Genotypic Categorization of Loeys-Dietz Syndrome Based on 24 Novel Families and Literature Data


 ,
,  , ,
, , 
 , , and
, , and
Abstract
:1. Introduction
2. Patients and Methods
2.1. Patients
2.2. Molecular Investigations
2.3. Genotype-Phenotype Analysis and Literature Review
2.4. Ethical Compliance
3. Results
3.1. Demographic Data and Genotype-Phenotype Analysis of LDS Patient’s Cohort
3.2. Molecular Findings
4. Discussion
4.1. TGFBR1/2 Genes
4.2. SMAD3 Gene
4.3. TGFB2 Gene
4.4. Other Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Loeys, B.L.; Chen, J.; Neptune, E.R.; Judge, D.P.; Podowski, M.; Holm, T.; Meyers, J.; Leitch, C.C.; Katsanis, N.; Sharifi, N.; et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat. Genet. 2005, 37, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Loeys, B.L.; Schwarze, U.; Holm, T.; Callewaert, B.L.; Thomas, G.H.; Pannu, H.; De Backer, J.F.; Oswald, G.L.; Symoens, S.; Manouvrier, S.; et al. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N. Engl. J. Med. 2006, 355, 788–798. [Google Scholar] [CrossRef] [PubMed]
- Schepers, D.; Tortora, G.; Morisaki, H.; MacCarrick, G.; Lindsay, M.; Liang, D.; Mehta, S.G.; Hague, J.; Verhagen, J.; van de Laar, I.; et al. A mutation update on the LDS-associated genes TGFB2/3 and SMAD2/3. Hum. Mutat. 2018, 39, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Van de Laar, I.M.; Oldenburg, R.A.; Pals, G.; Roos-Hesselink, J.; de Graaf, B.; Verhagen, J.; Hoedemaekrs, Y.; Willemsen, R.; Severijnen, L.; Venselaar, H.; et al. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat. Genet. 2011, 43, 121–126. [Google Scholar] [CrossRef] [PubMed]
- MacCarrick, G.; Black, J.H., 3rd; Bowdin, S.; El-Hamamsy, I.; Frischmeyer-Guerrerio, P.A.; Guerrerio, A.L.; Sponseller, P.D.; Loeys, B.; Dietz, H.C., 3rd. Loeys-Dietz syndrome: A primer for diagnosis and management. Genet. Med. 2014, 16, 576–587. [Google Scholar] [CrossRef] [PubMed]
- Loeys, B.L.; Dietz, H.C. Loeys-Dietz Syndrome. In GeneReviews®; University of Washington: Seattle, WA, USA, 2008. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1133/ (accessed on 21 August 2019).
- The Genome Aggregation Database (gnomAD) v2.1.1. Available online: https://gnomad.broadinstitute.org (accessed on 21 August 2019).
- Leiden Open Variation Database (LOVD). Available online: https://databases.lovd.nl/shared/genes (accessed on 21 August 2019).
