat. Rev. Endocrinol. 2013, 9, 522–536. [Google Scholar] [CrossRef] [PubMed]
- Penna, S.; Capo, V.; Palagano, E.; Sobacchi, C.; Villa, A. One disease, many genes: Implications for the treatment of osteopetroses. Front. Endocrinol. 2019, 10, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Superti-Furga, A.; Unger, S. Nosology and classification of genetic skeletal disorders: 2006 revision. Am. J. Med. Genet. 2007, 143A, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Askmyr, M.K.; Fasth, A.; Richter, J. Towards a better understanding and new therapeutics of osteopetrosis. Br. J. Haematol. 2008, 140, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Villa, A.; Guerrini, M.M.; Cassani, B.; Pangrazio, A.; Sobacchi, C. Infantile Malignant, Autosomal Recessive Osteopetrosis: The Rich and The Poor. Calcif. Tissue. Int. 2008, 84, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Pangrazio, A.; Pusch, M.; Caldana, E.; Frattini, A.; Lanino, E.; Tamhankar, P.M.; Phadke, S.; Lopez, A.G.; Orchard, P.; Mihci, E.; et al. Molecular and clinical heterogeneity in CLCN7-dependent osteopetrosis: Report of 20 novel mutations. Hum. Mutat. 2010, 31, E1071–E1080. [Google Scholar] [CrossRef] [PubMed]
