
p66Shc in Cardiovascular Pathology


1
Biochemistry and Molecular Biology Department, College of Medicine, University of Oklahoma Health Sciences Center, Oklahoma City, OK 73104, USA
2
Stephenson Center, University of Oklahoma Health Sciences Center, Oklahoma City, OK 73104, USA
3
Harold Hamm Diabetes Center, University of Oklahoma Health Sciences Center, Oklahoma City, OK 73104, USA
*
Author to whom correspondence should be addressed.
Cells 2022, 11(11), 1855; https://doi.org/10.3390/cells11111855
Submission received: 21 April 2022
/
Revised: 30 May 2022
/
Accepted: 1 June 2022
/
Published: 6 June 2022
(This article belongs to the Special Issue Redox Control of Cell Signaling in Cardiac and Skeletal Muscle)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:p66Shc is a widely expressed protein that governs a variety of cardiovascular pathologies by generating, and exacerbating, pro-apoptotic ROS signals. Here, we review p66Shc’s connections to reactive oxygen species, expression, localization, and discuss p66Shc signaling and mitochondrial functions. Emphasis is placed on recent p66Shc mitochondrial function discoveries including structure/function relationships, ROS identity and regulation, mechanistic insights, and how p66Shc-cyt c interactions can influence p66Shc mitochondrial function. Based on recent findings, a new p66Shc mitochondrial function model is also put forth wherein p66Shc acts as a rheostat that can promote or antagonize apoptosis. A discussion of how the revised p66Shc model fits previous findings in p66Shc-mediated cardiovascular pathology follows.
Keywords:
mitochondrial dysfunction; p66Shc; apoptosis; ischemia/reperfusion; myocardial infarction; ROS; ShcA1. Introduction
The p66Shc adaptor protein is a broadly expressed canonical scaffold protein involved in modulating intracellular signal response [1,2,3]. In addition to adaptor function, p66Shc is an oxidoreductase that produces reactive oxygen species (ROS) in a mitochondria-dependent manner [4,5,6,7]. p66Shc governs outcomes for many pathologies, including endothelial dysfunction, coronary artery disease (CAD), and ischemia/reperfusion injuries (IRI) [8,9,10]. During cardiovascular insults, p66Shc exacerbates ROS accumulation, which increases pro-apoptotic responses and worsens clinical outcomes. IRI clinical severity is closely tied to p66Shc expression and activity, such that p66Shc acts as a biomarker for CAD and IRIs [11,12,13]. Since ROS accumulation is the primary cause of p66Shc-mediated cardiovascular effects, its inhibition has high therapeutic potential in cardiovascular pathology. In this article we discuss, and review, the role that p66Shc plays in cardiovascular pathology.
2. Reactive Oxygen Species, Aging, and Oxidative Stress
p66Shc mitochondrial function is coupled to ROS and ROS-mediated conditions such as aging and cardiovascular disease [14,15]. Free radical aging and oxidative stress theories posit that accumulated ROS-induced oxidative damage is central to aging and its associated pathologies [16,17]. Free radical aging and oxidative stress processes are characterized by consistent fitness loss as cellular processes become incrementally less efficient, leading to protein expression and functional alterations that cause cellular dysregulation [18]. However, this view of aging, and pathology etiology, was adapted because of the role ROS plays as a signaling molecule in routine cell functions [19]. Thus, excessive ROS or ROS dysregulation is key to free radical aging and oxidative stress.
In general, ROS are molecules that contain oxygen with higher reactivity profiles than O2 and are generated within cells by partial oxygen reduction [20]. ROS can exist as radicals (superoxide anion, hydroxyl radical) or as non-radical high-energy molecules capable of forming radicals (H2O2, organic peroxides). Radicals have unpaired/incomplete electron orbitals that increase their reactivity until stabilized by abstracting an electron from another molecule [21]. The distinction between radical and non-radical ROS is important as radicals are generally too reactive to travel outside of their production site, but non-radical ROS can [22,23,24]. Mobility differences between ROS forms can therefore affect and regulate subcellular redox signaling [25,26].
However, non-radical ROS can also generate radical ROS, which can affect intercompartmental signaling. A common example is known as Fenton chemistry, where H2O2 reacts with transition metal ions to produce hydroxyl radicals [27]. Excessive radicals cause damage by destabilizing or mismatching DNA, forming lipid peroxides or peroxyradicals, and oxidizing sulfur residues or carbonylating proteins [21,28,29,30]. These reactions can result in genomic, membrane, and protein instability that accumulate with age, which makes them aging biomarkers [31,32,33,34,35,36,37].
Within cells, there are several ROS sources. ROS are created by myeloperoxidase and NADPH oxidase (NOX) which form hypochlorous acid and superoxide anion, respectively. These proteins are associated with immune cells and have high activity in respiratory bursts that target pathogens [38,39]. However, non-immune cells have NOX homologues involved in ROS-mediated survival, growth, and apoptosis signaling [40]. Other ROS-generating proteins include: xanthine oxidoreductase (purine base breakdown), cytochrome p450 proteins (xenobiotic detoxification), and monoamine oxidase (dopamine breakdown) [41,42,43]. Peroxisomal fatty acid β-oxidation also produces H2O2, which enters the cytoplasm and may contribute to NF-kB and mTORC1 regulation [39,44]. Other common ROS generation sites include peroxisomes, lysosomes, endoplasmic reticulum, and the plasma membrane [45]. However, the primary cellular ROS source is mitochondrial respiration. This occurs when electrons leak between electron transport chain (ETC) complexes I and II or complexes II and III, reacting with oxygen to form superoxide anion [46,47,48,49]. Lastly, p66Shc is an oxidoreductase that directly contributes to pro-apoptotic mitochondrial ROS activity and indirectly to cytoplasmic ROS levels, which will be discussed in detail below [7]. p66hc-mediated ROS activity plays a central role in cardiovascular pathology through a variety of interacting pathways and is still emerging as a research field and potential therapeutic target in cardiovascular pathology, making it the focus of this review.
3. Reactive Oxygen Species—Neutralization, Necessity, and Pathology
Proper ROS function requires homeostatic balance. Excess ROS is damaging and promotes apoptosis, while deficiency prevents normal cell signaling required for growth and survival [50]. Although Excess ROS is pro-apoptotic, it is balanced by antioxidants which can be enzymes or small molecules. Known enzyme antioxidants can work independent of regenerating molecules but often require reducing equivalents from interacting proteins. Enzyme antioxidants that work independently include: catalase (reduces H2O2), glutathione S-transferases (reduce hydroperoxides), superoxide dismutases (reduce superoxide anion to H2O2), and peroxidase-active cytochrome c (oxidizes superoxide anion to O2). Enzyme antioxidants that work with interacting proteins include: glutaredoxins with glutathione (reduce disulfide bonds), thioredoxins with thioredoxin reductase (reduce disulfide bonds), peroxiredoxins with glutathione or thioredoxins (reduce peroxides), and glutathione peroxidases with glutathione/glutathione reductase (reduce H2O2 and peroxides) [51,52,53,54,55,56,57,58,59,60,61,62,63,64,65]. Most enzyme antioxidants have subcellular location-dependent isozymes with at least one form present in mitochondria [57,59,64,66,67,68,69].
Small molecules that contribute to redox homeostasis include vitamin C, vitamin E, and N-acetyl cysteine (NAC). Vitamin C is a water-soluble antioxidant that protects against cholesterol oxidation, scavenges dissolved O2, and neutralizes radical ROS, while vitamin E is a lipid soluble radical scavenger that can be regenerated by vitamin C [68,70,71,72]. NAC is a glutathione precursor with a redox active thiol that neutralizes ROS and can reverse deleterious disulfide formations [73].
Although ROS overproduction plays an important role in many pathologies, moderate ROS levels are required for optimal cell function. Technical limitations have prevented precise quantification of beneficial and detrimental ROS concentrations but it is widely accepted that ROS effects exist as a spectrum. Beneficial levels lie at an intermediate position in the spectrum but can fluctuate based on cell type and ROS activating stimuli, while excessive decreases and increases in ROS are harmful [20,24].
ROS play an important role in transducing environmental signals that dictate cell fate [19]. These redox signals are often transferred to downstream effectors via reversible protein modifications such as cysteine residue oxidation, affecting an array of proteins including phosphatases, kinases, transcription factors, and others [74]. Although, in theory, protein oxidation signaling reactions could be random, kinetics and solvent accessibility cause specific oxidation patterns in target proteins [50]. Since free radicals are restricted to their origination sites, H2O2 performs the majority of ROS signaling between cell compartments, often using peroxiporins to facilitate transfers [22,23,25,75,76,77,78,79]. However, it should be noted that ROS dysregulation (radical or non-radical) in a single compartment can affect whole-cell health, emphasizing the importance of subcellular compartmental ROS regulation [80,81].
ROS-activated pathways include nuclear factor-κB (NF-κB), phosphoinositide 3-kinase/AKT (PI3K/AKT), nuclear factor-erythroid factor 2-related factor 2/Kelch-like ECH-associated protein 1 (NRF2/KEAP1), and mitogen-activated protein kinase (MAPK) [25,82,83,84,85,86,87]. These pathways demonstrate the diverse effects of ROS signaling and provide insight into how ROS dysregulation can affect cells as these pathways regulate responses in inflammation, immunity, growth, survival, and apoptosis. In addition, downstream ROS effects differ by ROS concentration and stimulus source with low, moderate, and high ROS levels respectively favoring proliferation, differentiation, and cell death [19,88,89]. However, these are generalized trends and must be taken within the context of upstream signaling and local area or compartmental concentrations [80].
These pathways dictate many cell- and tissue-specific roles in pathology, making ROS-targeting therapies central to treatment strategies for many conditions. Some examples include neurodegenerative disease, aging, diabetes, organ failure, cancer, chronic obstructive pulmonary disease (COPD), endothelial dysfunction, sepsis, wound healing, and ischemia/reperfusion (I/R) injuries [8,14,18,31,90,91,92,93,94,95,96,97,98,99,100,101]. However, most treatments have not been directed towards location- or protein-specific ROS sources but rather whole-cell redox status and have been met with recurrent clinical failure. In fact, some treatments were harmful in cardiovascular pathologies [102]. Clinical failure of non-specific antioxidant administration is considered a result of neutralizing both physiological and pathological ROS [80,81,103,104,105]. Thus, emerging treatments to ROS-mediated pathology have shifted toward selective modulation of ROS sources dysregulated during pathology [16,81]. One of the proteins associated with dysregulated ROS in pathology is p66Shc, which was discovered as a longevity modifying protein that can promote apoptosis by generating ROS within mitochondria while also acting as a signal adapter at cell membranes.
4. p66Shc Discovery—Aging Research
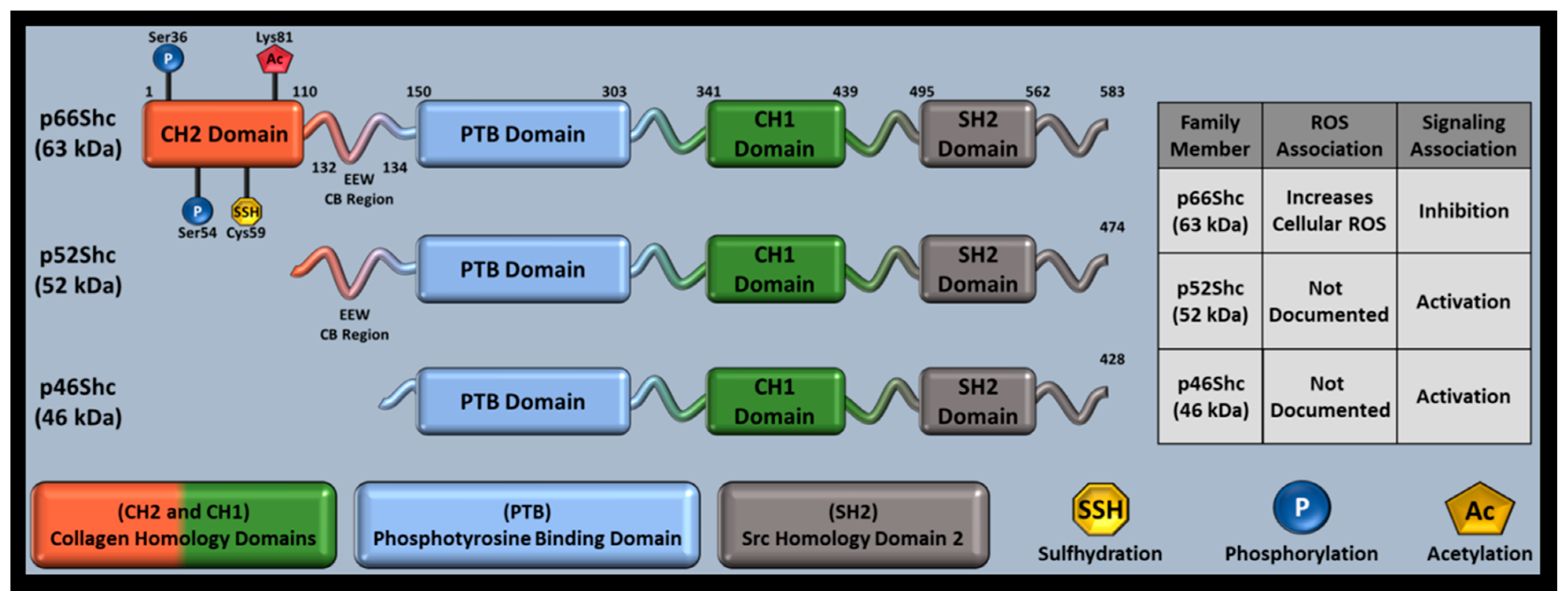
Interest in how Src homology 2 (SH2) domains mediate tyrosine receptor signaling led to the ShcA family’s discovery, which contains an SH2 domain in all family members (Figure 1) [106]. Tyrosine kinase receptors utilize adaptor proteins with SH2 domains at their cytoplasmic tails near receptor phosphorylation sites. SH2 domains can then recruit Grb2, another adaptor protein that promotes cell differentiation and growth via mitogen activated protein kinase (MAPK). ShcA transgene overexpression increased tumor incidence in mice, suggesting that ShcA proteins may activate MAPK. Although MAPK activation was later observed in response to p46Shc and p52Shc, p66Shc decreased mitogen signaling and was associated with increased ROS, separating p66Shc from p46Shc and p56Shc as a redox protein [107]. Further investigation identified that p46Shc and p52Shc share a transcript, while p66Shc has a unique transcript and N-terminal CH2 domain capable of replicating many p66Shc functions absent of its remaining domains [108].
When tested against UV and oxidative stress or increased tyrosine kinase signaling in mouse embryonic fibroblasts (MEFs), p66Shc was associated with cell death and altered electrophoretic movement, indicating that a post-translational modification (PTM), Ser36 phosphorylation, was associated with cell death [4]. Mouse p66Shc knockout (KO) studies corroborated p66Shc’s role as a redox protein that governs apoptosis and cell fate as p66Shc genetic removal increased mouse lifespan and tolerance to oxidative damage in stressed or pathological settings but had the opposite effect in unstressed settings [4,109,110,111,112]. These dichotomous effects may be explained by p66Shc ROS dysregulation since increased p66Shc-mediated ROS is consistent with worsened outcomes for many pathologies, but complete ShcA KO is embryonic lethal and moderate ROS is required for normal cell activity [19,113]. Promising results in lifespan and ROS attenuation have since led to many discoveries regarding p66Shc adaptor and oxidoreductase functions.
The original p66Shc KO mice experiments showed an around 30% increase in lifespan over WT mice and oxidative stress resistance, with no apparent negative effects [4]. Lifespan effects also appeared to be dose dependent as p66Shc+/− mice live longer than WT but not as long as KO mice [4,111]. Upon further characterization, p66Shc KO mice also had decreased adipocyte triglyceride accumulation, increased metabolism, improved behavioral plasticity as adults, and have improved health at older ages [114,115]. These mice also show increased resistance to obesity, diabetes, ischemic insults, and atherosclerosis [115,116,117,118,119,120,121,122]. However, when p66Shc KO mice were moved to natural conditions, subjected to winter weather and food competition for one year, p66Shc KO mice had shortened lifespans [111]. This observation was consistent when low temperature and starvation conditions were mimicked within a controlled laboratory environment, killing 50% of the KO mice, and no WT mice. These results were accompanied with findings indicating adipose dysfunction [111]. Thus, p66Shc plays an important role in cardiovascular health, energy metabolism, and gerontology.
5. The ShcA Family
The ShcA family consists of three members: p46Shc, p52Shc, and p66Shc. ShcA members are named by their approximate mass (~63 kDa, ~52 kDa, ~46 kDa) and domain composition, as well as Src Homology (SH) and Collagen homology (CH) domains. Despite their namesake only indicating SH and CH domains, all ShcA proteins also contain a phosphotyrosine binding (PTB) domain (Figure 1). P66Shc is the longest family member (583 residues), has its own set of promoters, and is primarily associated with oxidative stress and RAS inhibition while p52Shc (474 residues) and p46Shc (429 residues) share a set of promoters and are primarily associated with cell cycle progression and differentiation [3,108,123,124]. The family shares a single locus (1q21), producing different family members from 13 exons via alternative splicing and different start codons [125]. This results in ShcA proteins that share a single sequence, only differing by N-terminal amino acid length. The ShcA N-terminal domain is known as the CH2 (collagen homology 2) domain. P66Shc contains a full CH2 domain, p52Shc has a truncated CH2 domain, and p46Shc has no CH2 domain [125]. Functional differences between ShcA proteins are therefore CH2-mediated. All ShcA proteins function as signal adapters and undergo post-translational modifications (PTMs) that alter function, but p66Shc is the only protein member shown to produce reactive oxygen species (ROS), potentially explaining the divergent gene structure within the family and making p66Shc the most studied family member [7].
6. p66Shc Expression and Localization
p66Shc is expressed differentially throughout organs, but is expressed in adipocytes, lymphocytes, spleen, kidney, liver, lung, brain, and heart [114,115,126,127,128,129]. p66Shc’s differential expression led to p66Shc transcription and post-translation investigations that could explain expression patterns. Expressing p66Shc in cell lines not natively expressing p66Shc revealed that demethylating agents and deacetylases cause dose-dependent increases in p66Shc expression, suggesting that p66Shc’s promoter could be regulated via cysteine methylation or histone deacetylation [130]. Later studies revealed that low-density lipoprotein (LDL) could also increase p66Shc transcription via p66Shc promoter methylation at a CpG site [131]. p53, a transcriptional regulator and tumor repressor whose apoptotic function requires p66Shc, is associated with vascular disorders and increases p66Shc expression by binding p66Shc’s promoter region [132,133,134].
SIRT1 (histone deacetylase) also binds p66Shc’s promoter, decreasing p66Shc transcription via histone H3 Lys9 deacetylation. Similarly, SIRT1 overexpression in mice decreased p66Shc transcript and protein levels. However, SIRT1-mediated repression caused by SIRT1 overexpression can be overcome with high glucose and oxidized LDL in cell lines [135]. Further investigation indicated that prolonged high glucose causes a memory effect (sustained hypomethylation) on p66Shc’s promoter. This results in increased p66Shc and histone acetyl transferase (GCN5) expression, even after the original high glucose stimulus is normalized, while worsening diabetic endothelial dysfunction [136].
p66Shc’s expression is also affected by obesity-induced SUV39H1 (methyltransferase) downregulation. In these conditions acetyl transferase SRC-1 and demethylase JMJD2C are upregulated, resulting in decreased p66Shc promoter methylation and increased acetylation that increases p66Shc expression [137]. Of note, SIRT1 also protects SUV39H1 from ubiquitin-mediated degradation and may affect p66Shc expression through this interaction [138]. Thus, p66Shc expression is governed by transcriptional regulators and diet-inducible epigenetic factors.
In cancerous cells, nuclear erythroid 2-related factor 2 (Nrf2) can bind demethylated p66Shc promoter, increasing p66Shc transcription [4,139,140]. This finding is interesting because Nrf2 normally increases antioxidant responses and p66Shc’s traditional mitochondrial role is ROS production that leads to apoptosis; however, in this report, Ser36 phosphorylation did not increase mitochondrial trafficking as it has in other reported cell types [141]. These results suggest that p66Shc transcription and signaling may be cell dependent and that Ser36 phosphorylation may not be an exclusive signal for p66Shc mitochondrial translocation.
PTMs also affect p66Shc expression. Oxidative stress can cause Rac1-mediated p66Shc phosphorylation at Ser54 and Thr386, increasing p66Shc stability by decreasing proteasome degradation [142]. Since adapter proteins function primarily in cytosol, p66Shc cellular localization may have been limited to cytoplasm; however, p66Shc was found unevenly distributed between mitochondria, cytoplasm, and endoplasmic reticulum [143,144]. The other ShcA members are found in the cytosol and either the endoplasmic reticulum (p52Shc) or mitochondrial matrix (p46Shc) [143,145]. In non-stressed cells that express p66Shc, p66Shc localizes throughout cellular compartments with the following subcellular pattern: endoplasmic reticulum (24%), cytoplasmic complex with Peroxiredoxin 1 (Prx1, 32%), and mitochondria (44%) [146,147]. However, a stressed cellular environment causes CH2 phosphorylation and increased mitochondrial trafficking (discussed in the following sections).
7. p66Shc Signaling Overview
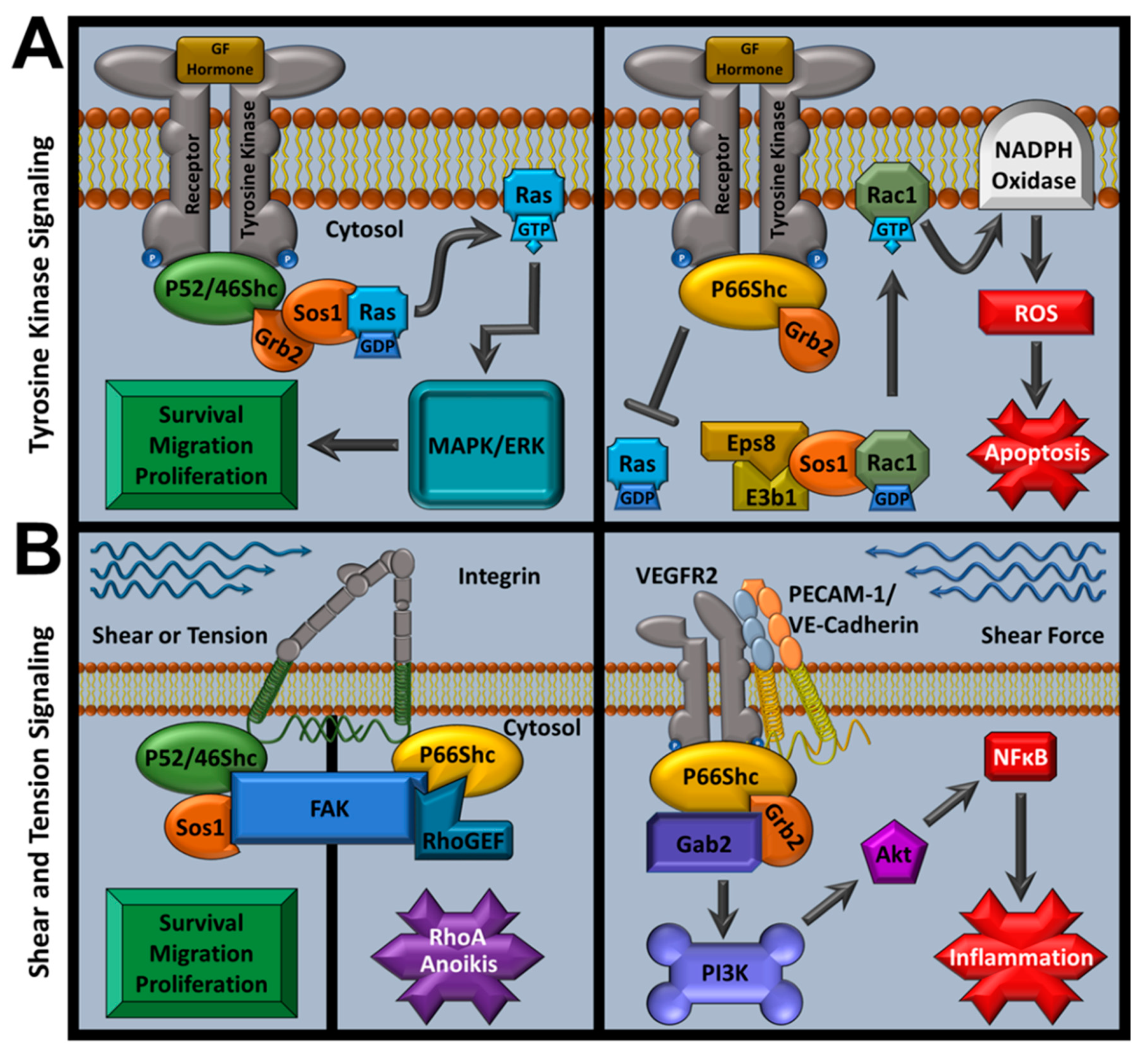
As an adaptor family, ShcA proteins are associated with signal transduction from varied stimuli (e.g., integrins, cytokines, and growth factors) and regulate downstream signaling for both physiological and pathological responses [148]. These observations support previous suggestions that p66Shc may provide a dual regulatory role as it can function as a proto-oncogene via growth factor signaling, or as an apoptosis regulator through pro-apoptotic mitochondrial ROS production (reviewed in detail in subsequent sections) [149]. Activated receptor tyrosine kinases recruit ShcA members with their cytoplasmic tails. When p52Shc or p46Shc bind receptor tails, they recruit the growth factor receptor-bound protein 2 and son of sevenless 1 (Grb2-Sos1) complex at p52Shc’s or p46Shc’s SH2 domain, activating Ras/MAPK mitogenic signaling [108,125]. However, p66Shc fails to induce mitogenic signaling and competes against the other ShcA members for Grb2, inhibiting potential MAPK signals [108,150,151]. p66Shc also inhibits extracellular-signal regulated kinase (ERK) signaling from insulin growth factor (IGF-1) and p52Shc is required for polyoma middle T antigen-induced hemangioma transformation in rats by promoting PI3K activation [143,152]. p66Shc Ser36 phosphorylation causes Grb2-Sos1 complex dissolution that leads to p66Shc/RAC1-mediated NADPH oxidase activation (via a Sos1/Eps8/E3b1 complex) and terminates Ras-MAPK and Ras-ERK signaling [108,153,154].
In addition, p66Shc increases the guanine exchange factor activity of Sos1 by competitively inhibiting Sos1-Grb2 interactions and increasing Sos1/Eps8/E3b1 complex formation to selectively promote Rac1 activity [155]. This action is primarily mediated by interactions between a PPLP motif in p66Shc’s CH2 domain and Grb2′s COOH-terminal src homology 3 domain [155]. In vitro assays did not directly assess other residues or PTM effects, but they may further promote Rac1 activation. p66Shc-mediated Rac1 activation is also associated with increased intracellular H2O2 production, likely via NADPH oxidase, and showed CH2 domain PPLP dependency [155]. These findings suggest that p66Shc may modulate many cellular functions regulated by Sos1 and Grb2, including cytosolic ROS production. Rac1 overexpression in PC-12 lines increased p66Shc expression, phosphorylation, and protein stability (half-life increased from 4.5 to 8 h) while decreasing ubiquitination [156]. These effects were reversed when p38MAPK was inhibited in PC-12 cells with SB203580 or SP600125, indicating that Rac1-mediated overexpression is governed by p38MAPK [156]. p38MAPK-dependent p66Shc overexpression was dependent on S54 (CH2 domain) and, to a lesser extent, T386 (CH1 domain) phosphorylation [156]. Intracellular Rac1-mediated ROS activity and oxidative stress-mediated apoptosis were also dependent on S54 and T386 hydroxyl group chemistry as cell lines coexpressing WT p66Shc and a p66Shc S54A/T386A double mutant generated less ROS than those overproducing p66Shc. Rac1 overexpression appeared to only affect p66Shc, not p52Shc or p46Shc [156]. However, Rac1 overexpression increased cellular ROS independent of p66Shc expression, suggesting that p66Shc serves as a non-essential adaptor protein that governs NADPH oxidase activity in these tests [156]. These functions are important in p66Shc cardiovascular pathology because overactive NADPH oxidase is associated with worsened IRI outcomes, decreased post-MI heart function, and heart failure [157].
Since phosphorylation patterns have ShcA signaling effects, phosphatase effects were also examined and showed that PTP-PEST interacts with p66Shc’s and p52Shc’s PTB domain, reducing MAPK activation under conditions that stimulate insulin signaling and antigen receptor stimulated lymphocyte activation [142,158]. Phosphatase and tensin homologue deleted at chromosome 10 (PTEN) is another phosphatase and tumor suppressor that dephosphorylates ShcA members and prevents ShcA-Grb2 binding, decreasing MAPK signaling and ShcA-mediated cell migration [159,160]. Protein phosphatase 2A (PP2A) and protein tyrosine phosphatase ε (PTPε) can also cause ShcA dephosphorylation and inhibit MAPK signaling [160]. Thus, p66Shc can inhibit mitogenic signaling or promote NADPH ROS-mediated apoptosis via its roles at the cell membrane.
Yet, ROS also regulates Wnt signaling and the Wnt3a ligand causes p66Shc phosphorylation in endothelial cells and β-catenin dephosphorylation [161]. In these cells, p66Shc KD caused decreased β-catenin-dependent transcription but p66Shc overexpression decreased β-catenin dephosphorylation while increasing β-catenin-dependent transcription. Exogenous H2O2 also showed increased dephosphorylation and was inhibited by the non-specific antioxidant N-acetyl cysteine or catalase, collectively suggesting that p66Shc has an important role in ROS–Wnt signal integration [161].