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Micha, D.; Guo, D.C.; Hilhorst-Hofstee, Y.; van Kooten, F.; Atmaja, D.; Overwater, E.; Cayami, F.K.; Regalado, E.S.; van Uffelen, R.; Venselaar, H.; et al. SMAD2 mutations are associated with arterial aneurysms and dissections. Hum. Mutat. 2015, 36, 1145–1149. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, M.E.; Schepers, D.; Bolar, N.A.; Doyle, J.J.; Gallo, E.; Fert-Bober, J.; Kempers, M.J.; Fishman, E.K.; Chen, Y.; Myers, L.; et al. Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat. Genet. 2012, 44, 922–927. [Google Scholar] [CrossRef] [PubMed]
- Bertoli-Avella, A.M.; Gillis, E.; Morisaki, H.; Verhagen, J.M.A.; de Graaf, B.M.; van de Beek, G.; Gallo, E.; Kruithof, B.P.T.; Venselaar, H.; Myers, L.A.; et al. Mutations in a TGF-β ligand, TGFB3, cause syndromic aortic aneurysms and dissections. J. Am. Coll. Cardiol. 2015, 65, 1324–1336. [Google Scholar] [CrossRef] [PubMed]
- Cannaerts, E.; Kempers, M.; Maugeri, A.; Marcelis, C.; Gardeitchik, T.; Richer, J.; Micha, D.; Beauchesne, L.; Timmermans, J.; Vermeersch, P.; et al. Novel pathogenic SMAD2 variants in five families with arterial aneurysm and dissection: Further delineation of the phenotype. J. Med. Genet. 2019, 56, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, B.M.; Braverman, A.C.; Anadkat, M.J. Multiple facial milia in patients with Loeys-Dietz syndrome. Arch. Dermatol. 2011, 147, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Morkmued, S.; Hemmerle, J.; Mathieu, E.; Laugel-Haushalter, V.; Dabovic, B.; Rifkin, D.B.; Dollé, P.; Niederreither, K.; Bloch-Zupan, A. Enamel and dental anomalies in latent-transforming growth factor beta-binding protein 3 mutant mice. Eur. J. Oral Sci. 2017, 125, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Wischmeijer, A.; Van