Abstract
Kernel-related traits are important agronomic traits that have a significant impact on maize yield, and the general combining ability (GCA) is one of the most crucial factors in maize breeding. Analyzing the genetic basis of kernel traits and their GCA is essential for maize yield improvement. In this study, a natural population including 205 inbred lines and an F1 population comprising 400 hybrids derived from four testers (Mo17, Zheng58, Chang7-2, and E28) were planted at two locations for two consecutive years, and four kernel-related traits were measured. A genome-wide association study (GWAS) was conducted using six multi-locus methods with 76,492 single-nucleotide polymorphism (SNP) markers to detect the SNPs related to four kernel-related traits and the corresponding GCA. A total of 66 and 52 SNPs significantly associated with kernel-related traits and the GCA were identified based on the threshold value of the logarithm of odds (LOD) = 3.0, respectively. Among them, eight SNPs related to the kernel traits and eight SNPs related to the GCA were simultaneously identified by using at least three methods. Finally, a total of 75 candidate genes were identified within the interval of the SNPs co-detected, of which 46 were annotated. The genes and significant SNPs identified in this study will deepen the understanding of the genetic foundation of kernel-related traits and GCAs, and also serve as molecular tools for improving kernel-related traits in maize.
1. Introduction
Maize is the most important crop for food, animal feed, and industrial materials in the world. Therefore, improving the yield of maize is essential to ensure food security [1]. Due to the complexity of its genetic basis and low heritability, there are constraints on the direct genetic enhancement of yield. Yield is significantly affected by kernel-related traits, such as kernel thickness (KT), kernel depth (KD), kernel width (KW) and 100 kernel weights (KWEI) [2,3]. The traits of KD and KW were significantly positively correlated within a single year’s yield and unit yield according to a previous study [4]. Furthermore, regression analysis also revealed that kernel-related traits had a positive contribution to yield [5]. Consequently, elucidating of the genetic basis of kernel traits will aid the comprehension of the genetic mechanism of grain yield and the development of strategies to improve yield in maize [6].
Additionally, maize is one of the crops that effectively exhibit the natural phenomenon of heterosis [7]. Increases in maize yield over the decades can largely be attributed to the introduction of hybrids, especially single-cross hybrids [8]. The GCA is essential in the utilization of heterosis in breeding [9]. The GCA of a line indicates the average performance of its hybrid progeny. GCA evaluation is a crucial step in maize hybrid development [10]. High GCA has been widely used in screening parental lines for developing hybrids with strong heterosis [11]. Previous studies of GCA in maize concentrated primarily phenotypic analysis. In recent years, with the development of molecular marker technologies, quantitative trait local (QTL) mapping of the combining ability’s genetic basis has flourished. Huang et al. [12] dissected 25 significant loci for the GCA of seven yield-related traits across a variety of environments. Wang et al. [9] used hybrids crossed by doubled haploid lines and detected 14 QTLs associated with the GCA for the ear height, the kernel moisture content, the kernel ratio, and the yield per plant. Using hybrids derived from recombinant inbred lines and two testers, Zhou et al. [13] dissected the genetic basis underlying the combining ability for plant height and demonstrated that only seven QTLs could be simultaneously detected for height and their GCA. GCA has become the subject of some research, but the genetic basis of GCA still remains unclear.
The genetic basis of complex traits in maize has been revealed using genome-wide association studies (GWASs), such as peduncle vascular bundle-related traits [14], leaf architecture [15], inflorescence [16], the zinc and iron content of kernels [17], the accumulation of cadmium in leaves [18], and the plant types [19]. Liu et al. [1] used an association panel of 310 maize inbred lines to identify genetic loci and candidate genes for kernel length (KL), KW, and KT in four environments using GWAS, detecting 21 significant SNPs and 73 candidate genes. Dong et al. [20] utilized 1283 maize inbred lines to investigate KL, KW, and KWEI, and conducted a GWAS on three kernel-related traits, resulting in the identification of 29 significantly associated SNPs and 6 candidate genes. The majority of GWAS models have focused on a single locus. In recent years, Zhang et al. [21] developed an R package called mrMLM 1.0, including six multi-locus GWAS methods (ISIS EM-BLASSO [22], pLARmEB [21], FASTmrEMMA [23], FASTmrMLM [24], pKWmEB [25], and mrMLM [26]) that have been recommended for GWAS due to their higher statistical power and low false positive rate (FPR) [27,28]. The basic idea behind multi-locus is to put the significant markers obtained from the whole genome using the single-locus method according to a low critical value (such as p = 0.01) into the multi-locus model to obtain the final significant loci without the correction of the p value. Zhang et al. [29] conducted in a population of 257 inbred lines, with 48,193 single-nucleotide polymorphisms, identified 423 significant quantitative trait nucleotides (QTNs) associated with stalk lodging resistance in maize by four multi-locus GWAS methods. Xu et al. [30] carried out a GWAS with a panel of 230 inbred lines and 145,232 SNPs and identified 60 QTNs related to starch pasting properties in maize by four multi-locus GWAS methods. Zheng et al. [31] utilized an F1 population of 240 hybrids to perform association mapping and detected 20 quantitative trait loci associated with grain yield, plant height, ear height, and GCA. Ma et al. [32] used 537 F1 hybrid combinations derived from the NCII mating design as experimental material and conducted a study utilizing seven multi-locus models. A total of eighteen SNPs significantly associated with ear diameter GCA were identified, among which six loci were concurrently identified using two to five multi-locus GWAS models, and three loci were consistently detected across two distinct environments. However, there are rare reports on SNPs related to kernel-related traits and GCA in maize by a multiple-locus GWAS model.
In this study, we used a natural population of maize inbred lines and an F1 hybrid population, genotyped with 76,492 polymorphic SNPs, to identify significant SNPs associated with kernel-related traits and GCA using six multi-locus GWAS models. This study aims to offer valuable insights into elucidating the genetic basis of traits related to kernels, as well as their GCA. Additionally, it will provide recommendations for the selection of a multi-locus GWAS model.
2. Materials and Methods
2.1. Genetic Population Development
A natural population consisting of 205 maize inbred lines with diverse origins and significant genetic variation was employed to investigate kernel-related traits. A total of one hundred inbred lines, randomly selected from the natural population, were used as female parents in the study. Additionally, four elite inbred lines, specifically Mo17, Chang7-2, E28, and Zheng58, were utilized as testers to assess the GCA of kernel-related traits (Supplementary Table S1). Zheng58 and Chang7-2, belonging to the Reid and Sipingtou heterotic group, are the parental inbred lines of ZhengDan 958, which is known for its large planting area in the country. An elite inbred line, Mo17, developed from the Lancaster subgroup has been used extensively in commercial hybrid production [13]. E28 is a high GCA line derived from the Lvda red cob subgroup. A total of 400 cross combinations were generated using the NCII mating design. The crosses were made during the winter seasons of 2014 and 2015 in Sanya, Hainan Province, China.
