
In-Situ Synthesis of TiO2@GO Nanosheets for Polymers Degradation in a Natural Environment

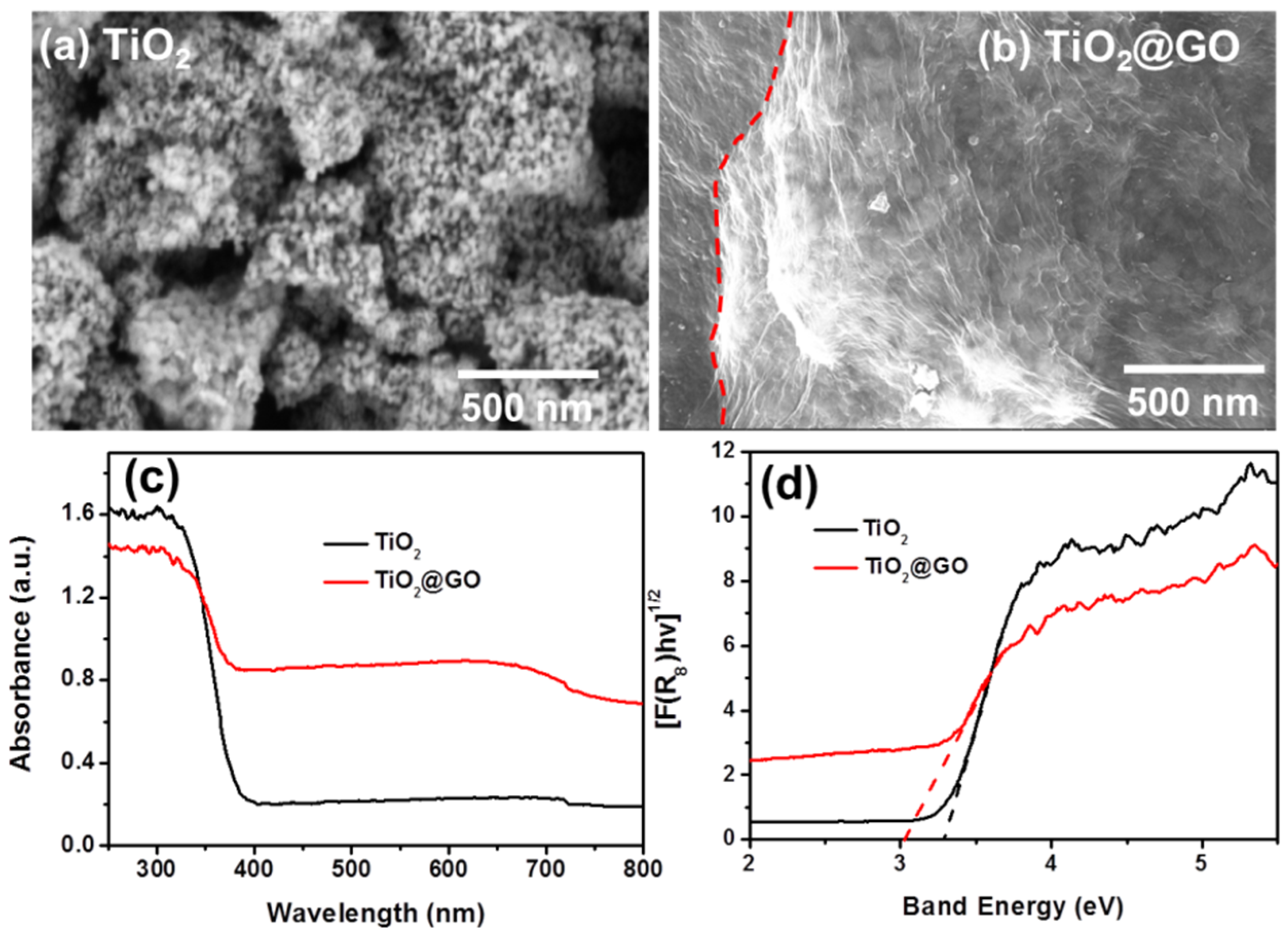{kind=link}
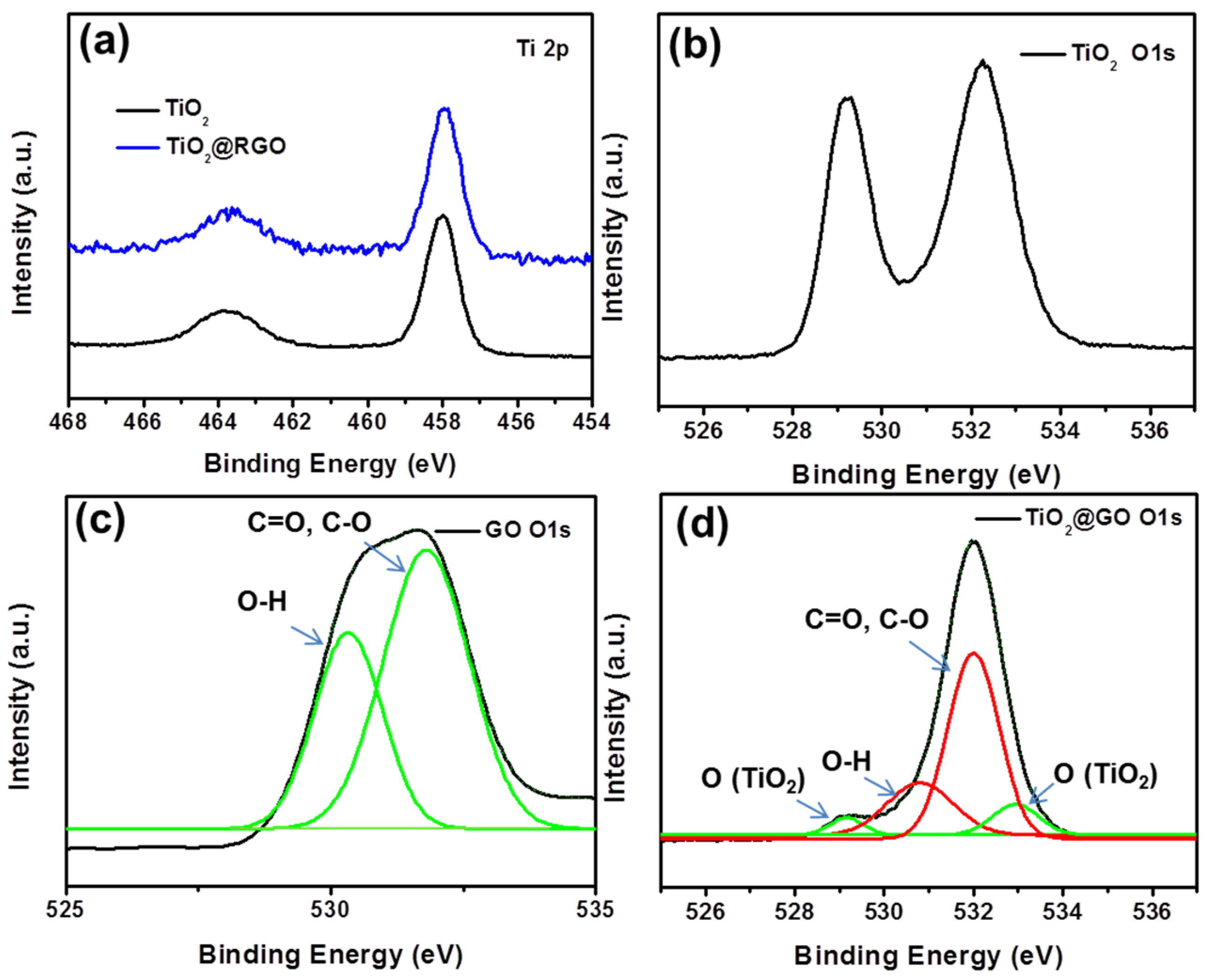{kind=link}
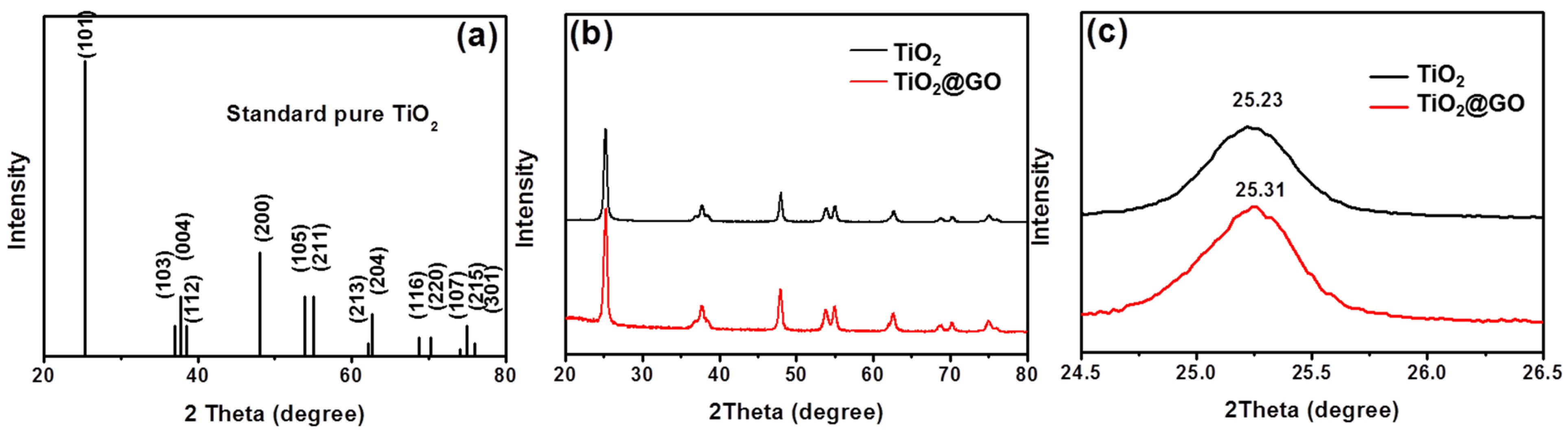{kind=link}
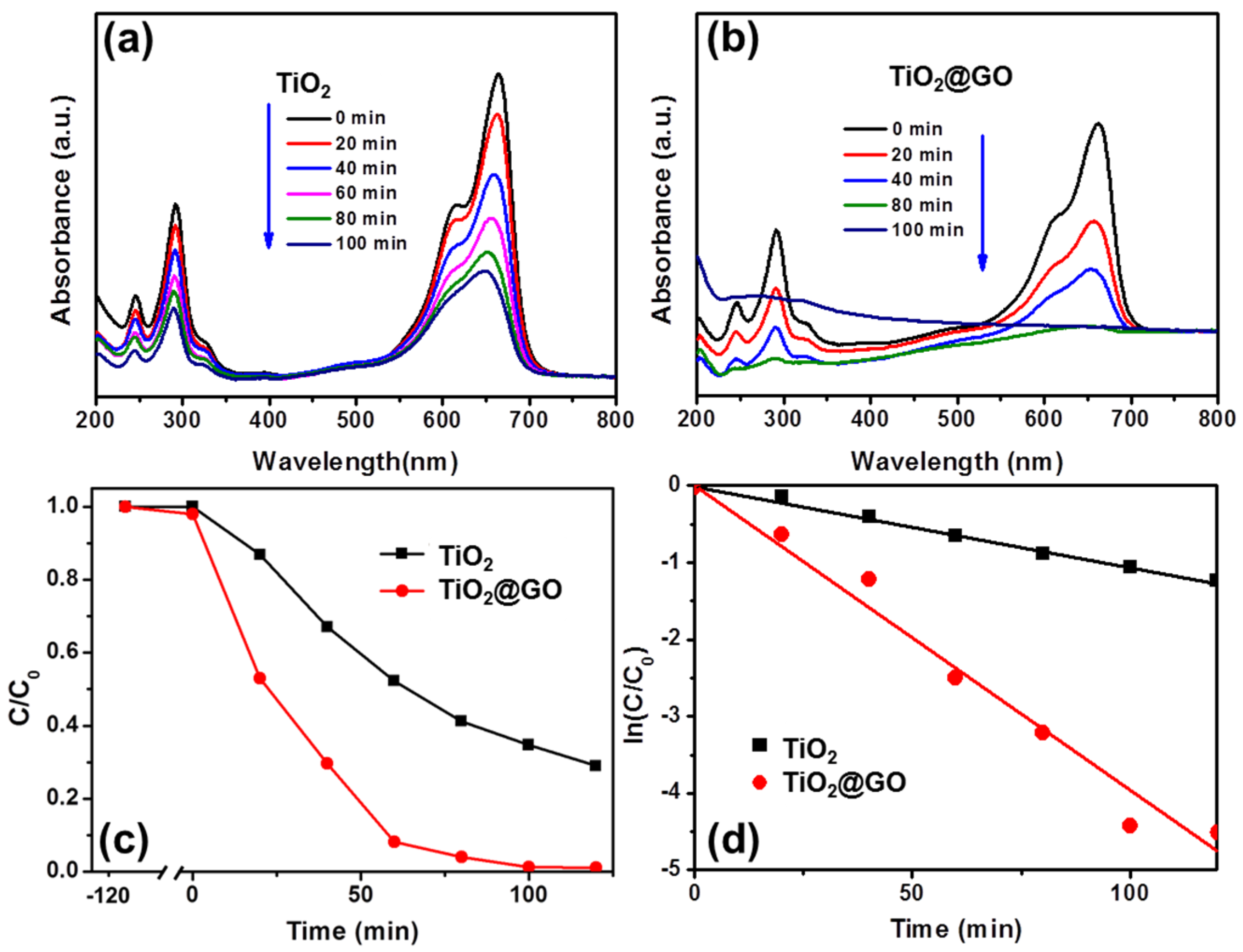{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Procedure
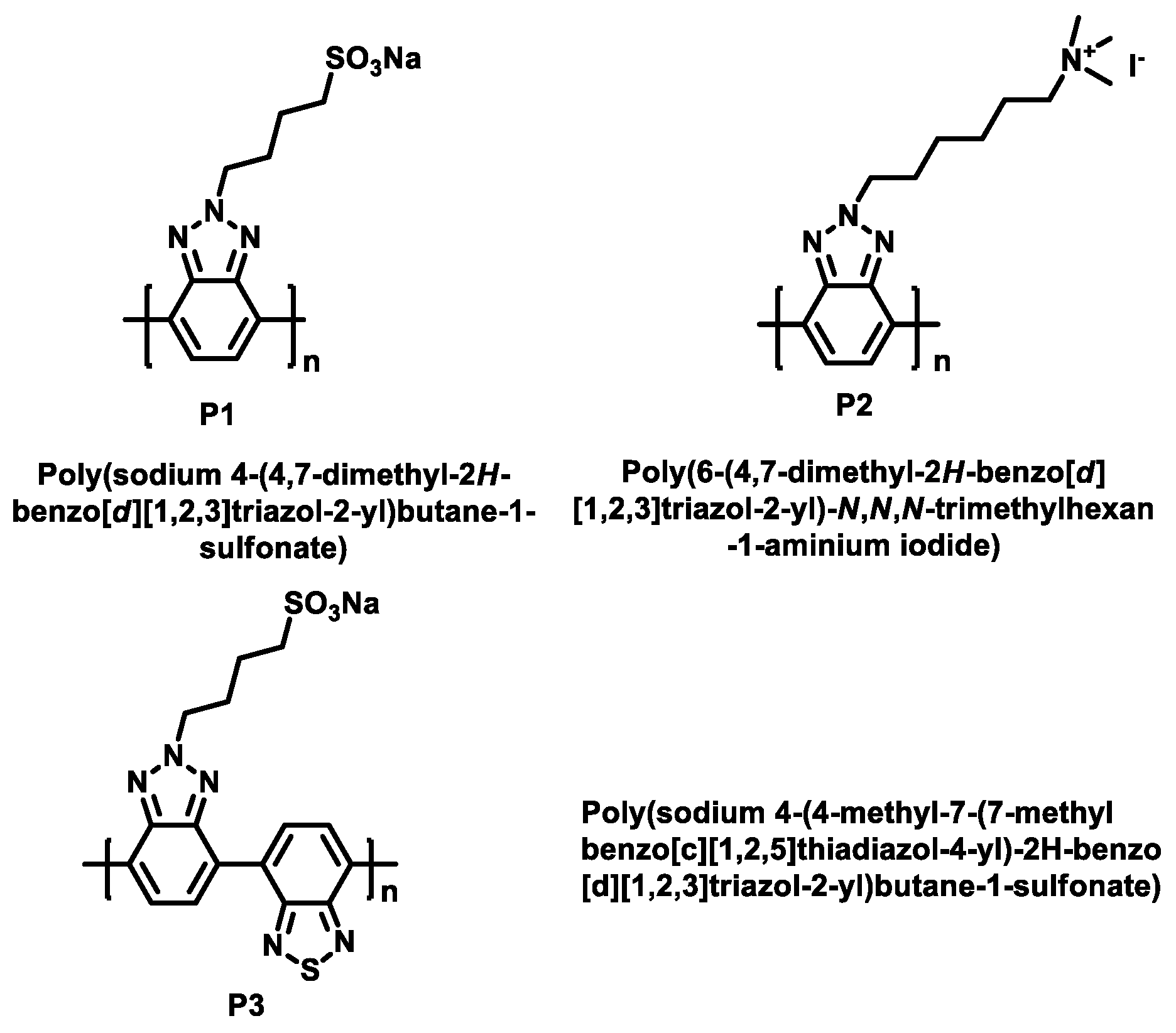
2.1. Synthesis of All Samples
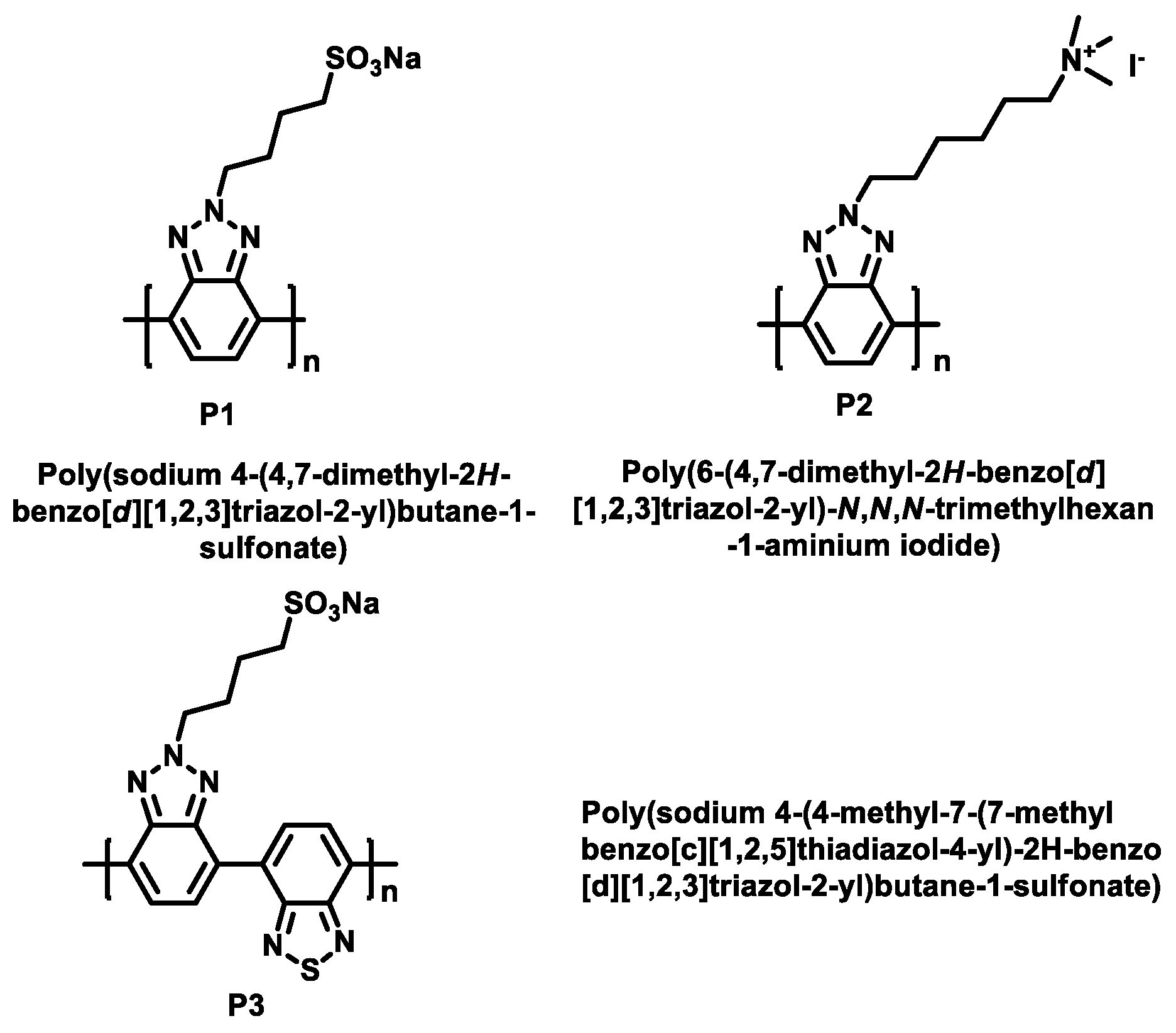
2.1.1. The Polymers’ Synthesis
2.1.2. Yamamoto Polymerizations of PBTz-SO3Na (P1) and PBTz-TMAI (P2)
2.1.3. Suzuki Coupling Polymerization of PBTBTz-SO3Na (P3)
2.2. Characterizations
2.3. Photocatalytic Experiments of Methylene Blue
2.4. Photocatalytic Degradation of Polymers
2.5. Hydroxyl Radical (•OH) Measurement
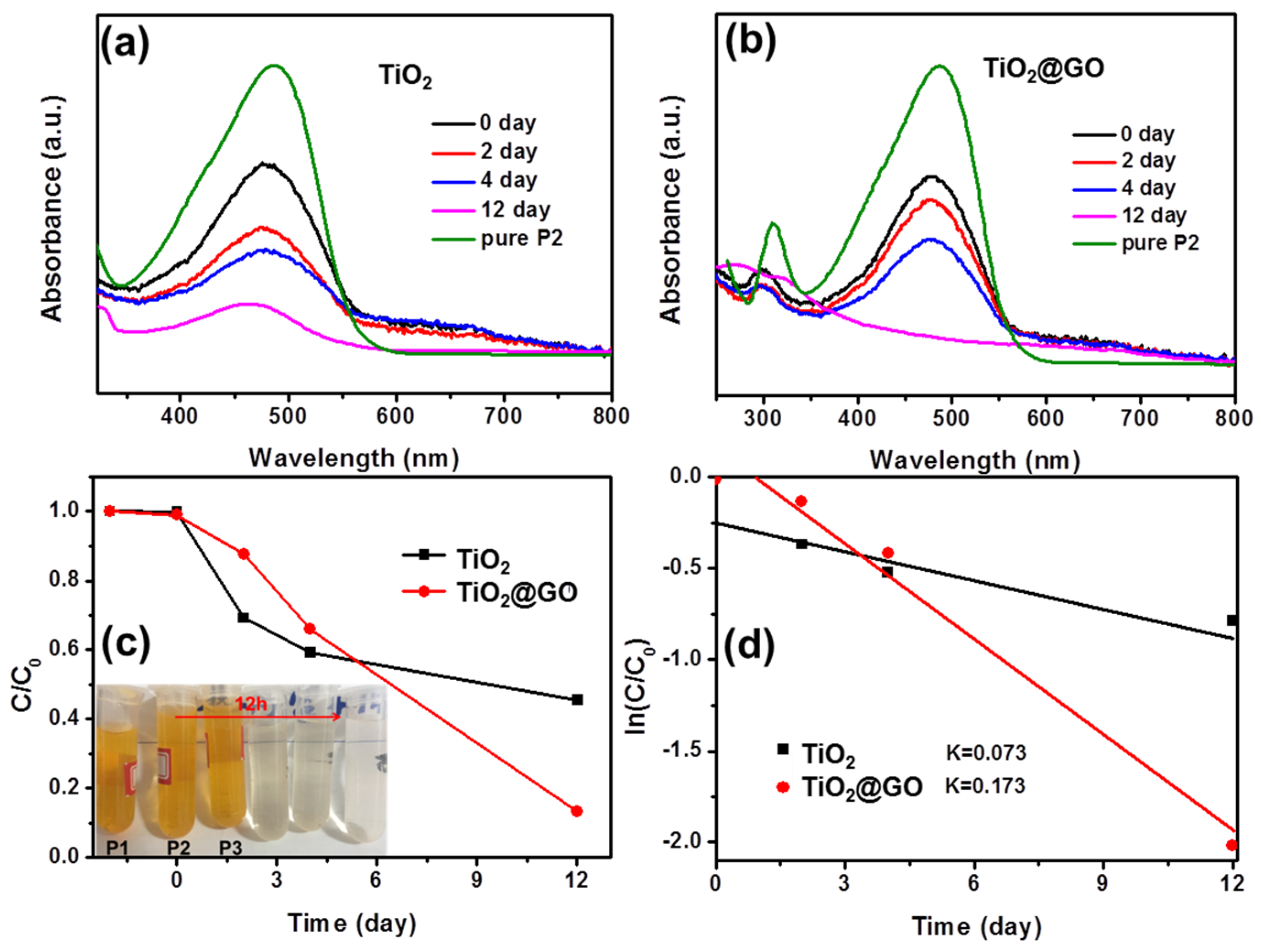
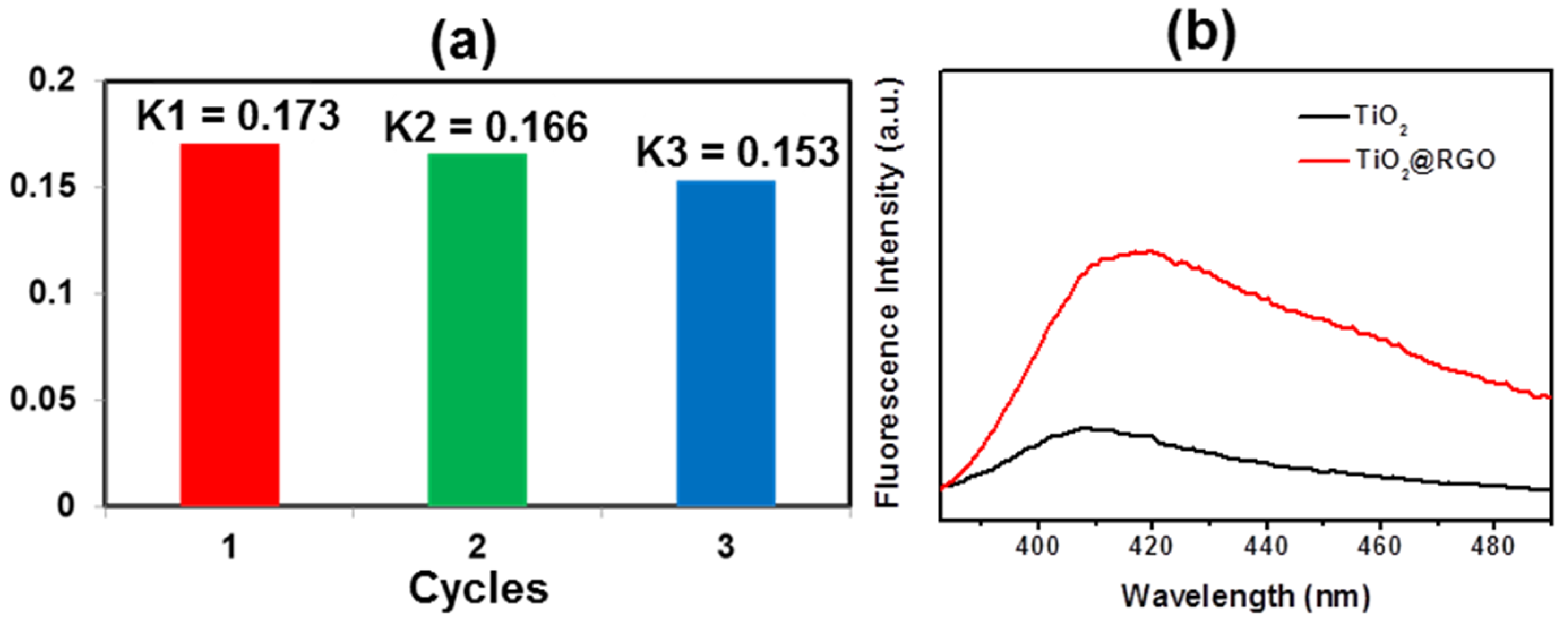
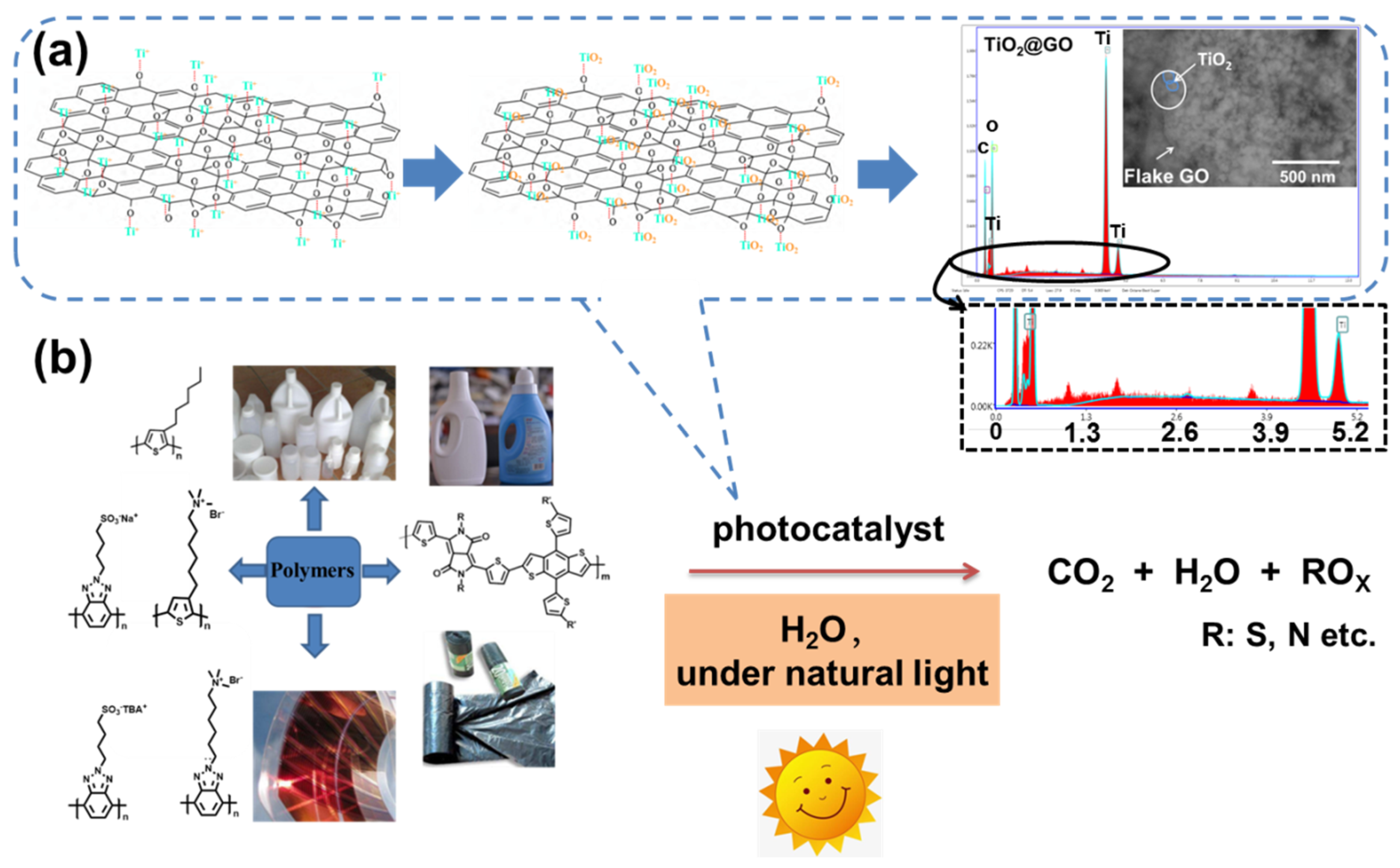
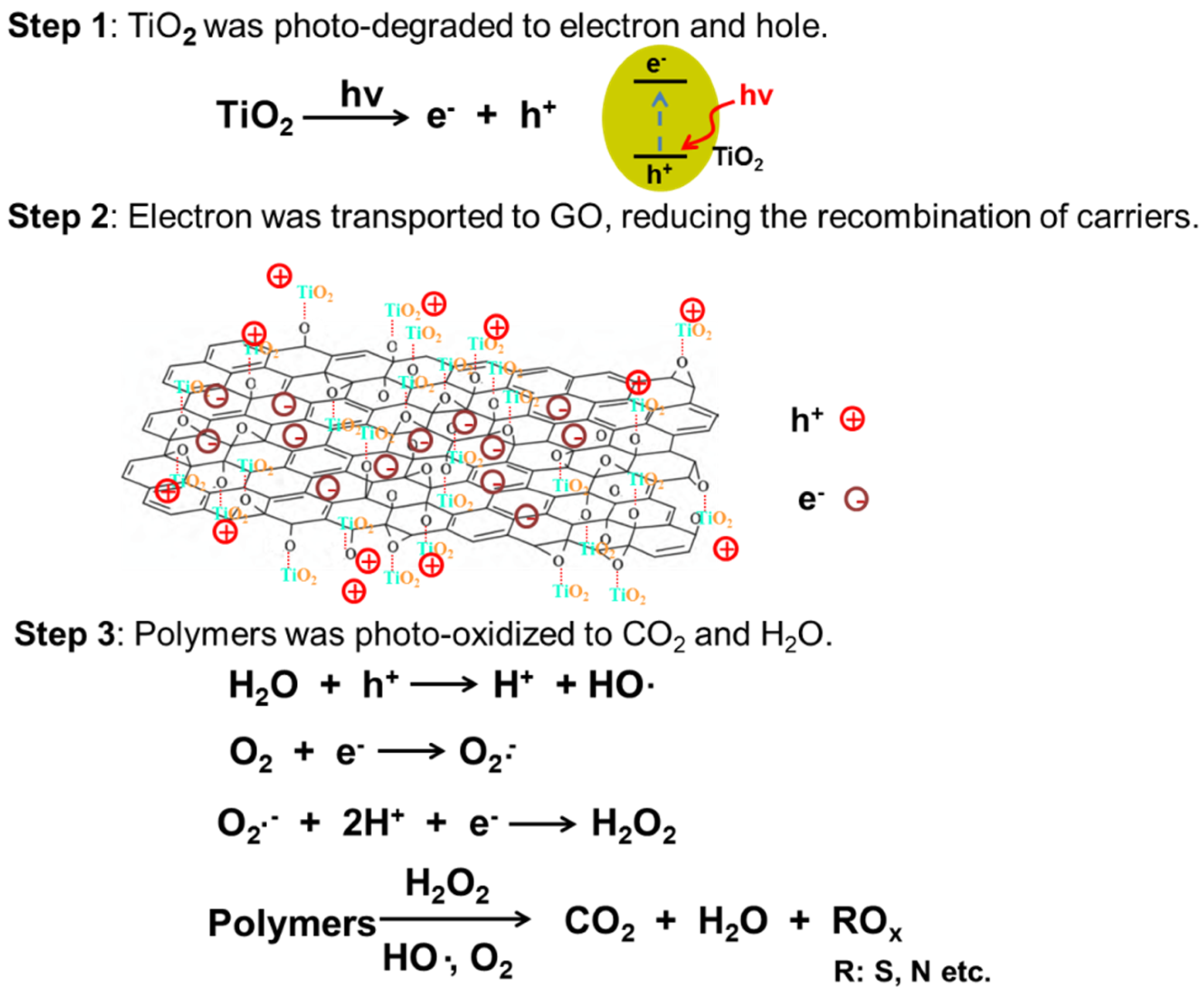
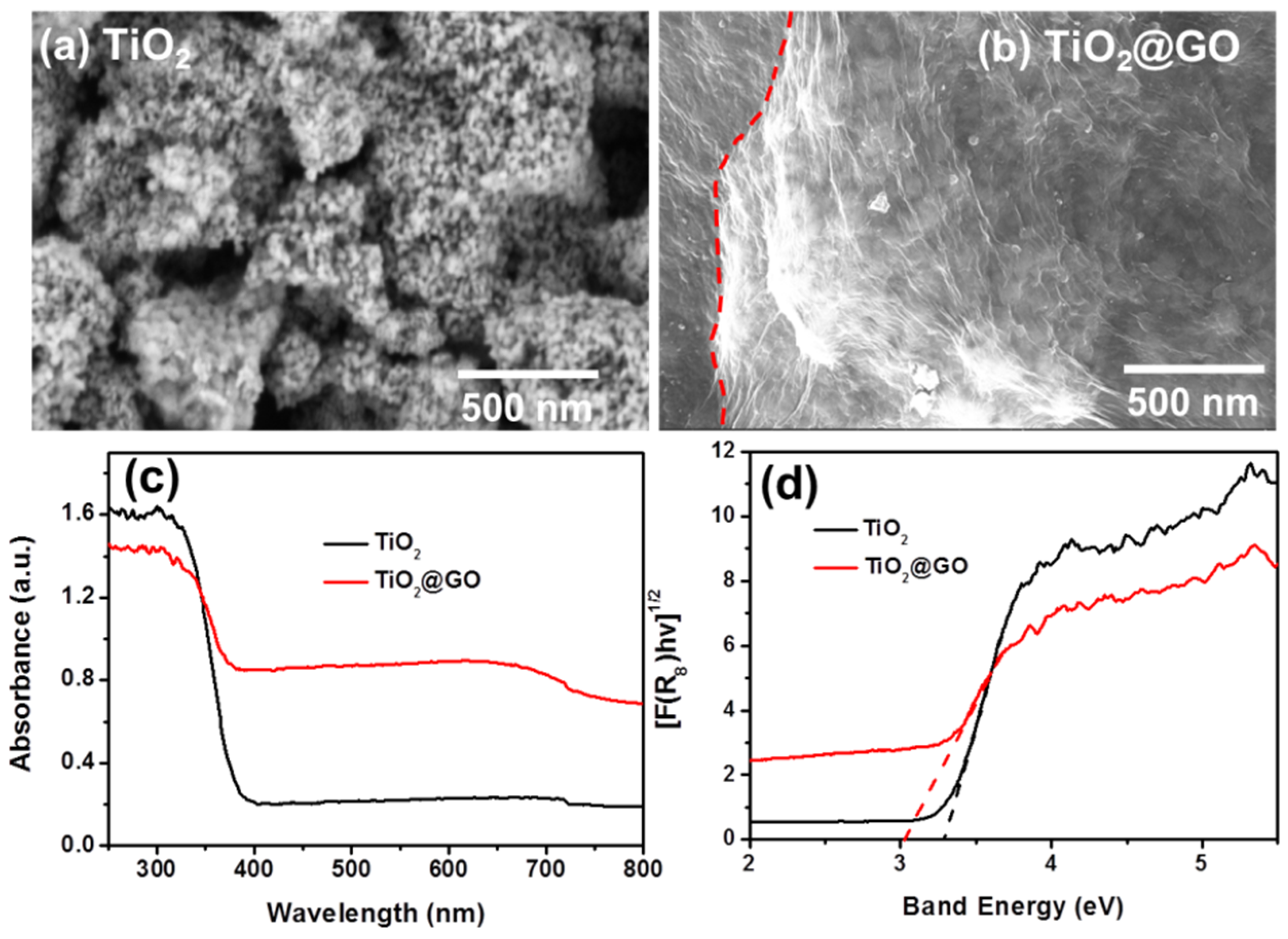
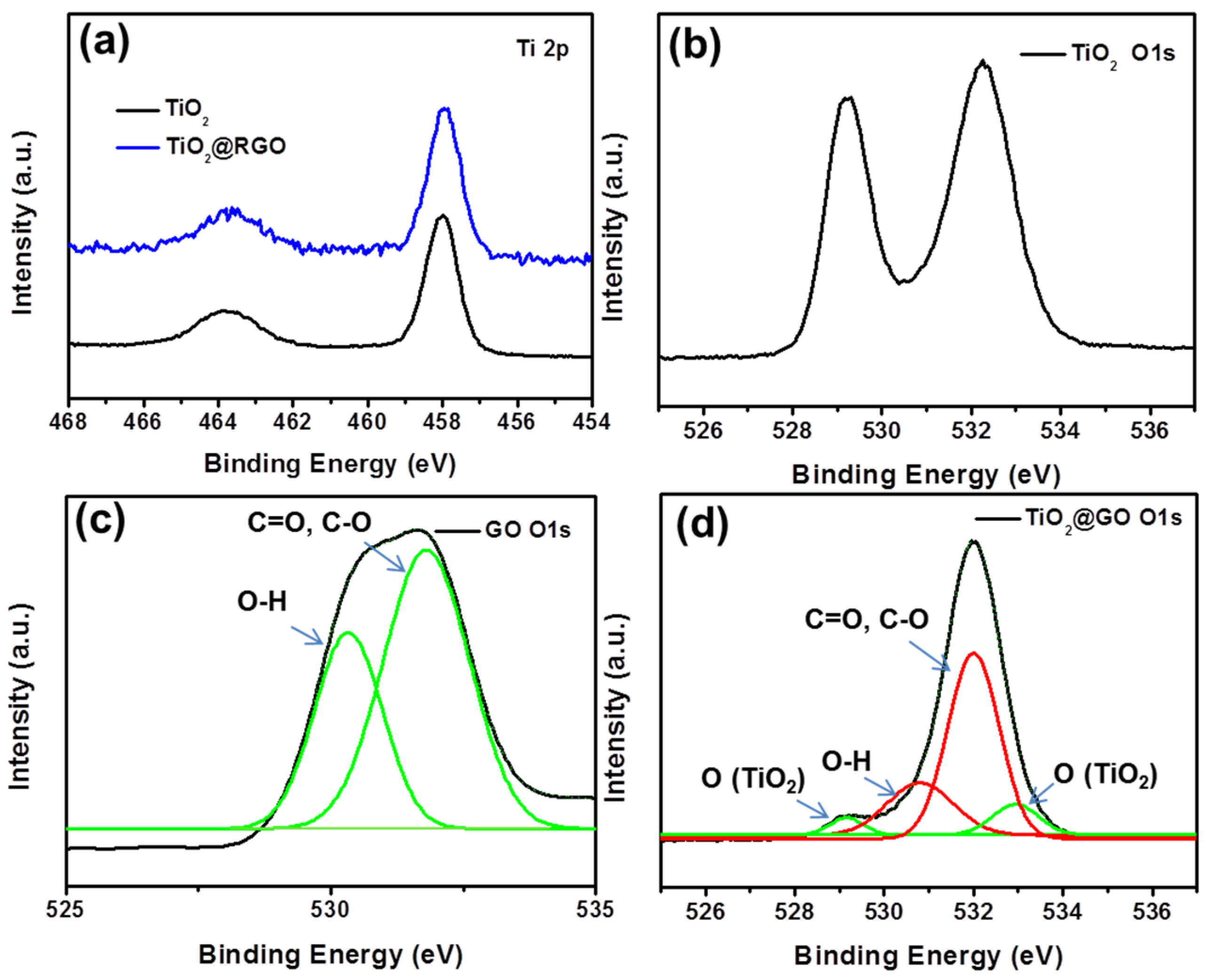
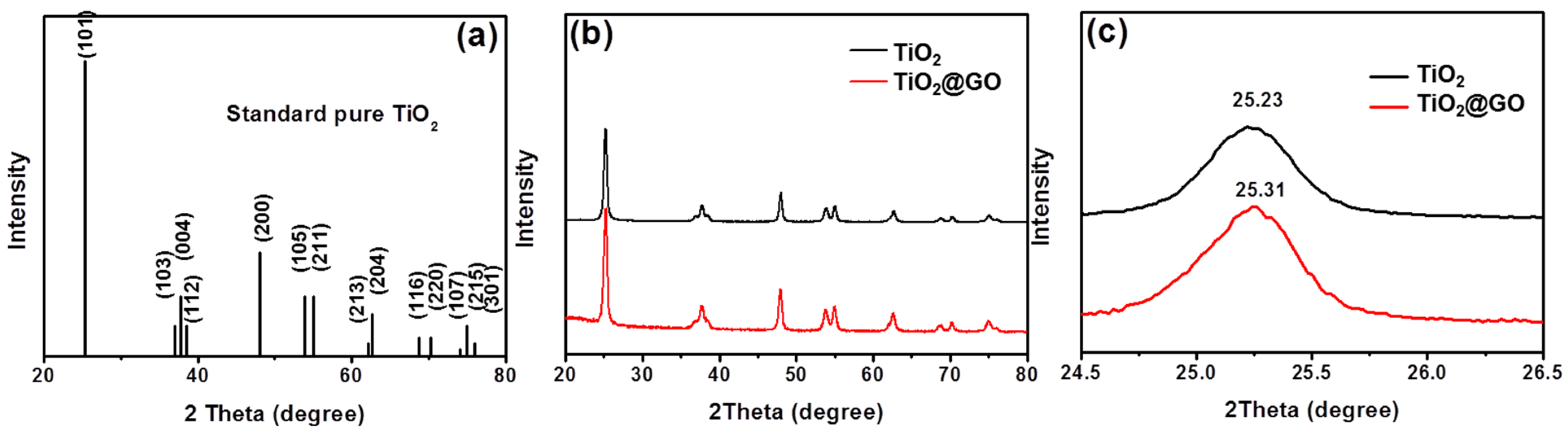
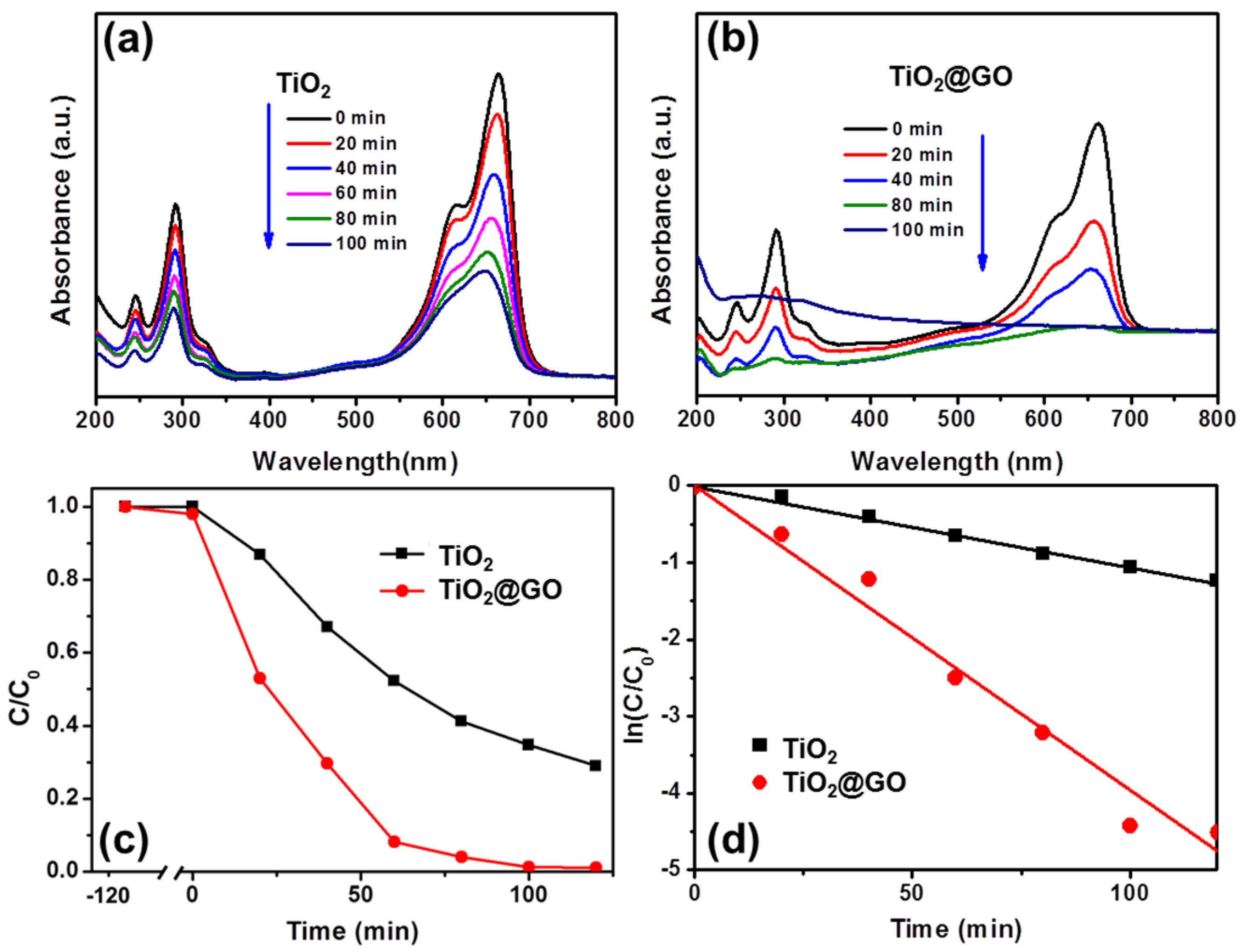
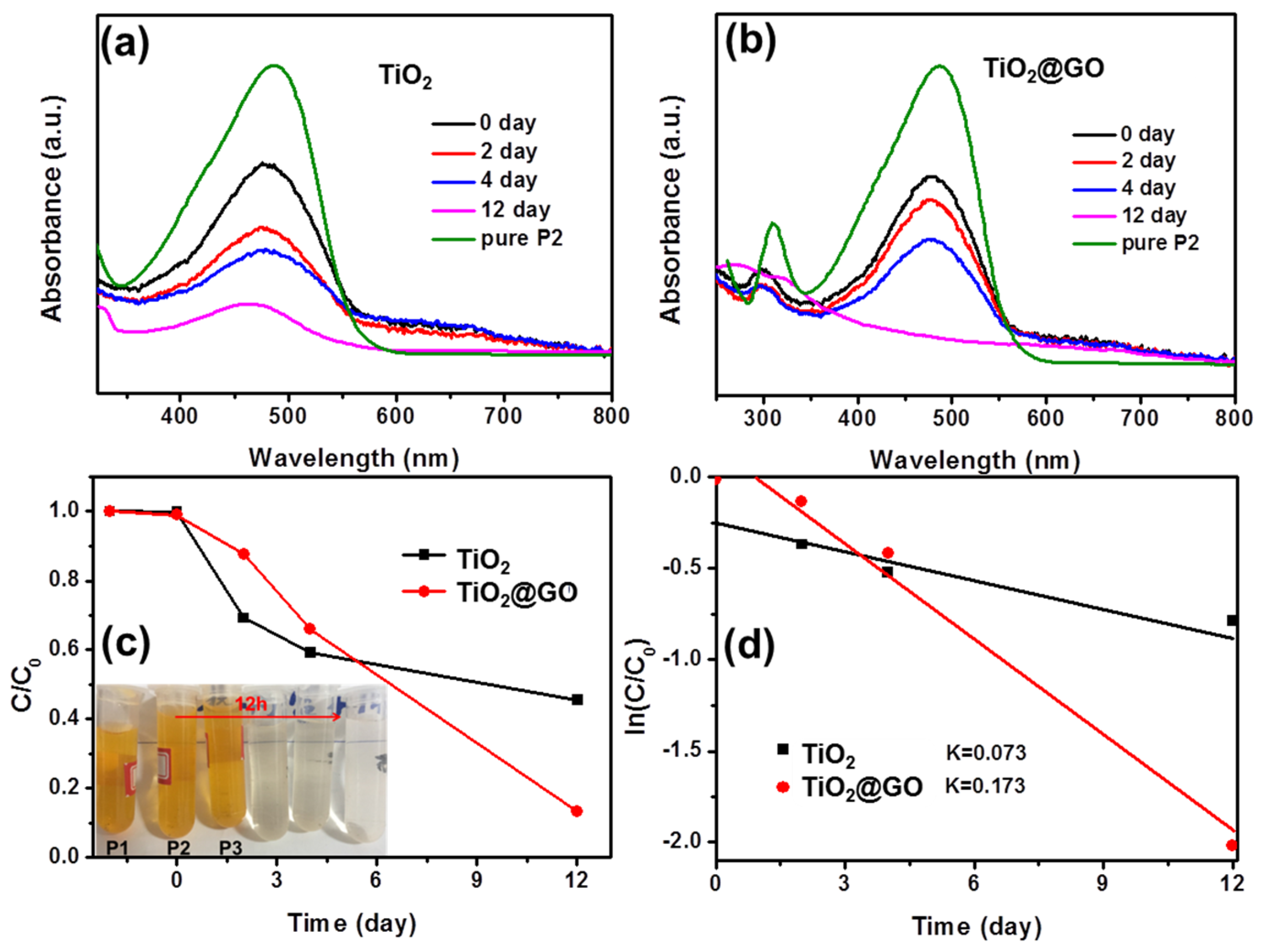
