Roles of Calcium Regulating MicroRNAs in Cardiac Ischemia-Reperfusion Injury

{kind=link}
{kind=link}
Abstract
:1. Introduction
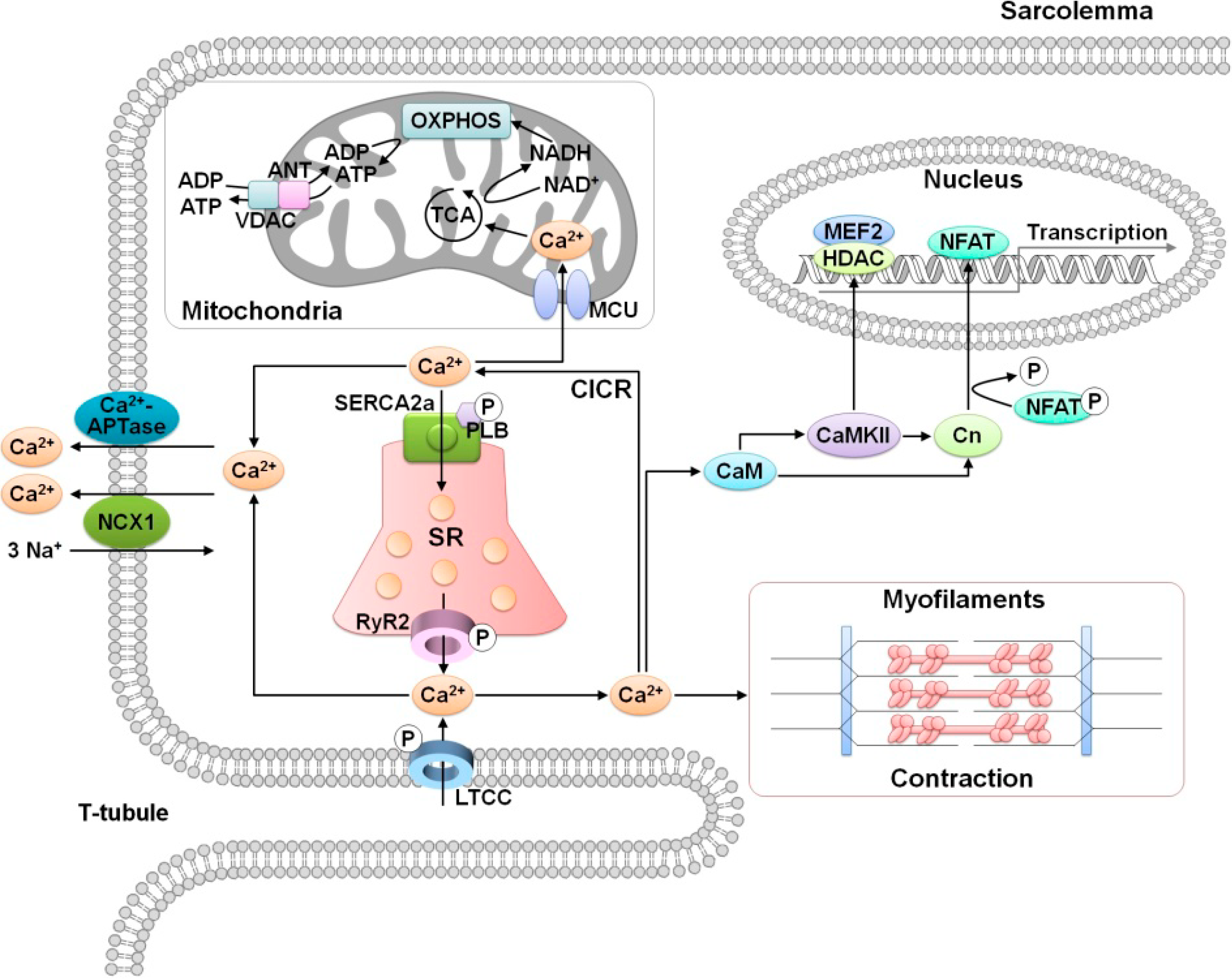
2. Role of Calcium in Cardiac Function
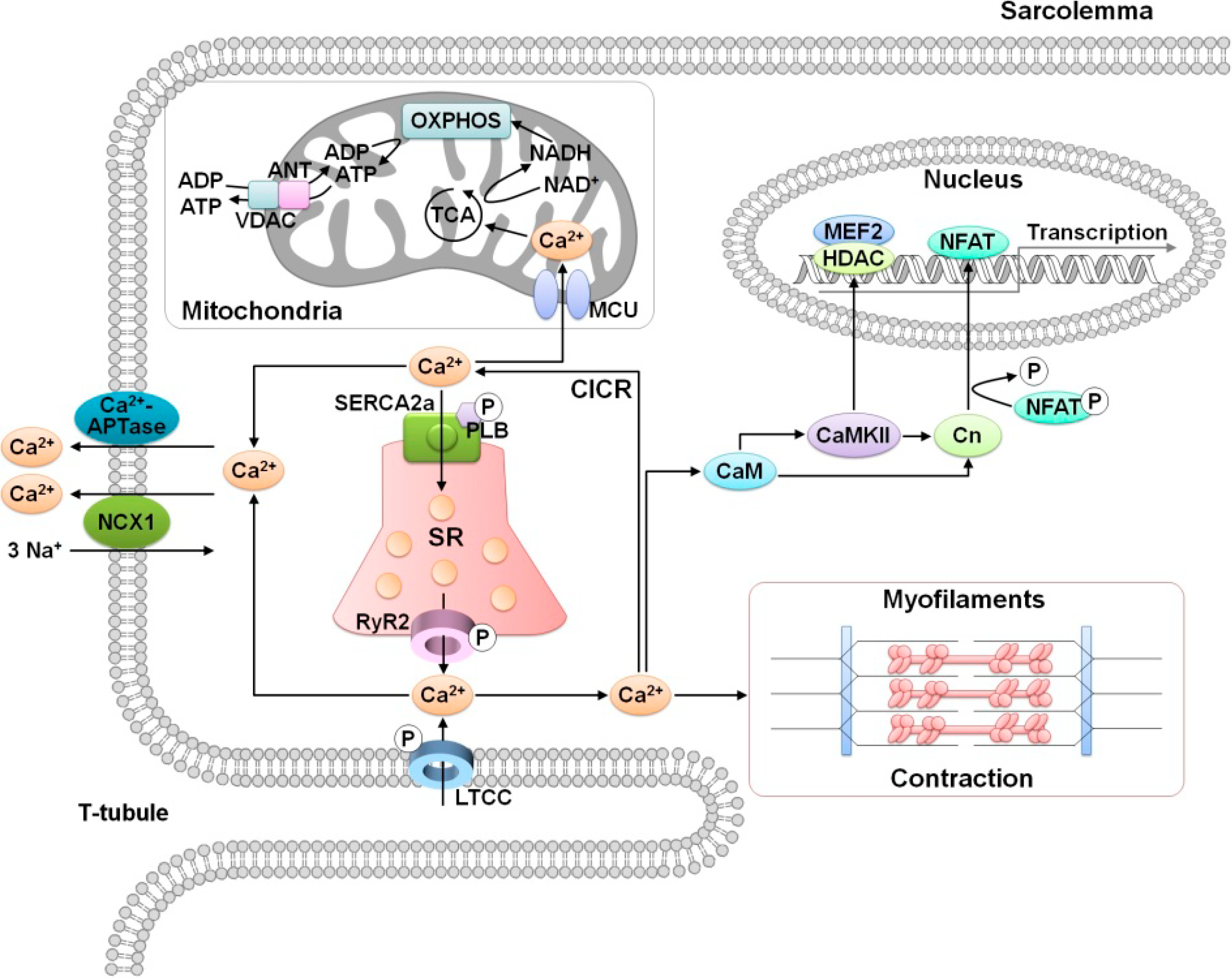
3. MicroRNA Regulation of Calcium Signaling in Myocardial Ischemia-Reperfusion Injury
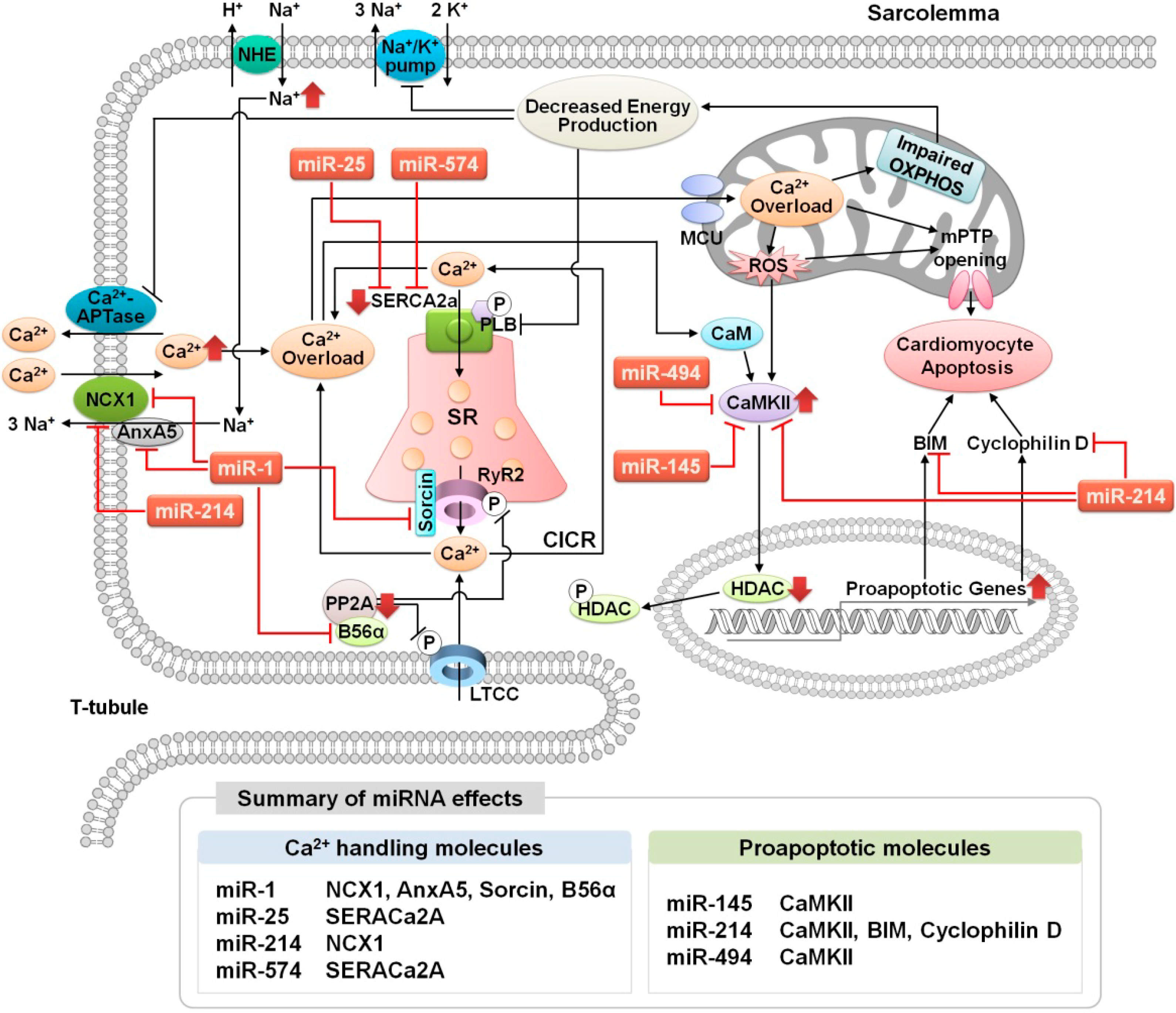
3.1. Altered Ca2+ Homeostasis During Cardiac Ischemia-Reperfusion Injury
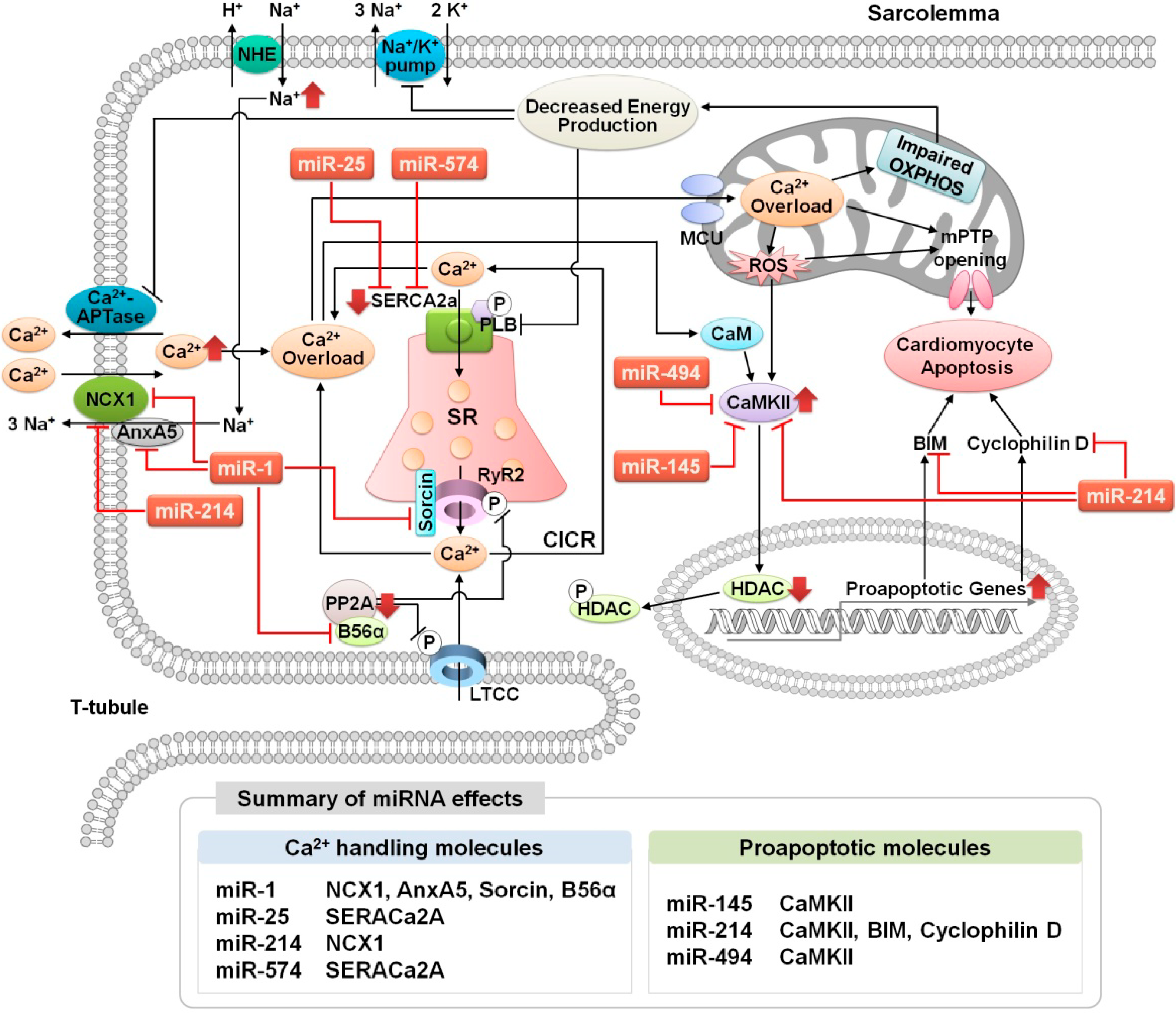
3.2. MicroRNA Regulation of Ca2+ Signaling During Cardiac Ischemia-Reperfusion Injury
3.2.1. miR-25
3.2.2. miR-1
3.2.3. miR-145
3.2.4. miR-214
3.2.5. miR-494
3.2.6. miR-574-3p
3.2.7. miRNAs Related to Cardiomyocyte Cell Death: miR-15, miR-21, and miR-144/451 Cluster
3.3. Circulating MicroRNAs as New Biomarkers
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Luo, M.; Anderson, M.E. Mechanisms of altered Ca2+ handling in heart failure. Circ. Res. 2013, 113, 690–708. [Google Scholar] [CrossRef] [PubMed]
- Buja, L.M. Myocardial ischemia and reperfusion injury. Cardiovasc. Pathol. 2005, 14, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Viola, H.M.; Hool, L.C. How does calcium regulate mitochondrial energetics in the heart?—New insights. Heart Lung Circ. 2014, 23, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Kho, C.; Lee, A.; Hajjar, R.J. Altered sarcoplasmic reticulum calcium cycling—targets for heart failure therapy. Nat. Rev. Cardiol. 2012, 9, 717–733. [Google Scholar] [CrossRef] [PubMed]
- Peterson, S.M.; Thompson, J.A.; Ufkin, M.L.; Sathyanarayana, P.; Liaw, L.; Congdon, C.B. Common features of microRNA target prediction tools. Front. Genet. 2014, 5, 23. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.; Choi, E.; Hwang, K.C. MicroRNAs as novel regulators of stem cell fate. World J. Stem Cell. 2013, 5, 172–187. [Google Scholar] [CrossRef]
- Macfarlane, L.A.; Murphy, P.R. MicroRNA: Biogenesis, function and role in cancer. Curr. Genom. 2010, 11, 537–561. [Google Scholar] [CrossRef]
- Elramah, S.; Landry, M.; Favereaux, A. MicroRNAs regulate neuronal plasticity and are involved in pain mechanisms. Front. Cell. Neurosci. 2014, 8. [Google Scholar] [CrossRef]
- Di Leva, G.; Garofalo, M.; Croce, C.M. MicroRNAs in cancer. Annu. Rev. Pathol. 2014, 9, 287–314. [Google Scholar]
- Winter, J.; Jung, S.; Keller, S.; Gregory, R.I.; Diederichs, S. Many roads to maturity: MicroRNA biogenesis pathways and their regulation. Nat. Cell Biol. 2009, 11, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Harada, M.; Luo, X.; Murohara, T.; Yang, B.; Dobrev, D.; Nattel, S. MicroRNA regulation and cardiac calcium signaling: Role in cardiac disease and therapeutic potential. Circ. Res. 2014, 114, 689–705. [Google Scholar] [CrossRef] [PubMed]