2.2. The Experimental Design and Phenotypic Data Collection
In the years of 2015 and 2016, a total of 205 inbred lines and 400 F1 hybrids were planted in the Experimental Station of Hebei Agricultural University in Baoding (BD, 115.48° and 38.85°) and Xinji (XJ, 115.12° and 37.54°), Hebei province, China. Field trials were designed using randomized complete blocks with two replicates. The inbred lines were planted at a density of 75,000 plants per hectare. Each plot contained a single row with a length of 3 m and a distance of 0.6 m between rows. On the other hand, the planting density of F1 hybrids was 60,000 plants per hectare, each plot contained 3 rows, with each row being 4 m long and spaced 0.6 m apart. Field management was the same as the local maize field. The phenotypic data were collected from two locations in two years. Upon reaching maturity, the individuals located at both the initial and terminal positions of the row were removed. For the inbred lines planted in single rows, ears were harvested from the central plants of each row and dried naturally. For the F1 hybrids, ears were collected from the central row of each plot and dried naturally. A total of ten uniform ears in each plot were selected to measure the kernel-related traits. The KT was measured using an electronic digital caliper with a precision of 0.1 mm. After threshing, the KWEI was measured as the average weight from three repeated measurements of 100 kernels using an electronic balance. The KD and KW were determined using WSeen’s Automatic seeds test system.
2.3. Phenotypic Data Analysis
Phenotypic data in the four environments were collated in Excel 2021, and the average value of 10 individuals per line was used as the phenotypic value. The formula utilized by Qi et al. [33] involves the calculation of the general combining ability (GCA) for the kernel-related traits of individual lines.
, where gi represents the GCA of the inbred line i; xi is the average value of the hybrid of the inbred line i and the four testers; and u is the average of the performance of all combinations. A descriptive statistical analysis was made with SPSS statistics V21.0 software. The boxplot was drawn using the “ggplot2” package in R (version 4.3.2). Using the GLM module of the SAS8.02 software, a joint analysis of variance was performed on each trait. The broad-sense heritability for each trait was estimated using the following formula:
where σg2 and σe2 represent the genetic variance and error variance, σge2 represents the interaction variance of genotype by environment, while n and r represent the number of environments and replications, respectively [34]. The best linear unbiased estimation (BLUE) values for each line were computed across four environments using the lme4 package and then we used the emmeans package to calculate the predicted means. The predicted means were used as the as phenotypic values for GWAS analysis.
2.4. Genotyping Data
Genomic DNA was extracted from the fresh leaves of each line using the CTAB method [35]. The sequencing libraries of 205 inbred lines were constructed and sequenced by genotyping-by-sequencing (GBS). The qualified library was sequenced by the second-generation sequencer, Illumina Hiseq2000 (Illumina Inc., San Diego, CA, USA) [36]. After the sequencing was completed, the BWA software (version 0.7.13) was used to compare with the reference genome of the fourth version of B73, and the SNP markers were screened by SAM tools software (version 1.0). We used PLINK (version 1.9) to obtain 76,492 SNPs with a missing values ratio < 0.2 and minor allele frequencies (MAFs) of >5% to perform the GWAS [31].
2.5. Genome-Wide Association Study
We conducted the GWAS using six multi-locus models, namely mrMLM, FASTmrMLM, FASTmrEMMA, pLARmEB, pKWmEB, and ISIS EM-BLASSO, in the mrMLM.GUI V4.02 software package [27]. The analysis involved 76,492 SNPs and utilized the BLUE values to identify significant SNPs in association with kernel-related traits and the GCA. The software used the Q + K model, wherein the population structure value Q was calculated by Admixture 1.30 software and the kinship value K was calculated by the mrMLM. In the study by Zhang et al. [28], the GUI software package (version 1.2) was used with default parameter values.
2.6. Haplotype Analysis
Haplotype analysis was conducted on SNPs co-detected by at least three multi-locus GWAS models. A two-tailed T-test was then performed to assess the significance of inter-group differences, with the BLUE values of the respective haplotypes. Subsequently, box plots of BLUE values were constructed to identify superior allelic variant types.
2.7. Candidate Gene Mining
The linkage disequilibrium (LD) decay rate between each pair of SNPs on each chromosome was analyzed with the r2 value by the PLINK software (version 1.9) and plotted by the R software (version 4.3.2). To mine candidate genes based on the SNPs which were detected by at least three multi-locus GWAS methods, the genome sequence of the maize line B73 (RefGenV4) in the MaizeGDB (https://www.maizegdb.org/, accessed on 14 November 2024) was used as the reference genome. Then, the annotation and ontology information of the candidate genes was searched using database websites NCBI (https://www.ncbi.nlm.nih.gov/, accessed on 14 November 2024) and DAVID (https://davidbioinformatics.nih.gov/, accessed on 14 November 2024). Analysis of the expression patterns of seed and endosperm-related candidate genes used publicly available transcriptome data from maize B73 [37]. We generated gene tissue-specific expression heatmaps by utilizing the Chi Plot website (https://www.chiplot.online/, accessed on 15 November 2024).
3. Results
3.1. The Performance of Kernel-Related Traits and GCA Across Environments
The descriptive statistics of four kernel-related traits for the natural population under two locations for two years are presented in Table 1. The coefficient of variation for the same trait varies with location and year. For KT, KD, KW, and KWEI, the phenotypic ranges were 10.88~12.38, 6.98~15.33, 6.68~14.23, and 9.75~17.25 (%), respectively. All traits’ coefficients of variation were highly variable, indicating that the population had a high genetic variation. The absolute kurtosis and skewness of all traits were close to zero, indicating that these traits were in a normal distribution. After removing the abnormal data, the means of the GCA for all the traits were near to zero. The GCA of all of the traits was in a normal distribution, which indicated that the GCA of kernel-related traits was controlled by micro-effective polygene (Figure 1).

Table 1.
Descriptive statistical analysis of kernel-related traits in different environments.