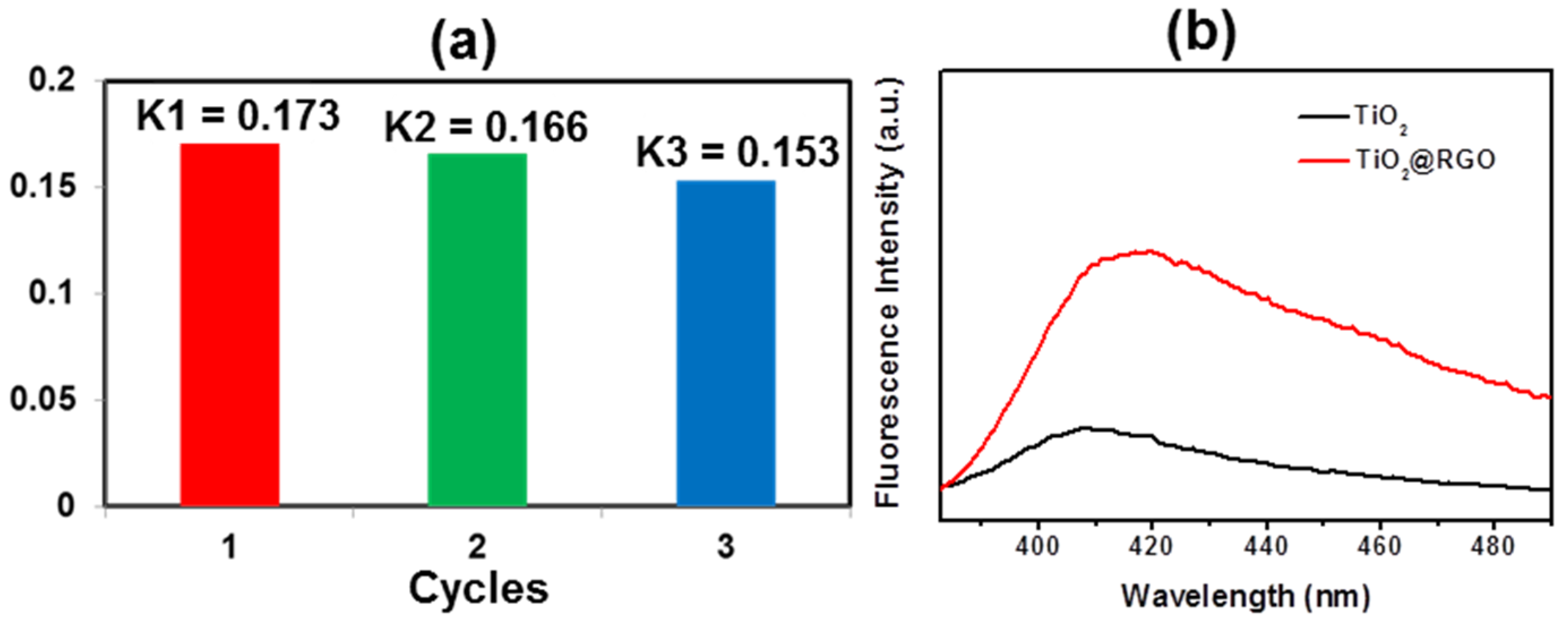
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Garcia, J.M.; Robertson, M.L. The future of plastics recycling. Science 2017, 358, 870–872. [Google Scholar] [CrossRef] [PubMed]
- Uekert, T.; Kasap, H.; Reisner, E. Photoreforming of nonrecyclable plastic waste over a carbon nitride/nickel phosphide catalyst. J. Am. Chem. Soc. 2019, 141, 15201–15210. [Google Scholar] [CrossRef] [Green Version]
- Seh, Z.W.; Kibsgaard, J.; Dickens, C.F.; Chorkendorff, I.B.; Nørskov, J.K.; Jaramillo, T.F. Combining theory and experiment in electrocatalysis: Insights into materials design. Science 2017, 355, 4998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eagan, J.M.; Xu, J.; Di Girolamo, R.; Thurber, C.M.; Macosko, C.W.; LaPointe, A.M.; Bates, F.S.; Coates, G.W. Combining polyethylene and polypropylene: Enhanced performance with PE/iPP multiblock polymers. Science 2017, 355, 814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niether, C.; Faure, S.; Bordet, A.; Deseure, J.; Chatenet, M.; Carrey, J.; Chaudret, B.; Rouet, A. Improved water electrolysis using magnetic heating of FeC-Ni core-shell nanoparticles. Nat. Energy 2018, 3, 476. [Google Scholar] [CrossRef]