- Aghabozorg Afjeh, S.S.; Ghaderian, S.M. The role of microRNAs in cardiovascular disease. Int. J. Mol. Cell. Med. 2013, 2, 50–57. [Google Scholar]
- Ling, H.; Fabbri, M.; Calin, G.A. MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nat. Rev. Drug Discov. 2013, 12, 847–865. [Google Scholar] [CrossRef] [PubMed]
- Vickers, K.C.; Rye, K.A.; Tabet, F. MicroRNAs in the onset and development of cardiovascular disease. Clin. Sci. 2014, 126, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Liu, Y.; Prater, K.; Zheng, Y.; Cai, L. Roles of microRNAs in pressure overload- and ischemia-related myocardial remodeling. Life Sci. 2013, 93, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Aronsen, J.M.; Swift, F.; Sejersted, O.M. Cardiac sodium transport and excitation-contraction coupling. J. Mol. Cell.Cardiol. 2013, 61, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Bodi, I.; Mikala, G.; Koch, S.E.; Akhter, S.A.; Schwartz, A. The l-type calcium channel in the heart: The beat goes on. J. Clin. Invest. 2005, 115, 3306–3317. [Google Scholar] [CrossRef] [PubMed]
- Katz, A.M.; Lorell, B.H. Regulation of cardiac contraction and relaxation. Circulation 2000, 102, 69–74. [Google Scholar] [CrossRef]
- Balaban, R.S. Cardiac energy metabolism homeostasis: Role of cytosolic calcium. J. Mol. Cell. Cardiol. 2002, 34, 1259–1271. [Google Scholar] [CrossRef] [PubMed]
- Glancy, B.; Balaban, R.S. Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry 2012, 51, 2959–2973. [Google Scholar] [CrossRef] [PubMed]
- Tarasov, A.I.; Griffiths, E.J.; Rutter, G.A. Regulation of ATP production by mitochondrial Ca2+. Cell Calcium 2012, 52, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Porrello, E.R. Micrornas in cardiac development and regeneration. Clin. Sci. 2013, 125, 151–166. [Google Scholar] [CrossRef] [PubMed]
- Condorelli, G.; Latronico, M.V.; Cavarretta, E. Micrornas in cardiovascular diseases: Current knowledge and the road ahead. J. Am. Coll. Cardiol. 2014, 63, 2177–2187. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.B.; Eisenhardt, S.U.; Stark, G.B.; Bode, C.; Moser, M.; Grundmann, S. MicroRNAs in ischemia-reperfusion injury. Am. J. Cardiovasc. Dis. 2012, 2, 237–247. [Google Scholar] [PubMed]
- Ye, Y.; Perez-Polo, J.R.; Qian, J.; Birnbaum, Y. The role of microRNA in modulating myocardial ischemia-reperfusion injury. Physiol. Genom. 2011, 43, 534–542. [Google Scholar] [CrossRef]