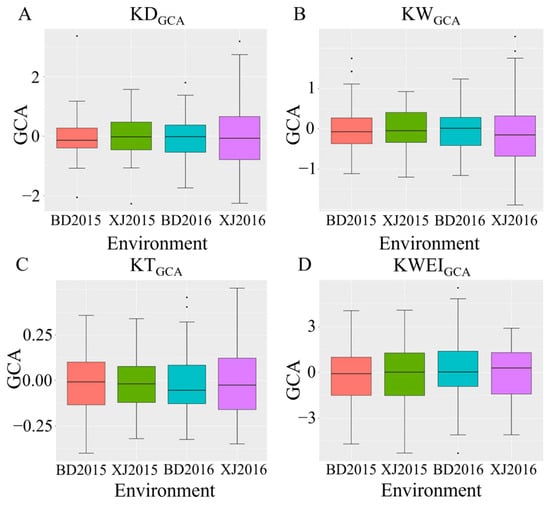
Figure 1.
Box plots of the GCA of maize kernel-related traits under four environments (A–D). Note: BD2015 means Baoding in 2015, XJ2015 means Xinji in 2015, BD2016 means Baoding in 2016, and XJ2016 means Xinji in 2016.
The ANOVA for kernel-related traits and the GCA across four environments are summarized in Table 2. The results showed that there were significant differences between genotypes (p < 0.05) for all the traits. It indicated that there were significant genetic differences among genotypes in terms of KT, KD, KW, KWEI, and their GCA. The heritability for these traits ranges from 25.49% to 86.46%, with KTGCA having the highest heritability and KDGCA having the lowest.
Table 2.
The joint variance analysis of kernel-related traits and GCA.
3.2. Result of SNPs Associated with Kernel-Related Traits and GCA Using the Multi-Locus GWAS
A total of 66 and 52 SNPs were detected to separately associate with kernel-related traits and GCAs according to the threshold value of LOD ≥ 3.0 using six multi-locus GWAS models from the mrMLM.GUI software package (version 1.2) (Figure 2). Among the 66 SNPs, 16, 28, 10, and 12 SNPs were found to be associated with the KD, KW, KT, and KWEI, respectively. For the trait KD, the sixteen SNPs distributed on eight chromosomes had phenotypic variation explained (PVE) values ranging from 2.50% to 10.23%. Three of the sixteen SNPs, Chr6_95626517, Chr6_117044211, and Chr2_199947075, were simultaneously identified in three models, with the average PVE ranging from 4.88% to 7.1%. For the trait KW, the detected twenty-eight SNPs were located on eight chromosomes, with PVE values ranging from 2.67% to 11.95%. Among the twenty-eight SNPs, nine SNPs were simultaneously identified in two models. Chr6_117044211 was simultaneously detected in four methods with an average PVE of 6.18%, which was also detected in relation to KD. Ten SNPs were identified in relation to KT, with PVE values ranging from 2.25% to 12.89%, of which four SNPs were simultaneously detected in two models. For the trait KWEI, the identified twelve SNPs located on eight chromosomes had PVE values from 1.83% to 16.56%. Two SNPs called Chr2_15696722 and Chr6_159607970 were simultaneously detected in three models with an average PVE of 5.77% and 6.0%, respectively. Two SNPs named Chr3_30058124 and Chr5_47996610 were simultaneously found by four models with the average PVE from 8.99% and 9.73%, respectively (Supplementary Table S2).
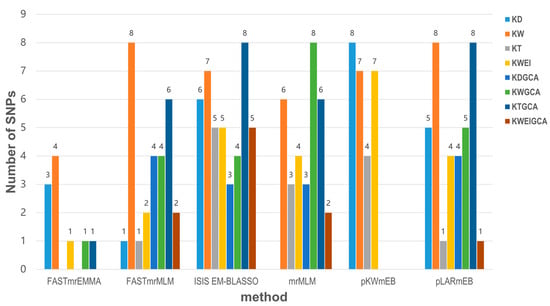
Figure 2.
Comparison of the number of SNPs identified by six multi-locus GWAS models for kernel-related traits and GCA in maize.
Among the detected 52 SNPs, 7 SNPs in relation to KDGCA were detected on four chromosomes with the PVE values ranged from 4.76% to 33.46%; 19 SNPs related to KWGCA were identified on 10 chromosomes with PVE values from 3.60% to 25.64%; 18 SNPs associated with KTGCA were found on 8 chromosomes carrying PVE values from 2.46% to 33.96%; 8 SNPs related to KWGCA were identified on 6 chromosomes carrying the PVE values from 3.09% to 6.16%. For the trait KDGCA, Chr4_225246099 was simultaneously detected in three methods with an average PVE of 30.22%, and Chr4_35278162 was simultaneously detected in four models with an average PVE of 10.29%. For KWGCA, Chr2_7095525 was simultaneously detected in two models, and Chr1_252385219 was simultaneously detected in three models with an average PVE of 7.95%. Two SNPs named Chr10_43507687 and Chr7_165731619 were simultaneously detected to be associated with KTGCA in two models. Three SNPs, Chr1_35777029, Chr1_58952116, and Chr4_198307583, were simultaneously detected in three models with average PVE values from 5.30% to 21.63%. Chr7_20835381, related to KWEIGCA, was simultaneously detected in four models with an average PVE of 8.64% (Supplementary Table S3). A total of eight SNPs associated with kernel-related traits and eight SNPs related to the GCAs of kernel-related traits were co-detected by at least three models (Figure 3).
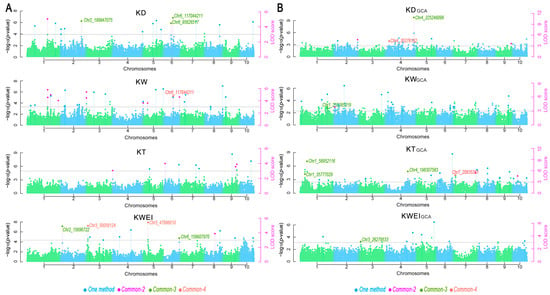
Figure 3.
Manhattan plots based on six multi-locus GWAS models of kernel-related traits and GCAs in maize (A,B). Light blue and light green indicate non-significant loci.
3.3. Allelic Variation Effects
Allelic variation effects analysis was conducted on 16 significant associated SNPs detected by more than three models simultaneously. The BLUE values of the two genotypes at the 16 loci displayed extremely significant differences in the t-test (p-value < 0.01), except for one locus associated with KTGCA on Chr4_198307583 (p = 0.013) and another locus associated with KD on chr6-117044211 (p = 0.015) (Figure 4).