Laer, L.; Tortora, G.; Bolar, N.A.; Van Camp, G.; Fransen, E.; Peeters, N.; Di Bartolomeo, R.; Pacini, D.; Gargiulo, G.; et al. Thoracic aortic aneurysm in infancy in aneurysms-osteoarthritis syndrome due to a novel SMAD3 mutation: Further delineation of the phenotype. Am. J. Med. Genet. A 2013, 161, 1028–1035. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Horie, M.; Nagase, T. TGF-β Signaling in Lung Health and Disease. Int. J. Mol. Sci. 2018, 19, 2460. [Google Scholar] [CrossRef] [PubMed]
- Ritelli, M.; Chiarelli, N.; Dordoni, C.; Quinzani, S.; Venturini, M.; Maroldi, R.; Calzavara-Pinton, P.; Colombi, M. Further delineation of Loeys-Dietz syndrome type 4 in a family with mild vascular involvement and a TGFB2 splicing mutation. BMC Med. Genet. 2014, 15, 91. [Google Scholar] [CrossRef] [PubMed]
- Mazzella, J.M.; Frank, M.; Collignon, P.; Langeois, M.; Legrand, A.; Jeunemaitre, X.; Albuisson, J. Phenotypic variability and diffuse arterial lesions in a family with Loeys-Dietz syndrome type 4. Clin. Genet. 2017, 91, 458–462. [Google Scholar] [CrossRef] [PubMed]
- Sanford, L.P.; Ormsby, I.; Gittenberger-de Groot, A.C.; Sariola, H.; Friedman, R.; Boivin, G.P.; Cardell, E.L.; Doetschman, T. TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development 1997, 124, 2659–2670. [Google Scholar] [PubMed]
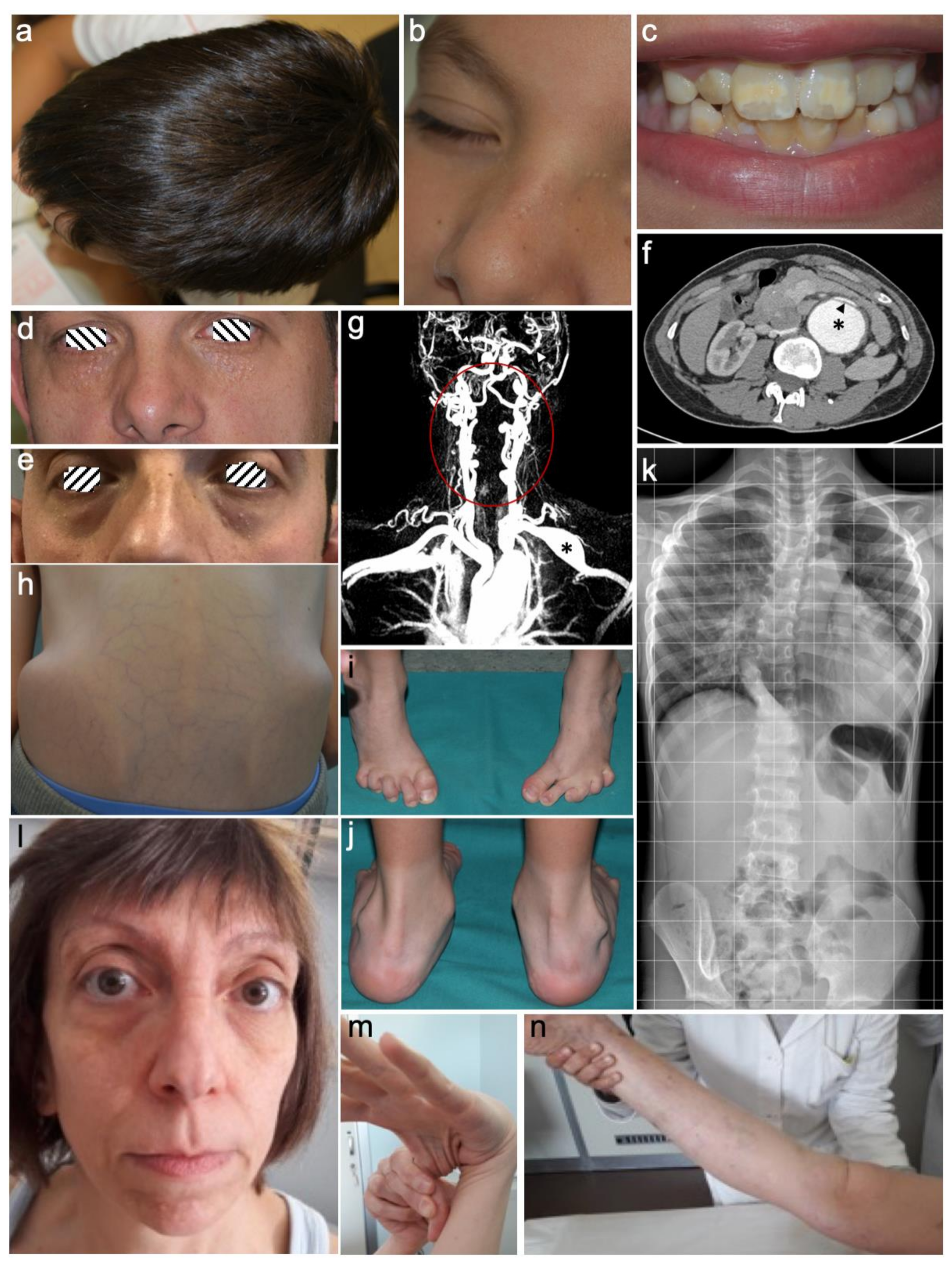

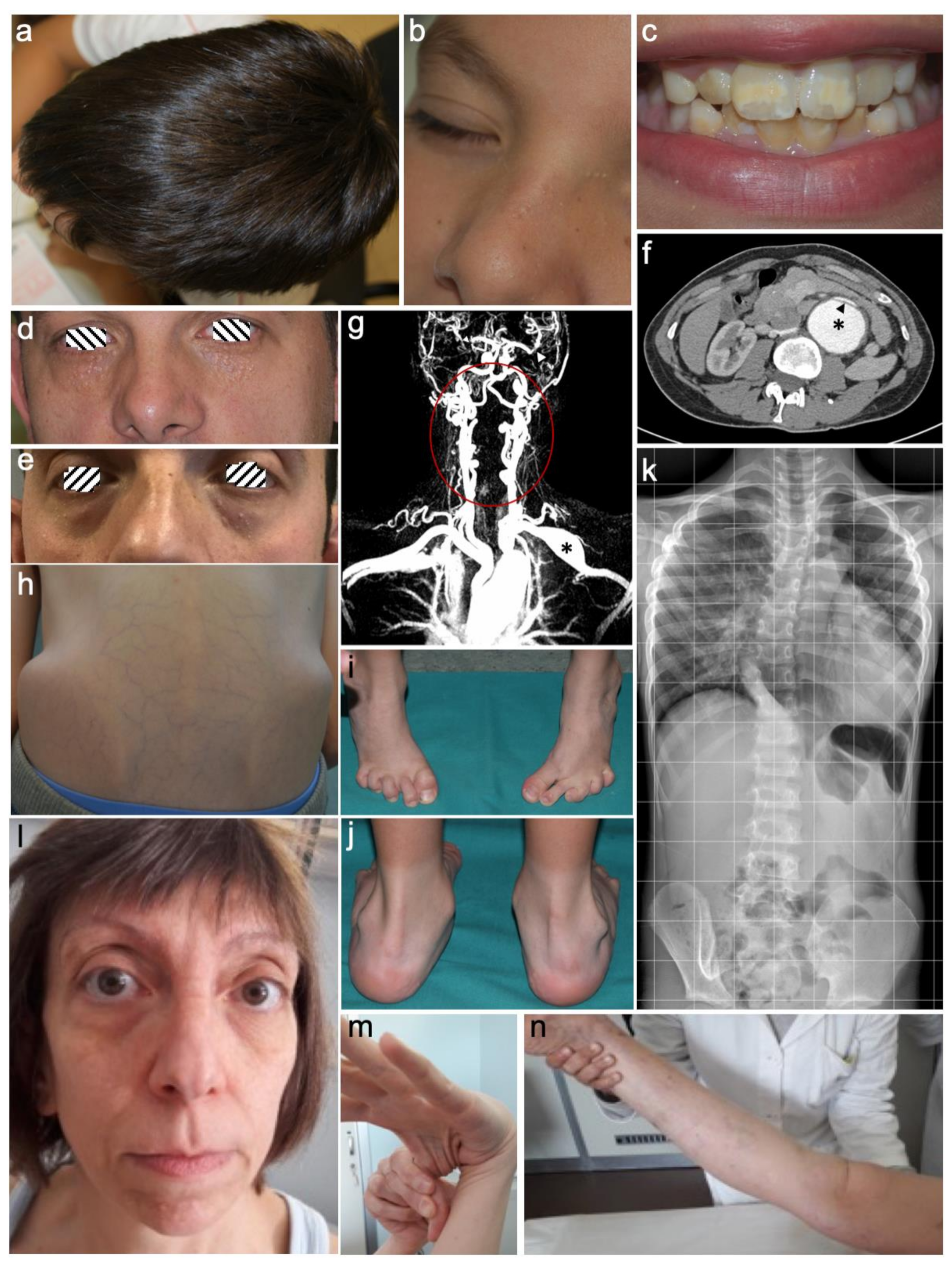{kind=link}
| Clinical Features | TGFBR1 | TGFBR2 | SMAD3 | TGFB2 | SMAD2 | TGFB3 | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Lit. | This cohort n = 12 (%) | Lit. | This cohort n = 12 (%) | Lit. | This cohort n = 9 (%) | Lit. | This cohort n = 1 | Lit. | Lit. | |
| Hypertelorism | ++++ | 10/12 (84) | ++++ | 6/12 (50) | ++ | 4/9 (44) | ++ | 1/1 | + | ++ |