- Gladka, M.M.; da Costa Martins, P.A.; de Windt, L.J. Small changes can make a big difference - microRNA regulation of cardiac hypertrophy. J. Mol. Cell. Cardiol. 2012, 52, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Dorn, G.W., Jr. Therapeutic potential of microRNAs in heart failure. Curr. Cardiol. Rep. 2010, 12, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Shintani-Ishida, K.; Inui, M.; Yoshida, K. Ischemia-reperfusion induces myocardial infarction through mitochondrial Ca2+ overload. J. Mol. Cell. Cardiol. 2012, 53, 233–239. [Google Scholar]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Cell biology of ischemia/reperfusion injury. Int. Rev. Cell Mol. Biol. 2012, 298, 229–317. [Google Scholar] [PubMed]
- Griffiths, E.J. Mitochondria and heart disease. Adv. Exp. Med. Biol. 2012, 942, 249–267. [Google Scholar] [PubMed]
- Walters, A.M.; Porter, G.A., Jr.; Brookes, P.S. Mitochondria as a drug target in ischemic heart disease and cardiomyopathy. Circul. Res. 2012, 111, 1222–1236. [Google Scholar] [CrossRef]
- Mozaffari, M.S.; Liu, J.Y.; Abebe, W.; Baban, B. Mechanisms of load dependency of myocardial ischemia reperfusion injury. Am. J. Cardiovasc. Dis. 2013, 3, 180–196. [Google Scholar] [PubMed]
- Kadenbach, B.; Ramzan, R.; Moosdorf, R.; Vogt, S. The role of mitochondrial membrane potential in ischemic heart failure. Mitochondrion 2011, 11, 700–706. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Dorado, D.; Ruiz-Meana, M.; Inserte, J.; Rodriguez-Sinovas, A.; Piper, H.M. Calcium-mediated cell death during myocardial reperfusion. Cardiovasc. Res. 2012, 94, 168–180. [Google Scholar]
- Webster, K.A. Mitochondrial membrane permeabilization and cell death during myocardial infarction: Roles of calcium and reactive oxygen species. Fut. Cardiol. 2012, 8, 863–884. [Google Scholar] [CrossRef]
- Ong, S.B.; Gustafsson, A.B. New roles for mitochondria in cell death in the reperfused myocardium. Cardiovasc. Res. 2012, 94, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, P.D.; Purohit, A.; Hund, T.J.; Anderson, M.E. Calmodulin-dependent protein kinase ii: Linking heart failure and arrhythmias. Circ. Res. 2012, 110, 1661–1677. [Google Scholar] [CrossRef] [PubMed]
- Westenbrink, B.D.; Edwards, A.G.; McCulloch, A.D.; Brown, J.H. The promise of camkii inhibition for heart disease: Preventing heart failure and arrhythmias. Expert Opin. Ther. Tar. 2013, 17, 889–903. [Google Scholar] [CrossRef]