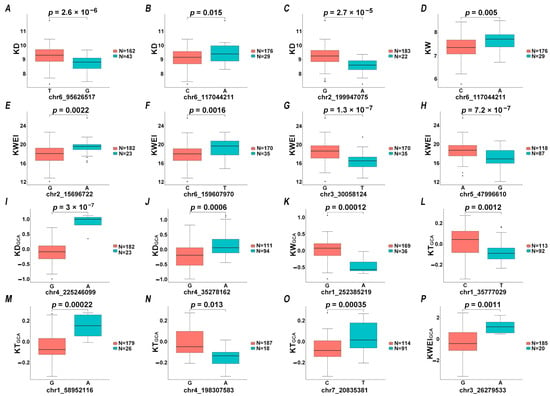
Figure 4.
Analysis of phenotypic difference in different alleles of significant SNPs (A–P). N represents the number of that haplotype.
3.4. Candidate Gene Mining
The LD values of the whole genome of 205 maize inbred lines were estimated using the 76,492 SNPs. The results indicate that when the cut-off value r2 = 0.2, the average LD decay distance is approximately 220 kb (Figure 5).
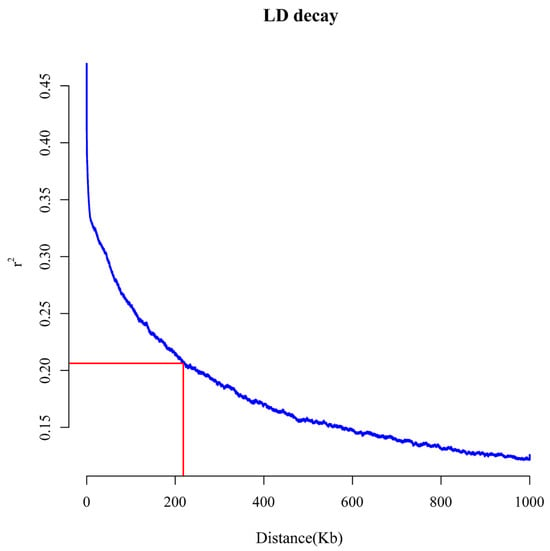
Figure 5.
Population linkage disequilibrium (LD). The horizontal axis represents the physical distance between the SNP on the same chromosome, and the vertical axis represents the linkage disequilibrium parameter r2 value.
Candidate genes were searched within the interval of 220 kb upstream and downstream of the SNPs co-detected by at least three multi-locus models. According to the genome reference map of Zm-B73-REFERENCE-GRAMENE-4.0 and databases such as NCBI, a total of 75 candidate genes were identified, among which 46 candidate genes have explicit annotations (Supplementary Table S4).
The IDs of the above 75 candidate genes were uploaded to the DAVID website for GO (Gene Ontology) analysis. The top 20 items with the largest number of enrichments were screened for plotting (Figure 6). The results of GO analysis revealed that 14 candidate genes were enriched in four GO terms within the biological processes, with a primary focus on methylation, proteolysis, and phosphorylation. Meanwhile, 34 candidate genes were enriched in five GO terms among the cellular components, predominantly concentrating on cell membranes, cytoplasm, and nuclei. Additionally, 27 candidate genes were enriched in 11 GO terms within the molecular functions, mainly centered on ATP binding, protein binding, and metal ion binding.
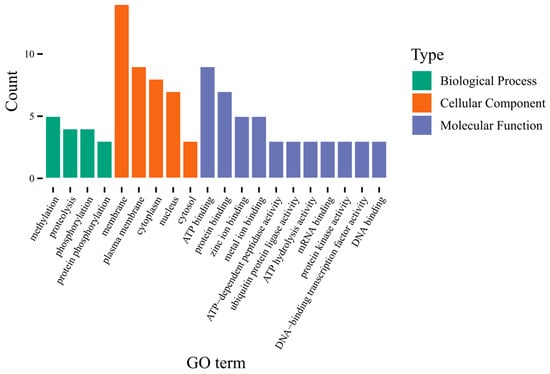
Figure 6.
GO analysis of candidate genes.
A further specific analysis was conducted on the expression levels of the 75 candidate genes in the endosperm and seeds of maize B73 from 6 to 36 days after pollination (Figure 7), and they were all expressed in different parts at various stages. Among them, there are nine candidate genes with relatively high expression levels in different parts at various stages, which were Zm00001d036626, Zm00001d006160, Zm00001d037234, Zm00001d038544, Zm00001d053308, Zm00001d033159, Zm00001d029120, Zm00001d029124, and Zm00001d052709.
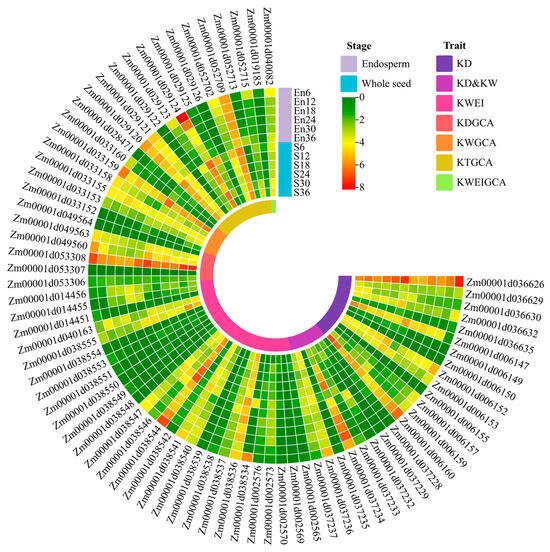
Figure 7.
Heatmap of gene expression for candidate genes in endosperm and whole seed across developmental stages.
4. Discussion
4.1. Combining Ability Shared the Different Genetic Basis with the Kernel-Related Traits Per Se
The application of heterosis has made a breakthrough in maize production, leading to increased focus from breeders on research concerning combining ability. The GCA is determined by the additive effect of the gene and is the part that can be inherited. The more favorable alleles an inbred line carries, the higher its GCA [38]. Therefore, improving the GCA through genetic improvement is a hotspot for current breeders. In this study, no common SNPs were detected for GCA and corresponding kernel-related traits when LOD = 3.0 was used as the threshold value. A similar result has been reported by Lv et al. [39]. Furthermore, Qi et al. [33] conducted a study on the genetic analysis of maize’s combining ability using introgression lines (ILs). Their results showed that there were no significant genetic correlations between the GCA of yield-related traits and the corresponding traits. Therefore, the genetic basis of GCA and its corresponding traits are somewhat different. It is clear that the performance of inbred lines per se in maize breeding cannot be used as the only indicator to evaluate the quality of the inbred lines. In the evaluation and assessment of inbred lines, the performance of hybrid offspring and their combination serves as a crucial benchmark.