- Wu, J.; Zhang, Q.; Wang, J.; Huang, X.; Bai, H. A self-assembly route to porous polyaniline/reduced graphene oxide composite materials with molecular-level uniformity for high-performance supercapacitors. Energy Environ. Sci. 2018, 11, 1280. [Google Scholar] [CrossRef]
- Geyer, R.; Jambeck, J.R.; Law, K.L. Production, use, and fate of all plastics ever made. Sci. Adv. 2017, 3, e1700782. [Google Scholar] [CrossRef] [Green Version]
- Wakerley, D.W.; Kuehnel, M.F.; Orchard, K.L.; Ly, K.H.; Rosser, T.E.; Reisner, E. Solar-driven reforming of lignocellulose to H2 with a CdS/CdOx photocatalyst. Nat. Energy 2017, 2, 17021. [Google Scholar] [CrossRef] [Green Version]
- Law, K.L.; Thompson, R.C. Microplastics in the seas. Science 2014, 345, 144–145. [Google Scholar] [CrossRef]
- Kuehnel, M.F.; Reisner, E. Solar hydrogen generation from lignocellulose. Angew. Chem. Int. Ed. 2018, 57, 3290–3296. [Google Scholar] [CrossRef]
- Lin, L.; Yu, Z.; Wang, X. Crystalline carbon nitride semiconductors for photocatalytic water splitting. Angew. Chem. Int. Ed. 2019, 58, 6164–6175. [Google Scholar] [CrossRef]