Given the connection between oxidative stress and aging, p66Shc-mediated ROS production likely contributes to aging, but p66Shc’s ROS production is strongly associated with many other pathologies, particularly cardiovascular and ischemia/reperfusion pathology [162]. While the CH1 domain governs adaptor protein function, CH2 Ser36 (only present in p66Shc) phosphorylation is associated with p66Shc-mediated mitochondrial ROS generation [4]. Ser36 phosphorylation is primarily regulated by protein kinase β or protein kinase C (PKC-β, PKC), other proteins such as lectin-like oxidized LDL receptor 1 and PKC-β2, c-Jun N-terminal kinase (JNK) can also phosphorylate p66Shc’s Ser36 residue [153,163,164].
After p66Shc is phosphorylated at Ser36, the prolyl isomerase peptidyl-prolyl cis-trans isomerase NIMA-interacting 1 (PIN1) induces a p66Shc conformational change and, after a dephosphorylation event (potentially mediated by PP2a or PTPε), p66Shc enters the intermitochondrial space and can form complexes with the heat shock protein HSPA9 (mtHSP70, mortalin) [146,159,160]. The mortalin-p66Shc complex dissociates under high stress conditions, resulting in cyt c release, caspase 3 activation, and apoptosis [146].
Investigating p66Shc-mediated apoptosis revealed that p66Shc utilizes its unique CH2 domain to produce ROS, governed by possible dimer/tetramer formation that involves thiol-disulfide exchanges at Cys59 and cyt c interactions [5,124]. Tests with H2S supplementation or overexpression of proteins involved in H2S synthesis also showed that Cys59 S-sulfhydration can decrease p66Shc-PKCβII interactions, decreasing downstream ROS production, and indicated that sulfur-based therapeutics may affect p66Shc function via thiol-disulfide manipulations [165]. Surprisingly, the CH2 domain was able to induce ROS and cause mitochondrial swelling, independent of p66Shc’s other domains [124].
Since p66Shc increased mitochondrial ROS in an oxidative stress dependent manner, p66Shc’s definition expanded to include redox sensitive oxidoreductase apart from its adaptor protein role. Investigating SIRT1′s potential direct effects on p66Shc found that Lys81 acetylation (CH2 domain), which is increased in diabetic conditions or with SIRT1 knockdown (KD), is also required for p66Shc’s oxidoreductase function in hyperglycemic endothelium [166]. However, human umbilical vein endothelial cells (HUVECs) stimulated with vascular endothelial growth factor (VEGF) were still able to cause Ser36 phosphorylation, indicating that Lys81 acetylation effects are stimulus dependent. Preventing Lys81 acetylation via p66Shc Lys81Arg mutations inhibited Ser36 phosphorylation and mitochondrial ROS production, improving endothelial dysfunction and linking p66Shc PTMs to endothelial health.
Apart from linking Ras to receptor stimuli and oxidoreductase function, the ShcA family also plays an important role in transducing mechanical stimuli. Complete ShcA KO is embryonic lethal (E11.5) and promotes severe cardiovascular tissue dysfunction with decreased contacts between neighboring cells and with extracellular matrix (ECM) [113]. This corroborates additional localization studies that identified p52Shc and p66Shc recruit focal adhesion kinase (FAK) at focal adhesion sites via their PTB domain [167,168]. Complete ShcA KO also causes decreased ability to spread on fibronectin, a sign of dysfunctional mechanotransduction [113].
Within endothelial cells, p52Shc interacts with a variety of integrins that increase ERK and Rac1 activity which promotes adhesion-dependent survival and all ShcA members respond to endothelial shear stress by interacting with αvβ3 and β1 integrins [169,170]. Mouse models show that increased aortic arch shear stress leads to ShcA phosphorylation via EGFR2 signaling and interactions with the mechanosensory complex PECAM-1/VE-Cadherin/VEGFR2, leading to inflammatory responses [2]. Myocardial-specific ShcA KO in mice further explored ShcA’s role in mechanical coupling. Myocardial ShcA KO impairs systolic function while increasing myocardial contractility and causes ECM and collagen alterations, which was replicated by selectively inhibiting myocardial ShcA PTB domain [171]. Further evidence of ShcA’s involvement in mechanosensory functions was provided when investigators performed knock-in mouse studies that demonstrated that muscle spindles, which govern motor behavior, require phosphorylated ShcA CH1 domains to form [172].
p66Shc also contributes to ShcA mechanosensory function. p66Shc antisense blocking prevents the myoblast–myotube transition, a process that requires distinct cellular mechanical changes. The same researchers also observed decreased stress fiber structure and cell rounding in their study, indicative of severe internal tension loss [173]. In other studies, p66Shc KD showed decreased epithelial differentiation and increased proliferation, but decreased functions governed by mechanical stimuli, such as morphogenesis and mitosis arrest [174,175]. These findings suggested that p66Shc governs anoikis, which was later confirmed as a p66Shc-dependent process that is regulated by RhoA signaling [168,176]. On the contrary, anchorage-independent growth has been observed with RhoA suppression [177]. Tumor metastasis and cell line transformations resistant to anoikis are associated with decreased p66Shc expression, with ectopic p66Shc expression returning the cells to normal anoikis levels but only if focal adhesion targeting is restored [176]. Meanwhile, cells with high dependence on anoikis and mechanosensory information for normal function, such as endothelium and bronchial epithelium, also require p66Shc for sustained anoikis but lose anoikis function with p66Shc KD [176].
Both anoikis and cell proliferation after adhesion loss are mediated by the p66Shc/FAK/RhoGEF complex and are governed by matrix stiffness [167,178]. Similar to tyrosine kinase signaling, p52Shc and p66Shc have different effects in their mechanosensory roles. The p66Shc-FAK complex recruits Rho GEFs, while the p52Shc-FAK complex recruits Ras GEFs, altering Rho and Ras downstream signaling, respectively [179]. This provides context for how decreased p66Shc in test conditions or metastatic cancers can cause both increased cell survival and decreased anoikis. Despite p66Shc’s role in cell–cell adhesion and cytoskeletal signaling, which could influence cardiovascular pathologies such as angina perctoris or cardiac arrest, this aspect of p66Shc research is limited in the context of cardiovascular disease. However, it is plausible that p66Shc’s mechanosensory function contributes to atherosceloris, MI, and other pathologies as p66Shc KO improves their outcomes, but these outcomes are also associated with decreased p66Shc-mediated ROS activity and deconvoluting the interactions between p66Shc mechanical and ROS-activating stimuli is difficult [118,121,180,181,182]. p66Shc signaling pathways are summarized in Figure 2.
8. Classical p66Shc Mitochondrial ROS Activity Pathway
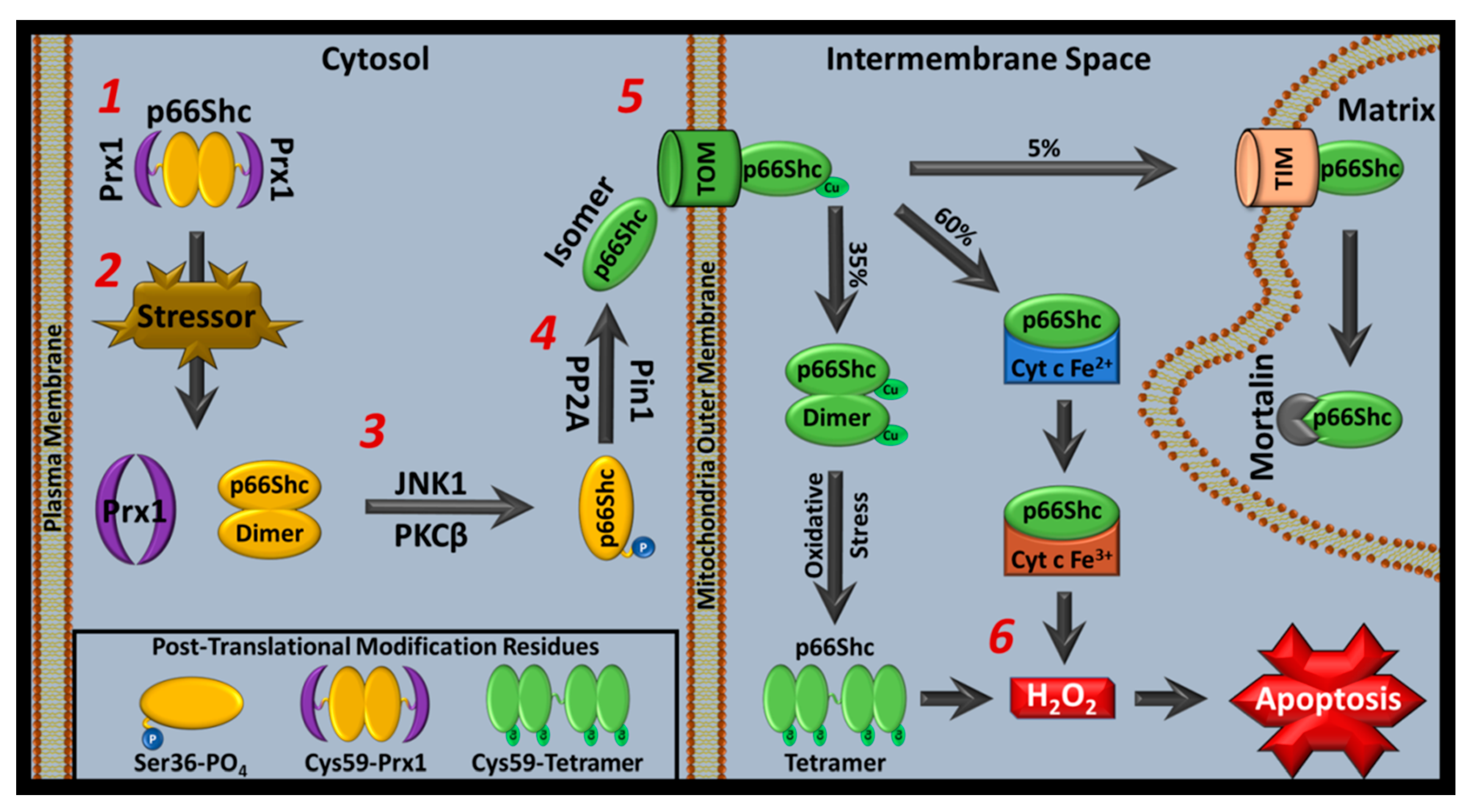
In response to cellular stress, p66Shc translocates into mitochondria. Once inside, p66Shc contributes to pro-apoptotic ROS accumulation causing caspase activation and apoptosis. However, several proteins influence p66Shc ROS activity by regulating p66Shc’s mitochondrial translocation [7]. In this section, we describe the classical pro-apoptotic p66Shc pathway and background on p66Shc ROS activity influencers (Figure 3).
8.1. Absent of Cell Stress, Peroxiredoxin 1 Prevents p66Shc Mitochondrial Translocation via Cytoplasmic Sequestration
Prx1’s role in p66Shc mitochondrial function is to sequester p66Shc within cytoplasm via direct binding interactions, mediated by p66Shc’s Cys59 residue. This prevents mitochondrial translocation until cell stress signals lead to p66Shc-Prx1 complex dissolution [147]. Peroxiredoxins are ubiquitous ROS regulators with location-dependent functions and are major contributors to metastasis progression. Peroxiredoxin 1 (Prx1) primarily resides in the cytoplasm and contributes to antioxidant processes via thiol-disulfide exchange where Prx1’s Cys52 and Cys173 become oxidized, producing an intermolecular disulfide bond and detoxified peroxides [183,184]. Reduced Prx1 can then be regenerated through various mechanisms [183,185]. In a common route for Prx1-mediated peroxide detoxification, thioredoxin (Trx) provides reducing equivalents to Prx1 that allows Prx1 to suppress ROS-mediated apoptosis driven by various proteins, including p66Shc [147,186,187,188]. However, oxidized Prx1 replaces peroxidase with other functions, including: chaperone, oncogene suppressing, immune enhancement, and ROS-dependent signaling [189,190,191,192,193,194,195,196,197,198,199,200].
In addition, Prx1 acts as a c-Abl tyrosine kinase inhibitor, an important cell death regulator that modulates p53 signaling and responds to oxidative stress, increasing JNK and MAPK pathway activation [192,194,201,202]. Prx1 also contributes to pro-apoptotic signaling by preventing PTEN inactivation caused by oxidation [183]. Furthermore, Prx1 associates with transcription factors important in cell death and growth such as NF-κB, androgen receptors, and p53 [203,204]. p53 is a key regulator of oxidative stress-induced apoptosis. Once activated, p53 increases pro-apoptotic protein expression, including p66Shc, causing caspase activation and mitochondrial apoptosis signals [205]. Prx1 mediates p53-regulated ROS-induced apoptosis, downstream of mammalian Ste20-like kinase-1 (MST1), which is phosphorylated during oxidative stress and causes increased proapoptotic signaling via MST1-forkhead box O3 (MST-FOXO3) interactions [206,207]. Thus, Prx1 contributes to the balance between pro-apoptotic and physiological ROS levels by direct ROS neutralization, inhibition of pro-apoptotic proteins that produce ROS (e.g., p66Shc), and transcriptional regulation over ROS pathways.
8.2. Cell Stress Causes Peroxiredoxin 1-p66Shc Dissociation, Allowing Protein Kinase c Family Members to Transduce Cell Stress Signals by Phosphorylating p66Shc at Ser36
The PKC family regulates p66Shc mitochondrial translocation and its activity is considered necessary for p66Shc to become ROS-active. In general, PKCs prime p66Shc for mitochondrial translocation via phosphorylation of p66Shc’s Ser36 in the CH2 domain. The PKC family has ten members that transduce many signals across a variety of pathways, including energy homeostasis and ROS production [208,209]. PKCs are categorized by their response to diacylglycerol (DAG) and other ligands downstream of GPCR-mediated phospholipase C activation. Their categories are conventional (requiring DAG, calcium, and phosphatidylserine), novel (requiring DAG and phosphatidylserine), and atypical PKCs (not affected by phosphatidyl serine or DAG) [208,210,211].
PKCβ is the PKC member most closely governing p66Shc’s ROS activity. PKCβ is a conventional PKC that is unique from other PKC family members as two isoforms are generated from a single locus via c-terminal exon alternative splicing, and each isoform (PKCβI and PKCβII) specializes in different roles [212]. PKCβI and PKCβII have similar primary structures but have differing C-terminal domains. PKCβ’s C-terminal domain is a catalytic center that is inactivated when its regulatory N-terminal domain binds the catalytic domain (resting state). When DAG binds PKCβ, these interactions are lost and PKCβ becomes active [213,214]. Once activated, PKCβ moves from cytosol to cell particulate fractions, which are mediated by lipid binding interactions that favor PKCβ’s active conformation [212,215,216,217].
Apart from DAG, PKCβ is also activated by PTMs including cysteine oxidation and tyrosine phosphorylation [218,219]. PKCβ has three constitutive phosphorylation sites. Their phosphorylation mechanisms are dependent on upstream kinase regulators, mTORC2, and PDK-1 [220,221]. After phosphorylation, PKCβ can interact with secondary messenger lipids but sustained activity results in PKCβ ubiquitination and degradation [222,223]. In addition, oxidative stress can activate PKCβ and may alter PKCβ cysteine chemistry, independent of calcium and DAG [224,225,226,227,228].
Since nutrient excess and obesity cause mitochondrial dysfunction, and because mitochondrial dysfunction causes ROS formation that activates PKCβ and increases p66Shc mitochondrial activity, obesity and p66Shc mitochondrial functions are linked [164,209,229,230,231,232,233,234]. PKCβ and p66Shc KO mice have increased metabolism, less insulin resistance, and heightened obesity resistance [114,115,120]. Along with causing p66Shc mitochondrial translocation, PKCβ activation inhibits autophagy and PKCβ KO or inhibition increases autophagy, a process that mitigates inflammation and must occur at low levels to maintain homeostasis [235,236,237,238,239]. Interestingly, exercise studies showed that exercise does not mitigate high fat diet-induced mitochondrial dysfunction, fat deposition, and insulin resistance in PKCβ KO mice while it does in WT mice [240]. Other reports suggest that PKCβ is downregulated with exercise, which decreases insulin resistance [241,242]. Since PKCβ and p66Shc function are coupled, it is not surprising that p66Shc also contributes to diabetic conditions [243].
Another PKC isoform, PKCδ, alters p66Shc mitochondrial activity. PKCδ is primarily located, in an inactive form, within cytoplasm, but a portion of PKCδ translocates to mitochondrial intermembrane spaces (IMS) where it can associate with p66Shc [143,244]. PKCδ is classified as a novel PKC isoform that binds p66Shc’s SH2 domain via a phosphorylated Tyr [245]. p66Shc-cyt c interactions activate PKCδ by facilitating site-specific PKCδ cysteine oxidation, independent of DAG [5,244]. Of note, this reaction causes p66Shc-mediated cyt c reduction and PKCδ oxidation. The reaction is catalyzed by vitamin A, which can bind PKCδ and cyt c in an orientation that facilitates this electron transfer [246,247,248,249,250]. However, PKCδ redox activation requires ferric cyt c, as respiration was only restored when ferric, but not ferrous, cyt c and retinol were added to isolated mitoplasts. Furthermore, respiration was not restored if p66Shc mutations preventing cyt c binding were present [246]. p66Shc KO MEFs also show lower glycolytic flux than controls, suggesting that p66Shc may alter metabolic pathways and contribute to ETC activity via cyt c reduction [122].
Further PKCδ experiments suggested a link between in vivo oxygen consumption and ATP synthesis upregulation via pyruvate dehydrogenase complex (PDHC) activation [246]. Although PKCε causes PDHC inhibition and localizes to mitochondrial matrices, it does not appear to directly interact with p66Shc [246,251]. p66Shc-mediated PKCδ redox-dependent activation may be reversible and would provide logical regulatory conditions for PKCδ activation via respiration activity [244]. High respiration increases ferrous cyt c pools while low respiration increases ferric cyt c pools, causing decreased or increased P66Shc-mediated PKCδ redox-dependent activation, respectively. Disrupting the p66Shc-vitamin A-PKCδ complex by decreasing p66Shc-cyt c or p66Shc-PKCδ interactions attenuates downstream signaling [245]. PKCδ also binds Raf near its DAG binding site and, as with its redox activation, requires vitamin A for Raf function [249,252,253,254]. Although other retinoids can bind in the same manner as vitamin A, vitamin A appears to the primary modulator of PKCδ function, with sustained activation from other retinoids causing cytotoxicity [249,251,255]. PKCδ KO and overexpression models are consistent with p66Shc KO models, where PKCδ overexpression caused insulin resistance and obesity while PKCδ KO mice were lean and had decreased insulin resistance [256,257]. This indicates that p66Shc ROS activity may govern PKCδ-related phenotypes.
8.3. Prolyl Isomerase 1 Interacts with p66Shc, Causing Conformational Changes That Prime p66Shc for Mitochondrial Translocation
After p66Shc’s Ser36 is phosphorylated, it can interact with prolyl isomerase 1 (Pin1), which induces phosphorylation-dependent cis–trans isomerization [164,258]. This step is a prerequisite to interactions with proteins that permit p66Shc IMS translocation. Pin1 is found in cytoplasm and nuclei but does not have a nuclear localization signal [259,260,261,262]. Pin1 expression correlates with increasing cell division but its regulation is not clear [259,261,263]. Pin1-induced transformational changes have strong biochemical effects on many proteins which may play an important role regulating cell growth and ROS-mediated diseases, such as Alzheimer’s and cancer [263]. Disrupting p66Shc–Pin1 interactions via Pin1 KO in MEFs decreases p66Shc translocation and shows increased resistance toward oxidative stress [164]. Similarly, Pin1 inhibition attenuates p66Shc ROS-mediated ischemia/reperfusion injuries in rat intestines [264].
Pin1 is also linked to mitosis regulation, with genetic constructs that decrease Pin1 function demonstrating mitotic arrest, cell cycle effects, or apoptosis [265,266,267,268,269,270,271]. These Pin1 depletion effects may be due to decreased phosphoprotein interactions, as Pin1 normally interacts with many pro-mitotic phosphoproteins, contributing to disorders like Alzheimer’s disease and cancer [263]. Protein phosphatase 2A (PP2A) specifically dephosphorylates the trans motif induced by Pin1 isomerization and has reciprocal effects to those observed by Pin1 in yeast studies [263,272,273].
8.4. p66Shc Is Dephosphorylated by Protein Phosphatase 2A to Allow Mitochondrial Translocation
In canonical p66Shc oxidoreductase mitochondrial function, Pin1-mediated isomerization is followed by dephosphorylation via PP2A interactions [7]. This set of reactions prepares p66Shc for mitochondrial entry by providing the required geometry for subsequent transport protein interactions. Although localization varies, in general, PP2A is dynamically localized between nuclei and cytoplasm. PP2A is a trimeric holoenzyme that can also function as dimer, with isoforms of each monomer determining localization and activity [274,275,276]. PP2A regulates oxidative stress, with increased PP2A activity improving oxidative stress-related outcomes in cardiovascular pathology, inflammation, and cancer [275]. PP2A is ubiquitously expressed and, in conjunction with PP1, represents ~90% of heart phosphatase activity [277,278]. It is also a therapeutic target for various diseases, including cardiovascular disease [279].
Decreased PP2A activity activates the ERK 1/2 signaling pathway (via increased ERK1/2, Akt, and glycogen synthase kinase 3β phosphorylation), an important predictor of IRI outcomes [280,281]. Yet, some IRI models have also demonstrated that these pathways are associated with increased PP2A activity [282,283,284]. One interpretation of these results is that PP2A activity fluctuates with time, as models collecting samples within 1 h and 6 h suggested that PP2A inhibition increases with time [285]. Potentially in agreement, PP2A therapeutic inhibition improved cardiac function when administered during reperfusion, but worsened outcomes if given during preconditioning [286].
Several reports suggest that PP2A activity is regulated by ROS, but they conflict in whether PP2A activity is upregulated or downregulated by ROS. An emerging view on these findings is that PP2A activity has tissue-dependent differential responses that vary in response to ROS concentration and time [275]. Data regarding PP2A during IRI is representative of this view as PP2A activity can be increased or decreased in IRIs, which may be caused by ROS fluctuations [275,287,288,289,290,291]. Furthermore, antioxidants and anti-inflammatory agents preserved PP2A expression in brain IRI models, improving outcomes and re-emphasizing ROS’s role in PP2A regulation [292]. In addition, PP2A inhibitors decrease infarct size more than preconditioning in rabbit ischemic heart models [293].
Since p66Shc produces ROS downstream of p66Shc-PP2A interactions, p66Shc mitochondrial translocation and ROS activity may have a causal link to PP2A activity, acting as a regulatory feedback loop that responds to cellular environments. PP2A activity and abundance decreases with increasing apoptosis, and excessive p66Shc mitochondrial activation causes ROS-mediated apoptosis.
8.5. p66Shc Enters the Intermembrane Space via Interactions with Mitochondrial Translocase of the Outer Membrane
The final step in p66Shc mitochondrial translocation is an interaction with the outer membrane translocase (TOM). p66Shc–TOM interactions place p66Shc in the IMS. Mitochondria have four distinct areas to which proteins can associate: mitochondrial matrix, inner mitochondrial membrane (IMM), intermembrane space (IMS), and outer mitochondrial membrane (OMM). Accurate protein targeting to each area can require specific machinery [294]. p66Shc utilizes TOM to reach p66Shc’s primary destination, the IMS, but a small portion of p66Shc interacts with the inner membrane translocase (TIM) to enter mitochondrial matrices where p66Shc forms complexes with mortalin that are destabilized by stress [146,295]. Although p66Shc reaches matrices, its matrix function(s) are not well-studied, and its ROS functions are tied to IMS localization.
TOM is a complex of seven subunits and two associated receptor subunits that form a channel in the OMM [296]. Of these subunits, p66Shc binds to Tom22 and Tom20 [295]. Tom22 and Tom20 play important roles in mitochondrial homeostasis, apoptosis, and mitochondrial health [297,298,299,300,301,302,303]. In general, proteins imported via TOM are recognized by Tom20, before interacting with Tom22, which permits IMS access [294]. Although IRI effects on TOM are not well-studied, researchers have shown that ischemia decreases Tom20 expression and that expression is rescued with ischemic preconditioning [304].
8.6. p66Shc Interacts with Cytochrome c, Causing H2O2 Accumulation, Permeability Transition Pore Formation, and Apoptosis
Once inside mitochondria, p66Shc traditional mitochondria function involves redox chemistry with cyt c, causing pro-apoptotic H2O2 accumulation. However, an alternative method for H2O2 accumulation mediated by p66Shc-cyt c interactions is discussed later in this review. Cyt c is a small, water soluble, heme c-containing protein that associates with the IMM outer leaflet and transports single electrons between complex III and complex IV of the ETC. This action promotes electron flux, ATP generation, and homeostasis [305]. In addition, cyt c can promote cell survival by decreasing mitochondrial ROS via superoxide anion scavenging or peroxidase activity, which is gained after interactions with cardiolipin (CL, an IMM lipid component comprising ~20–25% of the IMM) cause cyt c conformational changes that permit peroxide breakdown [51,52,53,54,55,56,306,307].
However, cyt c can also have pro-apoptotic functions. Under severe pro-apoptotic stress (e.g., ROS dysregulation) the IMM reorganizes, and endogenous reductant concentrations drop which causes cyt c to act as a CL-specific oxygenase [308,309,310,311]. This illustrates that CL-interactions govern cyt c’s dichotomous activity profile. CL oxidation is a critical step in pro-apoptotic signaling that produces various cell fate deciding saturated fatty acids, contributing to permeability transition pore (PTP) formation and cyt c release [311,312,313,314]. Released cyt c activates apoptotic protease activating factor – 1 (APAF-1) and associates with pro-caspase 9 to form the apoptosome, which activates caspase 3 and causes cell degradation [305,315,316].
According to one report, using overexpressed mouse CH2 domain, p66Shc exists primarily as a dimer that becomes pro-apoptotic after stress-induced Cys59-mediated dimer-tetramer transitions within mitochondria [124]. However, other studies suggest that a monomeric state is favored [146,295]. Regardless, stressed conditions lead to p66Shc pro-apoptotic H2O2 accumulation and permeability transition pore (PTP) formation, which is followed by cyt c outflow and caspase-mediated apoptosis [4,5,124,317]. Stressed conditions that increase p66Shc-derived ROS also increase p53 expression. In this state, PKCβ and JNK are activated to further increase p66Shc pro-apoptotic mitochondrial activity and glucose transport/actin polymerization is altered [5,318,319,320,321,322,323,324,325].
p66Shc also drives apoptosis by inhibiting antioxidant defenses, including superoxide dismutase (MnSOD, CuSOD, and ZnSOD), REF-1, and glutathione peroxidase via FOXO3a transcription factor downregulation [115,326,327,328,329,330]. Continued p53-mediated upregulation during high stress events may increase previously reported β1Pix/p66Shc/FOXO3a complex formation, sequestering FOXO3a to further promote apoptosis [331]. Although p66Shc-mediated apoptosis is associated with decreased antioxidant levels, decreases are not always observed [5,146,332,333]. Despite traditional p66Shc mitochondrial function being associated with H2O2 accumulation, some reports suggest that p66Shc produces superoxide anion, not H2O2 [90,334,335].
In total, previous studies suggest that p66Shc is a cell stress sensor that governs apoptosis, with Cys59 and Prx1 interactions inhibiting ROS activity in low-stress environments that are overcome by high stress which causes adverse effects in a variety of pathologies. Beyond potential Cys59 mediated interactions and cyt c effects, little was known about p66Shc’s ROS-generating mechanism or how parameters (e.g., pH) influence p66Shc ROS activity until recently (below).
9. Recent Advances in p66Shc Mitochondrial Function
9.1. p66Shc Oligomerization Status and Structure
p66Shc ROS activity models were previously built around the observation that overexpressed mouse-derived CH2 domains oligomerize [124]. It was suggested that a ROS-inactive cytoplasmic p66Shc dimer population was complexed with Prx1, which are monomerized and conformationally altered through several protein-mediated reactions before p66Shc monomers enter mitochondria. Once inside mitochondria, p66Shc was hypothesized to re-oligomerize via Cys59 interactions, and produce ROS, in a copper-dependent manner [7,124]. In this model, p66Shc could switch between dimer (reduced form) and tetramer (oxidized form), with normal conditions maintaining reduced dimeric p66Shc. Stress-associated antioxidant loss would then trigger tetramerization and subsequent ROS generation. However, these studies were performed with isolated CH2 domain, not full-length p66Shc (FLp66Shc). Furthermore, in vivo studies have not consistently reported p66Shc dimers or tetramers in their Western blot analyses, but monomers have been reported.
Although p66Shc’s primary sequence and modular domain identities have long been known, little structural data has been collected on FLp66Shc despite its importance in understanding p66Shc activity. A recent study used analytical ultracentrifugation on a variety of human FLp66Shc and CH2 constructs, as opposed to mouse-derived CH2 domain. All constructs were monomeric and reducing agents did not alter oligomer status. However, sedimentation coefficient differences were observed between reducing and non-reducing conditions, indicating disulfide bond presence. Mass spectroscopy experiments identified that although Cys59 does not mediate intermolecular disulfide bonding, it plays a central role within an interdomain thiol-disulfide exchange network that governs p66Shc ROS production (below). Interestingly, CH2 (amino acids 1-110) was aggregation-prone and polydisperse, but including distal cyt c binding domain residues reversed this trend. These results suggest that FLp66Shc conformational dynamics could be regulated by distal CH2 amino acids. Molecular dynamics simulations and in silico structural models supported these findings, indicating that p66Shc has high conformational variability and is intrinsically disordered with interspersed regions of organization [182].