4.2. Comparison of Results in the Present Study with Previously Reported QTLs
To improve the reliability of GWAS results, the SNPs detected by at least three methods were selected as the final results. In the process of studying maize kernel size traits, the known locus regions for kernel size traits were bin 1.07, bin 4.05, bin 4.08, bin 6.02, and bin 7.02 [40,41]. In our study, the SNP Chr4_35278162, related to KDGCA, is located the same region as bin 4.05. The SNP Chr6_95626517, related to KD, is located in the interval of bin 6.02. The SNP Chr7_20835381, associated with KTGCA, is located in the same region as bin 7.02. Zhang et al. [42] also found that a gene called qKW7a, related to KW, is located in the region of bin7.02--7.03. Previous research reported that the qGW4.05 located in bin 4.05 affects the size of grains, and it was discussed that bin 4.05 is the main factor affecting grain-related traits [43]. Many studies reported that using different populations to detect QTNs related to grain size at bin 7.02 and were detected in multiple populations and multiple environments [44,45,46]. This indicates that these intervals may be the main region affecting the kernel size traits.
4.3. Comparison of the Detection Effectiveness of Different GWAS Methods
Bonferroni correction is commonly used to control the false positive rate in GWASs. Nonetheless, this correction is overly stringent, resulting in the exclusion of loci that have less influence on characteristics but may be significant [29]. In this study, we also performed a GWAS using six single-locus models, including GLM, MLM, MLMM, SUPER, FarmCPU, and BLINK (Supplementary Table S5). Most of the traits cannot be identified with their associated QTNs, except for KWEI. It has been suggested that a multi-locus GWAS model without Bonferroni correction is close to the true genetic model of plants and has advantages over single-locus GWAS models in terms of higher statistical power and lower FPR [47]. Even though the multi-locus GWAS methods, such as FASTmrEMMA, ISIS EM-BLASSO, mrMLM, and pLARmEB, detect SNPs more effectively and precisely [21,23,48], the detection powers for different traits are varied. In our study, the number of SNPs detected for kernel-related traits was larger than for their GCA. Furthermore, the SNPs identified for kernel traits did not overlap with those identified for their corresponding GCA traits. This finding suggests that GCA traits may exhibit greater complexity in terms of gene expression, with a diminished influence of genes controlling the GCA. FASTmrEMMA detected the smallest number of SNPs compared to the other six methods. The number of SNPs detected by the remaining five methods did not show any significant difference. These findings are consistent with those of Zhang (2018) [29] but contrast with the results reported by Xu (2018) [30] regarding starch pasting. The potential connection between the various target traits of the investigation has been noted in previous studies. The results of GCA demonstrated a significant reduction in the detection efficiency of all six methods.
4.4. Candidate Genes Analysis
In this study, six GWAS models were used to mine candidate loci. The SNPs were simultaneously identified by at least three methods to further excavate the genes and query their functions. This involved gene annotation and GO analysis to determine the primary functions of the proteins encoded by these genes. Most of these candidate genes encode proteins, mainly enzymes and transcription factors.
It was found that once phosphorylated, ZmBES1/BZR1-5 binds to the co-factor ZmFdx2 and inhibits the transcription of AP2/EREBP genes by binding to their E-box and BRRE elements, thus reducing the negative regulation of kernel development by AP2/EREBP proteins [49]. The results indicated that the binding effect would affect the kernel size of maize.
The research has revealed that the gene Zm00001d036630 has an impact on the synthesis of Abscisic Acid (ABA). Many studies have reported lower levels of seed storage products due to defects in ABA signaling, including effects on oil content and starch [50,51,52].
We found a candidate gene Zm00001d037234 that is co-related to KD and KW. It encodes soluble starch synthase 2-3 and participates in the biosynthesis process of starch. The biosynthesis of starch is a highly complex metabolic process that requires the synergistic action of multiple enzymes. Existing studies have shown that SSIII acts as the core and regulatory enzyme of maize starch synthases, with several important functions, including synthesizing highly ordered branched starch, influencing the structure of branched starch, and catalyzing the formation of longer sugar chains [53].
We also discovered a candidate gene Zm00001d053308 that is related to KDGCA. It encodes peroxisomal fatty acid beta-oxidation multifunctional protein (peroxisomal FAO MFP). Primarily, it catalyzes the beta-oxidation of fatty acids in peroxisomes. Possessing multiple enzymatic activities, it can catalyze several steps in the fatty acid beta-oxidation process, including the activities of 2-enoyl-CoA hydratase, L-3-hydroxyacyl-CoA dehydrogenase, enoyl-CoA reductase, and thioesterase. In maize, fatty acid beta-oxidation plays an important role in hormone synthesis and seed germination, as well as providing energy and carbon skeletons [54].
We also discovered three candidate genes related to KTGCA (Zm00001d029120, Zm00001d029123, and Zm00001d052709). Among them, the candidate gene Zm00001d029120 encodes AAA-type ATPase family protein, which is a type of protein that plays multiple functions in cells. It converts the chemical energy of ATP hydrolysis into mechanical energy to drive various biological processes, including protein degradation, membrane fusion, microtubule severing, peroxisome biosynthesis, signal transduction, and regulation of gene expression, etc. [55]; the candidate gene Zm00001d029123 encodes auxin response factor 75. By binding to chlorophyll, it participates in regulating the growth and development processes of plants, including embryo pattern formation, vascular tissue formation, flower development, and phototropism, etc., and regulates the expression of auxin-responsive genes, playing a key role in auxin signal transduction [56]; The candidate gene Zm00001d052709 encodes probable E3 ubiquitin-protein ligase ARI8 in plants. It is an E3 ubiquitin ligase and plays an important role in a series of life activities such as plant growth and development, stress response, and signal transduction. Different types of E3 ubiquitin ligases play key roles in plant stress responses, hormone signal transduction, and reproductive development, etc. For example, AtPUB4 in Arabidopsis is a U-box type E3 ubiquitin ligase, and its mutants affect the normal development and dispersal of pollen, thereby affecting the fertility of plants [57,58].
5. Conclusions
In this study, six multi-locus GWAS methods were used to detect SNPs associated with kernel-related traits and GCA in maize. A total of 66 and 52 SNPs associated with kernel-related traits and their GCAs were identified, with 16 SNPs co-detected by at least three methods. Moreover, 75 genes were identified in the vicinity of the 16 SNPs based on functional annotations. These findings will be useful for understanding the genetic foundation of kernel-related traits and the corresponding GCAs.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/agronomy15122806/s1, Table S1: The information of F1 individuals; Table S2: Significant kernel-related traits detected by six multi-locus GWAS methods; Table S3: Significant kernel-related traits of GCA detected by six multi-locus GWAS methods; Table S4: Seventy-five genes annotated in the NCBI database; Table S5: The SNPs results using six single-locus methods from the gapit package.