- Zhang, J.; Fu, W.; Xi, J.; He, H.; Zhao, S.; Lu, H.; Ji, Z. N-doped rutile TiO2 nano-rods show tunable photocatalytic selectivity. Alloys Comp. 2013, 575, 40–47. [Google Scholar] [CrossRef]
- Zhang, J.; Qian, L.; Fu, W.; Xi, J.; Ji, Z. Alkaline-Earth Metal Ca and N Codoped TiO2 with Exposed {001} Facets for Enhancing Visible Light Photocatalytic Activity. J. Am. Ceram. Soc. 2014, 97, 2615–2622. [Google Scholar] [CrossRef]
- Yuan, K.; Lu, C.; Sfaelou, S.; Liao, X.; Zhuang, X.; Chen, Y.; Scherf, U.; Feng, X. In situ nanoarchitecturing and active-site engineering toward highly efficient carbonaceous electrocatalysts. Nano Energy 2019, 59, 207–215. [Google Scholar] [CrossRef]
- Rabady, R.I. Solar spectrum management for effective hydrogen production by hybrid thermo-photovoltaic water electrolysis. Int. J. Hydrogen. Energy 2014, 39, 6827–6836. [Google Scholar] [CrossRef]
- Shi, Y.; Kong, Y.; Song, L.; Zhang, J.; Ji, Z.; Ge, Z. Synthesis and characterization of polyelectrolytes based on benzotriazole backbone. Colloid Polym. Sci. 2018, 296, 1–9. [Google Scholar] [CrossRef]