- Jentsch, T.J.; Stein, V.; Weinreich, F.; Zdebik, A.A. Molecular structure and physiological function of chloride channels. Physiol. Rev. 2002, 82, 503–568. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Gupta, N. Femoral fractures in osteopetrosis: Case reports. J. Trauma. 2001, 51, 997–999. [Google Scholar] [CrossRef] [PubMed]
- Kornak, U.; Kasper, D.; Bösl, M.R.; Kaiser, E.; Schweizer, M.; Schulz, A.; Friedrich, W.; Delling, G.; Jentsch, T.J. Loss of the CLC-7 chloride channel leads to osteopetrosis in mice and man. Cell 2001, 104, 205–215. [Google Scholar] [CrossRef] [Green Version]
- Frattini, A.; Pangrazio, A.; Susani, L.; Sobacchi, C.; Mirolo, M.; Abinun, M.; Andolina, M.; Flanagan, A.; Horwitz, E.M.; Mihci, E.; et al. Chloride channel CLCN7 mutations are responsible for severe recessive, dominant, and intermediate osteopetrosis. J. Bone Miner. Res. 2003, 18, 1740–1747. [Google Scholar] [CrossRef] [PubMed]
- Adebali, O.; Ortega, D.R.; Zhulin, I.B. CDvist: A webserver for identification and visualization of conserved domains in protein sequences. Bioinformatics 2014, 31, 1475–1477. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.U.; Zoete, V.; Michielin, O.; Guex, N. Defining and searching for structural motifs using DeepView/Swiss-PDBViewer. BMC Bioinformatics 2012, 13, 173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, L.; Campbell, E.B.; Hsiung, Y.; MacKinnon, R. Structure of a eukaryotic CLC transporter defines an intermediate state in the transport cycle. Science 2010, 330, 635–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2-a multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; ACMG Laboratory Quality Assurance Committee; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Adebali, O.; Reznik, A.O.; Ory, D.S.; Zhulin, I.B. Establishing the precise evolutionary history of a gene improves prediction of disease-causing missense mutations. Genet. Med. 2016, 18, 1029–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
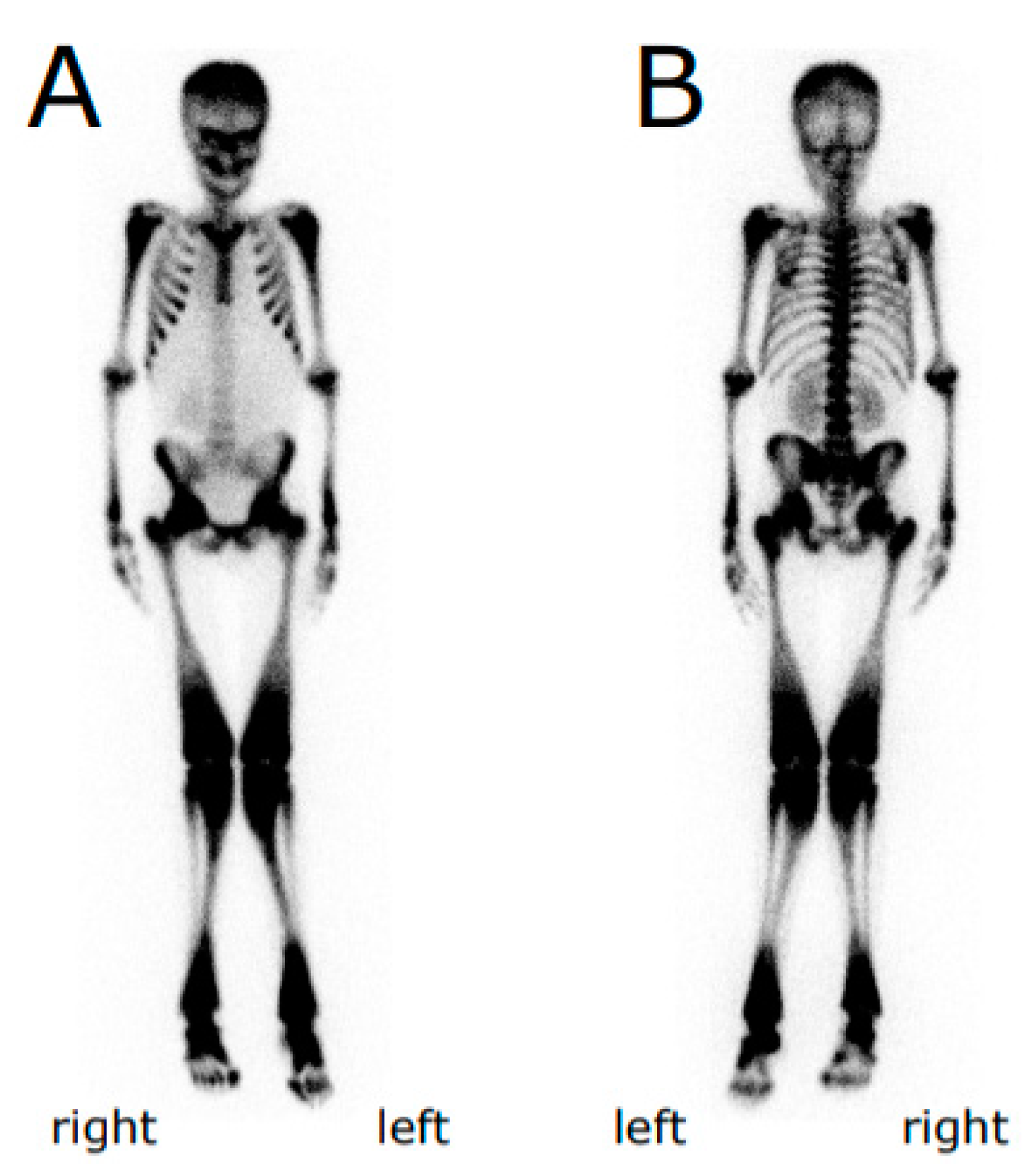
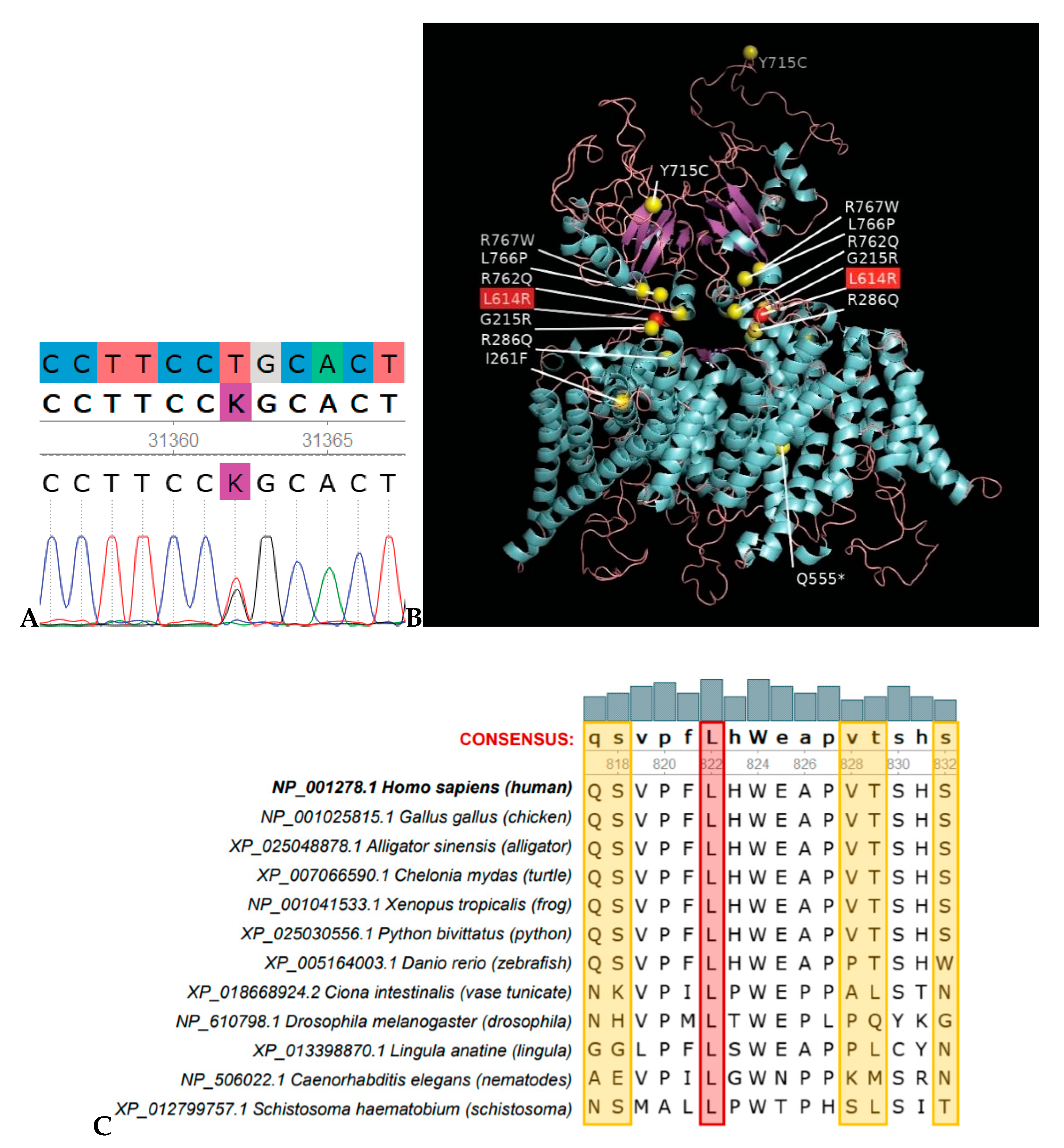

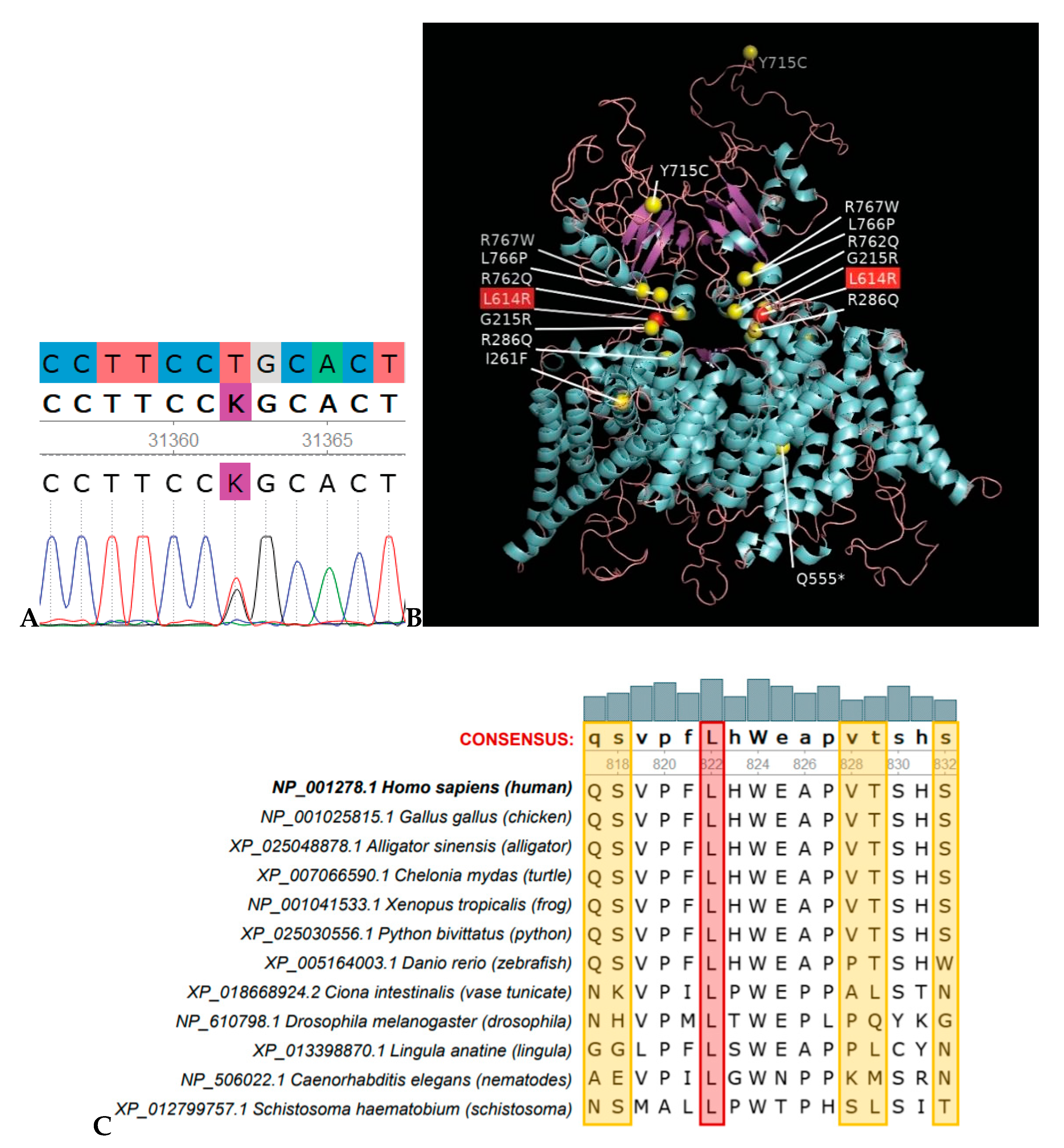
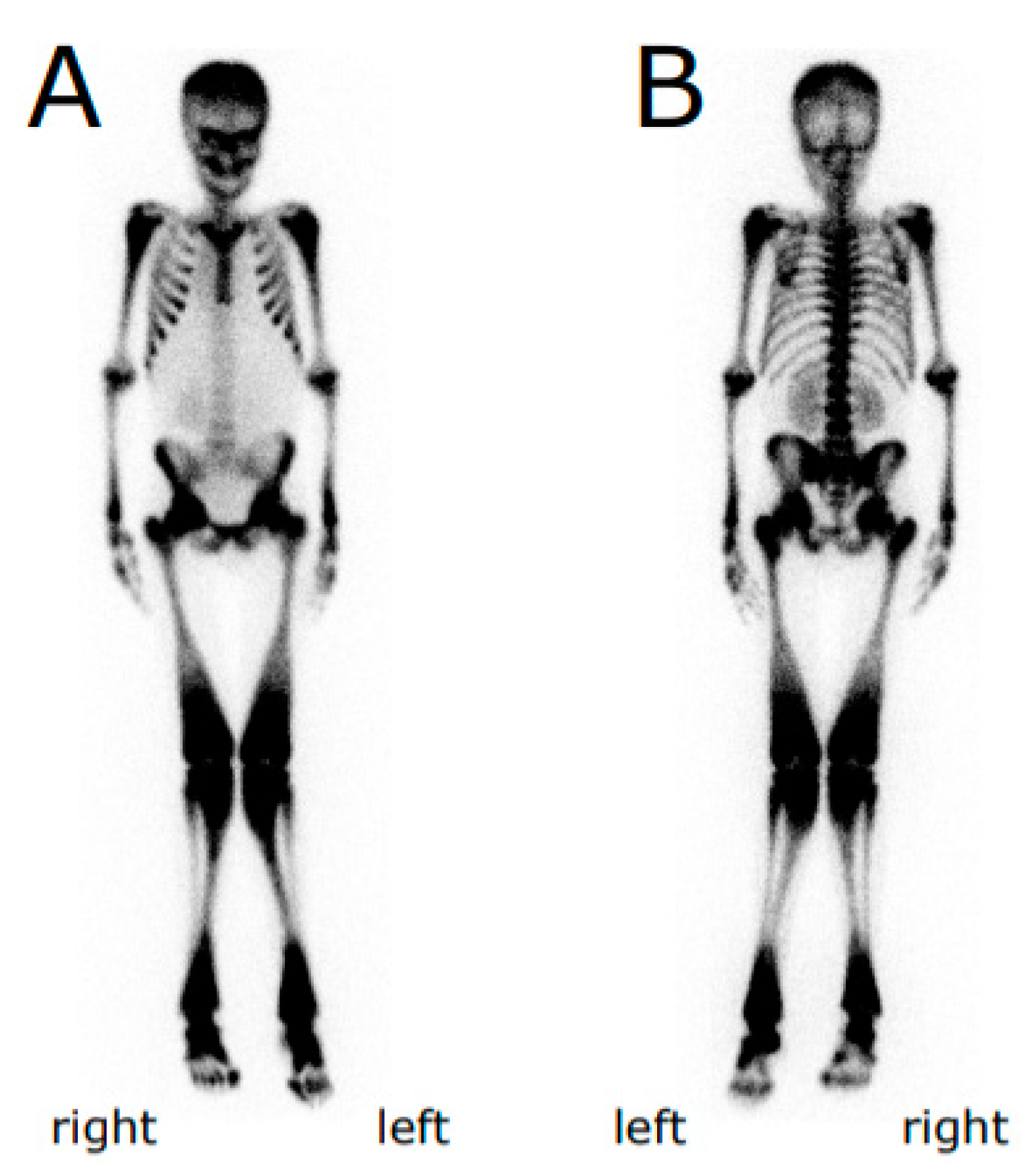{kind=link}
{kind=link}