- Anderson, M.E. Camkii and a failing strategy for growth in heart. J. Clin. Invest. 2009, 119, 1082–1085. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.E.; Brown, J.H.; Bers, D.M. Camkii in myocardial hypertrophy and heart failure. J. Mol. Cell. Cardiol. 2011, 51, 468–473. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Brown, J.H. Role of Ca2+/calmodulin-dependent protein kinase II in cardiac hypertrophy and heart failure. Cardiovasc. Res. 2004, 63, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Backs, J.; Song, K.; Bezprozvannaya, S.; Chang, S.; Olson, E.N. Cam kinase II selectively signals to histone deacetylase 4 during cardiomyocyte hypertrophy. J. Clin. Invest. 2006, 116, 1853–1864. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Kohlhaas, M.; Backs, J.; Mishra, S.; Phillips, W.; Dybkova, N.; Chang, S.; Ling, H.; Bers, D.M.; Maier, L.S.; et al. Camkiidelta isoforms differentially affect calcium handling but similarly regulate HDAC/MEF2 transcriptional responses. J. Biol. Chem. 2007, 282, 35078–35087. [Google Scholar]
- Shintani-Ishida, K.; Yoshida, K. Ischemia induces phospholamban dephosphorylation via activation of calcineurin, PKC-alpha, and protein phosphatase 1, thereby inducing calcium overload in reperfusion. Biochim. Biophys. Acta 2011, 1812, 743–751. [Google Scholar]
- Gorbe, A.; Giricz, Z.; Szunyog, A.; Csont, T.; Burley, D.S.; Baxter, G.F.; Ferdinandy, P. Role of CGMP-PKG signaling in the protection of neonatal rat cardiac myocytes subjected to simulated ischemia/reoxygenation. Basic Res. Cardiol. 2010, 105, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Vila-Petroff, M.; Salas, M.A.; Said, M.; Valverde, C.A.; Sapia, L.; Portiansky, E.; Hajjar, R.J.; Kranias, E.G.; Mundina-Weilenmann, C.; Mattiazzi, A. Camkii inhibition protects against necrosis and apoptosis in irreversible ischemia-reperfusion injury. Cardiovasc. Res. 2007, 73, 689–698. [Google Scholar]
- Tani, M.; Hasegawa, H.; Suganuma, Y.; Shinmura, K.; Kayashi, Y.; Nakamura, Y. Protection of ischemic myocardium by inhibition of contracture in isolated rat heart. Am. J. Physiol. 1996, 271, H2515–H2519. [Google Scholar] [PubMed]
- Dou, Y.; Arlock, P.; Arner, A. Blebbistatin specifically inhibits actin-myosin interaction in mouse cardiac muscle. Am. J. Physiol. Cell Physiol. 2007, 293, C1148–C1153. [Google Scholar] [CrossRef] [PubMed]
- Inserte, J.; Hernando, V.; Garcia-Dorado, D. Contribution of calpains to myocardial ischaemia/reperfusion injury. Cardiovasc. Res. 2012, 96, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Khalil, P.N.; Neuhof, C.; Huss, R.; Pollhammer, M.; Khalil, M.N.; Neuhof, H.; Fritz, H.; Siebeck, M. Calpain inhibition reduces infarct size and improves global hemodynamics and left ventricular contractility in a porcine myocardial ischemia/reperfusion model. Eur. J. Pharmacol. 