- Shi, Y.; Mai, C.K.; Fronk, S.L.; Chen, Y.; Bazan, G.C. Optical properties of benzotriazole-based conjugated polyelectrolytes. Macromolecules 2016, 49, 6343–6349. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Zhang, Q.; Zhou, A.; Huang, Z.; Bai, H.; Li, L. Phase-separated polyaniline/graphene composite electrodes for high-rate electrochemical supercapacitors. Adv. Mater. 2016, 28, 10211–10216. [Google Scholar] [CrossRef]
- Zhang, J.; Xi, J.; Ji, Z. Mo plus N codoped TiO2 sheets with dominant {001} facets for enhancing visible-light photocatalytic activity. J. Mater. Chem. 2012, 22, 17700–17708. [Google Scholar] [CrossRef]
- Yadav, H.M.; Kim, J.S. Solvothermal synthesis of anatase TiO2-graphene oxide nanocomposites and their photocatalytic performance. J. Alloy. Compd. 2016, 688, 123–129. [Google Scholar] [CrossRef]
- Wei, Q.; Pei, S.; Qian, X.; Liu, H.; Liu, Z.; Zhang, W.; Zhou, T.; Zhang, Z.; Zhang, X.; Cheng, H.M.; et al. Superhigh electromagnetic interference shielding of ultrathin aligned pristine graphene nanosheets film. Adv. Mater. 2020, 32, 1907411. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.H.; Li, J.G.; Kamiyama, H.; Moriyoshi, Y.; Ishigaki, T. Wavelength-sensitive photocatalytic degradation of methyl orange in aqueous suspension over iron(III)-doped TiO2 nanopowders under UV and visible light irradiation. J. Phys. Chem. B 2006, 110, 6804–6812. [Google Scholar] [CrossRef]
- Jiao, X.; Zheng, K.; Chen, Q.; Li, X.; Li, Y.; Shao, W.; Xu, J.; Zhu, J.; Pan, Y.; Sun, Y.; et al. Photocatalytic conversion of Waste Plastics into C2 Fuels under Simulated Natural Environment Conditions. Angew. Chem. Int. Ed. 2020, 59, 15497–15501. [Google Scholar] [CrossRef] [PubMed]
- Fujishima, A.; Zhang, X.; Tryk, D.A. TiO2 photocatalysis and related surface phenomena. Surf. Sci. Rep. 2008, 63, 515–582. [Google Scholar] [CrossRef]
- Williams, G.; Seger, B.; Kamat, P.V. TiO2-graphene nanocomposites. UV-assisted photocatalytic reduction of graphene oxide. ACS Nano 2008, 2, 1487–1491. [Google Scholar]
- Kamat, P.V. Photochemistry on nonreactive and reactive (semiconductor) surfaces. Chem. Rev. 1993, 93, 267–300. [Google Scholar] [CrossRef]