Author Contributions
L.Z. conceived the ideas and designed the methodology; Y.C. and J.G. revised the manuscript; L.J. conducted the field trials and collected the data; X.L. and F.H. analyzed the data and drafted the manuscript; X.J., Y.Z. (Yongfeng Zhao) and Y.Z. (Yunxiao Zheng) took part in the experiment. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by grants from the Key Research and Development Program of Hebei Province (21326325D), S&T program of Hebei (24466301D), the National Key Research and Development Program of China (2016YFD0101204) and the National Natural Science Foundation of China (32100496).
Data Availability Statement
The original contributions presented in this study are included in the article/Supplementary Materials. Further inquiries can be directed to the corresponding author.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Liu, M.; Tan, X.L.; Yang, Y.; Liu, P.; Zhang, X.X.; Zhang, Y.C.; Wang, L.; Hu, Y.; Ma, L.L.; Li, Z.L.; et al. Analysis of the genetic architecture of maize kernel size traits by combined linkage and association mapping. Plant Biotechnol. J. 2020, 18, 207–221. [Google Scholar] [CrossRef]
- Li, Y.X.; Wang, Y.; Shi, Y.S.; Song, Y.C.; Wang, T.Y.; Li, Y. Correlation analysis and QTL mapping for traits of kernel structure and yield components in maize. Sci. Agric. Sin. 2009, 42, 408–418. [Google Scholar]
- Zhu, X.M.; Shao, X.Y.; Pei, Y.H.; Guo, X.M.; Li, J.; Song, X.Y.; Zhao, M.A. Genetic Diversity and Genome-Wide Association Study of Major Ear Quantitative Traits Using High-Density SNPs in Maize. Front. Plant Sci. 2018, 9, 966. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhou, Z.; Ding, J.; Wu, Y.; Zhou, B.; Wang, R.; Ma, J.; Wang, S.; Zhang, X.; Xia, Z.; et al. Combined Linkage and Association Mapping Reveals QTL and Candidate Genes for Plant and Ear Height in Maize. Front. Plant Sci. 2016, 7, 833. [Google Scholar] [CrossRef]
- Peng, B.; Li, Y.X.; Wang, Y.; Liu, C.; Liu, Z.Z.; Tan, W.W.; Zhang, Y.; Wang, D.; Shi, Y.S.; Sun, B.C.; et al. QTL analysis for yield components and kernel-related traits in maize across multi-environments. Theor. Appl. Genet. 2011, 122, 1305–1320. [Google Scholar] [CrossRef]
- Yang, C.; Zhang, L.; Jia, A.M.; Rong, T.Z. Identification of QTL for maize grain yield and kernel-related traits. J. Genet. 2016, 95, 239–247. [Google Scholar] [CrossRef]
- Xiao, Y.J.; Jiang, S.Q.; Cheng, Q.; Wang, X.Q.; Yan, J.; Zhang, R.Y.; Qiao, F.; Ma, C.; Luo, J.Y.; Li, W.Q.; et al. The genetic mechanism of heterosis utilization in maize improvement. Genome Biol. 2021, 22, 148. [Google Scholar] [CrossRef]
- Tollenaar, M. Genetic Improvement in Grain Yield of Commercial Maize Hybrids Grown in Ontario from 1959 to 1988. Crop Sci. 1989, 29, 1365–1371. [Google Scholar] [CrossRef]
- Wang, H.; He, Y.; Wang, S.C. QTL mapping of general combining abilities of four traits in maize using a high-density genetic map. J. Integr. Agric. 2017, 16, 1700–1707. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, Z.L.; Xu, Y.; Li, P.C.; Zhang, X.C.; Xu, C.W. Using genomic data to improve the estimation of general combining ability based on sparse partial diallel cross designs in maize. Crop J. 2020, 8, 819–829. [Google Scholar] [CrossRef]
- Xiang, C.; Zhang, H.J.; Wang, H.; Wang, J.; Wang, W.S.; Xia, J.F.; Gao, Y.M.; Ye, G.Y. Dissection of combining ability for yield and related traits using introgression lines in the background of a key restorer line in rice (Oryza sativa L.). Field Crops Res. 2016, 193, 154–163. [Google Scholar] [CrossRef]
- Huang, J.; Qi, H.H.; Feng, X.M.; Huang, Y.Q.; Zhu, L.Y.; Yue, B. General combining ability of most yield-related traits had a genetic basis different from their corresponding traits per se in a set of maize introgression lines. Genetica 2013, 141, 453–461. [Google Scholar] [CrossRef]
- Zhou, Z.Q.; Zhang, C.S.; Lu, X.H.; Wang, L.W.; Hao, Z.F.; Li, M.S.; Zhang, D.G.; Yong, H.J.; Zhu, H.Y.; Weng, J.F.; et al. Dissecting the Genetic Basis Underlying Combining Ability of Plant Height Related Traits in Maize. Front. Plant Sci. 2018, 9, 1117. [Google Scholar] [CrossRef]
- Sun, G.Y.; Zhang, X.H.; Duan, H.Y.; Gao, J.H.; Li, N.; Su, P.P.; Xie, H.L.; Li, W.H.; Fu, Z.Y.; Huang, Y.B.; et al. Dissection of the genetic architecture of peduncle vascular bundle-related traits in maize by a genome-wide association study. Plant Biotechnol. J. 2022, 20, 1042–1053. [Google Scholar] [CrossRef]
- Tian, F.; Bradbury, P.J.; Brown, P.J.; Hung, H.; Sun, Q.; Flint-Garcia, S.; Rocheford, T.R.; McMullen, M.D.; Holland, J.B.; Buckler, E.S. Genome-wide association study of leaf architecture in the maize nested association mapping population. Nat. Genet. 2011, 43, 159–162. [Google Scholar] [CrossRef]
- Wu, X.; Li, Y.X.; Shi, Y.S.; Song, Y.C.; Zhang, D.F.; Li, C.H.; Buckler, E.S.; Li, Y.; Zhang, Z.W.; Wang, T.Y. Joint-linkage mapping and GWAS reveal extensive genetic loci that regulate male inflorescence size in maize. Plant Biotechnol. J. 2016, 14, 1551–1562. [Google Scholar] [CrossRef]