9.2. p66Shc ROS Identity and Regulation
Despite the traditional model that p66Shc produces H2O2, studies have also associated p66Shc with increased superoxide anion levels [90,334,335]. As discussed above, radical and non-radical ROS have different reactivity and signaling profiles, with radical ROS primarily functioning at its origination site and non-radical ROS responsible for inter-organelle signaling [21,22,23]. ROS composition differences can therefore govern compartmental and inter-compartmental redox signaling, altering cell fate [25,26]. A variety of functional assays have shown that p66Shc does not directly produce H2O2, but rather superoxide anion [182]. Further investigation determined that neither metal cations, nor cyt c, were needed for superoxide anion activity, even though both were previously considered requirements [5,90]. Purified CH2 domain did not bind copper of either valency. However, an alternative route for p66Shc-cyt c mediated H2O2 accumulation was identified that resolves discrepancies in observed ROS forms generated by p66Shc mitochondrial activity (below).
ROS therapy is undergoing paradigmatic shifts from increasing cellular reductant concentrations that change whole-cell redox state to targeted approaches focusing on compartmental ROS dysregulation [80,81,103,104]. Since p66Shc is dysregulated with increased ROS activity during several pathologies, understanding how p66Shc ROS activity is regulated could improve outcomes for p66Shc ROS-mediated pathology. Several environmental factors regulate p66Shc superoxide anion production during in vitro assays, such as: pH, temperature, shear, and cardiolipin presence. p66Shc ROS production is highest in acidic, warm, cardiolipin-rich environments but is also increased with direct shear (indirect shear on cell membranes also increases p66Shc mitochondrial translocation) [2,182]. Of note, CL is essential for normal electron transport and promotes respiratory complex assembly and could therefore affect structural characteristics of other proteins [336,337,338].
Conditions increasing superoxide anion activity were associated with increased α-helical secondary structure and the CH2 domain shows a larger increase in structure than FLp66Shc under high-activity conditions. Since conditions with optimal p66Shc pro-apoptotic ROS activity are commonly found in pathology (e.g., IRI), these observations suggested that p66Shc acts as a stress biosensor whose activity is upregulated by pathological environmental effects. Furthermore, p66Shc’s mitochondrial-specific ROS activity may be a result of non-IMS conditions having a relative inhibitory effect.
The first p66shc-selective ROS inhibitors have also been identified: idebenone and carvedilol. Carvedilol is a non-selective adrenergic blocker that is prescribed for heart failure and is a World Health Organization essential medicine that improves post-myocardial infarction (post-MI) remodeling; however, how carvedilol improves remodeling is not well-understood [80,339,340,341,342,343]. Carvedilol binds and inhibits p66Shc at nM concentrations (Kd ~ 48 nM, IC50 ~ 118 nM), which agrees with previous carvedilol-mediated lipid peroxidation inhibition values [344,345]. Of note, carvedilol is thought to provide added benefit over other adrenergic blockers because of its ability to inhibit ROS [344,346,347]. With the understanding that non-specific ROS inhibition fails to improve or worsens cardiovascular outcomes and that targeted ROS inhibition can improve outcomes, carvedilol-mediated p66Shc ROS inhibition may play a role in observed cardiovascular clinical outcomes [80,81,103,104]. On the other hand, idebenone has antioxidant properties and acts as a coenzyme Q mimetic. It is prescribed in Europe for mitochondrial diseases with p66Shc associations [348,349]. At high concentrations (40–80 µM), idebenone sensitizes PTP opening and inhibits ETC complex I, but p66Shc-idebenone affinity has a reported Kd of ~229 nM (IC50 ~ 328 nM) and idebenone’s low concentration mitochondrial mechanism of action has not previously been characterized [182]. Idebenone has also been shown to improve insulin sensitivity via p52Shc signaling activity [350]. These compounds were tested in a fish p66Shc KD cryoinjury MI model, and show promising p66Shc-mediated improvements in many parameters [182].
p66Shc superoxide anion activity is also modified by protein binding partners. Mortalin forms complexes with p66Shc, which is released as a monomer in a stress-dependent manner and is upregulated with oxidative stress. Therefore, mortalin may prevent p66Shc-mediated apoptosis via p66Shc binding and inhibition [146,351,352,353]. Mortalin-p66Shc binding affinity (Kd ~ 858 nM) and ROS inhibition was recently confirmed, corroborating previous findings where p66Shc release from its mortalin complex increases PTP formation, ROS, and cyt c release [146]. These findings also support reports where mortalin overexpression reduces ROS and provides IRI protection [354]. Although originally viewed as a mitochondrial-specific protein, mortalin localization studies demonstrate that mortalin is found throughout cells, including within cytoplasm [355]. In addition, mortalin inhibits p53 transcription, decreasing p66Shc expression via cytoplasmic p53 sequestration [132,133,134,356,357]. Since Prx1 governs p66Shc via cytoplasmic sequestration, and mortalin regulates p66Shc activity via p53 sequestration (decreasing p66Shc transcription), it follows that mortalin could have a second regulatory level over p66Shc involving cytoplasmic sequestration, such that mortalin works with Prx1 to prevent dysregulated p66Shc mitochondrial translocation [147].
9.3. p66Shc Superoxide Anion Mechanism
Although p66Shc’s superoxide anion production mechanism is not fully characterized, recent insights into p66Shc function have been published. Cys59-mediated thiol-disulfide interactions were previously implicated in p66Shc’s oxidoreductase activity as a potential ROS inhibiting interaction (p66Shc-Prx1 translocation inhibitory complex), but were also associated with intermolecular disulfide bond formation between purified mouse CH2 domains [124,147]. However, human FLp66Shc mass spectrometry studies indicate that Cys59 is the center of an intramolecular interdomain thiol-disulfide exchange network [182]. Both reports indicate that Cys59 is critical to conformational alterations that affect ROS production.
In FLp66Shc, Cys59 (CH2 domain) interacts with either Cys287 or Cys292 (PTB domain). Cys287 can also interact with Cys196 (PTB domain) while Cys292 also interacts with Cys574 (SH2 domain). Mutating any single Cys residue was less effective at inhibiting ROS production than a combination of C287/C292S mutation. This double mutant prevents Cys59 from interacting with either of its partners and shows precipitous p66Shc ROS inhibition. These findings are consistent with earlier Cys59 investigations that identified Cys59 as a sulfhydration site that decreases p66Shc-derived ROS upon sulfhydration. Cys59 sulfhydration was observed with H2S administration or when overexpressing proteins contributing to H2S synthesis [165]. The previously reported Cys59S/E132Q/E133Q mutant with decreased pro-apoptotic activity also showed stark ROS inhibition in these FLp66Shc constructs [5,182]. Since shear increases thiol-disulfide interchange, shear may increase superoxide anion production via enhanced intermolecular thiol recycling [358,359]. Furthermore, increased p66Shc superoxide anion production in acidic conditions suggests that thiol-disulfide interchanges may be a rate limiting step in ROS generation and regulated by electrophilic strength of leaving groups during interchange [360].
Cys59S/E132Q/E133Q mutations did not appear to prevent ROS activity based on lost thiol–disulfide interaction partners alone as molecular dynamics simulations using the triple mutant found that the EEW motif in the CH2 domain, or cyt c binding domain, form salt bridges and cation-π interactions with Arg177, Arg285, and Lys279. These interactions cause partial folding that alters the PTB’s N-terminal orientation, which also affects CH2 orientation. Since these salt bridges could be affected by environmental effects that alter p66Shc activity, they may regulate environmental effects on superoxide anion production [182]. Consistent with this notion, ShcA constructs showed increased alpha helical secondary structure when exposed to CL, which increased p66Shc superoxide anion production. CL also protected p66Shc from thermal denaturation and increased post-thermal melt secondary structure recovery, relative to controls.
The same study also identified a crucial amino acid for p66Shc-mediated superoxide anion generation, Tyr10. When CH2 constructs were reversibly acetylated at Tyr10 or if FLp66Shc constructs were point mutated to FLp66Shc Tyr10Ala, superoxide anion production was abrogated. Acetyl group removal restored ROS activity. Thus, Tyr10 may participate in an electron relay, which is further supported by molecular dynamics simulations that indicate close proximity between the CH2 domain and a putative electron sink in the PTB that is governed by interdomain interactions [361]. Of note, hypoxic conditions terminated ROS production, which may provide insight into why hypoxic conditions are beneficial in pathology associated with p66Shc ROS overproduction, such as MI and Leigh syndrome [362,363,364].
As Tyr radicals are important in many redox enzymatic reactions (e.g., cyt c oxidase) and because ROS-active ShcA constructs also displayed a 410 nm absorption peak (associated with Tyr radicals), p66Shc superoxide generation may be radical-mediated [365,366,367]. This hypothesis was supported by in vitro fluorescence assays conducted under conditions that increase radical formation, which showed increased p66Shc superoxide anion production.
9.4. p66Shc-Cytochrome c Interactions
p66Shc-cyt c reaction descriptions were originally limited and suggested that p66Shc produced H2O2 via copper-dependent cyt c oxidation, mediated by Cys59 oligomerization [4,5,124]. However, reported p66Shc and cyt c reduction potentials do not support spontaneous p66Shc-mediated cyt c oxidation and there are no known pathways energetically coupled to support this reaction [5,368]. In addition, p66Shc has been shown to reduce cyt c as part of the PKCδ/retinol signalosome. In PKCδ/retinol signalosomes, PKCδ is activated by p66Shc-mediated cyt c reduction that causes PKCδ oxidation, which is catalyzed by vitamin A. Testing oxidized or reduced cyt c addition, combined with vitamin A, showed that respiration was restored with oxidized but not reduced cyt c in a signalosome-dependent manner [244]. Similar tests, performed with only p66Shc and cyt c, demonstrated that p66Shc reduces, not oxidizes, cyt c. In these in vitro experiments, human FLp66Shc or CH2 is capable of reducing but not oxidizing cyt c, independent of other proteins [182]. Since oxidized cyt c is required for normal cytoplasm-mitochondria transport via the mitochondrial disulfide relay function, p66Shc’s cyt c reductase function may also play a role in regulating mitochondrial protein import [369,370,371].
Several findings suggest that ShcA cyt c reductase function is independent of superoxide anion activity. p66Shc-mediated cyt c reduction was not inhibited by SOD’s presence nor by hypoxia, which abrogates superoxide anion fluorescent signals in superoxide anion production assays. p46Shc, which does not have reported ROS activity, also showed some reductase activity, though it was lower than p66Shc. In addition, when idebenone or carvedilol (p66Shc ROS inhibitors) are added to p66Shc-cyt c redox assays, cyt c reduction is increased [182].
Corroborating the above report where mitoplast respiration was dependent on PKCδ/retinol signalosome-mediated cyt c reduction, recent experiments demonstrated that p66Shc alone can increase complex IV and complex V activity when added to mitoplasts [182,244]. WT (ROS-active), Tyr10Ala (ROS-inactive), and Cys59Ser FLp66Shc mutants all increased Complex IV and Complex V ETC activity, corroborating a recent study that showed increased ETC activity with p66Shc activation in CNS cells [97]. Thus, p66Shc can increase ETC activity, independent of p66Shc’s superoxide anion activity. Although data suggest that Complex IV/V enhancement occurs through cyt c recycling, it is not clear whether enhancement occurs by direct p66Shc-mediated ETC redox chemistry or if electrons are passed from reduced cyt c to the ETC.
Yet, p66Shc-cyt c interactions have a well-documented H2O2 accumulation effect, which was thought to occur via cyt c oxidation [5,372]. If p66Shc reduces cyt c, there must be an alternative mechanism for p66Shc-mediated H2O2 accumulation. An alternative mechanism was described where human FLp66Shc is able to non-competitively inhibit WT cyt c peroxidase function (KI ~ 1.43 µM), resulting in decreased H2O2 degradation. Constitutively peroxidase active His26Tyr mutant cyt c was also inhibited by FLp66Shc (KI ~ 3.54 µM) and shows inhibition values similar to minocycline-mediated cyt c His26Tyr peroxidase inhibition (KI ~ 1.0 µM) [373]. In addition, purified human CH2 domain also caused a decrease in H2O2 breakdown (~48.9% signal decrease). Yet, p66Shc-cyt c binding experiments demonstrate that FLp66Shc has a ~7.7-fold higher binding affinity for WT cyt c than its peroxidase active His26Tyr mutant, suggesting that non-stressed conditions may favor an interaction that prevents cyt c peroxidase transformation and pro-apoptotic H2O2 accumulation [182].
p66Shc was also found to have another function that may influence apoptosis, caspase inhibition. p66Shc-mediated ROS overproduction leads to PTP formation and mitochondrial cyt c release, which ultimately causes caspase cascade activation and cellular destruction [5,151,372]. p66Shc-mediated caspase inhibition was tested in three cell lines: HS27, PCS-201, and WI-38. P66Shc significantly inhibited caspase 3/7 activity (~3-fold) and caspase 9 activity (~5-fold) but did not inhibit purified caspase 3 activity [182]. However, since p66Shc upregulation is associated with apoptosis these results suggest that caspase inhibition may be a cytoprotective effect against transient stress spikes and caspase leakage rather than a function that occurs during apoptosis.
In summary, p66Shc directly contributes to mitochondrial ROS accumulation via superoxide anion production, superoxide anion spontaneous dismutation to H2O2, and cyt c peroxidase inhibition. It should be noted that pro-apoptotic p66Shc ROS production is associated with stressed or pathological conditions, but not physiological conditions. P66Shc also performs mitochondrial functions associated with cell survival: ETC enhancement, caspase inhibition, and cyt c reduction. Thus, p66Shc appears to act as a biosensor promoting cell survival until stress thresholds are met, leading to pro-apoptotic signaling.
9.5. Revised p66Shc Mitochondrial Function Model
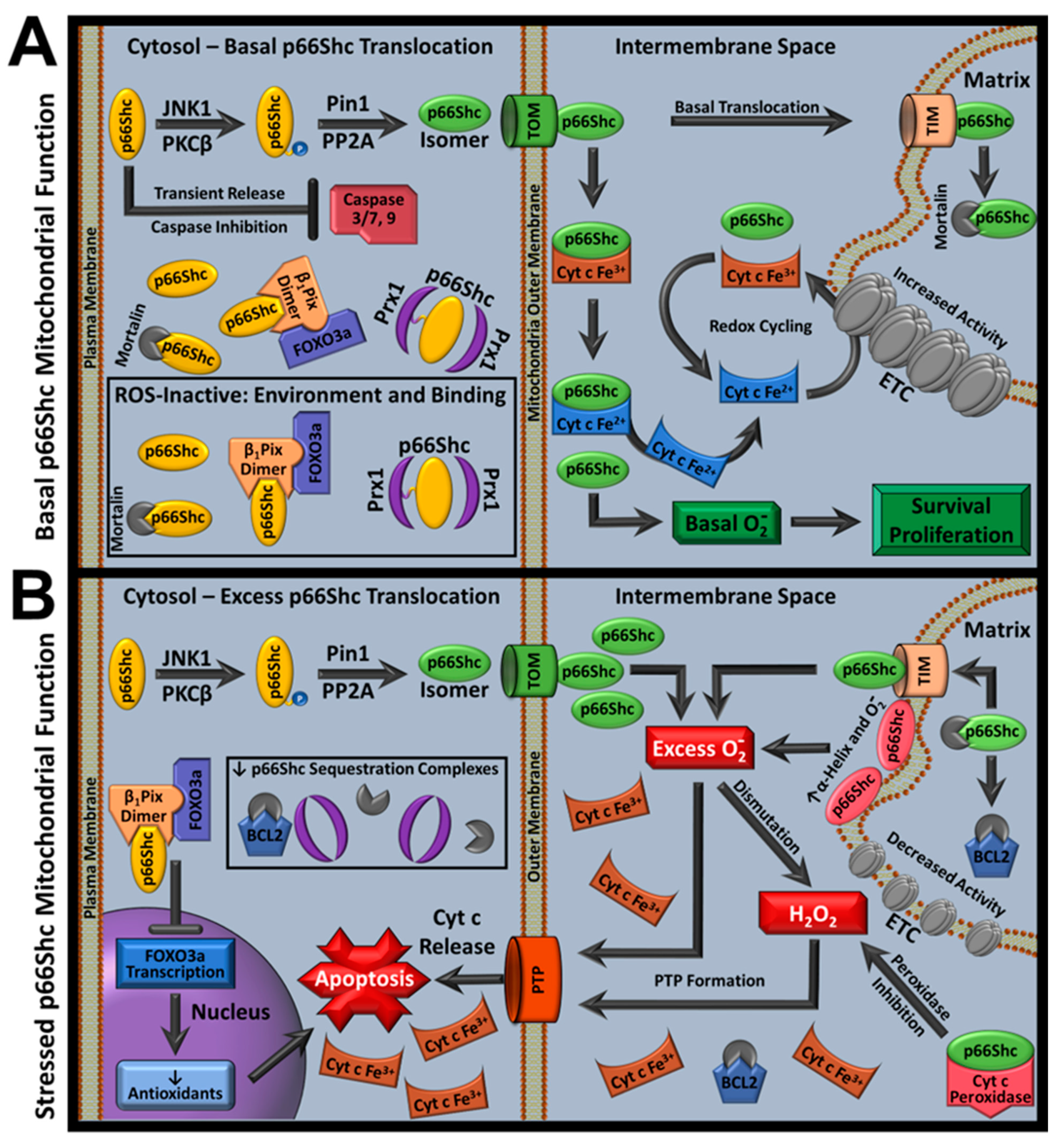
Given the above insights into p66Shc mitochondrial function, a new stress-dependent p66Shc model has been proposed that unifies previous and current findings (Figure 4). In the new model, during low-stress or physiological conditions, p66Shc favored functions are correlated with cell survival. Low-stress cytoplasmic p66Shc pools are ROS-inactivated by environmental factors and mortalin or Prx1 complexes. Meanwhile, mitochondrial p66Shc pools produce physiologic superoxide anion signals, prevent cyt c-mediated lipid peroxidation by stabilizing peroxidase-inactive cyt c conformations, and promote Complex IV/V activity in the ETC.
However, high-stress conditions favor p66Shc’s pro-apoptotic functions. High-stress releases p66Shc from Prx1 and mortalin complexes, leading to increased mitochondrial translocation. Translocation activates p66Shc superoxide anion production by introducing favorable environmental conditions for ROS production. Increased ROS-active mitochondrial p66Shc concentrations cause increased mitochondrial ROS accumulation. This contributes to antioxidant depletion, cyt c peroxidase formation, and CL oxidation. If the stress stimulus is not corrected, heightened CL oxidation causes cyt c IMM disassociation and CL depletion that disrupts ETC function while p53 mediates p66Shc upregulation. p66Shc upregulation propagates pro-apoptotic signals by inhibiting FOXO3a-mediated antioxidant transcription. In these conditions, p66Shc strictly promotes apoptosis, as ongoing processes associated with stress stimuli, such as acidosis in IRI, further enhance p66Shc ROS activity and decrease availability to pro-survival pathways as PTP formation begins (e.g., depleted IMM cyt c availability and cyt c–ETC interactions), leading to cellular degradation [182].
p66Shc has both pro-apoptotic and pro-survival functions linked to mitochondrial ROS activity. P66Shc pro-apoptotic functions are apparent in high-stress or pathological models but not in unstressed models. This matches general cellular ROS trends, where high stress is correlated with ROS dysregulation and pathology but moderate ROS levels are required for healthy cells. The coordination between cell stress, ROS, and p66Shc are better exemplified by the revised p66Shc mitochondrial function model than the previous model. This model also unites and resolves paradoxical p66Shc findings such as those demonstrating that decreased p66Shc expression or ROS production improves IRIs and increases mouse lifespan in non-stressed environments, but p66Shc KOt models decrease ischemic preconditioning benefits, increase short-term myocardial injuries, and decrease mouse lifespan in natural environments [4,13,90,109,110,111,374,375].
In addition, this model is supported by a set of experiments testing whether the balance between p66Shc’s pro-survival related functions and pro-apoptotic functions could be tipped to increase pro-survival functions in zebrafish cryoinjury MI models. The experiments utilized p66Shc WT fish that were given p66Shc selective ROS inhibitors and compared them to p66Shc KD fish. p66Shc KD and ROS inhibitor groups showed improvements in many parameters with a concomitant decrease in myocardial ROS production. Some improvements include increases in post-MI body mass and physical activity while demonstrating decreases in inflammatory cell recruitment, fibrotic tissue area, platelet aggregation, and injury area. Furthermore, proteome analysis was consistent with caspase cascade inhibition and energy metabolism alterations contributing to improved MI outcomes. Lastly, non-selective ROS inhibition via NAC administration proved detrimental, corroborating the proposed benefits of targeted ROS inhibition [80,81,103,104,182]. In addition, the revised p66Shc model is also under clinical investigation as a cancer treatment (NCT04928508). However, in this instance, p66Shc’s balance is pushed toward pro-apoptotic functions by disrupting the p66Shc–mortalin complex via SHetA2 administration [376,377,378].
10. p66Shc’s Mitochondrial ROS Activity Role in Cardiovascular Pathology
As noted above, p66Shc ROS production is negatively associated with many pathological states, indicating that inhibiting p66Shc ROS activity may improve their outcomes [118,119,379,380]. Some examples include neurodegenerative disease, aging, diabetes, organ failure, cancer, chronic obstructive pulmonary disease (COPD), endothelial dysfunction, sepsis, wound healing, and ischemia/reperfusion (I/R) injuries [31,90,91,92,93,96,97,98,100,101]. In this section, we review p66Shc-ROS mediated effects on cardiovascular pathology. Although p66Shc also plays an important role in metabolism, cytoskeleton/mechanosensory, and inflammatory functions, a detailed discussion of p66Shc’s role in these functions in cardiovascular pathology is beyond the scope of this review [113,114,115,120,169,170,174,175,176,177,178,179,182,350]. However, previous sections do include key interactions between p66Shc and these aspects of cardiovascular disease.
10.1. p66Shc in Ischemia/Reperfusion Injuries
Heart disease is a substantial global healthcare and financial burden. In the U.S. alone, heart disease is diagnosed in 10.6% of adults, attributed to 23.1% of deaths, costs USD 351.2 billion annually (2014–2015), and reports suggest that heart disease burdens are rising [381,382]. The most common heart disease is coronary artery disease (CAD) which narrows arterial diameter, decreasing blood flow beyond plaques. If poorly managed, CAD transitions into myocardial infarction (MI). MI pathology is characterized by dysfunctional mitochondria that reverse their ATP generating activity with ATP expenditure and that pump protons into the IMS under ischemic conditions [383]. Mitochondrial dysfunction can be reversed if myocardium is reperfused within ~20 min, but extended ischemic episodes cause permanent damage that is exacerbated by reperfusion and linked to ROS dysregulation, PTP formation, and apoptosis [162,383,384,385,386,387,388].
p66Shc’s role in ischemia reperfusion injuries (IRIs) varies depending on ischemia and reperfusion duration but has been traditionally viewed as pro-apoptotic independent of IRI location, and has almost exclusively been studied in p66Shc genetic KO models as no p66Shc-selective therapeutics had been described until recently. However, some studies analyze p66Shc in PKC-β inhibition or KO models. p66Shc deletion improved IRIs in hind limb ischemia models, showing improvements in tissue viability and capillary density via decreased p66Shc-mediated ROS accumulation [118]. Other hind limb ischemia models revealed that p66Shc KO improved wound healing in a ROS-dependent manner, indicating that p66Shc ROS balance may regulate ischemic wound healing [389].
In mouse cardiomyocytes, p66Shc KO increased resistance to angiotensin II-mediated apoptosis and showed decreases in IRI-induced oxidative stress which were not further decreased by antioxidant administration, indicating p66Shc ROS dysregulation in ischemic cardiac tissue [119,121]. Cardiac IRI models showed that p66Shc mitochondrial translocation did not increase with short-term ischemia alone or with short-term ischemia and reperfusion, but did increase with long-duration ischemia followed by reperfusion [380]. In addition, p66Shc ROS inhibition via pharmaceutical Complex I or PKC-β inhibition decreased p66Shc Ser36 phosphorylation and improved outcomes [380]. However, others found that short-term ischemia was worsened by p66Shc deletion, underscoring p66Shc’s dual role in cardiac IRI [110]. p66Shc’s protective role in short-duration ischemia, which could be viewed as a transient cell stress spike, is further supported by studies reporting reduced ERK/Akt-mediated growth factor signaling in p66Shc KO fibroblast lines that decreased normal fibroblast activity [390]. Furthermore, p66Shc IRI cytoprotective effects were also observed when p66Shc KO decreased survivor activating factor enhancement and reperfusion injury salvage kinase pathways, with concomitant increases in mitochondrial swelling and caspase-3 mediated apoptosis in mouse cardiac IRIs, indicating that p66Shc had a pro-survival role in these conditions [110].
Interestingly, mouse cardiac specific p66Shc KO studies revealed that p66Shc KO does not notably affect heart rate, heart mass, or blood pressure, but cardiomyocyte counts were increased despite having a similar ventricular wall size [121]. This may reflect p66Shc’s neonatal role in heart development, since neonatal p66Shc KO cardiac tissue has increased SOD activity and decreased ROS-mediated apoptosis that could manifest as increased cells in the adult heart [391].
p66Shc IRI has also been studied in stroke models and shows similar trends to cardiac IRIs. For example, p66Shc KD after ischemic insult in mice preserved vessel integrity at the blood–brain barrier and increased survival rates while decreasing ROS levels, neurological loss, and lesions [13]. The same study showed that acute ischemic stroke patients had increased p66Shc expression in peripheral blood monocytes that correlated with neurological outcomes. Yet, p66Shc is required for ischemic preconditioning neuroprotective effects [392].
p66Shc’s role in post-MI effects is less studied than initial infarct effects. However, p66Shc KO has shown improved post-MI healing with decreases in fibrosis measurements and cardiac rupture [374]. The same report demonstrated that these effects were mediated by increased collagen formation and decreased matrix metallopeptidase 2 (MMP2) activation. MMP2 expression has also been shown to increase or decrease with p66Shc overexpression or deletion, respectively [393]. Since MMP2 activates TGFβ, and because its activity also correlates with p66Shc expression, p66Shc may regulate TGFβ activation [394]. This potential connection is strengthened by TGFβ activation mechanisms including ROS exposure, ionizing radiation, shear, and acidic pH, which also increase p66Shc superoxide anion production and pro-apoptotic signaling [182,395,396]. Thus, improved post-MI remodeling effects from p66Shc KO may be secondary to decreased TGFβ activation, which is strongly activated in MI and regulates post-MI remodeling [397].
Due to its high morbidity and mortality, pharmaceutical treatments that can mitigate initial MI and post-infarct remodeling have been frequently researched. Yet, few drugs with pre-clinical effectiveness are beneficial in MI patients and none were previously described as a p66Shc ROS inhibitor [398,399]. Of the promising therapeutics, most are related to p66Shc’s functions. For example, Coenzyme Q (CoQ) and mitoquinone (mitoQ), a CoQ variant with increased mitochondrial localization, both target the respiratory chain and attenuate ATP loss during I/R while scavenging excess ROS. These treatments are associated with improvements in: post-MI contractility, cardiac function, oxidative damage, cell death, and I/R mitigation during cardiac grafts, potentially via decreased lipid peroxidation. CoQ and mitoQ treatment also decrease pro-apoptotic cyt c release [400,401,402,403,404,405]. In fact, mitoQ completed clinical trials that showed cardiovascular benefits and is undergoing clinical trials for a variety of ROS-related conditions, including heart failure and kidney disease. A functionally similar drug, mito-TEMPO, scavenges mitochondrial superoxide and shows improvements in heart failure and diabetic cardiomyopathy models by increasing contractility and decreasing heart dilation, cell death, and oxidative stress [406,407,408,409].
Bendavia (also known as MTP-131 or SS-31), is a tetra-peptide that binds CL, preventing ROS-induced CL oxidation and cyt c peroxidase formation [410,411]. Bendavia decreases oxidative stress, infarct expansion, apoptosis, and detrimental heart remodeling while increasing ETC Complex I/IV (other ETC complexes were not tested) activity and decreasing infarct sizes in sheep and guinea pig models [412,413,414]. Lastly, resveratrol treatment activates SIRT1/SIRT3 and reduces post I/R infarct size [415]. Since SIRT1 activation decreases p66Shc expression, these results suggest that decreased p66Shc expression is beneficial in IRI [90,135,416].
Thus, promising IRI therapeutics are tied to increasing p66Shc pro-survival signaling or decreasing p66Shc pro-apoptotic ROS activity. As noted above, the first-described p66Shc selective ROS inhibitors have already been tested in zebrafish MI models and show improvements in a variety of clinically translatable parameters [182]. Although consistency in mammals remains to be seen, these represent a novel approach to p66Shc ROS-mediated pathology, including MI treatment, that decreases initial infarct damage in MI models and may improve recurrent MI as fibrosis measurements were substantially decreased in treated fish.
In addition, targeted superoxide anion inhibition could improve post-MI heart failure because post-MI heart failure is accompanied by a significant increase in free radical-mediated lipid peroxidation and plasma superoxide anion levels, but decreased catalase, glutathione, and SOD plasma levels [417,418]. These trends are further enhanced with heart failure severity, which also correlate with decreased antioxidant pools and heart function in heart failure patients [417,419,420].
10.2. p66Shc in Endothelial Dysfunction
Age-induced endothelial dysfunction is characterized by decreased NO production and decreased ability to increase lumen diameter, which corresponds with oxidative stress; however, aged p66Shc KO mice do not exhibit these impairments [107,333,421,422,423,424,425]. p66Shc KO mice in this study also had decreased age-related superoxide anion and nitrotyrosine (formed when NO and superoxide anion react) levels. p66Shc also inhibits NO generation as p66Shc KD led to increased markers of endothelial NO synthase activity and aged p66Shc KO mice showed similar results [426,427]. Thus, p66Shc governs endothelial ROS and NO production, which can affect many other pathologies.