- Assayehegn, E.; Solaiappan, A.; Chebude, Y.; Alemayehu, E. Fabrication of tunable anatase/rutile heterojunction N/TiO2 nanophotocatalyst for enhanced visible light degradation activity. Appl. Surf. Sci. 2020, 515, 145966. [Google Scholar] [CrossRef]
- Elsellami, L.; Dappozze, F.; Fessi, N.; Houas, A.; Guillard, C. Highly photocatalytic activity of nanocrystalline TiO2 (anatase, rutile) powders prepared from TiCl4 by sol-gel method in aqueous solutions. Process Saf. Environ. Prot. 2018, 113, 109–121. [Google Scholar] [CrossRef]
- Sun, S.; Gao, P.; Yang, Y.; Yang, P.; Chen, Y.; Wang, Y. N-Doped TiO2 nanobelts with coexposed (001) and (101) facets and their highly efficient visible-light-driven photocatalytic hydrogen production. ACS Appl. Mater. Interfaces 2016, 8, 18126–18156. [Google Scholar] [CrossRef]
- Zhang, R.; Du, B.; Yin, L.; Miao, Y.; Cai, J.; Feng, G.; Wang, X. Molybdenum-doped ZnS sheets with dominant {111} facets for enhanced visible light photocatalytic activities. J. Colloid Interf. Sci. 2017, 507, 200–208. [Google Scholar] [CrossRef]
- Guo, F.; Shi, W.; Zhu, C.; Li, H.; Kang, Z. CoO and g-C3N4 complement each other for highly efficient overall water splitting under visible light. Appl. Catal. B Environ. 2018, 226, 412–420. [Google Scholar] [CrossRef]