2005, 528, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Sheiban, I.; Tonni, S.; Chizzoni, A.; Marini, A.; Trevi, G. Recovery of left ventricular function following early reperfusion in acute myocardial infarction: A potential role for the calcium antagonist nisoldipine. Cardiovasc. Drugs Ther. 1997, 11, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Theroux, P.; Gregoire, J.; Chin, C.; Pelletier, G.; de Guise, P.; Juneau, M. Intravenous diltiazem in acute myocardial infarction. Diltiazem as adjunctive therapy to activase (data) trial. J. Am. Coll. Cardiol. 1998, 32, 620–628. [Google Scholar]
- Bar, F.W.; Tzivoni, D.; Dirksen, M.T.; Fernandez-Ortiz, A.; Heyndrickx, G.R.; Brachmann, J.; Reiber, J.H.; Avasthy, N.; Tatsuno, J.; Davies, M.; et al. Results of the first clinical study of adjunctive caldaret (MCC-135) in patients undergoing primary percutaneous coronary intervention for ST-elevation myocardial infarction: The randomized multicentre castemi study. Eur. Heart J. 2006, 27, 2516–2523. [Google Scholar]
- Jang, I.K.; Weissman, N.J.; Picard, M.H.; Zile, M.R.; Pettigrew, V.; Shen, S.; Tatsuno, J.; Hibberd, M.G.; Tzivoni, D.; Wackers, F.J.; et al. A randomized, double-blind, placebo-controlled study of the safety and efficacy of intravenous MCC-135 as an adjunct to primary percutaneous coronary intervention in patients with acute myocardial infarction: Evaluation of MCC-135 for left ventricular salvage in acute myocardial infarction (evolve). Am. Heart J. 2008, 155, 113.e1–113.e8. [Google Scholar]
- Theroux, P.; Chaitman, B.R.; Danchin, N.; Erhardt, L.; Meinertz, T.; Schroeder, J.S.; Tognoni, G.; White, H.D.; Willerson, J.T.; Jessel, A. Inhibition of the sodium-hydrogen exchanger with cariporide to prevent myocardial infarction in high-risk ischemic situations. Main results of the guardian trial. Guard during ischemia against necrosis (guardian) investigators. Circulation 2000, 102, 3032–3038. [Google Scholar]
- Mentzer, R.M., Jr.; Bartels, C.; Bolli, R.; Boyce, S.; Buckberg, G.D.; Chaitman, B.; Haverich, A.; Knight, J.; Menasche, P.; Myers, M.L.; et al. Sodium-hydrogen exchange inhibition by cariporide to reduce the risk of ischemic cardiac events in patients undergoing coronary artery bypass grafting: Results of the expedition study. Anna. Thorac. Surg. 2008, 85, 1261–1270. [Google Scholar] [CrossRef]
- Zeymer, U.; Suryapranata, H.; Monassier, J.P.; Opolski, G.; Davies, J.; Rasmanis, G.; Linssen, G.; Tebbe, U.; Schroder, R.; Tiemann, R.; et al. The Na+/H+ exchange inhibitor eniporide as an adjunct to early reperfusion therapy for acute myocardial infarction. Results of the evaluation of the safety and cardioprotective effects of eniporide in acute myocardial infarction (escami) trial. J. Am. Coll. Cardiol. 2001, 38, 1644–1650. [Google Scholar]