| Strabismus | + | 1/12 (8) | + | 1/12 (8) | + | 0/9 | + | 0/1 | − | − |
| Malar hypoplasia | +++ | 8/12 (67) | +++ | 9/12 (75) | ++ | 9/9 (100) | ++ | 1/1 | ++++ | ++ |
| Bifid uvula/Cleft palate | ++++ | 3/12 (25) | ++++ | 5/12 (42) | ++ | 2/9 (22) | + | 1/1 | − | ++ |
| Dolichocephaly | +++ | 11/12 (92) | +++ | 7/12 (58) | + | 3/9 (33) | − | 0/1 | ++++ | − |
| Hernia | +++ | 4/12 (33) | +++ | 6/12 (50) | ++ | 5/9 (55) | ++ | 1/1 | ++++ | ++ |
| Striae | ++ | 5/12 (42) | ++ | 3/12 (25) | ++ | 3/8 (37) | ++ | 1/1 | ++ | + |
| Pectus deformity | +++ | 5/12 (42) | +++ | 7/12 (58) | ++ | 6/9 (66) | ++ | 1/1 | ++ | +++ |
| Scoliosis | +++ | 10/12 (84) | +++ | 8/12 (67) | ++ | 3/9 (33) | ++ | 1/1 | ++ | +++ |
| Arachnodactyly | +++ | 5/12 (42) | +++ | 6/12 (50) | ++ | 1/9 (11) | ++ | 1/1 | ++ | ++ |
| Talipes equinovarus | ++ | 1/12 (8) | ++ | 5/12 (42) | + | 1/9 (11) | + | 0/1 | − | ++ |
| Osteoarthritis | ++ | 0/11 | ++ | 0/10 | ++ | 3/6 (50) | + | 0/1 | ++++ | ++ |
| Cervical spine malformation/instability | + | 1/11 (9) | + | 2/9 (22) | + | 0/3 | − | 1/1 | − | + |
| Dural ectasia | ++ | 1/11 (9) | ++ | 3/8 (37) | +++ | 1/4 (25) | ++ | 1/1 | + | − |
| Mitral valve prolapse or insufficiency | ++ | 5/12 (42) | ++ | 7/10 (70) | ++ | 5/9 (55) | ++ | 1/1 | ++ | ++ |
| Arterial tortuosity | ++++ | 3/11 (27) | ++++ | 5/11 (45) | ++ | 1/6 (17) | ++ | 1/1 | + | + |
| Aortic root aneurysm | ++++ | 12/12 (100) | ++++ | 9/12 (75) | +++ | 7/9 (77) | +++ | 0/1 | ++++ | ++ |
| Arterial aneurysms | +++ | 5/12 (42) | +++ | 5/11 (45) | + | 2/9 (22) | + | 0/1 | + | + |
| Aortic dissection | ++++ | 3/12 (25) | ++++ | 2/12 (17) | ++ | 1/9 (11) | + | 0/1 | − | ++ |
| Gender | Age at diagnosis | Family history | Origin | Gene | HGVS | Protein | dbSNP | Patient ID (LOVD) | Variant ID (LOVD) |
|---|---|---|---|---|---|---|---|---|---|