- Hindu, V.; Palacios-Rojas, N.; Babu, R.; Suwarno, W.B.; Rashid, Z.; Usha, R.; Saykhedkar, G.R.; Nair, S.K. Identification and validation of genomic regions influencing kernel zinc and iron in maize. Theor. Appl. Genet. 2018, 131, 1443–1457. [Google Scholar] [CrossRef]
- Zhao, X.W.; Luo, L.X.; Cao, Y.H.; Liu, Y.J.; Li, Y.H.; Wu, W.M.; Lan, Y.Z.; Jiang, Y.W.; Gao, S.B.; Zhang, Z.M.; et al. Genome-wide association analysis and QTL mapping reveal the genetic control of cadmium accumulation in maize leaf. BMC Genom. 2018, 19, 91. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.; Zhang, X.H.; Sun, G.Y.; Yan, P.S.; Guo, H.P.; Chen, S.Y.; Xue, Y.D.; Guo, Z.Y.; Xie, H.L.; Tang, J.H.; et al. Genome-wide association studies of plant type traits in maize. Sci. Agric. Sin. 2018, 51, 821–834. [Google Scholar]
- Dong, H.L.; Zhuang, Z.L.; Bian, J.W.; Tang, R.; Ren, Z.P.; Peng, Y.L. Candidate Gene for Kernel-Related Traits in Maize Revealed by a Combination of GWAS and Meta-QTL Analyses. Plants 2025, 14, 959. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Feng, J.Y.; Ni, Y.L.; Wen, Y.J.; Niu, Y.; Tamba, C.L.; Yue, C.; Song, Q.; Zhang, Y.M. pLARmEB: Integration of least angle regression with empirical Bayes for multilocus genome-wide association studies. Heredity 2017, 118, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Tamba, C.L.; Ni, Y.L.; Zhang, Y.M. Iterative sure independence screening EM-Bayesian LASSO algorithm for multi-locus genome-wide association studies. PLOS Comput. Biol. 2017, 13, e1005357. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.J.; Zhang, H.W.; Ni, Y.L.; Huang, B.; Zhang, J.; Feng, J.Y.; Wang, S.B.; Dunwell, J.M.; Zhang, Y.M.; Wu, R.L. Methodological implementation of mixed linear models in multi-locus genome-wide association studies. Brief. Bioinform. 2018, 19, 700–712. [Google Scholar] [CrossRef] [PubMed]
- Tamba, C.L.; Zhang, Y.M. A fast mrMLM algorithm for multi-locus genome-wide association studies. bioRxiv 2018, 341784. [Google Scholar] [CrossRef]
- Ren, W.L.; Wen, Y.J.; Dunwell, J.M.; Zhang, Y.M. pKWmEB: Integration of Kruskal–Wallis test with empirical Bayes under polygenic background control for multi-locus genome-wide association study. Heredity 2018, 120, 208–218. [Google Scholar] [CrossRef]
- Wang, S.B.; Feng, J.Y.; Ren, W.L.; Huang, B.; Zhou, L.; Wen, Y.J.; Jin, Z.; Dunwell, J.M.; Xu, S.Z.; Zhang, Y.M. Improving power and accuracy of genome-wide association studies via a multi-locus mixed linear model methodology. Sci. Rep. 2016, 6, 19444. [Google Scholar] [CrossRef]
- Cui, Y.R.; Zhang, F.; Zhou, Y.L. The Application of Multi-Locus GWAS for the Detection of Salt-Tolerance Loci in Rice. Front. Plant Sci. 2018, 9, 1464. [Google Scholar] [CrossRef]
- Zhang, Y.M.; Jia, Z.Y.; Dunwell, J.M. Editorial: The Applications of New Multi-Locus GWAS Methodologies in the Genetic Dissection of Complex Traits. Front. Plant Sci. 2019, 10, 100. [Google Scholar] [CrossRef]
- Zhang, Y.L.; Liu, P.; Zhang, X.X.; Zheng, Q.; Chen, M.; Ge, F.; Li, Z.L.; Sun, W.T.; Guan, Z.R.; Liang, T.H.; et al. Multi-Locus Genome-Wide Association Study Reveals the Genetic Architecture of Stalk Lodging Resistance-Related Traits in Maize. Front. Plant Sci. 2018, 9, 611. [Google Scholar] [CrossRef]
- Xu, Y.; Yang, T.T.; Zhou, Y.; Yin, S.Y.; Li, P.C.; Liu, J.; Xu, S.H.; Yang, Z.F.; Xu, C.W. Genome-Wide Association Mapping of Starch Pasting Properties in Maize Using Single-Locus and Multi-Locus Models. Front. Plant Sci. 2018, 9, 1311. [Google Scholar] [CrossRef]
- Zheng, Y.X.; Hou, P.; Zhu, L.Y.; Song, W.B.; Liu, H.; Huang, Y.Q.; Wang, H.; Guo, J.J. Genome-Wide Association Study of Vascular Bundle-Related Traits in Maize Stalk. Front. Plant Sci. 2021, 12, 699486. [Google Scholar] [CrossRef]
- Ma, J.; Huang, L.; Yu, T.; Guo, G.J.; Zhu, W.H.; Liu, J.B. Multi-Locus Genome-Wide Association Study and Genomic Prediction for General Combining Ability of Maize Ear Diameter. Crops 2024, 1, 31–39. [Google Scholar]
- Qi, H.H.; Huang, J.; Zheng, Q.; Huang, Y.Q.; Shao, R.; Zhu, L.Y.; Zhang, Z.X.; Qiu, F.Z.; Zhou, G.C.; Zheng, Y.L.; et al. Identification of combining ability loci for five yield-related traits in maize using a set of testcrosses with introgression lines. Theor. Appl. Genet. 2013, 126, 369–377. [Google Scholar] [CrossRef]
- Knapp, S.J.; Stroup, W.W.; Ross, W.M. Exact Confidence Intervals for Heritability on a Progeny Mean Basis1. Crop Sci. 1985, 25, 192–194. [Google Scholar] [CrossRef]
- Murray, M.G.; Thompson, W.F. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 1980, 8, 4321–4326. [Google Scholar] [CrossRef] [PubMed]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A Robust, Simple Genotyping-by-Sequencing (GBS) Approach for High Diversity Species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zeng, B.; Zhang, M.; Xie, S.J.; Wang, G.K.; Hauck, A.; Lai, J.S. Dynamic transcriptome landscape of maize embryo and endosperm development. Plant Physiol. 2014, 166, 252–264. [Google Scholar] [CrossRef]