Diabetes increases p66Shc expression (see expression and localization section). This links p66Shc to many diabetes-related conditions, including diabetes-induced endothelial dysfunction. p66Shc KO mice exhibit protection against diabetic endothelial dysfunction, oxidative stress, cardiomyopathy, and glomerulopathy [116,332,428]. Although p66Shc did not protect against diabetes induction or hemoglobin glycosylation, KO mice had lower nitrotyrosine and lipid peroxidation levels than their WT counterparts. Diabetes also causes advanced glycation end product accumulation, which promotes inflammation and ROS production via p66Shc ser36 phosphorylation. p66Shc KO mice also have decreased renal damage, circulating oxidative stress markers, and oxidative stress associated with glycation products [429,430,431]. In addition, glycation products increased p66Shc Ser36 phosphorylation in HEK-293 cells, depleting antioxidants and suppressing FOXO transcription. Furthermore, hyperglycemia increases p66Shc expression and decreases signaling from IGF1-stimulated phosphoinositide-3 kinase, AKT, which decreased cell survival in smooth muscle cells [432]. Thus, hyperglycemic p66Shc upregulation may upregulate p66Shc beyond its beneficial basal activity such that decreasing p66Shc is beneficial at rest for some cells.
Hyperlipidemia promotes coronary artery disease (CAD), a condition that promotes endothelial dysfunction, and increases oxidative damage in vasculature [433,434,435,436,437,438,439]. p66Shc KO mice on an apoliprotein E KO background showed that p66Shc also contributes to atherosclerosis, a common MI risk factor and CAD precursor [180,181]. Dyslipidemia can further exacerbate p66Shc ROS-mediated endothelial dysfunction and other pathologies as LDL increases p66Shc transcription via p66Shc promoter hypomethylation in endothelial cells [131].
Interestingly, CAD severity correlates with peripheral blood monocyte p66Shc mRNA concentrations and oxidative stress in humans, and p66Shc expression is therefore a CAD biomarker [11,12]. CAD patients also have thicker carotids that are less able to dilate in response to blood flow and the severity of dilatation resistance increases with increasing p66Shc mRNA [440]. Although p66Shc KO did not alter lipid concentrations in mice fed a high fat diet, KO mice showed improvements in lesion area, with decreases in systemic ROS, tissue ROS, and apoptosis [120]. Of note, all ShcA proteins are phosphorylated by shear stress, which is perpetually higher in CAD, which leads to inflammatory signaling [2,441]. Thus, p66Shc KO models are also causing increased p52/46Shc shear signaling that promotes survival and growth that is no longer balanced by pro-apoptotic p66Shc shear signaling (Figure 2). Therefore, observed improvements in p66Shc KO CAD models cannot be viewed exclusively as results from decreased p66Shc mitochondrial ROS production.
11. Future Perspectives and Summary
Although p66Shc’s full mechanism for superoxide anion production remains unanswered, recent advances have provided critical insight that can be utilized to further investigate p66Shc mechanistic details. An important mechanistic detail requiring further investigation is identification of upstream p66Shc electron donors in cyt c reduction. Since p66Shc binds the ubiquinone mimetic, idebenone, ubiquinone is a potential electron donor. However, many potential electron donors with reduction potentials favoring cyt c reduction exist within cells and, given p66Shc’s binding promiscuity, this reaction may occur through a variety of donors.
Furthermore, there are questions concerning how p66Shc modulates Complex IV/V activity of the ETC. Although the data infer that ETC activity is enhanced via cyt c recycling, direct electron donation to Complex IV has not been negated. Detailed structure–function experiments are also missing that could explain the step-wise process for superoxide anion production and whether the cysteine network directly contributes to electron flow or just conformational dynamics that permit superoxide anion production. Despite correlations between metabolism and p66Shc inhibition, targeted metabolism studies and mitochondrial oxygen consumption studies are also required to determine their mechanistic impacts on pathology. Lastly, p66Shc-CL effects are similar to those seen with cyt c and CL because both promote ROS production, but this correlation may have broader impacts that could be revealed with detailed analysis.
Once elucidated, these details could provide the information required to inhibit mitochondrial pro-apoptotic p66Shc functions while keeping pro-survival functions intact. Current data suggest that specifically preventing cyt c peroxidase inhibition and superoxide anion production could provide substantial benefit to p66Shc-mediated cardiovascular disease. As indicated in previous sections, p66Shc-selective ROS inhibitors have been described and improve outcomes in zebrafish cryoinjury-induced MI. However, concomitant inhibition of p66Shc-mediated superoxide production and H2O2 accumulation, if administered at the proper time and concentration to avoid potential side effects, could provide greater cardioprotection than observed with p66Shc-selective ROS inhibition alone. On the contrary, increasing these functions can also provide benefits to many cancers. In addition, specifically increasing pro-survival functions in cardiovascular pathology may have similar benefits to p66Shc-selective ROS inhibition. The opposite approach can be taken with pro-survival functions as well, as decreasing p66Shc pro-survival signals could have a substantial impact on p66Shc-mediated cancers.
The potential health benefits of modulating both sides of p66Shc’s cell fate functions (pro-survival and pro-apoptotic) are substantial but many obstacles have prevented such innovation. For example, until carvedilol and idebenone were described as selective p66Shc inhibitors, the primary method of studying p66Shc effects was genetic ablation, (despite being a drug target since 1999), which removes both p66Shc signaling and mitochondrial functions and makes data interpretation difficult. The newly discovered p66Shc-selecitve ROS inhibitors also have potential convoluting effects. Although binding and inhibition constants indicate that these newly described inhibitors are selective for p66Shc, they may have other targets influencing their results. For example, carvedilol also acts as a non-selective adrenergic blocker that may overlap with p66Shc inhibition. Idebenone is less likely to have overlap with other functions (e.g., Complex I inhibition) as they require concentrations ~170-340-fold higher than required for p66Shc binding and inhibition, respectively. However, second-generation inhibitors could be designed around idebenone and carvedilol to provide greater specificity for p66Shc. A complete p66Shc crystal structure is critical to produce second-generation p66Shc ROS inhibitors, as well as first-generation p66Shc pro-survival activators, but p66Shc’s inherent conformational variation and intrinsically disordered regions present a significant hurdle to crystallization. Furthermore, complete characterization of p66Shc’s superoxide and ETC-enhancing mechanisms will require intensive physical biochemistry experiments whose design, execution, and analysis will be difficult. Lastly, p66Shc largely behaves like a membrane protein during purification and overexpression, increasing the difficulty of performing high-throughput methods.
Defining p66Shc activity is a complex process, given its broad range of functions, localization dynamics, location/environment effects on activity, and multiple isoforms. Yet, this complexity provides p66Shc with the flexibility required to act as a molecular rheostat of apoptosis. The refined p66Shc molecular rheostat model resolves previous discrepancies between research groups by uniting findings in a singular model that is supported by in vivo MI models and is under active clinical investigation for cancer studies. The revised model also provides new therapeutic targets with the potential to modulate a variety of pathological conditions by adjusting p66Shc’s rheostatic setpoint.
Author Contributions
Conceptualization, L.H. and F.A.H.; Resources, L.H. and F.A.H.; Writing—Original Draft Preparation, L.H.; Writing—Review and Editing, L.H., J.M.H. and F.A.H.; Visualization, L.H.; Supervision, F.A.H.; Project Administration, L.H. and F.A.H.; Funding Acquisition, L.H. and F.A.H. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the following awards: NIH awards from National Institute of General Medical Sciences: R01GM118599 (F.A.H.) and National Heart Lung and Blood Institute: F30HL149279 (L.H.), Pilot Project funding from an Institutional Development Award (IDeA): P20GM103650, and the Presbyterian Health Foundation (F.A.H.).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zheng, Y.; Zhang, C.; Croucher, D.R.; Soliman, M.A.; St-Denis, N.; Pasculescu, A.; Taylor, L.; Tate, S.A.; Hardy, W.R.; Colwill, K.; et al. Temporal regulation of EGF signalling networks by the scaffold protein Shc1. Nature 2013, 499, 166–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Sweet, D.T.; Irani-Tehrani, M.; Maeda, N.; Tzima, E. Shc coordinates signals from intercellular junctions and integrins to regulate flow-induced inflammation. J. Cell Biol. 2008, 182, 185–196. [Google Scholar] [CrossRef]
- Soliman, M.A.; Abdel Rahman, A.M.; Lamming, D.W.; Birsoy, K.; Pawling, J.; Frigolet, M.E.; Lu, H.; Fantus, I.G.; Pasculescu, A.; Zheng, Y.; et al. The adaptor protein p66Shc inhibits mTOR-dependent anabolic metabolism. Sci. Signal 2014, 7, ra17. [Google Scholar] [CrossRef] [Green Version]
- Migliaccio, E.; Giorgio, M.; Mele, S.; Pelicci, G.; Reboldi, P.; Pandolfi, P.P.; Lanfrancone, L.; Pelicci, P.G. The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature 1999, 402, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, M.; Migliaccio, E.; Orsini, F.; Paolucci, D.; Moroni, M.; Contursi, C.; Pelliccia, G.; Luzi, L.; Minucci, S.; Marcaccio, M.; et al. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell 2005, 122, 221–233. [Google Scholar] [CrossRef]
- Di Lisa, F.; Kaludercic, N.; Carpi, A.; Menabo, R.; Giorgio, M. Mitochondrial pathways for ROS formation and myocardial injury: The relevance of p66(Shc) and monoamine oxidase. Basic Res. Cardiol. 2009, 104, 131–139. [Google Scholar] [CrossRef]
- Galimov, E.R. The Role of p66shc in Oxidative Stress and Apoptosis. Acta Nat. 2010, 2, 44–51. [Google Scholar] [CrossRef]
- Cano Sanchez, M.; Lancel, S.; Boulanger, E.; Neviere, R. Targeting Oxidative Stress and Mitochondrial Dysfunction in the Treatment of Impaired Wound Healing: A Systematic Review. Antioxidants 2018, 7, 98. [Google Scholar] [CrossRef] [Green Version]
- D’Arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Silvia, B. Preconditioning and Shear Stress in the Microcirculation in Ischemia-Reperfusion Injury. Curr. Cardiol. Rev. 2006, 2, 237–245. [Google Scholar] [CrossRef]
- Franzeck, F.C.; Hof, D.; Spescha, R.D.; Hasun, M.; Akhmedov, A.; Steffel, J.; Shi, Y.; Cosentino, F.; Tanner, F.C.; von Eckardstein, A.; et al. Expression of the aging gene p66Shc is increased in peripheral blood monocytes of patients with acute coronary syndrome but not with stable coronary artery disease. Atherosclerosis 2012, 220, 282–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noda, Y.; Yamagishi, S.; Matsui, T.; Ueda, S.; Ueda, S.; Jinnouchi, Y.; Hirai, Y.; Imaizumi, T. The p66shc gene expression in peripheral blood monocytes is increased in patients with coronary artery disease. Clin. Cardiol. 2010, 33, 548–552. [Google Scholar] [CrossRef] [PubMed]
- Spescha, R.D.; Klohs, J.; Semerano, A.; Giacalone, G.; Derungs, R.S.; Reiner, M.F.; Rodriguez Gutierrez, D.; Mendez-Carmona, N.; Glanzmann, M.; Savarese, G.; et al. Post-ischaemic silencing of p66Shc reduces ischaemia/reperfusion brain injury and its expression correlates to clinical outcome in stroke. Eur. Heart J. 2015, 36, 1590–1600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bugger, H.; Pfeil, K. Mitochondrial ROS in myocardial ischemia reperfusion and remodeling. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165768. [Google Scholar] [CrossRef]
- Shaw, P.X.; Werstuck, G.; Chen, Y. Oxidative stress and aging diseases. Oxid. Med. Cell. Longev. 2014, 2014, 569146. [Google Scholar] [CrossRef] [PubMed]
- Ghezzi, P.; Jaquet, V.; Marcucci, F.; Schmidt, H. The oxidative stress theory of disease: Levels of evidence and epistemological aspects. Br. J. Pharm. 2017, 174, 1784–1796. [Google Scholar] [CrossRef]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [Green Version]
- Shields, H.J.; Traa, A.; Van Raamsdonk, J.M. Beneficial and Detrimental Effects of Reactive Oxygen Species on Lifespan: A Comprehensive Review of Comparative and Experimental Studies. Front. Cell Dev. Biol. 2021, 9, 628157. [Google Scholar] [CrossRef]
- Milkovic, L.; Cipak Gasparovic, A.; Cindric, M.; Mouthuy, P.A.; Zarkovic, N. Short Overview of ROS as Cell Function Regulators and Their Implications in Therapy Concepts. Cells 2019, 8, 793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, A.L.; Sinha, S.; Lindner, A.B. The Good, the Bad, and the Ugly of ROS: New Insights on Aging and Aging-Related Diseases from Eukaryotic and Prokaryotic Model Organisms. Oxid. Med. Cell Longev. 2018, 2018, 1941285. [Google Scholar] [CrossRef] [PubMed]
- Pham-Huy, L.A.; He, H.; Pham-Huy, C. Free radicals, antioxidants in disease and health. Int. J. Biomed. Sci. 2008, 4, 89–96. [Google Scholar]
- Phaniendra, A.; Jestadi, D.B.; Periyasamy, L. Free radicals: Properties, sources, targets, and their implication in various diseases. Indian J. Clin. Biochem. 2015, 30, 11–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winterbourn, C.C. Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol. 2008, 4, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Mittler, R. ROS Are Good. Trends Plant Sci. 2017, 22, 11–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Marzo, N.; Chisci, E.; Giovannoni, R. The Role of Hydrogen Peroxide in Redox-Dependent Signaling: Homeostatic and Pathological Responses in Mammalian Cells. Cells 2018, 7, 156. [Google Scholar] [CrossRef] [Green Version]
- Travasso, R.D.M.; Sampaio Dos Aidos, F.; Bayani, A.; Abranches, P.; Salvador, A. Localized redox relays as a privileged mode of cytoplasmic hydrogen peroxide signaling. Redox Biol. 2017, 12, 233–245. [Google Scholar] [CrossRef]
- Sutton, H.C.; Winterbourn, C.C. On the participation of higher oxidation states of iron and copper in Fenton reactions. Free Radic Biol Med. 1989, 6, 53–60. [Google Scholar] [CrossRef]
- Mylonas, C.; Kouretas, D. Lipid peroxidation and tissue damage. Vivo 1999, 13, 295–309. [Google Scholar]
- Sohal, R.S. Role of oxidative stress and protein oxidation in the aging process. Free Radic. Biol. Med. 2002, 33, 37–44. [Google Scholar] [CrossRef]
- Kregel, K.C.; Zhang, H.J. An integrated view of oxidative stress in aging: Basic mechanisms, functional effects, and pathological considerations. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R18–R36. [Google Scholar] [CrossRef] [PubMed]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syslova, K.; Bohmova, A.; Mikoska, M.; Kuzma, M.; Pelclova, D.; Kacer, P. Multimarker screening of oxidative stress in aging. Oxid. Med. Cell. Longev. 2014, 2014, 562860. [Google Scholar] [CrossRef] [Green Version]
- Park, C.B.; Larsson, N.G. Mitochondrial DNA mutations in disease and aging. J. Cell Biol. 2011, 193, 809–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jha, R.; Rizvi, S.I. Carbonyl formation in erythrocyte membrane proteins during aging in humans. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czech. Repub. 2011, 155, 39–42. [Google Scholar] [CrossRef] [Green Version]
- Moskovitz, J.; Oien, D.B. Protein carbonyl and the methionine sulfoxide reductase system. Antioxid. Redox Signal. 2010, 12, 405–415. [Google Scholar] [CrossRef]
- Moskalev, A.A.; Shaposhnikov, M.V.; Plyusnina, E.N.; Zhavoronkov, A.; Budovsky, A.; Yanai, H.; Fraifeld, V.E. The role of DNA damage and repair in aging through the prism of Koch-like criteria. Ageing Res. Rev. 2013, 12, 661–684. [Google Scholar] [CrossRef] [PubMed]
- Massudi, H.; Grant, R.; Braidy, N.; Guest, J.; Farnsworth, B.; Guillemin, G.J. Age-associated changes in oxidative stress and NAD+ metabolism in human tissue. PLoS ONE 2012, 7, e42357. [Google Scholar] [CrossRef]
- Nguyen, G.T.; Green, E.R.; Mecsas, J. Neutrophils to the ROScue: Mechanisms of NADPH Oxidase Activation and Bacterial Resistance. Front. Cell Infect. Microbiol. 2017, 7, 373. [Google Scholar] [CrossRef]
- Beckman, K.B.; Ames, B.N. The free radical theory of aging matures. Physiol. Rev. 1998, 78, 547–581. [Google Scholar] [CrossRef] [Green Version]
- Lambeth, J.D. NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 2004, 4, 181–189. [Google Scholar] [CrossRef]
- Shih, J.C.; Chen, K.; Ridd, M.J. Monoamine oxidase: From genes to behavior. Annu. Rev. Neurosci. 1999, 22, 197–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Battelli, M.G.; Polito, L.; Bortolotti, M.; Bolognesi, A. Xanthine Oxidoreductase in Drug Metabolism: Beyond a Role as a Detoxifying Enzyme. Curr. Med. Chem. 2016, 23, 4027–4036. [Google Scholar] [CrossRef] [Green Version]
- McDonnell, A.M.; Dang, C.H. Basic review of the cytochrome p450 system. J. Adv. Pract. Oncol. 2013, 4, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Lismont, C.; Nordgren, M.; Van Veldhoven, P.P.; Fransen, M. Redox interplay between mitochondria and peroxisomes. Front. Cell Dev. Biol. 2015, 3, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid. Med. Cell. Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef] [PubMed]
- Van Raamsdonk, J.M.; Hekimi, S. Reactive Oxygen Species and Aging in Caenorhabditis elegans: Causal or Casual Relationship? Antioxid. Redox Signal. 2010, 13, 1911–1953. [Google Scholar] [CrossRef]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Vazquez, E.J.; Moghaddas, S.; Hoppel, C.L.; Lesnefsky, E.J. Production of reactive oxygen species by mitochondria: Central role of complex III. J. Biol. Chem. 2003, 278, 36027–36031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kushnareva, Y.; Murphy, A.N.; Andreyev, A. Complex I-mediated reactive oxygen species generation: Modulation by cytochrome c and NAD(P)+ oxidation-reduction state. Biochem. J. 2002, 368, 545–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forman, H.J.; Ursini, F.; Maiorino, M. An overview of mechanisms of redox signaling. J. Mol. Cell. Cardiol. 2014, 73, 2–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korshunov, S.S.; Krasnikov, B.F.; Pereverzev, M.O.; Skulachev, V.P. The antioxidant functions of cytochrome c. FEBS Lett. 1999, 462, 192–198. [Google Scholar] [CrossRef] [Green Version]
- Sedlak, E.; Fabian, M.; Robinson, N.C.; Musatov, A. Ferricytochrome c protects mitochondrial cytochrome c oxidase against hydrogen peroxide-induced oxidative damage. Free Radic. Biol. Med. 2010, 49, 1574–1581. [Google Scholar] [CrossRef] [Green Version]
- Pasdois, P.; Parker, J.E.; Griffiths, E.J.; Halestrap, A.P. The role of oxidized cytochrome c in regulating mitochondrial reactive oxygen species production and its perturbation in ischaemia. Biochem. J. 2011, 436, 493–505. [Google Scholar] [CrossRef] [Green Version]
- Pereverzev, M.O.; Vygodina, T.V.; Konstantinov, A.A.; Skulachev, V.P. Cytochrome c, an ideal antioxidant. Biochem. Soc. Trans. 2003, 31, 1312–1315. [Google Scholar] [CrossRef] [PubMed]
- Atlante, A.; Calissano, P.; Bobba, A.; Azzariti, A.; Marra, E.; Passarella, S. Cytochrome c is released from mitochondria in a reactive oxygen species (ROS)-dependent fashion and can operate as a ROS scavenger and as a respiratory substrate in cerebellar neurons undergoing excitotoxic death. J. Biol. Chem. 2000, 275, 37159–37166. [Google Scholar] [CrossRef] [Green Version]
- Butler, J.; Jayson, G.G.; Swallow, A.J. The reaction between the superoxide anion radical and cytochrome c. Biochim. Biophys. Acta 1975, 408, 215–222. [Google Scholar] [CrossRef]
- Sharma, R.; Yang, Y.; Sharma, A.; Awasthi, S.; Awasthi, Y.C. Antioxidant role of glutathione S-transferases: Protection against oxidant toxicity and regulation of stress-mediated apoptosis. Antioxid. Redox Signal. 2004, 6, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Perkins, A.; Nelson, K.J.; Parsonage, D.; Poole, L.B.; Karplus, P.A. Peroxiredoxins: Guardians against oxidative stress and modulators of peroxide signaling. Trends Biochem. Sci. 2015, 40, 435–445. [Google Scholar] [CrossRef] [Green Version]
- Hanschmann, E.M.; Godoy, J.R.; Berndt, C.; Hudemann, C.; Lillig, C.H. Thioredoxins, glutaredoxins, and peroxiredoxins--molecular mechanisms and health significance: From cofactors to antioxidants to redox signaling. Antioxid. Redox Signal. 2013, 19, 1539–1605. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kim, S.M.; Lee, R.T. Thioredoxin and thioredoxin target proteins: From molecular mechanisms to functional significance. Antioxid. Redox Signal. 2013, 18, 1165–1207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ursini, F.; Maiorino, M.; Gregolin, C. The selenoenzyme phospholipid hydroperoxide glutathione peroxidase. Biochim. Biophys. Acta 1985, 839, 62–70. [Google Scholar] [CrossRef]
- Flohe, L. The glutathione peroxidase reaction: Molecular basis of the antioxidant function of selenium in mammals. Curr. Top. Cell. Regul. 1985, 27, 473–478. [Google Scholar] [CrossRef]
- Baud, O.; Greene, A.E.; Li, J.; Wang, H.; Volpe, J.J.; Rosenberg, P.A. Glutathione peroxidase-catalase cooperativity is required for resistance to hydrogen peroxide by mature rat oligodendrocytes. J. Neurosci. 2004, 24, 1531–1540. [Google Scholar] [CrossRef] [Green Version]
- Ighodaro, O.M.; Akinloye, O.A. First line defence antioxidants-superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPX): Their fundamental role in the entire antioxidant defence grid. Alex. J. Med. 2018, 54, 287–293. [Google Scholar] [CrossRef] [Green Version]
- McCord, J.M.; Fridovich, I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 1969, 244, 6049–6055. [Google Scholar] [CrossRef]
- Rhee, S.G. Overview on Peroxiredoxin. Mol. Cells 2016, 39, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Arthur, J.R. The glutathione peroxidases. Cell. Mol. Life Sci. 2000, 57, 1825–1835. [Google Scholar] [CrossRef] [PubMed]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ J. 2012, 5, 9–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukai, T.; Ushio-Fukai, M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef] [Green Version]
- Rizvi, S.; Raza, S.T.; Ahmed, F.; Ahmad, A.; Abbas, S.; Mahdi, F. The role of vitamin e in human health and some diseases. Sultan. Qaboos. Univ. Med. J. 2014, 14, e157–e165. [Google Scholar] [PubMed]
- Frei, B. Reactive oxygen species and antioxidant vitamins: Mechanisms of action. Am. J. Med. 1994, 97, 5S–13S. [Google Scholar] [CrossRef]
- Sharp, P.; Hand, D.; Guthrie, E. Quantitative Determination of Dissolved Oxygen: Ascorbic Acid Oxidase Method. Ind. Eng. Chem. Anal. Ed. 1941, 13, 593–597. [Google Scholar] [CrossRef]
- Aldini, G.; Altomare, A.; Baron, G.; Vistoli, G.; Carini, M.; Borsani, L.; Sergio, F. N-Acetylcysteine as an antioxidant and disulphide breaking agent: The reasons why. Free Radic. Res. 2018, 52, 751–762. [Google Scholar] [CrossRef]
- Laurindo, F.R.M. Chapter 10—Redox Cellular Signaling Pathways in Endothelial Dysfunction and Vascular Disease. In Endothelium and Cardiovascular Diseases; Da Luz, P.L., Libby, P., Chagas, A.C.P., Laurindo, F.R.M., Eds.; Academic Press: Cambridge, MA, USA, 2018; pp. 127–145. [Google Scholar]
- Bienert, G.P.; Chaumont, F. Aquaporin-facilitated transmembrane diffusion of hydrogen peroxide. Biochim. Biophys. Acta 2014, 1840, 1596–1604. [Google Scholar] [CrossRef]
- Lennicke, C.; Rahn, J.; Lichtenfels, R.; Wessjohann, L.A.; Seliger, B. Hydrogen peroxide—Production, fate and role in redox signaling of tumor cells. Cell Commun. Signal. 2015, 13, 39. [Google Scholar] [CrossRef] [Green Version]