- Kitakaze, M.; Asakura, M.; Kim, J.; Shintani, Y.; Asanuma, H.; Hamasaki, T.; Seguchi, O.; Myoishi, M.; Minamino, T.; Ohara, T.; et al. Human atrial natriuretic peptide and nicorandil as adjuncts to reperfusion treatment for acute myocardial infarction (J-WIND): Two randomised trials. Lancet 2007, 370, 1483–1493. [Google Scholar] [CrossRef] [PubMed]
- Wahlquist, C.; Jeong, D.; Rojas-Munoz, A.; Kho, C.; Lee, A.; Mitsuyama, S.; van Mil, A.; Park, W.J.; Sluijter, J.P.; Doevendans, P.A.; et al. Inhibition of miR-25 improves cardiac contractility in the failing heart. Nature 2014, 508, 531–535. [Google Scholar] [CrossRef] [PubMed]
- Wystub, K.; Besser, J.; Bachmann, A.; Boettger, T.; Braun, T. miR-1/133a clusters cooperatively specify the cardiomyogenic lineage by adjustment of myocardin levels during embryonic heart development. PLoS Genet. 2013, 9, e1003793. [Google Scholar] [CrossRef] [PubMed]
- Curcio, A.; Torella, D.; Iaconetti, C.; Pasceri, E.; Sabatino, J.; Sorrentino, S.; Giampa, S.; Micieli, M.; Polimeni, A.; Henning, B.J.; et al. MicroRNA-1 downregulation increases connexin 43 displacement and induces ventricular tachyarrhythmias in rodent hypertrophic hearts. PloS ONE 2013, 8, e70158. [Google Scholar] [CrossRef] [PubMed]
- Shan, H.; Zhang, Y.; Cai, B.; Chen, X.; Fan, Y.; Yang, L.; Chen, X.; Liang, H.; Zhang, Y.; Song, X.; et al. Upregulation of microRNA-1 and microRNA-133 contributes to arsenic-induced cardiac electrical remodeling. Int. J. Cardiol. 2013, 167, 2798–2805. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.; Sun, X.; Ren, J.; Li, X.; Gao, X.; Lu, C.; Zhang, Y.; Sun, H.; Wang, Y.; Wang, H.; et al. miR-1 exacerbates cardiac ischemia-reperfusion injury in mouse models. PloS One 2012, 7, e50515. [Google Scholar] [CrossRef] [PubMed]
- Tritsch, E.; Mallat, Y.; Lefebvre, F.; Diguet, N.; Escoubet, B.; Blanc, J.; De Windt, L.J.; Catalucci, D.; Vandecasteele, G.; Li, Z.; et al. An SRF/miR-1 axis regulates NCX1 and annexin A5 protein levels in the normal and failing heart. Cardiovasc. Res. 2013, 98, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Ali, R.; Huang, Y.; Maher, S.E.; Kim, R.W.; Giordano, F.J.; Tellides, G.; Geirsson, A. miR-1 mediated suppression of sorcin regulates myocardial contractility through modulation of Ca2+ signaling. J. Mol. Cell. Cardiol. 2012, 52, 1027–1037. [Google Scholar] [CrossRef] [PubMed]