- Qu, Z.; Li, L.Z.; Luo, J.Y.; Wang, P.; Yu, S.B.; Mou, T.M.; Zheng, X.F.; Hu, Z.L. QTL Mapping of Combining Ability and Heterosis of Agronomic Traits in Rice Backcross Recombinant Inbred Lines and Hybrid Crosses. PLoS ONE 2012, 7, e28463. [Google Scholar] [CrossRef]
- Lv, A.; Zhang, H.; Zhang, Z.X.; Tao, Y.S.; Yue, B.; Zheng, Y.L. Conversion of the Statistical Combining Ability into a Genetic Concept. J. Integr. Agric. 2012, 11, 43–52. [Google Scholar] [CrossRef]
- Gupta, P.K.; Rustgi, S.; Kumar, N. Genetic and molecular basis of grain size and grain number and its relevance to grain productivity in higher plants. Genome 2006, 49, 565–571. [Google Scholar] [CrossRef]
- Li, X.; Li, Y.X.; Chen, L.; Wu, X.; Qin, W.W.; Song, Y.C.; Zhang, D.F.; Wang, T.Y.; Li, Y.; Shi, Y.S. Fine mapping of qKW7, a major QTL for kernel weight and kernel width in maize, confirmed by the combined analytic approaches of linkage and association analysis. Euphytica 2016, 210, 221–232. [Google Scholar] [CrossRef]
- Zhang, Z.H.; Liu, Z.H.; Hu, Y.M.; Li, W.H.; Fu, Z.Y.; Ding, D.; Li, H.C.; Qiao, M.M.; Tang, J.H. QTL Analysis of Kernel-Related Traits in Maize Using an Immortalized F2 Population. PLoS ONE 2014, 9, e89645. [Google Scholar] [CrossRef]
- Chen, L.; Li, Y.X.; Li, C.H.; Wu, X.; Qin, W.W.; Li, X.; Jiao, F.C.; Zhang, X.J.; Zhang, D.F.; Shi, Y.S.; et al. Fine-mapping of qGW4.05, a major QTL for kernel weight and size in maize. BMC Plant Biol. 2016, 16, 81. [Google Scholar] [CrossRef]
- Li, Y.L.; Yang, M.L.; Dong, Y.B.; Wang, Q.L.; Zhou, Y.G.; Zhou, Q.; Shen, B.T.; Zhang, F.F.; Liang, X.J. Three main genetic regions for grain development revealed through QTL detection and meta-analysis in maize. Mol. Breed. 2012, 30, 195–211. [Google Scholar] [CrossRef]
- Alvarez Prado, S.; López, C.G.; Gambín, B.L.; Abertondo, V.J.; Borrás, L. Dissecting the genetic basis of physiological processes determining maize kernel weight using the IBM (B73 × Mo17) Syn4 population. Field Crops Res. 2013, 145, 33–43. [Google Scholar] [CrossRef]
- Peng, B.; Li, Y.X.; Wang, Y.; Liu, C.; Liu, Z.Z.; Zhang, Y.; Tan, W.W.; Wang, D.; Shi, Y.S.; Sun, B.C.; et al. Correlations and comparisons of quantitative trait loci with family per se and testcross performance for grain yield and related traits in maize. Theor. Appl. Genet. 2013, 126, 773–789. [Google Scholar] [CrossRef]
- Segura, V.; Vilhjálmsson, B.J.; Platt, A.; Korte, A.; Seren, Ü.; Long, Q.; Nordborg, M. An efficient multi-locus mixed-model approach for genome-wide association studies in structured populations. Nat. Genet. 2012, 44, 825–830. [Google Scholar] [CrossRef]
- Liu, X.L.; Huang, M.; Fan, B.; Buckler, E.S.; Zhang, Z.W. Iterative Usage of Fixed and Random Effect Models for Powerful and Efficient Genome-Wide Association Studies. PLoS Genet. 2016, 12, e1005767. [Google Scholar] [CrossRef]
- Sun, F.A.; Ding, L.; Feng, W.Q.; Cao, Y.; Lu, F.Z.; Yang, Q.Q.; Li, W.C.; Lu, Y.L.; Shabek, N.; Fu, F.L.; et al. Maize transcription factor ZmBES1/BZR1-5 positively regulates kernel size. J. Exp. Bot. 2021, 72, 1714–1726. [Google Scholar] [CrossRef]
- Bies-Etheve, N.; da Silva Conceicao, A.; Giraudat, J.; Koornneef, M.; Léon-Kloosterziel, K.; Valon, C.; Delseny, M. Importance of the B2 domain of the Arabidopsis ABI3 protein for Em and 2S albumin gene regulation. Plant Mol. Biol. 1999, 40, 1045–1054. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.F.; Xu, X.P.; Crosley, R.A.; Greenwalt, S.A.; Sun, Y.J.; Blakeslee, B.; Wang, L.Z.; Ni, W.T.; Sopko, M.S.; Yao, C.L.; et al. The Protein Kinase SnRK2.6 Mediates the Regulation of Sucrose Metabolism and Plant Growth in Arabidopsis. Plant Physiol. 2010, 153, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Ali, F.; Qanmber, G.; Li, F.G.; Wang, Z. Updated role of ABA in seed maturation, dormancy, and germination. J. Adv. Res. 2022, 35, 199–214. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.W.; Gao, Y.Y.Z.; Liu, L.; Shoaib, N.; Deng, Y.W.; Zhang, N.; Li, Y.P.; Huang, Y.B. The Structure, Function, and Regulation of Starch Synthesis Enzymes SSIII with Emphasis on Maize. Agronomy 2022, 12, 1359. [Google Scholar] [CrossRef]
- Chuong, S.D.X.; Park, N.I.; Freeman, M.C.; Mullen, R.T.; Muench, D.G. The peroxisomal multifunctional protein interacts with cortical microtubules in plant cells. BMC Cell Biol. 2005, 6, 40. [Google Scholar] [CrossRef]
- Hanson, P.I.; Whiteheart, S.W. AAA+ proteins: Have engine, will work. Nat. Rev. Mol. Cell Biol. 2005, 6, 519–529. [Google Scholar] [CrossRef]
- Song, X.Y.; Xiong, Y.L.; Kong, X.Z.; Huang, G.Q. Roles of auxin response factors in rice development and stress responses. Plant Cell Environ. 2023, 46, 1075–1086. [Google Scholar] [CrossRef]
- Kelley, D.R. E3 Ubiquitin Ligases: Key Regulators of Hormone Signaling in Plants. Mol. Cell. Proteom. MCP 2018, 17, 1047–1054. [Google Scholar] [CrossRef]
- Tian, A.M.; Yu, H.; Cao, J.S. Classification and Function of E3 Ubiquitin Ligase in Plants. Chin. J. Cell Biol. 2020, 42, 907–915. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).