| Autosomal Dominant Osteopetrosis Type 2 Clinical Features | Proband | Sibling | Relevance/Alternate Explanation |
|---|---|---|---|
| Autosomal dominant inheritance | Yes | Unknown | |
| Facial nerve palsy | No | Unknown | |
| Vision loss, severe, beginning in childhood | No | Unknown | |
| Osteosclerosis, diffuse symmetrical | Yes | Unknown | |
| Increased long bone fracture rate (75% of patients) | Yes | Yes | |
| Multiple fractures | Yes | Yes | |
| Pronounced skull base sclerosis | Unknown | Unknown | |
| Mandibular osteomyelitis | No | Unknown | |
| ‘Rugger-Jersey’ spine (vertebral endplate thickening) | Unknown | Unknown | |
| Endobones (bone within bone) | Unknown | Unknown | |
| Hip osteoarthritis | No | Unknown | |
| Facial palsy due to cranial nerve VII compression | No | Unknown | |
| Bone marrow failure | Yes | No | |
| Elevated serum acid phosphatase | Unknown | Unknown | |
| Onset in childhood | Yes | Yes | |
| Progressive sclerosis with age | Unknown | Unknown | |
| 20–40% patients are asymptomatic | No | No | |
| Other Clinical Features | |||
| Hepatomegaly | Yes | Unknown | Clinical feature of autosomal recessive osteopetrosis type 4 (OMIM* #611490) |