- Prata, C.; Hrelia, S.; Fiorentini, D. Peroxiporins in Cancer. Int. J. Mol. Sci. 2019, 20, 1371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, C.; Mósca, A.F.; Martins, A.P.; Nobre, T.; Prista, C.; Antunes, F.; Cipak Gasparovic, A.; Soveral, G. Rat Aquaporin-5 Is pH-Gated Induced by Phosphorylation and Is Implicated in Oxidative Stress. Int. J. Mol. Sci. 2016, 17, 2090. [Google Scholar] [CrossRef] [Green Version]
- Veal, E.A.; Day, A.M.; Morgan, B.A. Hydrogen peroxide sensing and signaling. Mol. Cell 2007, 26, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.H.; Stocker, R.; Vollbracht, C.; Paulsen, G.; Riley, D.; Daiber, A.; Cuadrado, A. Antioxidants in Translational Medicine. Antioxid. Redox Signal. 2015, 23, 1130–1143. [Google Scholar] [CrossRef] [Green Version]
- Casas, A.I.; Nogales, C.; Mucke, H.A.M.; Petraina, A.; Cuadrado, A.; Rojo, A.I.; Ghezzi, P.; Jaquet, V.; Augsburger, F.; Dufrasne, F.; et al. On the Clinical Pharmacology of Reactive Oxygen Species. Pharm. Rev. 2020, 72, 801–828. [Google Scholar] [CrossRef]
- Kitagishi, Y.; Matsuda, S. Redox regulation of tumor suppressor PTEN in cancer and aging (Review). Int. J. Mol. Med. 2013, 31, 511–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [Green Version]
- Lingappan, K. NF-κB in Oxidative Stress. Curr. Opin. Toxicol. 2018, 7, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Milkovic, L.; Zarkovic, N.; Saso, L. Controversy about pharmacological modulation of Nrf2 for cancer therapy. Redox Biol. 2017, 12, 727–732. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxid. Med. Cell. Longev. 2016, 2016, 4350965. [Google Scholar] [CrossRef] [Green Version]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef] [Green Version]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef] [PubMed]
- Mishra, M.; Duraisamy, A.J.; Bhattacharjee, S.; Kowluru, R.A. Adaptor Protein p66Shc: A Link between Cytosolic and Mitochondrial Dysfunction in the Development of Diabetic Retinopathy. Antioxid. Redox Signal. 2019, 30, 1621–1634. [Google Scholar] [CrossRef] [PubMed]
- Szocs, K. Endothelial dysfunction and reactive oxygen species production in ischemia/reperfusion and nitrate tolerance. Gen. Physiol. Biophys. 2004, 23, 265–295. [Google Scholar] [PubMed]
- Al Sabaani, N. Exendin-4 inhibits high glucose-induced oxidative stress in retinal pigment epithelial cells by modulating the expression and activation of p(66)Shc. Cutan. Ocul. Toxicol. 2021, 40, 175–186. [Google Scholar] [CrossRef]
- Mansouri, A.; Gattolliat, C.H.; Asselah, T. Mitochondrial Dysfunction and Signaling in Chronic Liver Diseases. Gastroenterology 2018, 155, 629–647. [Google Scholar] [CrossRef] [Green Version]
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vasc. Pharm. 2018, 100, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Oxidative stress-based therapeutics in COPD. Redox. Biol. 2020, 33, 101544. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, W.; Ijaz, B.; Shabbiri, K.; Ahmed, F.; Rehman, S. Oxidative toxicity in diabetes and Alzheimer’s disease: Mechanisms behind ROS/ RNS generation. J. Biomed. Sci. 2017, 24, 76. [Google Scholar] [CrossRef]
- Lone, A.; Harris, R.A.; Singh, O.; Betts, D.H.; Cumming, R.C. p66Shc activation promotes increased oxidative phosphorylation and renders CNS cells more vulnerable to amyloid beta toxicity. Sci. Rep. 2018, 8, 17081. [Google Scholar] [CrossRef]
- Irazabal, M.V.; Torres, V.E. Reactive Oxygen Species and Redox Signaling in Chronic Kidney Disease. Cells 2020, 9, 1342. [Google Scholar] [CrossRef]
- Nagar, H.; Piao, S.; Kim, C.S. Role of Mitochondrial Oxidative Stress in Sepsis. Acute Crit. Care 2018, 33, 65–72. [Google Scholar] [CrossRef]
- Kaneto, H.; Katakami, N.; Matsuhisa, M.; Matsuoka, T.A. Role of reactive oxygen species in the progression of type 2 diabetes and atherosclerosis. Mediat. Inflamm. 2010, 2010, 453892. [Google Scholar] [CrossRef] [Green Version]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Sugamura, K.; Keaney, J.F., Jr. Reactive oxygen species in cardiovascular disease. Free Radic. Biol. Med. 2011, 51, 978–992. [Google Scholar] [CrossRef] [Green Version]
- Daiber, A.; Di Lisa, F.; Ferdinandy, P. Pharmacology of oxidative stress: Translational opportunities. Br. J. Pharm. 2017, 174, 1511–1513. [Google Scholar] [CrossRef] [Green Version]
- Kornfeld, O.S.; Hwang, S.; Disatnik, M.H.; Chen, C.H.; Qvit, N.; Mochly-Rosen, D. Mitochondrial reactive oxygen species at the heart of the matter: New therapeutic approaches for cardiovascular diseases. Circ. Res. 2015, 116, 1783–1799. [Google Scholar] [CrossRef] [PubMed]
- Steinhubl, S.R. Why have antioxidants failed in clinical trials? Am. J. Cardiol. 2008, 101, 14d–19d. [Google Scholar] [CrossRef]
- Pawson, T.; Gish, G.D. SH2 and SH3 domains: From structure to function. Cell 1992, 71, 359–362. [Google Scholar] [CrossRef]
- Kumar, S. P66Shc and vascular endothelial function. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Migliaccio, E.; Mele, S.; Salcini, A.E.; Pelicci, G.; Lai, K.M.; Superti-Furga, G.; Pawson, T.; Di Fiore, P.P.; Lanfrancone, L.; Pelicci, P.G. Opposite effects of the p52shc/p46shc and p66shc splicing isoforms on the EGF receptor-MAP kinase-fos signalling pathway. EMBO J. 1997, 16, 706–716. [Google Scholar] [CrossRef]
- Heusch, G. Mitochondria at the heart of cardiovascular protection: p66shc-friend or foe? Eur. Heart J. 2015, 36, 469–471. [Google Scholar] [CrossRef] [Green Version]
- Akhmedov, A.; Montecucco, F.; Braunersreuther, V.; Camici, G.G.; Jakob, P.; Reiner, M.F.; Glanzmann, M.; Burger, F.; Paneni, F.; Galan, K.; et al. Genetic deletion of the adaptor protein p66Shc increases susceptibility to short-term ischaemic myocardial injury via intracellular salvage pathways. Eur. Heart J. 2015, 36, 516–526a. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giorgio, M.; Berry, A.; Berniakovich, I.; Poletaeva, I.; Trinei, M.; Stendardo, M.; Hagopian, K.; Ramsey, J.J.; Cortopassi, G.; Migliaccio, E.; et al. The p66Shc knocked out mice are short lived under natural condition. Aging Cell 2012, 11, 162–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramsey, J.J.; Tran, D.; Giorgio, M.; Griffey, S.M.; Koehne, A.; Laing, S.T.; Taylor, S.L.; Kim, K.; Cortopassi, G.A.; Lloyd, K.C.; et al. The influence of Shc proteins on life span in mice. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, 1177–1185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, K.M.; Pawson, T. The ShcA phosphotyrosine docking protein sensitizes cardiovascular signaling in the mouse embryo. Genes Dev. 2000, 14, 1132–1145. [Google Scholar] [CrossRef] [PubMed]
- Tomilov, A.A.; Ramsey, J.J.; Hagopian, K.; Giorgio, M.; Kim, K.M.; Lam, A.; Migliaccio, E.; Lloyd, K.C.; Berniakovich, I.; Prolla, T.A.; et al. The Shc locus regulates insulin signaling and adiposity in mammals. Aging Cell 2011, 10, 55–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berniakovich, I.; Trinei, M.; Stendardo, M.; Migliaccio, E.; Minucci, S.; Bernardi, P.; Pelicci, P.G.; Giorgio, M. p66Shc-generated oxidative signal promotes fat accumulation. J. Biol. Chem. 2008, 283, 34283–34293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menini, S.; Amadio, L.; Oddi, G.; Ricci, C.; Pesce, C.; Pugliese, F.; Giorgio, M.; Migliaccio, E.; Pelicci, P.; Iacobini, C.; et al. Deletion of p66Shc longevity gene protects against experimental diabetic glomerulopathy by preventing diabetes-induced oxidative stress. Diabetes 2006, 55, 1642–1650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fadini, G.P.; Ceolotto, G.; Pagnin, E.; de Kreutzenberg, S.; Avogaro, A. At the crossroads of longevity and metabolism: The metabolic syndrome and lifespan determinant pathways. Aging Cell 2011, 10, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Zaccagnini, G.; Martelli, F.; Fasanaro, P.; Magenta, A.; Gaetano, C.; Di Carlo, A.; Biglioli, P.; Giorgio, M.; Martin-Padura, I.; Pelicci, P.G.; et al. p66ShcA modulates tissue response to hindlimb ischemia. Circulation 2004, 109, 2917–2923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpi, A.; Menabo, R.; Kaludercic, N.; Pelicci, P.; Di Lisa, F.; Giorgio, M. The cardioprotective effects elicited by p66(Shc) ablation demonstrate the crucial role of mitochondrial ROS formation in ischemia/reperfusion injury. Biochim. Biophys. Acta 2009, 1787, 774–780. [Google Scholar] [CrossRef] [PubMed]
- Napoli, C.; Martin-Padura, I.; de Nigris, F.; Giorgio, M.; Mansueto, G.; Somma, P.; Condorelli, M.; Sica, G.; De Rosa, G.; Pelicci, P. Deletion of the p66Shc longevity gene reduces systemic and tissue oxidative stress, vascular cell apoptosis, and early atherogenesis in mice fed a high-fat diet. Proc. Natl. Acad. Sci. USA 2003, 100, 2112–2116. [Google Scholar] [CrossRef] [Green Version]
- Graiani, G.; Lagrasta, C.; Migliaccio, E.; Spillmann, F.; Meloni, M.; Madeddu, P.; Quaini, F.; Padura, I.M.; Lanfrancone, L.; Pelicci, P.; et al. Genetic deletion of the p66Shc adaptor protein protects from angiotensin II-induced myocardial damage. Hypertension 2005, 46, 433–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranieri Sofia, C.; Fusco, S.; Panieri, E.; Labate, V.; Mele, M.; Tesori, V.; Ferrara Anna, M.; Maulucci, G.; De Spirito, M.; Martorana Giuseppe, E.; et al. Mammalian life-span determinant p66shcA mediates obesity-induced insulin resistance. Proc. Natl. Acad. Sci. USA 2010, 107, 13420–13425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevenson, L.E.; Frackelton, A.R., Jr. Constitutively tyrosine phosphorylated p52 Shc in breast cancer cells: Correlation with ErbB2 and p66 Shc expression. Breast Cancer Res. Treat 1998, 49, 119–128. [Google Scholar] [CrossRef]
- Gertz, M.; Fischer, F.; Wolters, D.; Steegborn, C. Activation of the lifespan regulator p66Shc through reversible disulfide bond formation. Proc. Natl. Acad. Sci. USA 2008, 105, 5705–5709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelicci, G.; Lanfrancone, L.; Grignani, F.; McGlade, J.; Cavallo, F.; Forni, G.; Nicoletti, I.; Grignani, F.; Pawson, T.; Pelicci, P.G. A novel transforming protein (SHC) with an SH2 domain is implicated in mitogenic signal transduction. Cell 1992, 70, 93–104. [Google Scholar] [CrossRef]
- Berry, A.; Capone, F.; Giorgio, M.; Pelicci, P.G.; de Kloet, E.R.; Alleva, E.; Minghetti, L.; Cirulli, F. Deletion of the life span determinant p66Shc prevents age-dependent increases in emotionality and pain sensitivity in mice. Exp. Gerontol. 2007, 42, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Berry, A.; Greco, A.; Giorgio, M.; Pelicci, P.G.; de Kloet, R.; Alleva, E.; Minghetti, L.; Cirulli, F. Deletion of the lifespan determinant p66(Shc) improves performance in a spatial memory task, decreases levels of oxidative stress markers in the hippocampus and increases levels of the neurotrophin BDNF in adult mice. Exp. Gerontol. 2008, 43, 200–208. [Google Scholar] [CrossRef]
- Conti, L.; De Fraja, C.; Gulisano, M.; Migliaccio, E.; Govoni, S.; Cattaneo, E. Expression and activation of SH2/PTB-containing ShcA adaptor protein reflects the pattern of neurogenesis in the mammalian brain. Proc. Natl. Acad. Sci. USA 1997, 94, 8185–8190. [Google Scholar] [CrossRef] [Green Version]
- Lebiedzinska, M.; Duszynski, J.; Rizzuto, R.; Pinton, P.; Wieckowski, M.R. Age-related changes in levels of p66Shc and serine 36-phosphorylated p66Shc in organs and mouse tissues. Arch. Biochem. Biophys. 2009, 486, 73–80. [Google Scholar] [CrossRef]
- Ventura, A.; Luzi, L.; Pacini, S.; Baldari, C.T.; Pelicci, P.G. The p66Shc longevity gene is silenced through epigenetic modifications of an alternative promoter. J. Biol. Chem. 2002, 277, 22370–22376. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.R.; Kim, C.S.; Naqvi, A.; Kumar, A.; Kumar, S.; Hoffman, T.A.; Irani, K. Epigenetic upregulation of p66shc mediates low-density lipoprotein cholesterol-induced endothelial cell dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H189–H196. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.S.; Jung, S.B.; Naqvi, A.; Hoffman, T.A.; DeRicco, J.; Yamamori, T.; Cole, M.P.; Jeon, B.H.; Irani, K. p53 impairs endothelium-dependent vasomotor function through transcriptional upregulation of p66shc. Circ. Res. 2008, 103, 1441–1450. [Google Scholar] [CrossRef]
- Ihling, C.; Haendeler, J.; Menzel, G.; Hess, R.D.; Fraedrich, G.; Schaefer, H.E.; Zeiher, A.M. Co-expression of p53 and MDM2 in human atherosclerosis: Implications for the regulation of cellularity of atherosclerotic lesions. J. Pathol. 1998, 185, 303–312. [Google Scholar] [CrossRef]
- Trinei, M.; Giorgio, M.; Cicalese, A.; Barozzi, S.; Ventura, A.; Migliaccio, E.; Milia, E.; Padura, I.M.; Raker, V.A.; Maccarana, M.; et al. A p53-p66Shc signalling pathway controls intracellular redox status, levels of oxidation-damaged DNA and oxidative stress-induced apoptosis. Oncogene 2002, 21, 3872–3878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, S.; Chen, H.Z.; Wan, Y.Z.; Zhang, Q.J.; Wei, Y.S.; Huang, S.; Liu, J.J.; Lu, Y.B.; Zhang, Z.Q.; Yang, R.F.; et al. Repression of P66Shc expression by SIRT1 contributes to the prevention of hyperglycemia-induced endothelial dysfunction. Circ. Res. 2011, 109, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Paneni, F.; Mocharla, P.; Akhmedov, A.; Costantino, S.; Osto, E.; Volpe, M.; Luscher, T.F.; Cosentino, F. Gene silencing of the mitochondrial adaptor p66(Shc) suppresses vascular hyperglycemic memory in diabetes. Circ. Res. 2012, 111, 278–289. [Google Scholar] [CrossRef] [Green Version]
- Costantino, S.; Paneni, F.; Virdis, A.; Hussain, S.; Mohammed, S.A.; Capretti, G.; Akhmedov, A.; Dalgaard, K.; Chiandotto, S.; Pospisilik, J.A.; et al. Interplay among H3K9-editing enzymes SUV39H1, JMJD2C and SRC-1 drives p66Shc transcription and vascular oxidative stress in obesity. Eur. Heart J. 2019, 40, 383–391. [Google Scholar] [CrossRef] [Green Version]
- Bosch-Presegué, L.; Raurell-Vila, H.; Marazuela-Duque, A.; Kane-Goldsmith, N.; Valle, A.; Oliver, J.; Serrano, L.; Vaquero, A. Stabilization of Suv39H1 by SirT1 is part of oxidative stress response and ensures genome protection. Mol. Cell 2011, 42, 210–223. [Google Scholar] [CrossRef]
- Du, W.; Jiang, Y.; Zheng, Z.; Zhang, Z.; Chen, N.; Ma, Z.; Yao, Z.; Terada, L.; Liu, Z. Feedback loop between p66(Shc) and Nrf2 promotes lung cancer progression. Cancer Lett. 2013, 337, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Miyazawa, M.; Tsuji, Y. Evidence for a novel antioxidant function and isoform-specific regulation of the human p66Shc gene. Mol. Biol. Cell 2014, 25, 2116–2127. [Google Scholar] [CrossRef]
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal 2012, 24, 981–990. [Google Scholar] [CrossRef] [Green Version]
- Faisal, A.; el-Shemerly, M.; Hess, D.; Nagamine, Y. Serine/threonine phosphorylation of ShcA. Regulation of protein-tyrosine phosphatase-pest binding and involvement in insulin signaling. J. Biol. Chem. 2002, 277, 30144–30152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemoto, S.; Combs, C.A.; French, S.; Ahn, B.H.; Fergusson, M.M.; Balaban, R.S.; Finkel, T. The mammalian longevity-associated gene product p66shc regulates mitochondrial metabolism. J. Biol. Chem. 2006, 281, 10555–10560. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, S.B.M.; Prigent, S.A. Insights into the Shc Family of Adaptor Proteins. J. Mol. Signal. 2017, 12, 2. [Google Scholar] [CrossRef] [Green Version]
- Ventura, A.; Maccarana, M.; Raker, V.A.; Pelicci, P.G. A cryptic targeting signal induces isoform-specific localization of p46Shc to mitochondria. J. Biol. Chem. 2004, 279, 2299–2306. [Google Scholar] [CrossRef] [Green Version]
- Orsini, F.; Migliaccio, E.; Moroni, M.; Contursi, C.; Raker, V.A.; Piccini, D.; Martin-Padura, I.; Pelliccia, G.; Trinei, M.; Bono, M.; et al. The life span determinant p66Shc localizes to mitochondria where it associates with mitochondrial heat shock protein 70 and regulates trans-membrane potential. J. Biol. Chem. 2004, 279, 25689–25695. [Google Scholar] [CrossRef] [Green Version]
- Gertz, M.; Fischer, F.; Leipelt, M.; Wolters, D.; Steegborn, C. Identification of Peroxiredoxin 1 as a novel interaction partner for the lifespan regulator protein p66Shc. Aging (Albany NY) 2009, 1, 254–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, K.D.; Staruschenko, A.; Sorokin, A. Role of adaptor protein p66Shc in renal pathologies. Am. J. Physiol. Ren. Physiol. 2018, 314, F143–F153. [Google Scholar] [CrossRef]
- Bhat, S.S.; Anand, D.; Khanday, F.A. p66Shc as a switch in bringing about contrasting responses in cell growth: Implications on cell proliferation and apoptosis. Mol. Cancer 2015, 14, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, S.; Kao, A.W.; Ceresa, B.P.; Blaikie, P.; Margolis, B.; Pessin, J.E. The 66-kDa Shc isoform is a negative regulator of the epidermal growth factor-stimulated mitogen-activated protein kinase pathway. J. Biol. Chem. 1997, 272, 28042–28049. [Google Scholar] [CrossRef] [Green Version]
- Pacini, S.; Pellegrini, M.; Migliaccio, E.; Patrussi, L.; Ulivieri, C.; Ventura, A.; Carraro, F.; Naldini, A.; Lanfrancone, L.; Pelicci, P.; et al. p66SHC promotes apoptosis and antagonizes mitogenic signaling in T cells. Mol. Cell. Biol. 2004, 24, 1747–1757. [Google Scholar] [CrossRef] [Green Version]
- Ong, S.H.; Dilworth, S.; Hauck-Schmalenberger, I.; Pawson, T.; Kiefer, F. ShcA and Grb2 mediate polyoma middle T antigen-induced endothelial transformation and Gab1 tyrosine phosphorylation. EMBO J. 2001, 20, 6327–6336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arany, I.; Faisal, A.; Nagamine, Y.; Safirstein, R.L. p66shc inhibits pro-survival epidermal growth factor receptor/ERK signaling during severe oxidative stress in mouse renal proximal tubule cells. J. Biol. Chem. 2008, 283, 6110–6117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xi, G.; Shen, X.; Clemmons, D.R. p66shc negatively regulates insulin-like growth factor I signal transduction via inhibition of p52shc binding to Src homology 2 domain-containing protein tyrosine phosphatase substrate-1 leading to impaired growth factor receptor-bound protein-2 membrane recruitment. Mol. Endocrinol. 2008, 22, 2162–2175. [Google Scholar] [CrossRef] [Green Version]
- Khanday, F.A.; Santhanam, L.; Kasuno, K.; Yamamori, T.; Naqvi, A.; Dericco, J.; Bugayenko, A.; Mattagajasingh, I.; Disanza, A.; Scita, G.; et al. Sos-mediated activation of rac1 by p66shc. J. Cell Biol. 2006, 172, 817–822. [Google Scholar] [CrossRef] [Green Version]
- Khanday, F.A.; Yamamori, T.; Mattagajasingh, I.; Zhang, Z.; Bugayenko, A.; Naqvi, A.; Santhanam, L.; Nabi, N.; Kasuno, K.; Day, B.W.; et al. Rac1 leads to phosphorylation-dependent increase in stability of the p66shc adaptor protein: Role in Rac1-induced oxidative stress. Mol. Biol. Cell. 2006, 17, 122–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, F.; Simeone, M.; Patel, R. Inhibition of NADPH oxidase reduces myocardial oxidative stress and apoptosis and improves cardiac function in heart failure after myocardial infarction. Free Radic. Biol. Med. 2007, 43, 271–281. [Google Scholar] [CrossRef]
- Davidson, D.; Veillette, A. PTP-PEST, a scaffold protein tyrosine phosphatase, negatively regulates lymphocyte activation by targeting a unique set of substrates. EMBO J. 2001, 20, 3414–3426. [Google Scholar] [CrossRef]
- Schneider, E.; Keppler, R.; Prawitt, D.; Steinwender, C.; Roos, F.C.; Thuroff, J.W.; Lausch, E.; Brenner, W. Migration of renal tumor cells depends on dephosphorylation of Shc by PTEN. Int. J. Oncol. 2011, 38, 823–831. [Google Scholar] [CrossRef] [Green Version]
- Kraut-Cohen, J.; Muller, W.J.; Elson, A. Protein-tyrosine phosphatase epsilon regulates Shc signaling in a kinase-specific manner: Increasing coherence in tyrosine phosphatase signaling. J. Biol. Chem. 2008, 283, 4612–4621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vikram, A.; Kim, Y.R.; Kumar, S.; Naqvi, A.; Hoffman, T.A.; Kumar, A.; Miller, F.J., Jr.; Kim, C.S.; Irani, K. Canonical Wnt signaling induces vascular endothelial dysfunction via p66Shc-regulated reactive oxygen species. Arter. Thromb. Vasc. Biol. 2014, 34, 2301–2309. [Google Scholar] [CrossRef] [Green Version]
- Di Lisa, F.; Giorgio, M.; Ferdinandy, P.; Schulz, R. New aspects of p66Shc in ischaemia reperfusion injury and other cardiovascular diseases. Br. J. Pharm. 2017, 174, 1690–1703. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Cosentino, F.; Camici, G.G.; Akhmedov, A.; Vanhoutte, P.M.; Tanner, F.C.; Luscher, T.F. Oxidized low-density lipoprotein activates p66Shc via lectin-like oxidized low-density lipoprotein receptor-1, protein kinase C-beta, and c-Jun N-terminal kinase kinase in human endothelial cells. Arter. Thromb. Vasc. Biol. 2011, 31, 2090–2097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinton, P.; Rimessi, A.; Marchi, S.; Orsini, F.; Migliaccio, E.; Giorgio, M.; Contursi, C.; Minucci, S.; Mantovani, F.; Wieckowski, M.R.; et al. Protein kinase C beta and prolyl isomerase 1 regulate mitochondrial effects of the life-span determinant p66Shc. Science 2007, 315, 659–663. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.Z.; Shi, M.M.; Xie, L.; Wu, Z.Y.; Li, G.; Hua, F.; Bian, J.S. Sulfhydration of p66Shc at cysteine59 mediates the antioxidant effect of hydrogen sulfide. Antioxid. Redox Signal. 2014, 21, 2531–2542. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Kim, Y.R.; Vikram, A.; Naqvi, A.; Li, Q.; Kassan, M.; Kumar, V.; Bachschmid, M.M.; Jacobs, J.S.; Kumar, A.; et al. Sirtuin1-regulated lysine acetylation of p66Shc governs diabetes-induced vascular oxidative stress and endothelial dysfunction. Proc. Natl. Acad. Sci. USA 2017, 114, 1714–1719. [Google Scholar] [CrossRef] [Green Version]
- Wu, R.F.; Liao, C.; Fu, G.; Hayenga, H.N.; Yang, K.; Ma, Z.; Liu, Z.; Terada, L.S. p66(Shc) Couples Mechanical Signals to RhoA through Focal Adhesion Kinase-Dependent Recruitment of p115-RhoGEF and GEF-H1. Mol. Cell. Biol. 2016, 36, 2824–2837. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.; Myers, D.P.; Wu, R.F.; Nwariaku, F.E.; Terada, L.S. p66Shc mediates anoikis through RhoA. J. Cell Biol. 2007, 179, 23–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.D.; Li, Y.S.; Kim, M.; Li, S.; Yuan, S.; Chien, S.; Shyy, J.Y. Mechanotransduction in response to shear stress. Roles of receptor tyrosine kinases, integrins, and Shc. J. Biol. Chem. 1999, 274, 18393–18400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wary, K.K.; Mainiero, F.; Isakoff, S.J.; Marcantonio, E.E.; Giancotti, F.G. The adaptor protein Shc couples a class of integrins to the control of cell cycle progression. Cell 1996, 87, 733–743. [Google Scholar] [CrossRef] [Green Version]
- Vanderlaan, R.D.; Hardy, W.R.; Kabir, M.G.; Pasculescu, A.; Jones, N.; deTombe, P.P.; Backx, P.H.; Pawson, T. The ShcA phosphotyrosine docking protein uses distinct mechanisms to regulate myocyte and global heart function. Circ. Res. 2011, 108, 184–193. [Google Scholar] [CrossRef] [Green Version]
- Hardy, W.R.; Li, L.; Wang, Z.; Sedy, J.; Fawcett, J.; Frank, E.; Kucera, J.; Pawson, T. Combinatorial ShcA docking interactions support diversity in tissue morphogenesis. Science 2007, 317, 251–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natalicchio, A.; Laviola, L.; De Tullio, C.; Renna, L.A.; Montrone, C.; Perrini, S.; Valenti, G.; Procino, G.; Svelto, M.; Giorgino, F. Role of the p66Shc isoform in insulin-like growth factor I receptor signaling through MEK/Erk and regulation of actin cytoskeleton in rat myoblasts. J. Biol. Chem. 2004, 279, 43900–43909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Favetta, L.A.; Madan, P.; Mastromonaco, G.F.; St John, E.J.; King, W.A.; Betts, D.H. The oxidative stress adaptor p66Shc is required for permanent embryo arrest in vitro. BMC Dev. Biol. 2007, 7, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.K.; Smith, S.M.; Banerjee, M.M.; Li, C.; Minoo, P.; Volpe, M.V.; Nielsen, H.C. The p66Shc adapter protein regulates the morphogenesis and epithelial maturation of fetal mouse lungs. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 306, L316–L325. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.; Liu, Z.; Wu, R.F.; Terada, L.S. p66(Shc) restrains Ras hyperactivation and suppresses metastatic behavior. Oncogene 2010, 29, 5559–5567. [Google Scholar] [CrossRef] [Green Version]