| M | 7 years | − | Italy | TGFBR1 | c.1199A>G | p.(Asp400Gly) | rs121918711 | #00245208 | #0000498906 |
| F | 31 years | + | Italy | TGFBR1 | c.1120G>A | p.(Gly374Arg) § | #00245211 | #0000498909 | |
| F | 29 years | − | Italy | TGFBR1 | c.1052A>T | p.(Asp351Val) § | #00245212 | #0000498911 | |
| M | 29 years | + | Italy | TGFBR1 | c.812G>A | p.(Gly271Asp) § | #00245213 | #0000498912 | |
| M | 47 years | + | Italy | TGFBR1 | c.705_707del | p.(Ser236del) | rs863223830 | #00245343 | #0000499180 |
| F | 17 years | − | Philippines | TGFBR1 | c.650G>T | p.(Gly217Val) § | #00245345 | #0000499182 | |
| M | 23 years | − | Italy | TGFBR1 | c.1057G>C | p.(Gly353Arg) § | #00245346 | #0000499183 | |
| M | 17 years | − | Italy | TGFBR1 | c.1460G>A | p.(Arg487Gln) | rs113605875 | #00245347 | #0000499184 |
| M | 43 years | + | Italy | TGFBR1 | c.693_699delinsC | p.(Lys232_Ile233del) § | #00245348 | #0000499185 | |
| F | 51 years | + | Italy | TGFBR2 | c.1609C>T | p.(Arg537Cys) | rs104893809 | #00245350 | #0000499187 |
| F | 3 years | − | Sri Lanka | TGFBR2 | c.1582C>T | p.(Arg528Cys) | rs104893810 | #00245351 | #0000499232 |
| F | 3 years | − | Italy | TGFBR2 | c.1598G>T | p.(Cys533Phe) § | #00245396 | #0000499233 | |
| M | 9 years | − | Italy | TGFBR2 | c.1336G>T | p.(Asp446Tyr) § | #00245398 | #0000499234 | |
| F | 45 years | + | Italy | TGFBR2 | c.263+6C>T | r.263_264insguaa * p.(Arg114 *) § | rs758501054 | #00245408 | #0000499245 |
| F | 36 years | + | Italy | TGFBR2 | c.1564G>A | p.(Asp522Asn) | rs863223854 | #00245409 | #0000499246 |
| M | 1 year | − | Italy | TGFBR2 | c.1336G>A | p.(Asp446Asn) | rs886039551 | #00245410 | #0000499247 |
| M | 37 years | + | Italy | TGFBR2 | c.1187G>A | p.(Cys396Tyr) § | #00245411 | #0000499248 | |
| F | 3 years | − | Italy | TGFBR2 | c.1184T>C | p.(Leu395Pro) § | #00245412 | #0000499249 | |