| Splenomegaly | Yes | Unknown | Same as above |
| Anemia | Yes | Unknown | Same as above |
| Reticulocytosis | Yes | Unknown | Same as above |
| Thrombocytopenia | Yes | Unknown | Same as above |
| Failure to thrive | Yes | Unknown | Clinical feature of autosomal recessive osteopetrosis type 1 (OMIM #259700) |
| Hydrocephalus | Yes | Unknown | Same as above |
| Splayed metaphyses | Yes | Unknown | Same as above |
| Low serum calcium | Yes | Unknown | Same as above |
| Elevated alkaline phosphatase | Yes | Unknown | Same as above |
| Valgus deformity | Yes | Unknown | Clinical feature of autosomal recessive osteopetrosis type 2 (OMIM #259710) |
| Dental anomalies | Yes | Unknown | Same as above |
| Elevated serum lactate dehydrogenase | Yes | Unknown | Clinical feature of autosomal recessive osteopetrosis type 5 (OMIM #259720) |
| Lymphocytosis | Yes | Unknown | Assumed related |
| Skeletal effects | Yes | Unknown | Assumed related |
| Scoliosis | Yes | Unknown | Assumed related |
| Low hairline | Yes | Unknown | Assumed related |
| Double xiphoid process | Yes | Unknown | Assumed related |