- Dohn, M.R.; Brown, M.V.; Reynolds, A.B. An essential role for p120-catenin in Src- and Rac1-mediated anchorage-independent cell growth. J. Cell Biol. 2009, 184, 437–450. [Google Scholar] [CrossRef] [Green Version]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Terada, L.S. Shc and the mechanotransduction of cellular anchorage and metastasis. Small GTPases 2019, 10, 64–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutta, P.; Courties, G.; Wei, Y.; Leuschner, F.; Gorbatov, R.; Robbins, C.S.; Iwamoto, Y.; Thompson, B.; Carlson, A.L.; Heidt, T.; et al. Myocardial infarction accelerates atherosclerosis. Nature 2012, 487, 325–329. [Google Scholar] [CrossRef] [Green Version]
- Martin-Padura, I.; de Nigris, F.; Migliaccio, E.; Mansueto, G.; Minardi, S.; Rienzo, M.; Lerman, L.O.; Stendardo, M.; Giorgio, M.; De Rosa, G.; et al. p66Shc deletion confers vascular protection in advanced atherosclerosis in hypercholesterolemic apolipoprotein E knockout mice. Endothelium 2008, 15, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Haslem, L.; Hays, J.M.; Schmitz, H.; Matsuzaki, S.; Sjoelund, V.; Byrum, S.D.; Humphries, K.M.; Frazer, J.K.; Demeler, B.; Benbrook, D.M.; et al. p66Shc is an apoptotic rheostat whose targeted ROS inhibition improves MI outcomes. bioRxiv 2022, 2022.2004.2014.487897. [Google Scholar] [CrossRef]
- Neumann, C.A.; Cao, J.; Manevich, Y. Peroxiredoxin 1 and its role in cell signaling. Cell Cycle 2009, 8, 4072–4078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirotsu, S.; Abe, Y.; Okada, K.; Nagahara, N.; Hori, H.; Nishino, T.; Hakoshima, T. Crystal structure of a multifunctional 2-Cys peroxiredoxin heme-binding protein 23 kDa/proliferation-associated gene product. Proc. Natl. Acad. Sci. USA 1999, 96, 12333–12338. [Google Scholar] [CrossRef] [Green Version]
- Wood, Z.A.; Schroder, E.; Robin Harris, J.; Poole, L.B. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem. Sci. 2003, 28, 32–40. [Google Scholar] [CrossRef]
- Kim, S.Y.; Kim, T.J.; Lee, K.Y. A novel function of peroxiredoxin 1 (Prx-1) in apoptosis signal-regulating kinase 1 (ASK1)-mediated signaling pathway. FEBS Lett. 2008, 582, 1913–1918. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.J.; Lee, W.S.; Ip, C.; Chae, H.Z.; Park, E.M.; Park, Y.M. Prx1 suppresses radiation-induced c-Jun NH2-terminal kinase signaling in lung cancer cells through interaction with the glutathione S-transferase Pi/c-Jun NH2-terminal kinase complex. Cancer Res. 2006, 66, 7136–7142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Wang, Y.; Su, Y. Peroxiredoxins, a novel target in cancer radiotherapy. Cancer Lett 2009, 286, 154–160. [Google Scholar] [CrossRef]
- Lee, W.; Choi, K.S.; Riddell, J.; Ip, C.; Ghosh, D.; Park, J.H.; Park, Y.M. Human peroxiredoxin 1 and 2 are not duplicate proteins: The unique presence of CYS83 in Prx1 underscores the structural and functional differences between Prx1 and Prx2. J. Biol. Chem. 2007, 282, 22011–22022. [Google Scholar] [CrossRef]
- Shau, H.; Gupta, R.K.; Golub, S.H. Identification of a natural killer enhancing factor (NKEF) from human erythroid cells. Cell Immunol. 1993, 147, 1–11. [Google Scholar] [CrossRef]
- Rhee, S.G.; Woo, H.A. Multiple functions of peroxiredoxins: Peroxidases, sensors and regulators of the intracellular messenger H2O2, and protein chaperones. Antioxid. Redox Signal. 2011, 15, 781–794. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Warabi, E.; Yanagawa, T. Novel roles of peroxiredoxins in inflammation, cancer and innate immunity. J. Clin. Biochem. Nutr. 2012, 50, 91–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, J.C.; Hah, Y.S.; Kim, W.Y.; Jung, B.G.; Jang, H.H.; Lee, J.R.; Kim, S.Y.; Lee, Y.M.; Jeon, M.G.; Kim, C.W.; et al. Oxidative stress-dependent structural and functional switching of a human 2-Cys peroxiredoxin isotype II that enhances HeLa cell resistance to H2O2-induced cell death. J. Biol. Chem. 2005, 280, 28775–28784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, S.T.; Van Etten, R.A. The PAG gene product, a stress-induced protein with antioxidant properties, is an Abl SH3-binding protein and a physiological inhibitor of c-Abl tyrosine kinase activity. Genes Dev. 1997, 11, 2456–2467. [Google Scholar] [CrossRef] [Green Version]
- Mu, Z.M.; Yin, X.Y.; Prochownik, E.V. Pag, a putative tumor suppressor, interacts with the Myc Box II domain of c-Myc and selectively alters its biological function and target gene expression. J. Biol. Chem. 2002, 277, 43175–43184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riddell, J.R.; Bshara, W.; Moser, M.T.; Spernyak, J.A.; Foster, B.A.; Gollnick, S.O. Peroxiredoxin 1 controls prostate cancer growth through Toll-like receptor 4-dependent regulation of tumor vasculature. Cancer Res. 2011, 71, 1637–1646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, P.; Ye, H.; Dai, L.; Liu, M.; Liu, X.; Chai, Y.; Shao, Q.; Li, Y.; Lei, N.; Peng, B.; et al. Peroxiredoxin 1 is a tumor-associated antigen in esophageal squamous cell carcinoma. Oncol. Rep. 2013, 30, 2297–2303. [Google Scholar] [CrossRef] [Green Version]
- Karihtala, P.; Mantyniemi, A.; Kang, S.W.; Kinnula, V.L.; Soini, Y. Peroxiredoxins in breast carcinoma. Clin. Cancer Res. 2003, 9, 3418–3424. [Google Scholar] [PubMed]
- Deng, B.; Ye, N.; Luo, G.; Chen, X.; Wang, Y. Proteomics analysis of stage-specific proteins expressed in human squamous cell lung carcinoma tissues. Cancer Biomark 2005, 1, 279–286. [Google Scholar] [CrossRef]
- Jang, H.H.; Lee, K.O.; Chi, Y.H.; Jung, B.G.; Park, S.K.; Park, J.H.; Lee, J.R.; Lee, S.S.; Moon, J.C.; Yun, J.W.; et al. Two enzymes in one; two yeast peroxiredoxins display oxidative stress-dependent switching from a peroxidase to a molecular chaperone function. Cell 2004, 117, 625–635. [Google Scholar] [CrossRef]
- Levav-Cohen, Y.; Goldberg, Z.; Zuckerman, V.; Grossman, T.; Haupt, S.; Haupt, Y. C-Abl as a modulator of p53. Biochem. Biophys. Res. Commun. 2005, 331, 737–749. [Google Scholar] [CrossRef]
- Neumann, C.A.; Fang, Q. Are peroxiredoxins tumor suppressors? Curr. Opin. Pharm. 2007, 7, 375–380. [Google Scholar] [CrossRef]
- Hansen, J.M.; Moriarty-Craige, S.; Jones, D.P. Nuclear and cytoplasmic peroxiredoxin-1 differentially regulate NF-kappaB activities. Free Radic. Biol. Med. 2007, 43, 282–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chhipa, R.R.; Lee, K.S.; Onate, S.; Wu, Y.; Ip, C. Prx1 enhances androgen receptor function in prostate cancer cells by increasing receptor affinity to dihydrotestosterone. Mol. Cancer Res. 2009, 7, 1543–1552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budanov, A.V. The role of tumor suppressor p53 in the antioxidant defense and metabolism. Subcell Biochem. 2014, 85, 337–358. [Google Scholar] [CrossRef] [Green Version]
- Xiao, L.; Chen, D.; Hu, P.; Wu, J.; Liu, W.; Zhao, Y.; Cao, M.; Fang, Y.; Bi, W.; Zheng, Z.; et al. The c-Abl-MST1 signaling pathway mediates oxidative stress-induced neuronal cell death. J. Neurosci. 2011, 31, 9611–9619. [Google Scholar] [CrossRef]
- Morinaka, A.; Funato, Y.; Uesugi, K.; Miki, H. Oligomeric peroxiredoxin-I is an essential intermediate for p53 to activate MST1 kinase and apoptosis. Oncogene 2011, 30, 4208–4218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butterworth, P.J. Protein kinase C: Current concepts and future perspectives. Cell Biochem. Funct. 1993, 11, 292. [Google Scholar] [CrossRef]
- Mehta, N.K.; Mehta, K.D. Protein kinase C-beta: An emerging connection between nutrient excess and obesity. Biochim. Biophys. Acta 2014, 1841, 1491–1497. [Google Scholar] [CrossRef]
- Newton, A.C. Protein kinase C: Structure, function, and regulation. J. Biol. Chem. 1995, 270, 28495–28498. [Google Scholar] [CrossRef] [Green Version]
- Nishizuka, Y. Protein kinase C and lipid signaling for sustained cellular responses. FASEB J. 1995, 9, 484–496. [Google Scholar] [CrossRef]
- Ono, Y.; Kikkawa, U.; Ogita, K.; Fujii, T.; Kurokawa, T.; Asaoka, Y.; Sekiguchi, K.; Ase, K.; Igarashi, K.; Nishizuka, Y. Expression and properties of two types of protein kinase C: Alternative splicing from a single gene. Science 1987, 236, 1116–1120. [Google Scholar] [CrossRef]
- Leonard, T.A.; Różycki, B.; Saidi, L.F.; Hummer, G.; Hurley, J.H. Crystal structure and allosteric activation of protein kinase C βII. Cell 2011, 144, 55–66. [Google Scholar] [CrossRef] [Green Version]
- Grodsky, N.; Li, Y.; Bouzida, D.; Love, R.; Jensen, J.; Nodes, B.; Nonomiya, J.; Grant, S. Structure of the catalytic domain of human protein kinase C beta II complexed with a bisindolylmaleimide inhibitor. Biochemistry 2006, 45, 13970–13981. [Google Scholar] [CrossRef]
- Newton, A.C. Regulation of the ABC kinases by phosphorylation: Protein kinase C as a paradigm. Biochem. J. 2003, 370, 361–371. [Google Scholar] [CrossRef]
- Newton, A.C. Lipid activation of protein kinases. J. Lipid Res. 2009, 50, S266–S271. [Google Scholar] [CrossRef] [Green Version]
- Parker, P.J.; Parkinson, S.J. AGC protein kinase phosphorylation and protein kinase C. Biochem. Soc. Trans. 2001, 29, 860–863. [Google Scholar] [CrossRef]
- Konishi, H.; Tanaka, M.; Takemura, Y.; Matsuzaki, H.; Ono, Y.; Kikkawa, U.; Nishizuka, Y. Activation of protein kinase C by tyrosine phosphorylation in response to H2O2. Proc. Natl. Acad. Sci. USA 1997, 94, 11233–11237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosentino-Gomes, D.; Rocco-Machado, N.; Meyer-Fernandes, J.R. Cell signaling through protein kinase C oxidation and activation. Int. J. Mol. Sci. 2012, 13, 10697–10721. [Google Scholar] [CrossRef] [Green Version]
- Ikenoue, T.; Inoki, K.; Yang, Q.; Zhou, X.; Guan, K.L. Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. EMBO J. 2008, 27, 1919–1931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 2004, 14, 1296–1302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Gould, C.; Garza, R.; Gao, T.; Hampton, R.Y.; Newton, A.C. Amplitude control of protein kinase C by RINCK, a novel E3 ubiquitin ligase. J. Biol. Chem. 2007, 282, 33776–33787. [Google Scholar] [CrossRef] [Green Version]
- Gould, C.M.; Kannan, N.; Taylor, S.S.; Newton, A.C. The chaperones Hsp90 and Cdc37 mediate the maturation and stabilization of protein kinase C through a conserved PXXP motif in the C-terminal tail. J. Biol. Chem. 2009, 284, 4921–4935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scivittaro, V.; Ganz, M.B.; Weiss, M.F. AGEs induce oxidative stress and activate protein kinase C-beta(II) in neonatal mesangial cells. Am. J. Physiol. Ren. Physiol. 2000, 278, F676–F683. [Google Scholar] [CrossRef] [Green Version]
- Rimessi, A.; Rizzuto, R.; Pinton, P. Differential recruitment of PKC isoforms in HeLa cells during redox stress. Cell Stress Chaperones 2007, 12, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.; Han, L.; Ambrogini, E.; Bartell, S.M.; Manolagas, S.C. Oxidative stress stimulates apoptosis and activates NF-kappaB in osteoblastic cells via a PKCbeta/p66shc signaling cascade: Counter regulation by estrogens or androgens. Mol. Endocrinol. 2010, 24, 2030–2037. [Google Scholar] [CrossRef] [Green Version]
- Gopalakrishna, R.; Jaken, S. Protein kinase C signaling and oxidative stress. Free Radic. Biol. Med. 2000, 28, 1349–1361. [Google Scholar] [CrossRef]
- Giorgi, C.; Agnoletto, C.; Baldini, C.; Bononi, A.; Bonora, M.; Marchi, S.; Missiroli, S.; Patergnani, S.; Poletti, F.; Rimessi, A.; et al. Redox control of protein kinase C: Cell- and disease-specific aspects. Antioxid. Redox Signal. 2010, 13, 1051–1085. [Google Scholar] [CrossRef]
- Choo, H.J.; Kim, J.H.; Kwon, O.B.; Lee, C.S.; Mun, J.Y.; Han, S.S.; Yoon, Y.S.; Yoon, G.; Choi, K.M.; Ko, Y.G. Mitochondria are impaired in the adipocytes of type 2 diabetic mice. Diabetologia 2006, 49, 784–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Investig. 2004, 114, 1752–1761. [Google Scholar] [CrossRef]
- Keaney, J.F., Jr.; Larson, M.G.; Vasan, R.S.; Wilson, P.W.; Lipinska, I.; Corey, D.; Massaro, J.M.; Sutherland, P.; Vita, J.A.; Benjamin, E.J.; et al. Obesity and systemic oxidative stress: Clinical correlates of oxidative stress in the Framingham Study. Arter. Thromb. Vasc. Biol. 2003, 23, 434–439. [Google Scholar] [CrossRef] [Green Version]
- Kusminski, C.M.; Scherer, P.E. Mitochondrial dysfunction in white adipose tissue. Trends Endocrinol. Metab. 2012, 23, 435–443. [Google Scholar] [CrossRef] [Green Version]
- Patti, M.E.; Corvera, S. The role of mitochondria in the pathogenesis of type 2 diabetes. Endocr. Rev. 2010, 31, 364–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rong, J.X.; Qiu, Y.; Hansen, M.K.; Zhu, L.; Zhang, V.; Xie, M.; Okamoto, Y.; Mattie, M.D.; Higashiyama, H.; Asano, S.; et al. Adipose mitochondrial biogenesis is suppressed in db/db and high-fat diet-fed mice and improved by rosiglitazone. Diabetes 2007, 56, 1751–1760. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Goldman, S.; Baerga, R.; Zhao, Y.; Komatsu, M.; Jin, S. Adipose-specific deletion of autophagy-related gene 7 (atg7) in mice reveals a role in adipogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 19860–19865. [Google Scholar] [CrossRef] [Green Version]
- Jansen, H.J.; van Essen, P.; Koenen, T.; Joosten, L.A.; Netea, M.G.; Tack, C.J.; Stienstra, R. Autophagy activity is up-regulated in adipose tissue of obese individuals and modulates proinflammatory cytokine expression. Endocrinology 2012, 153, 5866–5874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunez, C.E.; Rodrigues, V.S.; Gomes, F.S.; Moura, R.F.; Victorio, S.C.; Bombassaro, B.; Chaim, E.A.; Pareja, J.C.; Geloneze, B.; Velloso, L.A.; et al. Defective regulation of adipose tissue autophagy in obesity. Int. J. Obes. 2013, 37, 1473–1480. [Google Scholar] [CrossRef] [Green Version]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 2011, 333, 1109–1112. [Google Scholar] [CrossRef] [Green Version]
- Patergnani, S.; Marchi, S.; Rimessi, A.; Bonora, M.; Giorgi, C.; Mehta, K.D.; Pinton, P. PRKCB/protein kinase C, beta and the mitochondrial axis as key regulators of autophagy. Autophagy 2013, 9, 1367–1385. [Google Scholar] [CrossRef] [Green Version]
- Goodyear, L.J.; Kahn, B.B. Exercise, glucose transport, and insulin sensitivity. Annu. Rev. Med. 1998, 49, 235–261. [Google Scholar] [CrossRef]
- Krssak, M.; Falk Petersen, K.; Dresner, A.; DiPietro, L.; Vogel, S.M.; Rothman, D.L.; Roden, M.; Shulman, G.I. Intramyocellular lipid concentrations are correlated with insulin sensitivity in humans: A 1H NMR spectroscopy study. Diabetologia 1999, 42, 113–116. [Google Scholar] [CrossRef] [Green Version]
- Hennige, A.M.; Heni, M.; Machann, J.; Staiger, H.; Sartorius, T.; Hoene, M.; Lehmann, R.; Weigert, C.; Peter, A.; Bornemann, A.; et al. Enforced expression of protein kinase C in skeletal muscle causes physical inactivity, fatty liver and insulin resistance in the brain. J. Cell. Mol. Med. 2010, 14, 903–913. [Google Scholar] [CrossRef]
- Mir, H.A.; Ali, R.; Mushtaq, U.; Khanday, F.A. Structure-functional implications of longevity protein p66Shc in health and disease. Ageing Res. Rev. 2020, 63, 101139. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Hammerling, U. The mitochondrial PKCdelta/retinol signal complex exerts real-time control on energy homeostasis. Biochim. Biophys. Acta Mol. Cell. Biol. Lipids 2020, 1865, 158614. [Google Scholar] [CrossRef]
- Morita, M.; Matsuzaki, H.; Yamamoto, T.; Fukami, Y.; Kikkawa, U. Epidermal growth factor receptor phosphorylates protein kinase C {delta} at Tyr332 to form a trimeric complex with p66Shc in the H2O2-stimulated cells. J. Biochem. 2008, 143, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Acin-Perez, R.; Hoyos, B.; Gong, J.; Vinogradov, V.; Fischman, D.A.; Leitges, M.; Borhan, B.; Starkov, A.; Manfredi, G.; Hammerling, U. Regulation of intermediary metabolism by the PKCdelta signalosome in mitochondria. FASEB J. 2010, 24, 5033–5042. [Google Scholar] [CrossRef] [Green Version]
- Hoyos, B.; Acin-Perez, R.; Fischman, D.A.; Manfredi, G.; Hammerling, U. Hiding in plain sight: Uncovering a new function of vitamin A in redox signaling. Biochim. Biophys. Acta 2012, 1821, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Imam, A.; Hoyos, B.; Swenson, C.; Levi, E.; Chua, R.; Viriya, E.; Hammerling, U. Retinoids as ligands and coactivators of protein kinase C alpha. FASEB J. 2001, 15, 28–30. [Google Scholar] [CrossRef] [PubMed]
- Hoyos, B.; Imam, A.; Chua, R.; Swenson, C.; Tong, G.X.; Levi, E.; Noy, N.; Hämmerling, U. The cysteine-rich regions of the regulatory domains of Raf and protein kinase C as retinoid receptors. J. Exp. Med. 2000, 192, 835–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, H.A.; Brudvig, G.W. Redox functions of carotenoids in photosynthesis. Biochemistry 2004, 43, 8607–8615. [Google Scholar] [CrossRef]
- Gong, J.; Hoyos, B.; Acin-Perez, R.; Vinogradov, V.; Shabrova, E.; Zhao, F.; Leitges, M.; Fischman, D.; Manfredi, G.; Hammerling, U. Two protein kinase C isoforms, δ and ε, regulate energy homeostasis in mitochondria by transmitting opposing signals to the pyruvate dehydrogenase complex. FASEB J. 2012, 26, 3537–3549. [Google Scholar] [CrossRef] [Green Version]
- Acin-Perez, R.; Hoyos, B.; Zhao, F.; Vinogradov, V.; Fischman, D.A.; Harris, R.A.; Leitges, M.; Wongsiriroj, N.; Blaner, W.S.; Manfredi, G.; et al. Control of oxidative phosphorylation by vitamin A illuminates a fundamental role in mitochondrial energy homoeostasis. FASEB J. 2010, 24, 627–636. [Google Scholar] [CrossRef] [Green Version]
- Hoyos, B.; Imam, A.; Korichneva, I.; Levi, E.; Chua, R.; Hammerling, U. Activation of c-Raf kinase by ultraviolet light. Regulation by retinoids. J. Biol. Chem. 2002, 277, 23949–23957. [Google Scholar] [CrossRef] [Green Version]
- Mott, H.R.; Carpenter, J.W.; Zhong, S.; Ghosh, S.; Bell, R.M.; Campbell, S.L. The solution structure of the Raf-1 cysteine-rich domain: A novel ras and phospholipid binding site. Proc. Natl. Acad. Sci. USA 1996, 93, 8312–8317. [Google Scholar] [CrossRef] [Green Version]
- Sakai, Y.; Meno, C.; Fujii, H.; Nishino, J.; Shiratori, H.; Saijoh, Y.; Rossant, J.; Hamada, H. The retinoic acid-inactivating enzyme CYP26 is essential for establishing an uneven distribution of retinoic acid along the anterio-posterior axis within the mouse embryo. Genes Dev. 2001, 15, 213–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leitges, M.; Gimborn, K.; Elis, W.; Kalesnikoff, J.; Hughes, M.R.; Krystal, G.; Huber, M. Protein kinase C-delta is a negative regulator of antigen-induced mast cell degranulation. Mol. Cell. Biol. 2002, 22, 3970–3980. [Google Scholar] [CrossRef] [Green Version]
- Bezy, O.; Tran, T.T.; Pihlajamäki, J.; Suzuki, R.; Emanuelli, B.; Winnay, J.; Mori, M.A.; Haas, J.; Biddinger, S.B.; Leitges, M.; et al. PKCδ regulates hepatic insulin sensitivity and hepatosteatosis in mice and humans. J. Clin. Investig. 2011, 121, 2504–2517. [Google Scholar] [CrossRef] [PubMed]
- Schutkowski, M.; Bernhardt, A.; Zhou, X.Z.; Shen, M.; Reimer, U.; Rahfeld, J.U.; Lu, K.P.; Fischer, G. Role of phosphorylation in determining the backbone dynamics of the serine/threonine-proline motif and Pin1 substrate recognition. Biochemistry 1998, 37, 5566–5575. [Google Scholar] [CrossRef] [PubMed]
- Liou, Y.C.; Ryo, A.; Huang, H.K.; Lu, P.J.; Bronson, R.; Fujimori, F.; Uchida, T.; Hunter, T.; Lu, K.P. Loss of Pin1 function in the mouse causes phenotypes resembling cyclin D1-null phenotypes. Proc. Natl. Acad. Sci. USA 2002, 99, 1335–1340. [Google Scholar] [CrossRef] [Green Version]
- Ryo, A.; Nakamura, M.; Wulf, G.; Liou, Y.C.; Lu, K.P. Pin1 regulates turnover and subcellular localization of beta-catenin by inhibiting its interaction with APC. Nat. Cell Biol. 2001, 3, 793–801. [Google Scholar] [CrossRef]
- Wulf, G.M.; Ryo, A.; Wulf, G.G.; Lee, S.W.; Niu, T.; Petkova, V.; Lu, K.P. Pin1 is overexpressed in breast cancer and cooperates with Ras signaling in increasing the transcriptional activity of c-Jun towards cyclin D1. Embo J. 2001, 20, 3459–3472. [Google Scholar] [CrossRef] [Green Version]
- Ranganathan, R.; Lu, K.P.; Hunter, T.; Noel, J.P. Structural and functional analysis of the mitotic rotamase Pin1 suggests substrate recognition is phosphorylation dependent. Cell 1997, 89, 875–886. [Google Scholar] [CrossRef] [Green Version]
- Lu, K.P.; Liou, Y.C.; Zhou, X.Z. Pinning down proline-directed phosphorylation signaling. Trends Cell Biol. 2002, 12, 164–172. [Google Scholar] [CrossRef]
- Feng, D.; Yao, J.; Wang, G.; Li, Z.; Zu, G.; Li, Y.; Luo, F.; Ning, S.; Qasim, W.; Chen, Z.; et al. Inhibition of p66Shc-mediated mitochondrial apoptosis via targeting prolyl-isomerase Pin1 attenuates intestinal ischemia/reperfusion injury in rats. Clin. Sci. 2017, 131, 759–773. [Google Scholar] [CrossRef]
- Lu, P.J.; Zhou, X.Z.; Liou, Y.C.; Noel, J.P.; Lu, K.P. Critical role of WW domain phosphorylation in regulating phosphoserine binding activity and Pin1 function. J. Biol. Chem. 2002, 277, 2381–2384. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Wilcox, C.B.; Devasahayam, G.; Hackett, R.L.; Arévalo-Rodríguez, M.; Cardenas, M.E.; Heitman, J.; Hanes, S.D. The Ess1 prolyl isomerase is linked to chromatin remodeling complexes and the general transcription machinery. Embo J. 2000, 19, 3727–3738. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.P.; Hanes, S.D.; Hunter, T. A human peptidyl-prolyl isomerase essential for regulation of mitosis. Nature 1996, 380, 544–547. [Google Scholar] [CrossRef]
- Hani, J.; Schelbert, B.; Bernhardt, A.; Domdey, H.; Fischer, G.; Wiebauer, K.; Rahfeld, J.U. Mutations in a peptidylprolyl-cis/trans-isomerase gene lead to a defect in 3’-end formation of a pre-mRNA in Saccharomyces cerevisiae. J. Biol. Chem. 1999, 274, 108–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rippmann, J.F.; Hobbie, S.; Daiber, C.; Guilliard, B.; Bauer, M.; Birk, J.; Nar, H.; Garin-Chesa, P.; Rettig, W.J.; Schnapp, A. Phosphorylation-dependent proline isomerization catalyzed by Pin1 is essential for tumor cell survival and entry into mitosis. Cell Growth Differ. 2000, 11, 409–416. [Google Scholar]
- Stukenberg, P.T.; Kirschner, M.W. Pin1 acts catalytically to promote a conformational change in Cdc25. Mol. Cell 2001, 7, 1071–1083. [Google Scholar] [CrossRef]
- Winkler, K.E.; Swenson, K.I.; Kornbluth, S.; Means, A.R. Requirement of the prolyl isomerase Pin1 for the replication checkpoint. Science 2000, 287, 1644–1647. [Google Scholar] [CrossRef]
- Zhou, X.Z.; Kops, O.; Werner, A.; Lu, P.J.; Shen, M.; Stoller, G.; Küllertz, G.; Stark, M.; Fischer, G.; Lu, K.P. Pin1-dependent prolyl isomerization regulates dephosphorylation of Cdc25C and tau proteins. Mol. Cell 2000, 6, 873–883. [Google Scholar] [CrossRef]
- Huang, H.K.; Forsburg, S.L.; John, U.P.; O’Connell, M.J.; Hunter, T. Isolation and characterization of the Pin1/Ess1p homologue in Schizosaccharomyces pombe. J. Cell. Sci. 2001, 114, 3779–3788. [Google Scholar] [CrossRef] [PubMed]
- Price, N.E.; Mumby, M.C. Effects of regulatory subunits on the kinetics of protein phosphatase 2A. Biochemistry 2000, 39, 11312–11318. [Google Scholar] [CrossRef]
- Elgenaidi, I.S.; Spiers, J.P. Regulation of the phosphoprotein phosphatase 2A system and its modulation during oxidative stress: A potential therapeutic target? Pharmacol. Ther. 2019, 198, 68–89. [Google Scholar] [CrossRef]
- Kremmer, E.; Ohst, K.; Kiefer, J.; Brewis, N.; Walter, G. Separation of PP2A core enzyme and holoenzyme with monoclonal antibodies against the regulatory A subunit: Abundant expression of both forms in cells. Mol. Cell. Biol. 1997, 17, 1692–1701. [Google Scholar] [CrossRef] [Green Version]
- Heijman, J.; Dewenter, M.; El-Armouche, A.; Dobrev, D. Function and regulation of serine/threonine phosphatases in the healthy and diseased heart. J. Mol. Cell. Cardiol. 2013, 64, 90–98. [Google Scholar] [CrossRef]
- Cohen, P.T.; Brewis, N.D.; Hughes, V.; Mann, D.J. Protein serine/threonine phosphatases; an expanding family. FEBS Lett. 1990, 268, 355–359. [Google Scholar] [CrossRef] [Green Version]
- Baskaran, R.; Velmurugan, B.K. Protein phosphatase 2A as therapeutic targets in various disease models. Life Sci. 2018, 210, 40–46. [Google Scholar] [CrossRef]
- Namura, S.; Iihara, K.; Takami, S.; Nagata, I.; Kikuchi, H.; Matsushita, K.; Moskowitz, M.A.; Bonventre, J.V.; Alessandrini, A. Intravenous administration of MEK inhibitor U0126 affords brain protection against forebrain ischemia and focal cerebral ischemia. Proc. Natl. Acad. Sci. USA 2001, 98, 11569–11574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alessandrini, A.; Namura, S.; Moskowitz, M.A.; Bonventre, J.V. MEK1 protein kinase inhibition protects against damage resulting from focal cerebral ischemia. Proc. Natl. Acad. Sci. USA 1999, 96, 12866–12869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szobi, A.; Lichy, M.; Carnicka, S.; Pancza, D.; Svec, P.; Ravingerova, T.; Adameova, A. Pleiotropic Effects of Simvastatin on Some Calcium Regulatory and Myofibrillar Proteins in Ischemic/Reperfused Heart: Causality of Statins Cardioprotection? Curr. Pharm. Des. 2016, 22, 6451–6458. [Google Scholar] [CrossRef]