| M | 12 years | − | Italy | TGFBR2 | c.1270T>G | p.(Tyr424Asp) § | #00245413 | #0000499250 | |
| F | 31 years | + | Italy | SMAD3 | c.1247C>T | p.(Ser416Phe) | #00245414 | #0000499251 | |
| M | 13 years | + | Italy | SMAD3 | c.1009 + 1G>A | r.872_1009del * p.(Arg292_Gly337del) § | #00245415 | #0000499252 | |
| F | 41 years | + | Italy | SMAD3 | c.803G>A | p.(Arg268His) | rs863223740 | #00245416 | #0000499253 |
| M | 23 years | + | Italy | SMAD3 | c.862_871+8del | p.(Arg288Glufs*50) § | #00245417 | #0000499254 | |
| F | 48 years | + | Italy | TGFB2 | c.480del | p.(Phe160Leufs*14) § | #00245418 | #0000499255 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Camerota, L.; Ritelli, M.; Wischmeijer, A.; Majore, S.; Cinquina, V.; Fortugno, P.; Monetta, R.; Gigante, L.; Marfan Syndrome Study Group Tor Vergata University Hospital; Sangiuolo, F.C.; et al. Genotypic Categorization of Loeys-Dietz Syndrome Based on 24 Novel Families and Literature Data. Genes 2019, 10, 764. https://doi.org/10.3390/genes10100764
Camerota L, Ritelli M, Wischmeijer A, Majore S, Cinquina V, Fortugno P, Monetta R, Gigante L, Marfan Syndrome Study Group Tor Vergata University Hospital, Sangiuolo FC, et al. Genotypic Categorization of Loeys-Dietz Syndrome Based on 24 Novel Families and Literature Data. Genes. 2019; 10(10):764. https://doi.org/10.3390/genes10100764
Chicago/Turabian StyleCamerota, Letizia, Marco Ritelli, Anita Wischmeijer, Silvia Majore, Valeria Cinquina, Paola Fortugno, Rosanna Monetta, Laura Gigante, Marfan Syndrome Study Group Tor Vergata University Hospital, Federica Carla Sangiuolo, and et al. 2019. "Genotypic Categorization of Loeys-Dietz Syndrome Based on 24 Novel Families and Literature Data" Genes 10, no. 10: 764. https://doi.org/10.3390/genes10100764