- Terentyev, D.; Belevych, A.E.; Terentyeva, R.; Martin, M.M.; Malana, G.E.; Kuhn, D.E.; Abdellatif, M.; Feldman, D.S.; Elton, T.S.; Gyorke, S. miR-1 overexpression enhances Ca2+ release and promotes cardiac arrhythmogenesis by targeting PP2A regulatory subunit b56alpha and causing camkii-dependent hyperphosphorylation of RyR2. Circ. Res. 2009, 104, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Belevych, A.E.; Sansom, S.E.; Terentyeva, R.; Ho, H.T.; Nishijima, Y.; Martin, M.M.; Jindal, H.K.; Rochira, J.A.; Kunitomo, Y.; Abdellatif, M.; et al. MicroRNA-1 and -133 increase arrhythmogenesis in heart failure by dissociating phosphatase activity from RyR2 complex. PloS One 2011, 6, e28324. [Google Scholar] [CrossRef] [PubMed]
- Cha, M.J.; Jang, J.K.; Ham, O.; Song, B.W.; Lee, S.Y.; Lee, C.Y.; Park, J.H.; Lee, J.; Seo, H.H.; Choi, E.; et al. Microrna-145 suppresses ros-induced Ca2+ overload of cardiomyocytes by targeting camkiidelta. Biochem. Biophys. Res. Commun. 2013, 435, 720–726. [Google Scholar] [CrossRef] [PubMed]
- Van Rooij, E.; Sutherland, L.B.; Liu, N.; Williams, A.H.; McAnally, J.; Gerard, R.D.; Richardson, J.A.; Olson, E.N. A signature pattern of stress-responsive micrornas that can evoke cardiac hypertrophy and heart failure. Proc. Natl. Acad. Sci. USA 2006, 103, 18255–18260. [Google Scholar]
- Aurora, A.B.; Mahmoud, A.I.; Luo, X.; Johnson, B.A.; van Rooij, E.; Matsuzaki, S.; Humphries, K.M.; Hill, J.A.; Bassel-Duby, R.; Sadek, H.A.; et al. MicroRNA-214 protects the mouse heart from ischemic injury by controlling Ca2+ overload and cell death. J. Clin. Invest. 2012, 122, 1222–1232. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, X.; Ren, X.P.; Chen, J.; Liu, H.; Yang, J.; Medvedovic, M.; Hu, Z.; Fan, G.C. MicroRNA-494 targeting both proapoptotic and antiapoptotic proteins protects against ischemia/reperfusion-induced cardiac injury. Circulation 2010, 122, 1308–1318. [Google Scholar] [CrossRef] [PubMed]
- Bostjancic, E.; Zidar, N.; Glavac, D. MicroRNAs and cardiac sarcoplasmic reticulum calcium ATPase-2 in human myocardial infarction: Expression and bioinformatic analysis. BMC Genomics 2012, 13, 552. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.F.; Liang, Z.; Lv, Z.R.; Liu, X.H.; Bai, J.; Chen, J.; Chen, C.; Wang, Y. MicroRNA-15a/b are up-regulated in response to myocardial ischemia/reperfusion injury. J. Geriatr. Cardiol. 2012, 9, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Hullinger, T.G.; Montgomery, R.L.; Seto, A.G.; Dickinson, B.A.; Semus, H.M.; Lynch, J.M.; Dalby, C.M.; Robinson, K.; Stack, C.; Latimer, P.A.; et al. Inhibition of miR-15 protects against cardiac ischemic injury. Circ. Res. 2012, 110, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Liu, X.; Zhang, S.; Lin, Y.; Yang, J.; Zhang, C. MicroRNA-21 protects against the H2O2-induced injury on cardiac myocytes via its target gene PDCD4. J. Mol. Cell. Cardiol. 