- Kronenbitter, A.; Funk, F.; Hackert, K.; Gorreßen, S.; Glaser, D.; Boknik, P.; Poschmann, G.; Stühler, K.; Isić, M.; Krüger, M.; et al. Impaired Ca(2+) cycling of nonischemic myocytes contributes to sarcomere dysfunction early after myocardial infarction. J. Mol. Cell. Cardiol. 2018, 119, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.P.; Qiu, T.Z.; Zhao, J.; Li, L.X.; Guo, J. Sphingomyelinase-induced ceramide production stimulate calcium-independent JNK and PP2A activation following cerebral ischemia. Brain Inj. 2009, 23, 1073–1080. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Wu, X.; Xu, J.; Zhou, J.; Han, X.; Guo, J. Src kinase up-regulates the ERK cascade through inactivation of protein phosphatase 2A following cerebral ischemia. BMC Neurosci. 2009, 10, 74. [Google Scholar] [CrossRef] [Green Version]
- Fan, W.J.; van Vuuren, D.; Genade, S.; Lochner, A. Kinases and phosphatases in ischaemic preconditioning: A re-evaluation. Basic Res. Cardiol. 2010, 105, 495–511. [Google Scholar] [CrossRef] [PubMed]
- Martín de la Vega, C.; Burda, J.; Toledo Lobo, M.V.; Salinas, M. Cerebral postischemic reperfusion-induced demethylation of the protein phosphatase 2A catalytic subunit. J. Neurosci. Res 2002, 69, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Penna, C.; Tullio, F.; Perrelli, M.G.; Moro, F.; Abbadessa, G.; Piccione, F.; Carriero, V.; Racca, S.; Pagliaro, P. Ischemia/reperfusion injury is increased and cardioprotection by a postconditioning protocol is lost as cardiac hypertrophy develops in nandrolone treated rats. Basic Res. Cardiol. 2011, 106, 409–420. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Kim, S.; Huh, J. Zinc plays a critical role in the cardioprotective effect of postconditioning by enhancing the activation of the RISK pathway in rat hearts. J. Mol. Cell. Cardiol. 2014, 66, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Qi, Y.; Chen, T. Long-term pre-treatment of antioxidant Ginkgo biloba extract EGb-761 attenuates cerebral-ischemia-induced neuronal damage in aged mice. Biomed. Pharm. 2017, 85, 256–263. [Google Scholar] [CrossRef]
- Zhu, M.; Wang, J.; Liu, M.; Du, D.; Xia, C.; Shen, L.; Zhu, D. Upregulation of protein phosphatase 2A and NR3A-pleiotropic effect of simvastatin on ischemic stroke rats. PLoS ONE 2012, 7, e51552. [Google Scholar] [CrossRef] [Green Version]
- Luo, D.; Fan, X.; Ma, C.; Fan, T.; Wang, X.; Chang, N.; Li, L.; Zhang, Y.; Meng, Z.; Wang, S.; et al. A Study on the Effect of Neurogenesis and Regulation of GSK3β/PP2A Expression in Acupuncture Treatment of Neural Functional Damage Caused by Focal Ischemia in MCAO Rats. Evid. Based Complement. Altern. Med. 2014, 2014, 962343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinbrenner, C.; Baines, C.P.; Liu, G.S.; Armstrong, S.C.; Ganote, C.E.; Walsh, A.H.; Honkanen, R.E.; Cohen, M.V.; Downey, J.M. Fostriecin, an inhibitor of protein phosphatase 2A, limits myocardial infarct size even when administered after onset of ischemia. Circulation 1998, 98, 899–905. [Google Scholar] [CrossRef] [Green Version]
- Pitt, A.S.; Buchanan, S.K. A Biochemical and Structural Understanding of TOM Complex Interactions and Implications for Human Health and Disease. Cells 2021, 10, 1164. [Google Scholar] [CrossRef]
- Orsini, F.; Moroni, M.; Contursi, C.; Yano, M.; Pelicci, P.; Giorgio, M.; Migliaccio, E. Regulatory effects of the mitochondrial energetic status on mitochondrial p66Shc. Biol. Chem. 2006, 387, 1405–1410. [Google Scholar] [CrossRef]
- Wang, W.; Chen, X.; Zhang, L.; Yi, J.; Ma, Q.; Yin, J.; Zhuo, W.; Gu, J.; Yang, M. Atomic structure of human TOM core complex. Cell Discov. 2020, 6, 67. [Google Scholar] [CrossRef]
- Große, L.; Wurm, C.A.; Brüser, C.; Neumann, D.; Jans, D.C.; Jakobs, S. Bax assembles into large ring-like structures remodeling the mitochondrial outer membrane in apoptosis. Embo J. 2016, 35, 402–413. [Google Scholar] [CrossRef]
- Cartron, P.F.; Bellot, G.; Oliver, L.; Grandier-Vazeille, X.; Manon, S.; Vallette, F.M. Bax inserts into the mitochondrial outer membrane by different mechanisms. FEBS Lett. 2008, 582, 3045–3051. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Ding, Y.; Ye, N.; Wild, C.; Chen, H.; Zhou, J. Direct Activation of Bax Protein for Cancer Therapy. Med. Res. Rev. 2016, 36, 313–341. [Google Scholar] [CrossRef] [PubMed]
- Geisler, S.; Holmström, K.M.; Treis, A.; Skujat, D.; Weber, S.S.; Fiesel, F.C.; Kahle, P.J.; Springer, W. The PINK1/Parkin-mediated mitophagy is compromised by PD-associated mutations. Autophagy 2010, 6, 871–878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiesel, F.C.; Caulfield, T.R.; Moussaud-Lamodière, E.L.; Ogaki, K.; Dourado, D.F.; Flores, S.C.; Ross, O.A.; Springer, W. Structural and Functional Impact of Parkinson Disease-Associated Mutations in the E3 Ubiquitin Ligase Parkin. Hum. Mutat. 2015, 36, 774–786. [Google Scholar] [CrossRef] [Green Version]
- Takano, T.; Kohara, M.; Kasama, Y.; Nishimura, T.; Saito, M.; Kai, C.; Tsukiyama-Kohara, K. Translocase of outer mitochondrial membrane 70 expression is induced by hepatitis C virus and is related to the apoptotic response. J. Med. Virol. 2011, 83, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Maio, R.; Barrett, P.J.; Hoffman, E.K.; Barrett, C.W.; Zharikov, A.; Borah, A.; Hu, X.; McCoy, J.; Chu, C.T.; Burton, E.A.; et al. α-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Sci. Transl. Med. 2016, 8, 342ra378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skulachev, V.P. Cytochrome c in the apoptotic and antioxidant cascades. FEBS Lett. 1998, 423, 275–280. [Google Scholar] [CrossRef] [Green Version]
- Daum, G. Lipids of mitochondria. Biochim. Biophys. Acta 1985, 822, 1–42. [Google Scholar] [CrossRef]
- Gasanoff, E.S.; Yaguzhinsky, L.S.; Garab, G. Cardiolipin, Non-Bilayer Structures and Mitochondrial Bioenergetics: Relevance to Cardiovascular Disease. Cells 2021, 10, 1721. [Google Scholar] [CrossRef]
- Hüttemann, M.; Pecina, P.; Rainbolt, M.; Sanderson, T.H.; Kagan, V.E.; Samavati, L.; Doan, J.W.; Lee, I. The multiple functions of cytochrome c and their regulation in life and death decisions of the mammalian cell: From respiration to apoptosis. Mitochondrion 2011, 11, 369–381. [Google Scholar] [CrossRef] [Green Version]
- Tyurin, V.A.; Tyurina, Y.Y.; Osipov, A.N.; Belikova, N.A.; Basova, L.V.; Kapralov, A.A.; Bayir, H.; Kagan, V.E. Interactions of cardiolipin and lyso-cardiolipins with cytochrome c and tBid: Conflict or assistance in apoptosis. Cell Death Differ. 2007, 14, 872–875. [Google Scholar] [CrossRef] [Green Version]
- Díaz-Quintana, A.; Pérez-Mejías, G.; Guerra-Castellano, A.; De la Rosa, M.A.; Díaz-Moreno, I. Wheel and Deal in the Mitochondrial Inner Membranes: The Tale of Cytochrome c and Cardiolipin. Oxid. Med. Cell. Longev. 2020, 2020, 6813405. [Google Scholar] [CrossRef] [Green Version]
- Kagan, V.E.; Tyurin, V.A.; Jiang, J.; Tyurina, Y.Y.; Ritov, V.B.; Amoscato, A.A.; Osipov, A.N.; Belikova, N.A.; Kapralov, A.A.; Kini, V.; et al. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat. Chem. Biol. 2005, 1, 223–232. [Google Scholar] [CrossRef]
- Tyurina, Y.Y.; Poloyac, S.M.; Tyurin, V.A.; Kapralov, A.A.; Jiang, J.; Anthonymuthu, T.S.; Kapralova, V.I.; Vikulina, A.S.; Jung, M.Y.; Epperly, M.W.; et al. A mitochondrial pathway for biosynthesis of lipid mediators. Nat. Chem. 2014, 6, 542–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrosillo, G.; Ruggiero, F.M.; Paradies, G. Role of reactive oxygen species and cardiolipin in the release of cytochrome c from mitochondria. Faseb J. 2003, 17, 2202–2208. [Google Scholar] [CrossRef] [Green Version]
- Shidoji, Y.; Hayashi, K.; Komura, S.; Ohishi, N.; Yagi, K. Loss of molecular interaction between cytochrome c and cardiolipin due to lipid peroxidation. Biochem. Biophys. Res. Commun. 1999, 264, 343–347. [Google Scholar] [CrossRef]
- Li, P.; Nijhawan, D.; Budihardjo, I.; Srinivasula, S.M.; Ahmad, M.; Alnemri, E.S.; Wang, X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997, 91, 479–489. [Google Scholar] [CrossRef] [Green Version]
- Zou, H.; Henzel, W.J.; Liu, X.; Lutschg, A.; Wang, X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell 1997, 90, 405–413. [Google Scholar] [CrossRef] [Green Version]
- Green, D.R.; Kroemer, G. The pathophysiology of mitochondrial cell death. Science 2004, 305, 626–629. [Google Scholar] [CrossRef]
- Attardi, L.D.; Reczek, E.E.; Cosmas, C.; Demicco, E.G.; McCurrach, M.E.; Lowe, S.W.; Jacks, T. PERP, an apoptosis-associated target of p53, is a novel member of the PMP-22/gas3 family. Genes Dev. 2000, 14, 704–718. [Google Scholar] [CrossRef]
- Le, S.; Connors, T.J.; Maroney, A.C. c-Jun N-terminal kinase specifically phosphorylates p66ShcA at serine 36 in response to ultraviolet irradiation. J. Biol. Chem. 2001, 276, 48332–48336. [Google Scholar] [CrossRef] [Green Version]
- Natalicchio, A.; De Stefano, F.; Perrini, S.; Laviola, L.; Cignarelli, A.; Caccioppoli, C.; Quagliara, A.; Melchiorre, M.; Leonardini, A.; Conserva, A.; et al. Involvement of the p66Shc protein in glucose transport regulation in skeletal muscle myoblasts. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E228–E237. [Google Scholar] [CrossRef] [Green Version]
- Natalicchio, A.; Tortosa, F.; Perrini, S.; Laviola, L.; Giorgino, F. p66Shc, a multifaceted protein linking Erk signalling, glucose metabolism, and oxidative stress. Arch. Physiol. Biochem. 2011, 117, 116–124. [Google Scholar] [CrossRef]
- Oda, E.; Ohki, R.; Murasawa, H.; Nemoto, J.; Shibue, T.; Yamashita, T.; Tokino, T.; Taniguchi, T.; Tanaka, N. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 2000, 288, 1053–1058. [Google Scholar] [CrossRef] [PubMed]
- Pronsato, L.; Milanesi, L. Effect of testosterone on the regulation of p53 and p66Shc during oxidative stress damage in C2C12 cells. Steroids 2016, 106, 41–54. [Google Scholar] [CrossRef]
- Xu, Y.; Li, N.; Xiang, R.; Sun, P. Emerging roles of the p38 MAPK and PI3K/AKT/mTOR pathways in oncogene-induced senescence. Trends Biochem. Sci. 2014, 39, 268–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.J.; Broz, D.K.; Noderer, W.L.; Ferreira, J.P.; Overton, K.W.; Spencer, S.L.; Meyer, T.; Tapscott, S.J.; Attardi, L.D.; Wang, C.L. p53 suppresses muscle differentiation at the myogenin step in response to genotoxic stress. Cell Death Differ. 2015, 22, 560–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haga, S.; Terui, K.; Fukai, M.; Oikawa, Y.; Irani, K.; Furukawa, H.; Todo, S.; Ozaki, M. Preventing hypoxia/reoxygenation damage to hepatocytes by p66(shc) ablation: Up-regulation of anti-oxidant and anti-apoptotic proteins. J. Hepatol. 2008, 48, 422–432. [Google Scholar] [CrossRef]
- Koch, O.R.; Fusco, S.; Ranieri, S.C.; Maulucci, G.; Palozza, P.; Larocca, L.M.; Cravero, A.A.; Farre, S.M.; De Spirito, M.; Galeotti, T.; et al. Role of the life span determinant P66(shcA) in ethanol-induced liver damage. Lab. Investig. 2008, 88, 750–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemoto, S.; Finkel, T. Redox regulation of forkhead proteins through a p66shc-dependent signaling pathway. Science 2002, 295, 2450–2452. [Google Scholar] [CrossRef]
- Smith, W.W.; Norton, D.D.; Gorospe, M.; Jiang, H.; Nemoto, S.; Holbrook, N.J.; Finkel, T.; Kusiak, J.W. Phosphorylation of p66Shc and forkhead proteins mediates Abeta toxicity. J. Cell Biol. 2005, 169, 331–339. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Rogers, B.; Kachi, S.; Hackett, S.F.; Sick, A.; Campochiaro, P.A. Reduction of p66Shc suppresses oxidative damage in retinal pigmented epithelial cells and retina. J. Cell. Physiol. 2006, 209, 996–1005. [Google Scholar] [CrossRef]
- Chahdi, A.; Sorokin, A. Endothelin-1 couples betaPix to p66Shc: Role of betaPix in cell proliferation through FOXO3a phosphorylation and p27kip1 down-regulation independently of Akt. Mol. Biol. Cell. 2008, 19, 2609–2619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camici, G.G.; Schiavoni, M.; Francia, P.; Bachschmid, M.; Martin-Padura, I.; Hersberger, M.; Tanner, F.C.; Pelicci, P.; Volpe, M.; Anversa, P.; et al. Genetic deletion of p66(Shc) adaptor protein prevents hyperglycemia-induced endothelial dysfunction and oxidative stress. Proc. Natl. Acad. Sci. USA 2007, 104, 5217–5222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francia, P.; delli Gatti, C.; Bachschmid, M.; Martin-Padura, I.; Savoia, C.; Migliaccio, E.; Pelicci, P.G.; Schiavoni, M.; Lüscher, T.F.; Volpe, M.; et al. Deletion of p66shc gene protects against age-related endothelial dysfunction. Circulation 2004, 110, 2889–2895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spescha, R.D.; Glanzmann, M.; Simic, B.; Witassek, F.; Keller, S.; Akhmedov, A.; Tanner, F.C.; Luscher, T.F.; Camici, G.G. Adaptor protein p66(Shc) mediates hypertension-associated, cyclic stretch-dependent, endothelial damage. Hypertension 2014, 64, 347–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vashistha, H.; Singhal, P.C.; Malhotra, A.; Husain, M.; Mathieson, P.; Saleem, M.A.; Kuriakose, C.; Seshan, S.; Wilk, A.; Delvalle, L.; et al. Null mutations at the p66 and bradykinin 2 receptor loci induce divergent phenotypes in the diabetic kidney. Am. J. Physiol. Ren. Physiol. 2012, 303, F1629–F1640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Mileykovskaya, E.; Dowhan, W. Gluing the respiratory chain together. Cardiolipin is required for supercomplex formation in the inner mitochondrial membrane. J. Biol. Chem. 2002, 277, 43553–43556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raja, V.; Greenberg, M.L. The functions of cardiolipin in cellular metabolism-potential modifiers of the Barth syndrome phenotype. Chem. Phys. Lipids 2014, 179, 49–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudek, J. Role of Cardiolipin in Mitochondrial Signaling Pathways. Front. Cell Dev. Biol. 2017, 5, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esposti, M.D.; Ngo, A.; Ghelli, A.; Benelli, B.; Carelli, V.; McLennan, H.; Linnane, A.W. The interaction of Q analogs, particularly hydroxydecyl benzoquinone (idebenone), with the respiratory complexes of heart mitochondria. Arch. Biochem. Biophys. 1996, 330, 395–400. [Google Scholar] [CrossRef]
- Fato, R.; Bergamini, C.; Bortolus, M.; Maniero, A.L.; Leoni, S.; Ohnishi, T.; Lenaz, G. Differential effects of mitochondrial Complex I inhibitors on production of reactive oxygen species. Biochim. Biophys. Acta 2009, 1787, 384–392. [Google Scholar] [CrossRef]
- Jaber, S.M.; Ge, S.X.; Milstein, J.L.; VanRyzin, J.W.; Waddell, J.; Polster, B.M. Idebenone Has Distinct Effects on Mitochondrial Respiration in Cortical Astrocytes Compared to Cortical Neurons Due to Differential NQO1 Activity. J. Neurosci. 2020, 40, 4609–4619. [Google Scholar] [CrossRef]
- Sugiyama, Y.; Fujita, T.; Matsumoto, M.; Okamoto, K.; Imada, I. Effects of idebenone (CV-2619) and its metabolites on respiratory activity and lipid peroxidation in brain mitochondria from rats and dogs. J. Pharm. 1985, 8, 1006–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walter, L.; Nogueira, V.; Leverve, X.; Heitz, M.P.; Bernardi, P.; Fontaine, E. Three classes of ubiquinone analogs regulate the mitochondrial permeability transition pore through a common site. J. Biol. Chem. 2000, 275, 29521–29527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, K.; Murakami, M.; Miura, D.; Yunoki, K.; Enko, K.; Tanaka, M.; Saito, Y.; Nishii, N.; Miyoshi, T.; Yoshida, M.; et al. Beta-Blockers and Oxidative Stress in Patients with Heart Failure. Pharmaceuticals 2011, 4, 1088–1100. [Google Scholar] [CrossRef] [Green Version]
- Tian-Li, Y.; Ruffolo, R.R., Jr.; Feuerstein, G. Antioxidant Action of Carvedilol: A Potential Role in Treatment of Heart Failure. Heart Fail. Rev. 1999, 4, 39–51. [Google Scholar]
- Dandona, P.; Karne, R.; Ghanim, H.; Hamouda, W.; Aljada, A.; Magsino, C.H., Jr. Carvedilol inhibits reactive oxygen species generation by leukocytes and oxidative damage to amino acids. Circulation 2000, 101, 122–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Packer, M.; Bristow, M.R.; Cohn, J.N.; Colucci, W.S.; Fowler, M.B.; Gilbert, E.M.; Shusterman, N.H. The effect of carvedilol on morbidity and mortality in patients with chronic heart failure. U.S. Carvedilol Heart Failure Study Group. N. Engl. J. Med. 1996, 334, 1349–1355. [Google Scholar] [CrossRef] [PubMed]
- Buyse, G.; Mertens, L.; Di Salvo, G.; Matthijs, I.; Weidemann, F.; Eyskens, B.; Goossens, W.; Goemans, N.; Sutherland, G.R.; Van Hove, J.L. Idebenone treatment in Friedreich’s ataxia: Neurological, cardiac, and biochemical monitoring. Neurology 2003, 60, 1679–1681. [Google Scholar] [CrossRef]
- Lyseng-Williamson, K.A. Idebenone: A Review in Leber’s Hereditary Optic Neuropathy. Drugs 2016, 76, 805–813. [Google Scholar] [CrossRef]
- Tomilov, A.; Allen, S.; Hui, C.K.; Bettaieb, A.; Cortopassi, G. Idebenone is a cytoprotective insulin sensitizer whose mechanism is Shc inhibition. Pharm. Res. 2018, 137, 89–103. [Google Scholar] [CrossRef]
- Gestl, E.E.; Anne Bottger, S. Cytoplasmic sequestration of the tumor suppressor p53 by a heat shock protein 70 family member, mortalin, in human colorectal adenocarcinoma cell lines. Biochem. Biophys. Res. Commun. 2012, 423, 411–416. [Google Scholar] [CrossRef]
- Lu, W.J.; Lee, N.P.; Kaul, S.C.; Lan, F.; Poon, R.T.; Wadhwa, R.; Luk, J.M. Mortalin-p53 interaction in cancer cells is stress dependent and constitutes a selective target for cancer therapy. Cell Death Differ. 2011, 18, 1046–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadekova, S.; Lehnert, S.; Chow, T.Y. Induction of PBP74/mortalin/Grp75, a member of the hsp70 family, by low doses of ionizing radiation: A possible role in induced radioresistance. Int. J. Radiat. Biol. 1997, 72, 653–660. [Google Scholar] [CrossRef]
- Xu, L.; Voloboueva, L.A.; Ouyang, Y.; Emery, J.F.; Giffard, R.G. Overexpression of mitochondrial Hsp70/Hsp75 in rat brain protects mitochondria, reduces oxidative stress, and protects from focal ischemia. J. Cereb. Blood Flow Metab. 2009, 29, 365–374. [Google Scholar] [CrossRef]
- Ran, Q.; Wadhwa, R.; Kawai, R.; Kaul, S.C.; Sifers, R.N.; Bick, R.J.; Smith, J.R.; Pereira-Smith, O.M. Extramitochondrial localization of mortalin/mthsp70/PBP74/GRP75. Biochem. Biophys. Res. Commun. 2000, 275, 174–179. [Google Scholar] [CrossRef]
- Yang, H.; Zhou, X.; Liu, X.; Yang, L.; Chen, Q.; Zhao, D.; Zuo, J.; Liu, W. Mitochondrial dysfunction induced by knockdown of mortalin is rescued by Parkin. Biochem. Biophys. Res. Commun. 2011, 410, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Havalova, H.; Ondrovicova, G.; Keresztesova, B.; Bauer, J.A.; Pevala, V.; Kutejova, E.; Kunova, N. Mitochondrial HSP70 Chaperone System-The Influence of Post-Translational Modifications and Involvement in Human Diseases. Int. J. Mol. Sci. 2021, 22, 8077. [Google Scholar] [CrossRef]
- Gogia, S.; Neelamegham, S. Role of fluid shear stress in regulating VWF structure, function and related blood disorders. Biorheology 2015, 52, 319–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganderton, T.; Berndt, M.C.; Chesterman, C.N.; Hogg, P.J. Hypothesis for control of von Willebrand factor multimer size by intra-molecular thiol-disulphide exchange. J. Thromb. Haemost. 2007, 5, 204–206. [Google Scholar] [CrossRef]
- Nagy, P. Kinetics and mechanisms of thiol-disulfide exchange covering direct substitution and thiol oxidation-mediated pathways. Antioxid. Redox Signal. 2013, 18, 1623–1641. [Google Scholar] [CrossRef] [Green Version]
- Giese, B.; Wang, M.; Gao, J.; Stoltz, M.; Müller, P.; Graber, M. Electron relay race in peptides. J. Org. Chem. 2009, 74, 3621–3625. [Google Scholar] [CrossRef]
- Wojtala, A.; Karkucinska-Wieckowska, A.; Sardao, V.A.; Szczepanowska, J.; Kowalski, P.; Pronicki, M.; Duszynski, J.; Wieckowski, M.R. Modulation of mitochondrial dysfunction-related oxidative stress in fibroblasts of patients with Leigh syndrome by inhibition of prooxidative p66Shc pathway. Mitochondrion 2017, 37, 62–79. [Google Scholar] [CrossRef]
- Pang, B.; Zhao, F.; Zhou, Y.; He, B.; Huang, W.; Zhang, F.; Long, Y.G.; Xia, X.; Liu, M.L.; Jiang, Y.H. Systematic Review and Meta-Analysis of the Impact of Hypoxia on Infarcted Myocardium: Better or Worse? Cell. Physiol. Biochem. 2018, 51, 949–960. [Google Scholar] [CrossRef] [PubMed]
- Jain, I.H.; Zazzeron, L.; Goldberger, O.; Marutani, E.; Wojtkiewicz, G.R.; Ast, T.; Wang, H.; Schleifer, G.; Stepanova, A.; Brepoels, K.; et al. Leigh Syndrome Mouse Model Can Be Rescued by Interventions that Normalize Brain Hyperoxia, but Not HIF Activation. Cell Metab. 2019, 30, 824–832.e823. [Google Scholar] [CrossRef]
- Lehrer, S.S.; Fasman, G.D. Ultraviolet irradiation effects in poly-L-tyrosine and model compounds. Identification of bityrosine as a photoproduct. Biochemistry 1967, 6, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Blomberg, M.R. Mechanism of Oxygen Reduction in Cytochrome c Oxidase and the Role of the Active Site Tyrosine. Biochemistry 2016, 55, 489–500. [Google Scholar] [CrossRef]
- Stubbe, J.; van Der Donk, W.A. Protein Radicals in Enzyme Catalysis. Chem. Rev. 1998, 98, 705–762. [Google Scholar] [CrossRef] [PubMed]
- Battistuzzi, G.; Borsari, M.; Cowan, J.A.; Ranieri, A.; Sola, M. Control of cytochrome C redox potential: Axial ligation and protein environment effects. J. Am. Chem. Soc. 2002, 124, 5315–5324. [Google Scholar] [CrossRef] [PubMed]
- Stojanovski, D.; Muller, J.M.; Milenkovic, D.; Guiard, B.; Pfanner, N.; Chacinska, A. The MIA system for protein import into the mitochondrial intermembrane space. Biochim. Biophys. Acta 2008, 1783, 610–617. [Google Scholar] [CrossRef] [Green Version]
- Habich, M.; Salscheider, S.L.; Riemer, J. Cysteine residues in mitochondrial intermembrane space proteins: More than just import. Br. J. Pharm. 2019, 176, 514–531. [Google Scholar] [CrossRef] [Green Version]
- Riemer, J.; Bulleid, N.; Herrmann, J.M. Disulfide formation in the ER and mitochondria: Two solutions to a common process. Science 2009, 324, 1284–1287. [Google Scholar] [CrossRef]
- Trinei, M.; Migliaccio, E.; Bernardi, P.; Paolucci, F.; Pelicci, P.; Giorgio, M. p66Shc, mitochondria, and the generation of reactive oxygen species. Methods Enzym. 2013, 528, 99–110. [Google Scholar] [CrossRef]
- Patriarca, A.; Polticelli, F.; Piro, M.C.; Sinibaldi, F.; Mei, G.; Bari, M.; Santucci, R.; Fiorucci, L. Conversion of cytochrome c into a peroxidase: Inhibitory mechanisms and implication for neurodegenerative diseases. Arch. Biochem. Biophys. 2012, 522, 62–69. [Google Scholar] [CrossRef] [Green Version]
- Baysa, A.; Sagave, J.; Carpi, A.; Zaglia, T.; Campesan, M.; Dahl, C.P.; Bilbija, D.; Troitskaya, M.; Gullestad, L.; Giorgio, M.; et al. The p66ShcA adaptor protein regulates healing after myocardial infarction. Basic Res. Cardiol. 2015, 110, 13. [Google Scholar] [CrossRef] [PubMed]
- Boengler, K.; Bornbaum, J.; Schluter, K.D.; Schulz, R. P66shc and its role in ischemic cardiovascular diseases. Basic Res. Cardiol. 2019, 114, 29. [Google Scholar] [CrossRef]
- Ramraj, S.K.; Elayapillai, S.P.; Pelikan, R.C.; Zhao, Y.D.; Isingizwe, Z.R.; Kennedy, A.L.; Lightfoot, S.A.; Benbrook, D.M. Novel ovarian cancer maintenance therapy targeted at mortalin and mutant p53. Int. J. Cancer 2020, 147, 1086–1097. [Google Scholar] [CrossRef] [PubMed]
- Chandra, V.; Rai, R.; Benbrook, D.M. Utility and Mechanism of SHetA2 and Paclitaxel for Treatment of Endometrial Cancer. Cancers 2021, 13, 2322. [Google Scholar] [CrossRef]
- Benbrook, D.M.; Nammalwar, B.; Long, A.; Matsumoto, H.; Singh, A.; Bunce, R.A.; Berlin, K.D. SHetA2 interference with mortalin binding to p66shc and p53 identified using drug-conjugated magnetic microspheres. Investig. New Drugs 2014, 32, 412–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zhao, J.; Yang, W.; Bi, Y.; Chi, J.; Tian, J.; Li, W. High-dose alcohol induces reactive oxygen species-mediated apoptosis via PKC-β/p66Shc in mouse primary cardiomyocytes. Biochem. Biophys. Res. Commun. 2015, 456, 656–661. [Google Scholar] [CrossRef]