| Gene | Genomic Location | DNA Reference | Protein Reference | Variant Type | Genotype | Origin | Observed Effect |
|---|---|---|---|---|---|---|---|
| CLCN7 | Chr16: 1498724 (GRCh37) | NM_001287.6: c.1841T>G | NP_001278.1: p.Leu614Arg | missense | heterozygous | Unknown | Deleterious |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bug, D.S.; Barkhatov, I.M.; Gudozhnikova, Y.V.; Tishkov, A.V.; Zhulin, I.B.; Petukhova, N.V. Identification and Characterization of a Novel CLCN7 Variant Associated with Osteopetrosis. Genes 2020, 11, 1242. https://doi.org/10.3390/genes11111242
Bug DS, Barkhatov IM, Gudozhnikova YV, Tishkov AV, Zhulin IB, Petukhova NV. Identification and Characterization of a Novel CLCN7 Variant Associated with Osteopetrosis. Genes. 2020; 11(11):1242. https://doi.org/10.3390/genes11111242
Chicago/Turabian StyleBug, Dmitrii S., Ildar M. Barkhatov, Yana V. Gudozhnikova, Artem V. Tishkov, Igor B. Zhulin, and Natalia V. Petukhova. 2020. "Identification and Characterization of a Novel CLCN7 Variant Associated with Osteopetrosis" Genes 11, no. 11: 1242. https://doi.org/10.3390/genes11111242