- Sun, S.; Watanabe, M.; Wu, J.; An, Q.; Ishihara, T. Ultrathin WO3·0.33 H2O nanotubes for CO2 photoreduction to acetate with high selectivity. J. Am. Chem. Soc. 2018, 140, 6474–6482. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, Y.; Yu, Z.; Li, Z.; Zhao, X.; Yuan, Y. In-Situ Synthesis of TiO2@GO Nanosheets for Polymers Degradation in a Natural Environment. Polymers 2021, 13, 2158. https://doi.org/10.3390/polym13132158
Shi Y, Yu Z, Li Z, Zhao X, Yuan Y. In-Situ Synthesis of TiO2@GO Nanosheets for Polymers Degradation in a Natural Environment. Polymers. 2021; 13(13):2158. https://doi.org/10.3390/polym13132158
Chicago/Turabian StyleShi, Yueqin, Zhanyang Yu, Zhengjun Li, Xiaodong Zhao, and Yongjun Yuan. 2021. "In-Situ Synthesis of TiO2@GO Nanosheets for Polymers Degradation in a Natural Environment" Polymers 13, no. 13: 2158. https://doi.org/10.3390/polym13132158
APA StyleShi, Y., Yu, Z., Li, Z., Zhao, X., & Yuan, Y. (2021). In-Situ Synthesis of TiO2@GO Nanosheets for Polymers Degradation in a Natural Environment. Polymers, 13(13), 2158. https://doi.org/10.3390/polym13132158