- Yang, M.; Stowe, D.F.; Udoh, K.B.; Heisner, J.S.; Camara, A.K. Reversible blockade of complex I or inhibition of PKCβ reduces activation and mitochondria translocation of p66Shc to preserve cardiac function after ischemia. PLoS ONE 2014, 9, e113534. [Google Scholar] [CrossRef] [Green Version]
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics 2014–2020 Update: A Report From the American Heart Association. Circulation 2020, 141, e139–e596. [Google Scholar] [CrossRef]
- Khavjou, O.; Phelps, D.; Leib, A. Projections of Cardiovascular Disease Prevalence and Costs: 2015–2035; RTI International: Research Triangle Park, NC, USA, 2016. [Google Scholar]
- Di Lisa, F.; Menabò, R.; Canton, M.; Petronilli, V. The role of mitochondria in the salvage and the injury of the ischemic myocardium. Biochim. Biophys. Acta 1998, 1366, 69–78. [Google Scholar] [CrossRef] [Green Version]
- Bernardi, P.; Di Lisa, F. The mitochondrial permeability transition pore: Molecular nature and role as a target in cardioprotection. J. Mol. Cell. Cardiol. 2015, 78, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Bolli, R.; Marbán, E. Molecular and cellular mechanisms of myocardial stunning. Physiol. Rev. 1999, 79, 609–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jennings, R.B.; Ganote, C.E.; Reimer, K.A. Ischemic tissue injury. Am J. Pathol. 1975, 81, 179–198. [Google Scholar] [PubMed]
- Ovize, M.; Baxter, G.F.; Di Lisa, F.; Ferdinandy, P.; Garcia-Dorado, D.; Hausenloy, D.J.; Heusch, G.; Vinten-Johansen, J.; Yellon, D.M.; Schulz, R. Postconditioning and protection from reperfusion injury: Where do we stand? Position paper from the Working Group of Cellular Biology of the Heart of the European Society of Cardiology. Cardiovasc. Res. 2010, 87, 406–423. [Google Scholar] [CrossRef] [Green Version]
- Yellon, D.M.; Hausenloy, D.J. Myocardial reperfusion injury. N. Engl. J. Med. 2007, 357, 1121–1135. [Google Scholar] [CrossRef] [PubMed]
- Fadini, G.P.; Albiero, M.; Menegazzo, L.; Boscaro, E.; Pagnin, E.; Iori, E.; Cosma, C.; Lapolla, A.; Pengo, V.; Stendardo, M.; et al. The redox enzyme p66Shc contributes to diabetes and ischemia-induced delay in cutaneous wound healing. Diabetes 2010, 59, 2306–2314. [Google Scholar] [CrossRef] [Green Version]
- Frijhoff, J.; Dagnell, M.; Augsten, M.; Beltrami, E.; Giorgio, M.; Östman, A. The mitochondrial reactive oxygen species regulator p66Shc controls PDGF-induced signaling and migration through protein tyrosine phosphatase oxidation. Free Radic. Biol. Med. 2014, 68, 268–277. [Google Scholar] [CrossRef]
- Obreztchikova, M.; Elouardighi, H.; Ho, M.; Wilson, B.A.; Gertsberg, Z.; Steinberg, S.F. Distinct signaling functions for Shc isoforms in the heart. J. Biol. Chem. 2006, 281, 20197–20204. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.E.; Zeiger, S.L.; Hettinger, J.C.; Brooks, J.D.; Holt, B.; Morrow, J.D.; Musiek, E.S.; Milne, G.; McLaughlin, B. Essential role of the redox-sensitive kinase p66shc in determining energetic and oxidative status and cell fate in neuronal preconditioning. J. Neurosci. 2010, 30, 5242–5252. [Google Scholar] [CrossRef] [Green Version]
- Lalu, M.M.; Csonka, C.; Giricz, Z.; Csont, T.; Schulz, R.; Ferdinandy, P. Preconditioning decreases ischemia/reperfusion-induced release and activation of matrix metalloproteinase-2. Biochem. Biophys. Res. Commun. 2002, 296, 937–941. [Google Scholar] [CrossRef]
- Wang, M.; Zhao, D.; Spinetti, G.; Zhang, J.; Jiang, L.Q.; Pintus, G.; Monticone, R.; Lakatta, E.G. Matrix metalloproteinase 2 activation of transforming growth factor-beta1 (TGF-beta1) and TGF-beta1-type II receptor signaling within the aged arterial wall. Arter. Thromb. Vasc. Biol. 2006, 26, 1503–1509. [Google Scholar] [CrossRef] [Green Version]
- Ahamed, J.; Burg, N.; Yoshinaga, K.; Janczak, C.A.; Rifkin, D.B.; Coller, B.S. In vitro and in vivo evidence for shear-induced activation of latent transforming growth factor-beta1. Blood 2008, 112, 3650–3660. [Google Scholar] [CrossRef] [Green Version]
- Annes, J.P.; Munger, J.S.; Rifkin, D.B. Making sense of latent TGFbeta activation. J. Cell Sci. 2003, 116, 217–224. [Google Scholar] [CrossRef] [Green Version]
- Bujak, M.; Frangogiannis, N.G. The role of TGF-beta signaling in myocardial infarction and cardiac remodeling. Cardiovasc. Res. 2007, 74, 184–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaludercic, N.; Mialet-Perez, J.; Paolocci, N.; Parini, A.; Di Lisa, F. Monoamine oxidases as sources of oxidants in the heart. J. Mol. Cell. Cardiol. 2014, 73, 34–42. [Google Scholar] [CrossRef] [Green Version]
- Kaludercic, N.; Di Lisa, F. Mitochondrial ROS Formation in the Pathogenesis of Diabetic Cardiomyopathy. Front. Cardiovasc. Med. 2020, 7, 12. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.P.; Smith, R.A. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu. Rev. Pharm. Toxicol. 2007, 47, 629–656. [Google Scholar] [CrossRef] [PubMed]
- Adlam, V.J.; Harrison, J.C.; Porteous, C.M.; James, A.M.; Smith, R.A.; Murphy, M.P.; Sammut, I.A. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. Faseb J. 2005, 19, 1088–1095. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.B.; Wander, G.S.; Rastogi, A.; Shukla, P.K.; Mittal, A.; Sharma, J.P.; Mehrotra, S.K.; Kapoor, R.; Chopra, R.K. Randomized, double-blind placebo-controlled trial of coenzyme Q10 in patients with acute myocardial infarction. Cardiovasc. Drugs 1998, 12, 347–353. [Google Scholar] [CrossRef]
- Maulik, N.; Yoshida, T.; Engelman, R.M.; Bagchi, D.; Otani, H.; Das, D.K. Dietary coenzyme Q(10) supplement renders swine hearts resistant to ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H1084–H1090. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, Y.; Hale, S.L.; Kloner, R.A. The effect of coenzyme Q10 on infarct size in a rabbit model of ischemia/reperfusion. Cardiovasc. Res. 1996, 32, 861–868. [Google Scholar] [CrossRef] [Green Version]
- Whitman, G.J.; Niibori, K.; Yokoyama, H.; Crestanello, J.A.; Lingle, D.M.; Momeni, R. The mechanisms of coenzyme Q10 as therapy for myocardial ischemia reperfusion injury. Mol. Asp. Med. 1997, 18, S195–S203. [Google Scholar] [CrossRef]
- Dikalova, A.E.; Bikineyeva, A.T.; Budzyn, K.; Nazarewicz, R.R.; McCann, L.; Lewis, W.; Harrison, D.G.; Dikalov, S.I. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ. Res. 2010, 107, 106–116. [Google Scholar] [CrossRef] [Green Version]
- Camara, A.K.; Bienengraeber, M.; Stowe, D.F. Mitochondrial approaches to protect against cardiac ischemia and reperfusion injury. Front. Physiol. 2011, 2, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dey, S.; DeMazumder, D.; Sidor, A.; Foster, D.B.; O’Rourke, B. Mitochondrial ROS Drive Sudden Cardiac Death and Chronic Proteome Remodeling in Heart Failure. Circ. Res. 2018, 123, 356–371. [Google Scholar] [CrossRef] [PubMed]
- Ni, R.; Cao, T.; Xiong, S.; Ma, J.; Fan, G.C.; Lacefield, J.C.; Lu, Y.; Le Tissier, S.; Peng, T. Therapeutic inhibition of mitochondrial reactive oxygen species with mito-TEMPO reduces diabetic cardiomyopathy. Free Radic. Biol. Med. 2016, 90, 12–23. [Google Scholar] [CrossRef] [Green Version]
- Birk, A.V.; Chao, W.M.; Bracken, C.; Warren, J.D.; Szeto, H.H. Targeting mitochondrial cardiolipin and the cytochrome c/cardiolipin complex to promote electron transport and optimize mitochondrial ATP synthesis. Br. J. Pharm. 2014, 171, 2017–2028. [Google Scholar] [CrossRef] [Green Version]
- Birk, A.V.; Liu, S.; Soong, Y.; Mills, W.; Singh, P.; Warren, J.D.; Seshan, S.V.; Pardee, J.D.; Szeto, H.H. The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. J. Am. Soc. Nephrol. 2013, 24, 1250–1261. [Google Scholar] [CrossRef]
- Kagan, V.E.; Bayir, H.A.; Belikova, N.A.; Kapralov, O.; Tyurina, Y.Y.; Tyurin, V.A.; Jiang, J.; Stoyanovsky, D.A.; Wipf, P.; Kochanek, P.M.; et al. Cytochrome c/cardiolipin relations in mitochondria: A kiss of death. Free Radic. Biol. Med. 2009, 46, 1439–1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kloner, R.A.; Hale, S.L.; Dai, W.; Gorman, R.C.; Shuto, T.; Koomalsingh, K.J.; Gorman, J.H., 3rd; Sloan, R.C.; Frasier, C.R.; Watson, C.A.; et al. Reduction of ischemia/reperfusion injury with bendavia, a mitochondria-targeting cytoprotective Peptide. J. Am. Heart Assoc. 2012, 1, e001644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, W.; Shi, J.; Gupta, R.C.; Sabbah, H.N.; Hale, S.L.; Kloner, R.A. Bendavia, a mitochondria-targeting peptide, improves postinfarction cardiac function, prevents adverse left ventricular remodeling, and restores mitochondria-related gene expression in rats. J. Cardiovasc. Pharm. 2014, 64, 543–553. [Google Scholar] [CrossRef]
- Mao, Z.J.; Lin, H.; Hou, J.W.; Zhou, Q.; Wang, Q.; Chen, Y.H. A Meta-Analysis of Resveratrol Protects against Myocardial Ischemia/Reperfusion Injury: Evidence from Small Animal Studies and Insight into Molecular Mechanisms. Oxid. Med. Cell. Longev. 2019, 2019, 5793867. [Google Scholar] [CrossRef]
- Chen, H.Z.; Wan, Y.Z.; Liu, D.P. Cross-talk between SIRT1 and p66Shc in vascular diseases. Trends Cardiovasc. Med. 2013, 23, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Ghatak, A.; Brar, M.J.; Agarwal, A.; Goel, N.; Rastogi, A.K.; Vaish, A.K.; Sircar, A.R.; Chandra, M. Oxy free radical system in heart failure and therapeutic role of oral vitamin E. Int. J. Cardiol. 1996, 57, 119–127. [Google Scholar] [CrossRef]
- McMurray, J.; Chopra, M.; Abdullah, I.; Smith, W.E.; Dargie, H.J. Evidence of oxidative stress in chronic heart failure in humans. Eur. Heart J. 1993, 14, 1493–1498. [Google Scholar] [CrossRef] [PubMed]
- Keith, M.; Geranmayegan, A.; Sole, M.J.; Kurian, R.; Robinson, A.; Omran, A.S.; Jeejeebhoy, K.N. Increased oxidative stress in patients with congestive heart failure. J. Am. Coll. Cardio.l 1998, 31, 1352–1356. [Google Scholar] [CrossRef] [Green Version]
- Belch, J.J.; Bridges, A.B.; Scott, N.; Chopra, M. Oxygen free radicals and congestive heart failure. Br. Heart J. 1991, 65, 245–248. [Google Scholar] [CrossRef] [Green Version]
- Fleisch, J.H.; Spaethe, S.M. Vasodilation and aging evaluated in the isolated perfused rat mesenteric vascular bed: Preliminary observations on the vascular pharmacology of dobutamine. J. Cardiovasc. Pharm. 1981, 3, 187–196. [Google Scholar] [CrossRef]
- Lüscher, T.F.; Tanner, F.C.; Tschudi, M.R.; Noll, G. Endothelial dysfunction in coronary artery disease. Annu. Rev. Med. 1993, 44, 395–418. [Google Scholar] [CrossRef]
- Moritoki, H.; Hosoki, E.; Ishida, Y. Age-related decrease in endothelium-dependent dilator response to histamine in rat mesenteric artery. Eur. J. Pharm. 1986, 126, 61–67. [Google Scholar] [CrossRef]
- Shirasaki, Y.; Su, C.; Lee, T.J.; Kolm, P.; Cline, W.H., Jr.; Nickols, G.A. Endothelial modulation of vascular relaxation to nitrovasodilators in aging and hypertension. J. Pharm. Exp. 1986, 239, 861–866. [Google Scholar]
- Tschudi, M.R.; Barton, M.; Bersinger, N.A.; Moreau, P.; Cosentino, F.; Noll, G.; Malinski, T.; Lüscher, T.F. Effect of age on kinetics of nitric oxide release in rat aorta and pulmonary artery. J. Clin. Investig. 1996, 98, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Savarese, G.; Perrone-Filardi, P.; Lüscher, T.F.; Camici, G.G. Enhanced age-dependent cerebrovascular dysfunction is mediated by adaptor protein p66Shc. Int. J. Cardiol. 2014, 175, 446–450. [Google Scholar] [CrossRef]
- Yamamori, T.; White, A.R.; Mattagajasingh, I.; Khanday, F.A.; Haile, A.; Qi, B.; Jeon, B.H.; Bugayenko, A.; Kasuno, K.; Berkowitz, D.E.; et al. P66shc regulates endothelial NO production and endothelium-dependent vasorelaxation: Implications for age-associated vascular dysfunction. J. Mol. Cell. Cardiol. 2005, 39, 992–995. [Google Scholar] [CrossRef]
- Rota, M.; LeCapitaine, N.; Hosoda, T.; Boni, A.; De Angelis, A.; Padin-Iruegas, M.E.; Esposito, G.; Vitale, S.; Urbanek, K.; Casarsa, C.; et al. Diabetes promotes cardiac stem cell aging and heart failure, which are prevented by deletion of the p66shc gene. Circ. Res. 2006, 99, 42–52. [Google Scholar] [CrossRef] [Green Version]
- Cai, W.; He, J.C.; Zhu, L.; Chen, X.; Striker, G.E.; Vlassara, H. AGE-receptor-1 counteracts cellular oxidant stress induced by AGEs via negative regulation of p66shc-dependent FKHRL1 phosphorylation. Am. J. Physiol. Cell Physiol. 2008, 294, C145–C152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menini, S.; Iacobini, C.; Ricci, C.; Oddi, G.; Pesce, C.; Pugliese, F.; Block, K.; Abboud, H.E.; Giorgio, M.; Migliaccio, E.; et al. Ablation of the gene encoding p66Shc protects mice against AGE-induced glomerulopathy by preventing oxidant-dependent tissue injury and further AGE accumulation. Diabetologia 2007, 50, 1997–2007. [Google Scholar] [CrossRef] [Green Version]
- Alikhani, M.; Maclellan, C.M.; Raptis, M.; Vora, S.; Trackman, P.C.; Graves, D.T. Advanced glycation end products induce apoptosis in fibroblasts through activation of ROS, MAP kinases, and the FOXO1 transcription factor. Am. J. Physiol. Cell Physiol. 2007, 292, C850–C856. [Google Scholar] [CrossRef] [PubMed]
- Xi, G.; Shen, X.; Radhakrishnan, Y.; Maile, L.; Clemmons, D. Hyperglycemia-induced p66shc inhibits insulin-like growth factor I-dependent cell survival via impairment of Src kinase-mediated phosphoinositide-3 kinase/AKT activation in vascular smooth muscle cells. Endocrinology 2010, 151, 3611–3623. [Google Scholar] [CrossRef] [Green Version]
- Diaz, M.N.; Frei, B.; Vita, J.A.; Keaney, J.F., Jr. Antioxidants and atherosclerotic heart disease. N. Engl. J. Med. 1997, 337, 408–416. [Google Scholar] [CrossRef]
- Keaney, J.F., Jr.; Shwaery, G.T.; Xu, A.; Nicolosi, R.J.; Loscalzo, J.; Foxall, T.L.; Vita, J.A. 17 beta-estradiol preserves endothelial vasodilator function and limits low-density lipoprotein oxidation in hypercholesterolemic swine. Circulation 1994, 89, 2251–2259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, S.M.; Morrow, J.D.; Roberts, L.J., 2nd; Frei, B. Formation of non-cyclooxygenase-derived prostanoids (F2-isoprostanes) in plasma and low density lipoprotein exposed to oxidative stress in vitro. J. Clin. Investig. 1994, 93, 998–1004. [Google Scholar] [CrossRef] [PubMed]
- Zeiher, A.M.; Schächlinger, V.; Hohnloser, S.H.; Saurbier, B.; Just, H. Coronary atherosclerotic wall thickening and vascular reactivity in humans. Elevated high-density lipoprotein levels ameliorate abnormal vasoconstriction in early atherosclerosis. Circulation 1994, 89, 2525–2532. [Google Scholar] [CrossRef] [Green Version]
- Napoli, C.; Ackah, E.; De Nigris, F.; Del Soldato, P.; D’Armiento, F.P.; Crimi, E.; Condorelli, M.; Sessa, W.C. Chronic treatment with nitric oxide-releasing aspirin reduces plasma low-density lipoprotein oxidation and oxidative stress, arterial oxidation-specific epitopes, and atherogenesis in hypercholesterolemic mice. Proc. Natl. Acad. Sci. USA 2002, 99, 12467–12470. [Google Scholar] [CrossRef] [Green Version]
- Mertens, A.; Verhamme, P.; Bielicki, J.K.; Phillips, M.C.; Quarck, R.; Verreth, W.; Stengel, D.; Ninio, E.; Navab, M.; Mackness, B.; et al. Increased low-density lipoprotein oxidation and impaired high-density lipoprotein antioxidant defense are associated with increased macrophage homing and atherosclerosis in dyslipidemic obese mice: LCAT gene transfer decreases atherosclerosis. Circulation 2003, 107, 1640–1646. [Google Scholar] [CrossRef] [Green Version]
- Ludmer, P.L.; Selwyn, A.P.; Shook, T.L.; Wayne, R.R.; Mudge, G.H.; Alexander, R.W.; Ganz, P. Paradoxical vasoconstriction induced by acetylcholine in atherosclerotic coronary arteries. N. Engl. J. Med. 1986, 315, 1046–1051. [Google Scholar] [CrossRef] [PubMed]
- Miao, Q.; Wang, Q.; Dong, L.; Wang, Y.; Tan, Y.; Zhang, X. The expression of p66shc in peripheral blood monocytes is increased in patients with coronary heart disease and correlated with endothelium-dependent vasodilatation. Heart Vessel. 2015, 30, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Samady, H.; Eshtehardi, P.; McDaniel, M.C.; Suo, J.; Dhawan, S.S.; Maynard, C.; Timmins, L.H.; Quyyumi, A.A.; Giddens, D.P. Coronary artery wall shear stress is associated with progression and transformation of atherosclerotic plaque and arterial remodeling in patients with coronary artery disease. Circulation 2011, 124, 779–788. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
ShcA family modular structure and CH2 domain post-translational modifications affecting ROS output. All ShcA proteins have SH2, CH1, and PTB domains but differ by CH2 domain length. p66Shc has a unique set of promoters and is tied to oxidative stress or Ras inhibition. p52/46Shc share a set of promoters, do not have documented ROS activity, and promote cell cycle progression and differentiation. Important ROS-related PTMs include Ser36 phosphorylation (mitochondrial translocation), Ser54 phosphorylation (decreased proteasome degradation), Cys59 sulfhydration (decreased Ser36 phosphorylation), and Lys81 acetylation (increased Ser36 phosphorylation).
Figure 1.
ShcA family modular structure and CH2 domain post-translational modifications affecting ROS output. All ShcA proteins have SH2, CH1, and PTB domains but differ by CH2 domain length. p66Shc has a unique set of promoters and is tied to oxidative stress or Ras inhibition. p52/46Shc share a set of promoters, do not have documented ROS activity, and promote cell cycle progression and differentiation. Important ROS-related PTMs include Ser36 phosphorylation (mitochondrial translocation), Ser54 phosphorylation (decreased proteasome degradation), Cys59 sulfhydration (decreased Ser36 phosphorylation), and Lys81 acetylation (increased Ser36 phosphorylation).
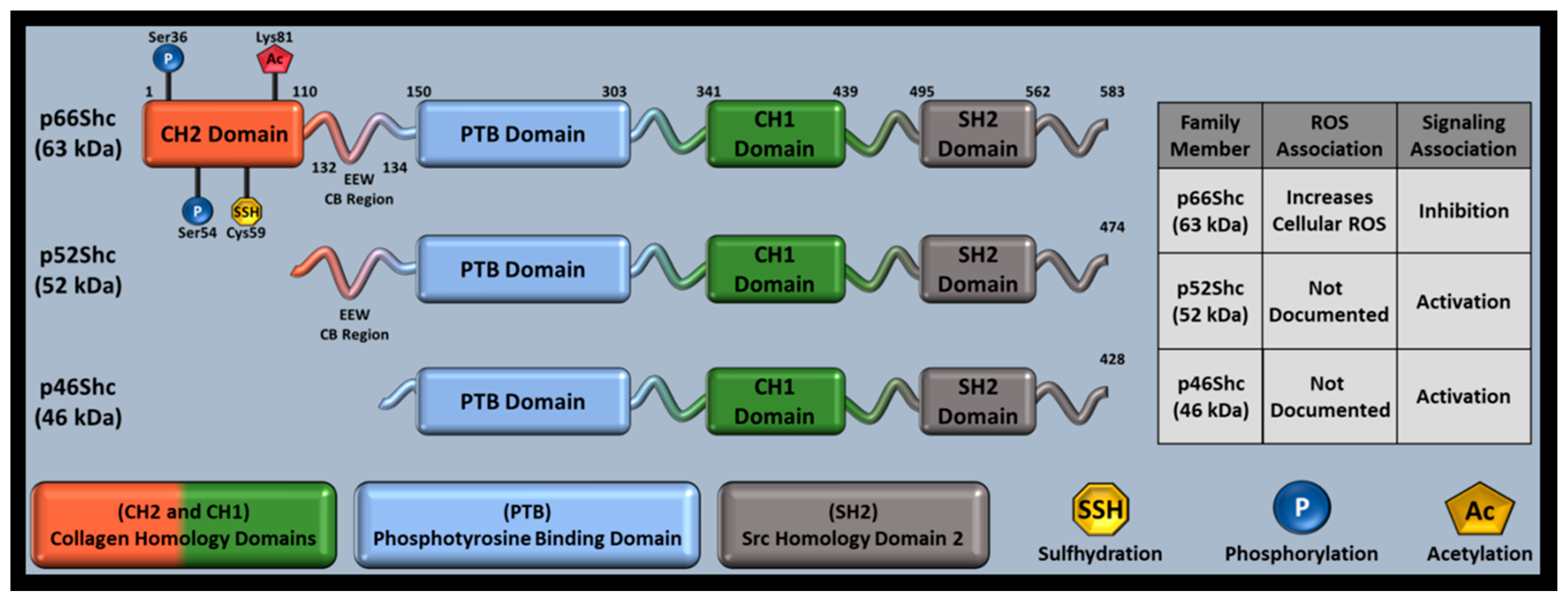
Figure 2.
ShcA signaling summary. (A) ShcA responses to tyrosine kinase growth factor (GF) and hormone signals. P52/46Shc causes Ras-dependent MAPK/ERK signaling that promotes survival, migration, and proliferation; left panel. p66Shc inhibits Ras signaling by competitively increasing Rac1-mediated ROS generation via NADPH oxidase activation; right panel. (B) ShcA-mediated integrin and VEGFR2-Cadherin-PECAM responses to shear force or tension signals. Integrin shear or tension signals result in p52/46Shc pro-survival signaling via FAK-Sos1 while p66Shc leads to anoikis via RhoGEF-RhoA signaling; left panel. Endothelial VEGFR2-VE-Cadherin-PECAM-1 complexes respond to shear by increasing p66Shc-mediated inflammation via Gab2-PI3K signaling; right panel.
Figure 2.
ShcA signaling summary. (A) ShcA responses to tyrosine kinase growth factor (GF) and hormone signals. P52/46Shc causes Ras-dependent MAPK/ERK signaling that promotes survival, migration, and proliferation; left panel. p66Shc inhibits Ras signaling by competitively increasing Rac1-mediated ROS generation via NADPH oxidase activation; right panel. (B) ShcA-mediated integrin and VEGFR2-Cadherin-PECAM responses to shear force or tension signals. Integrin shear or tension signals result in p52/46Shc pro-survival signaling via FAK-Sos1 while p66Shc leads to anoikis via RhoGEF-RhoA signaling; left panel. Endothelial VEGFR2-VE-Cadherin-PECAM-1 complexes respond to shear by increasing p66Shc-mediated inflammation via Gab2-PI3K signaling; right panel.
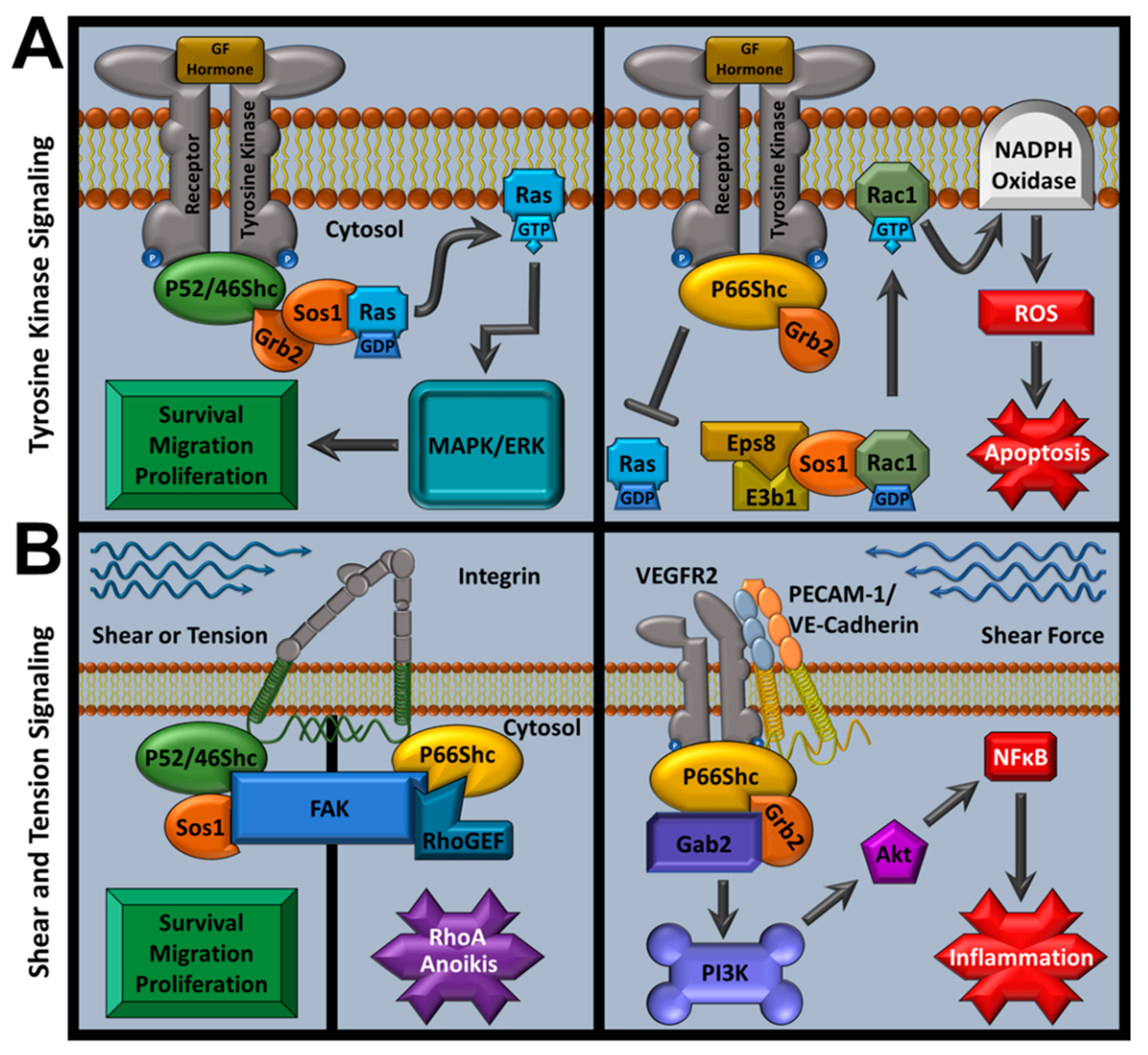
Figure 3.
Classical p66Shc mitochondrial ROS activity pathway. (1) Until stressors are present, p66Shc remains inactive via Prx1-mediated cytosol sequestration. (2) Once stressed, Prx1 releases p66Shc as a dimer. (3) Free p66Shc dimers are phosphorylated by JNK1 or PKCβ, resulting in phosphorylated p66Shc monomers. (4) p66Shc monomers are isomerized and dephosphorylated via Pin1 and PP2a, respectively. (5) p66Shc isomers are granted IMS access via TOM interactions that cause copper to associate with p66Shc. (6) p66Shc oxidizes cyt c to produce H2O2. Oligomerization status is unclear during ROS generation, but purified mouse CH2 experiments and mouse embryonic fibroblast studies implicate monomers, dimers, and tetramers.
Figure 3.
Classical p66Shc mitochondrial ROS activity pathway. (1) Until stressors are present, p66Shc remains inactive via Prx1-mediated cytosol sequestration. (2) Once stressed, Prx1 releases p66Shc as a dimer. (3) Free p66Shc dimers are phosphorylated by JNK1 or PKCβ, resulting in phosphorylated p66Shc monomers. (4) p66Shc monomers are isomerized and dephosphorylated via Pin1 and PP2a, respectively. (5) p66Shc isomers are granted IMS access via TOM interactions that cause copper to associate with p66Shc. (6) p66Shc oxidizes cyt c to produce H2O2. Oligomerization status is unclear during ROS generation, but purified mouse CH2 experiments and mouse embryonic fibroblast studies implicate monomers, dimers, and tetramers.

Figure 4.
Revised p66Shc mitochondrial function model. (A) Basal p66Shc mitochondrial function. Within cytoplasm, p66Shc is ROS-inactivated by environmental effects and complexes that sequester p66Shc by preventing Ser36 phosphorylation (mortalin-p66Shc, β1Pix-FOXO3a-p66Shc, and Prx1-p66Shc complexes). Free cytoplasmic p66Shc can also inhibit caspase 3/7 and caspase 9 activity, promoting survival against transient caspase leaks from short-term stress. Meanwhile, p66Shc reduces cyt c and enhances ETC activity, potentially via redox cycling. In addition, p66Shc is ROS-activated by the IMS environment, producing basal superoxide anion levels that are required for normal cell survival and proliferation functions. (B) p66Shc in stressed cytoplasm and mitochondrial matrix is released from sequestration complexes and enters the IMS at increased amounts through p66Shc’s classical translocation pathway (as illustrated in Figure 3), as a monomer, until p66Shc reaches the IMS. Increased p66Shc translocation causes excess superoxide anion production, which is increased when p66Shc undergoes conformational changes (increased α-helix) by interacting with cardiolipin. Accumulating ROS leads to increased cyt c peroxidase levels, which p66Shc inhibits, causing further H2O2 accumulation that is exacerbated by superoxide anion spontaneous dismutation. Once a threshold is reached, PTP formation occurs, leading to cyt c release and apoptosis. Overall cell antioxidant pools are also exhausted during this process as p66Shc prevents FOXO3a antioxidant transcription by retaining FOXO3a outside of nuclei.
Figure 4.
Revised p66Shc mitochondrial function model. (A) Basal p66Shc mitochondrial function. Within cytoplasm, p66Shc is ROS-inactivated by environmental effects and complexes that sequester p66Shc by preventing Ser36 phosphorylation (mortalin-p66Shc, β1Pix-FOXO3a-p66Shc, and Prx1-p66Shc complexes). Free cytoplasmic p66Shc can also inhibit caspase 3/7 and caspase 9 activity, promoting survival against transient caspase leaks from short-term stress. Meanwhile, p66Shc reduces cyt c and enhances ETC activity, potentially via redox cycling. In addition, p66Shc is ROS-activated by the IMS environment, producing basal superoxide anion levels that are required for normal cell survival and proliferation functions. (B) p66Shc in stressed cytoplasm and mitochondrial matrix is released from sequestration complexes and enters the IMS at increased amounts through p66Shc’s classical translocation pathway (as illustrated in Figure 3), as a monomer, until p66Shc reaches the IMS. Increased p66Shc translocation causes excess superoxide anion production, which is increased when p66Shc undergoes conformational changes (increased α-helix) by interacting with cardiolipin. Accumulating ROS leads to increased cyt c peroxidase levels, which p66Shc inhibits, causing further H2O2 accumulation that is exacerbated by superoxide anion spontaneous dismutation. Once a threshold is reached, PTP formation occurs, leading to cyt c release and apoptosis. Overall cell antioxidant pools are also exhausted during this process as p66Shc prevents FOXO3a antioxidant transcription by retaining FOXO3a outside of nuclei.
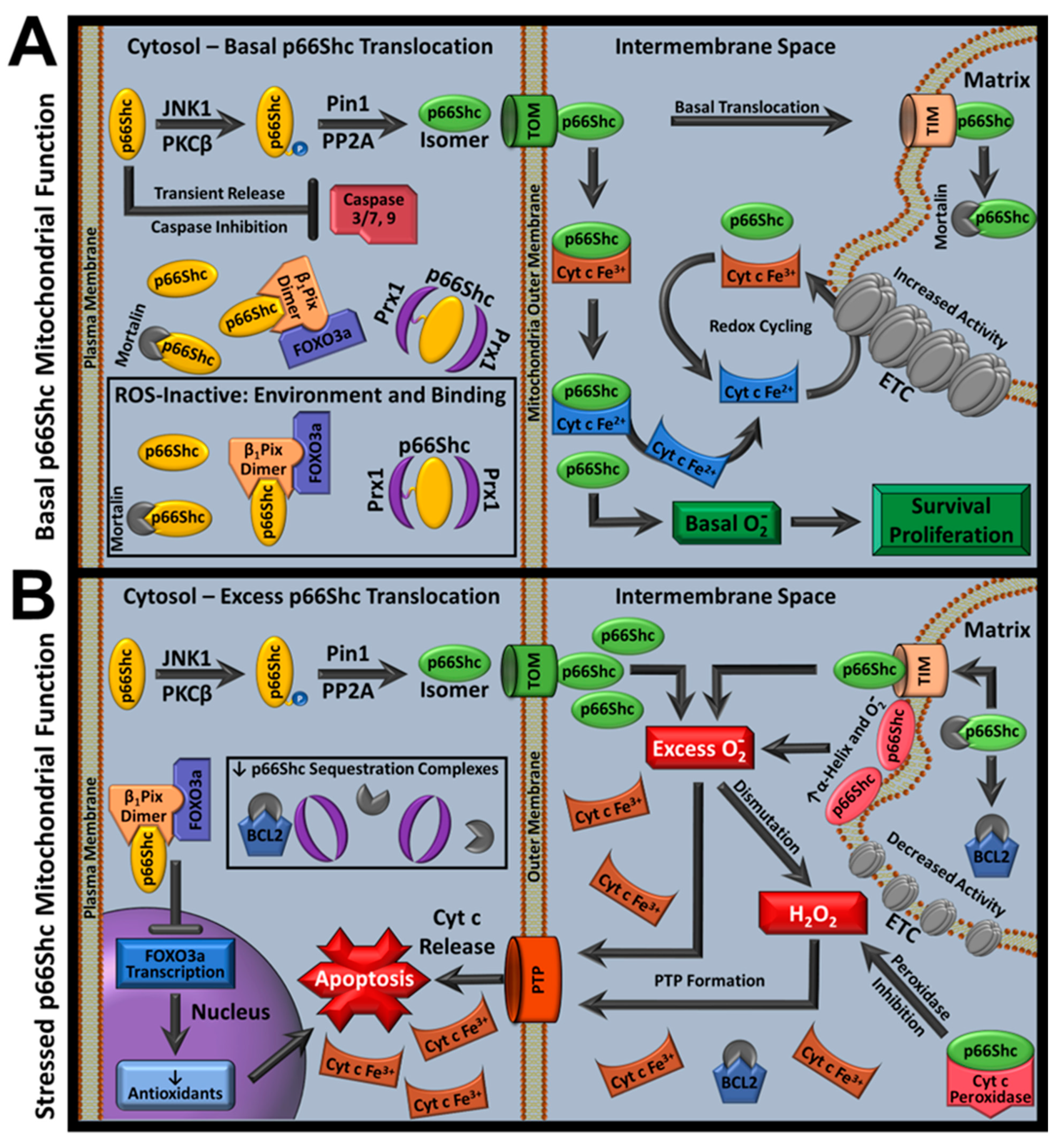
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Haslem, L.; Hays, J.M.; Hays, F.A. p66Shc in Cardiovascular Pathology. Cells 2022, 11, 1855. https://doi.org/10.3390/cells11111855
AMA Style
Haslem L, Hays JM, Hays FA. p66Shc in Cardiovascular Pathology. Cells. 2022; 11(11):1855. https://doi.org/10.3390/cells11111855
Chicago/Turabian StyleHaslem, Landon, Jennifer M. Hays, and Franklin A. Hays. 2022. "p66Shc in Cardiovascular Pathology" Cells 11, no. 11: 1855. https://doi.org/10.3390/cells11111855
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.