2009, 47, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, X.; Zhu, H.; Zhu, C.; Wang, Y.; Pu, W.T.; Jegga, A.G.; Fan, G.C. Synergistic effects of the GATA-4-mediated miR-144/451 cluster in protection against simulated ischemia/reperfusion-induced cardiomyocyte death. J. Mol. Cell. Cardiol. 2010, 49, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Salic, K.; De Windt, L.J. Micrornas as biomarkers for myocardial infarction. Curr. Atherosclerosis Rep. 2012, 14, 193–200. [Google Scholar] [CrossRef]
- Reid, G.; Kirschner, M.B.; van Zandwijk, N. Circulating micrornas: Association with disease and potential use as biomarkers. Crit. Rev. Oncol. Hematol. 2011, 80, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Pegtel, D.M.; Cosmopoulos, K.; Thorley-Lawson, D.A.; van Eijndhoven, M.A.; Hopmans, E.S.; Lindenberg, J.L.; de Gruijl, T.D.; Wurdinger, T.; Middeldorp, J.M. Functional delivery of viral mirnas via exosomes. Proc. Natl. Acad. Sci. USA 2010, 107, 6328–6333. [Google Scholar] [CrossRef] [PubMed]
- Creemers, E.E.; Tijsen, A.J.; Pinto, Y.M. Circulating micrornas: Novel biomarkers and extracellular communicators in cardiovascular disease? Circ. Res. 2012, 110, 483–495. [Google Scholar]
- Kuwabara, Y.; Ono, K.; Horie, T.; Nishi, H.; Nagao, K.; Kinoshita, M.; Watanabe, S.; Baba, O.; Kojima, Y.; Shizuta, S.; et al. Increased microRNA-1 and microRNA-133a levels in serum of patients with cardiovascular disease indicate myocardial damage. Circ. Cardiovasc. Genet. 2011, 4, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Adachi, T.; Nakanishi, M.; Otsuka, Y.; Nishimura, K.; Hirokawa, G.; Goto, Y.; Nonogi, H.; Iwai, N. Plasma microrna 499 as a biomarker of acute myocardial infarction. Clin. Chem. 2010, 56, 1183–1185. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Choi, E.; Cha, M.-J.; Hwang, K.-C. Roles of Calcium Regulating MicroRNAs in Cardiac Ischemia-Reperfusion Injury. Cells 2014, 3, 899-913. https://doi.org/10.3390/cells3030899
Choi E, Cha M-J, Hwang K-C. Roles of Calcium Regulating MicroRNAs in Cardiac Ischemia-Reperfusion Injury. Cells. 2014; 3(3):899-913. https://doi.org/10.3390/cells3030899
Chicago/Turabian StyleChoi, Eunhyun, Min-Ji Cha, and Ki-Chul Hwang. 2014. "Roles of Calcium Regulating MicroRNAs in Cardiac Ischemia-Reperfusion Injury" Cells 3, no. 3: 899-913. https://doi.org/10.3390/cells3030899