Effect of Radiation Crosslinking and Surface Modification of Cellulose Fibers on Properties and Characterization of Biopolymer Composites




Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Biopolymer Composites
2.3. Methods
2.3.1. Uniaxial Tensile Testing
2.3.2. Flexural Testing
2.3.3. Charpy Impact
2.3.4. Scanning Electron Microscopy (SEM)
3. Results and Discussion
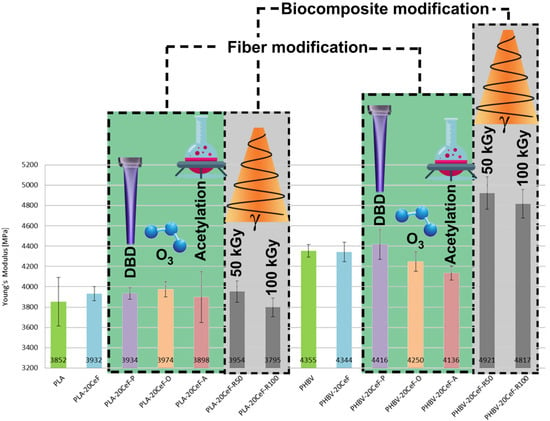
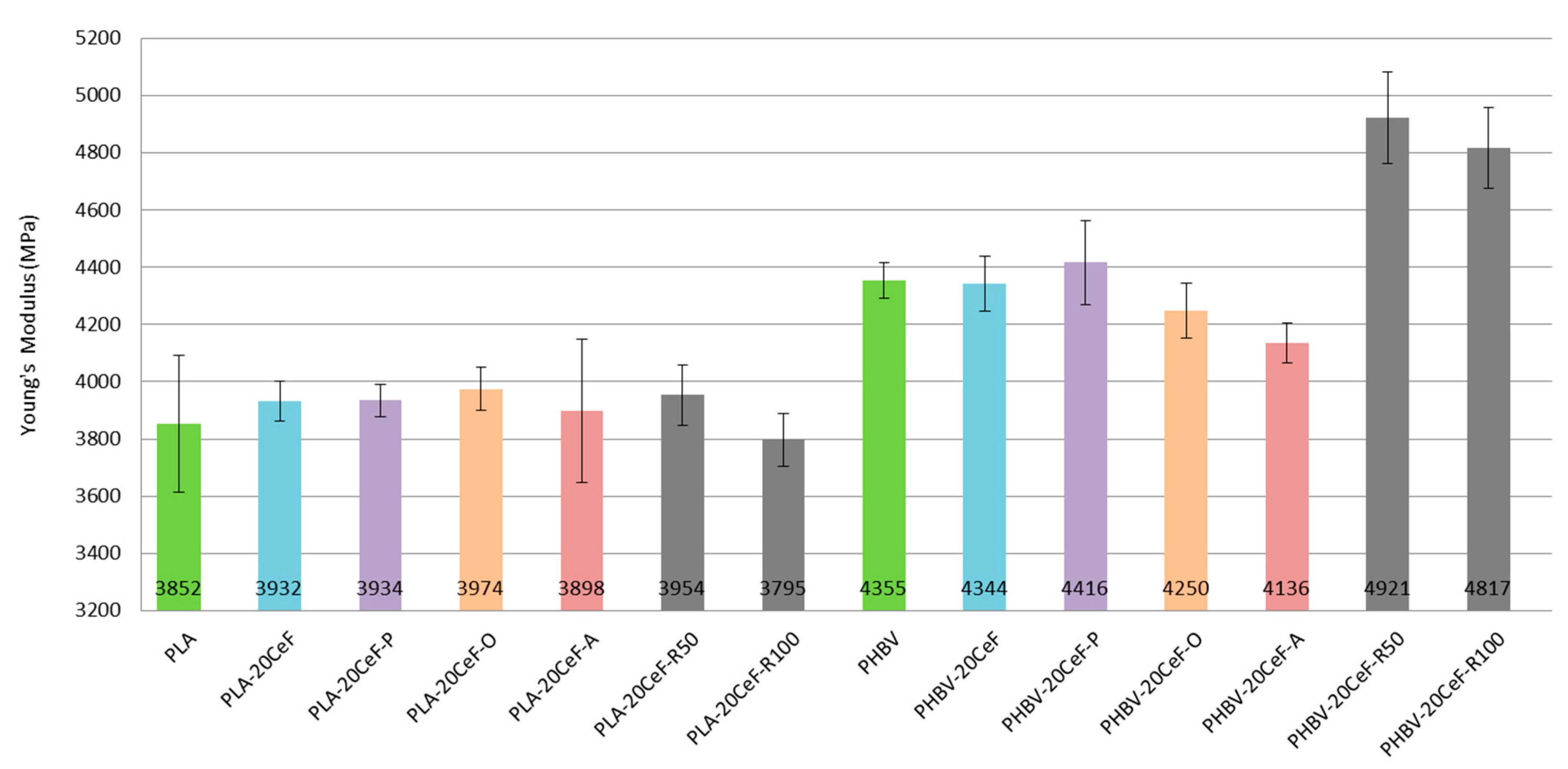
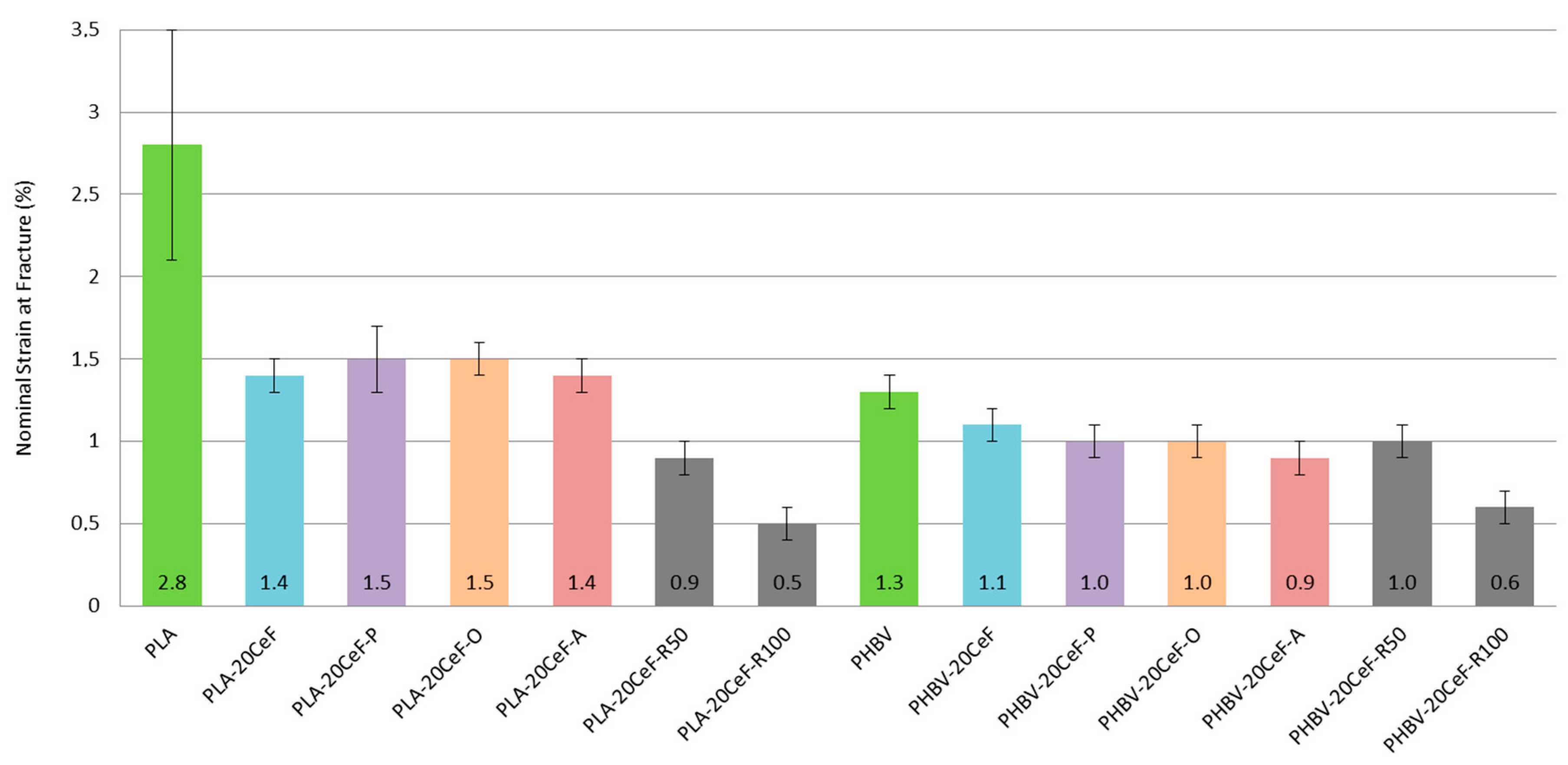
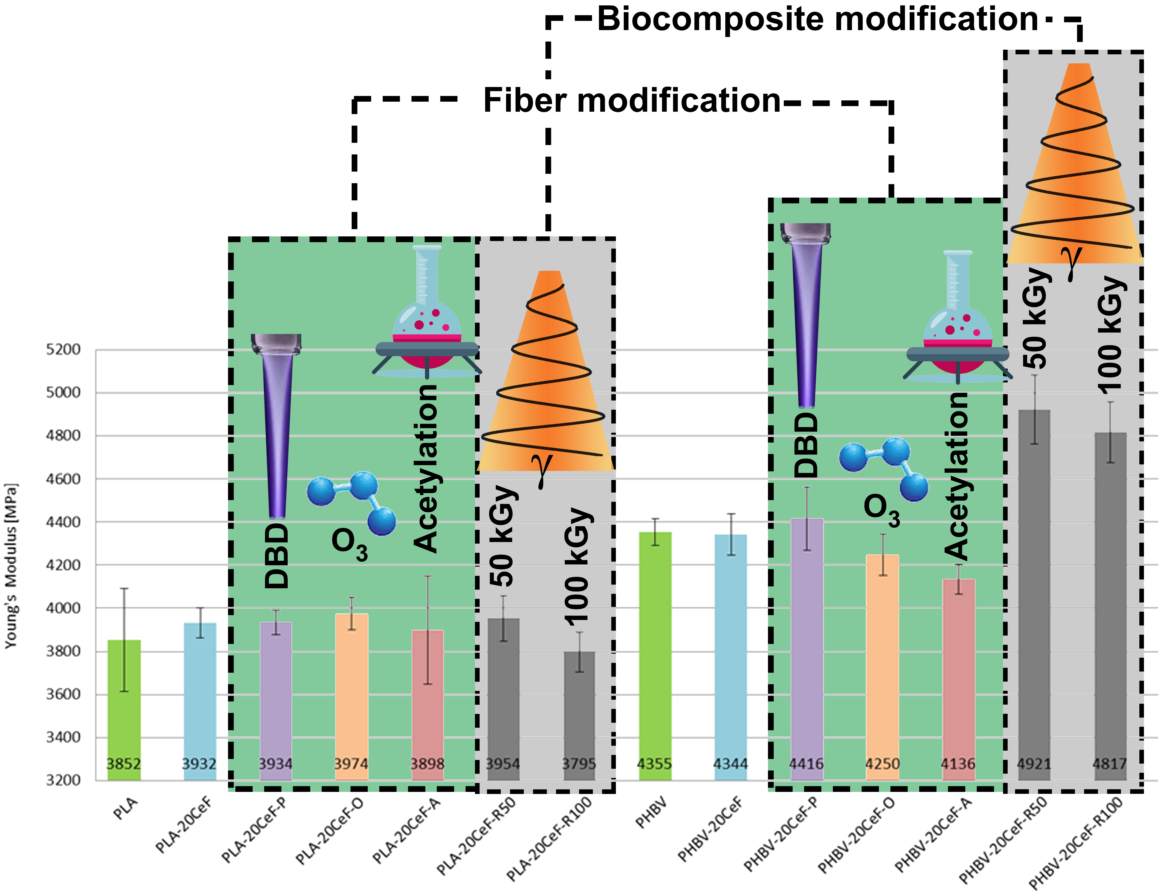
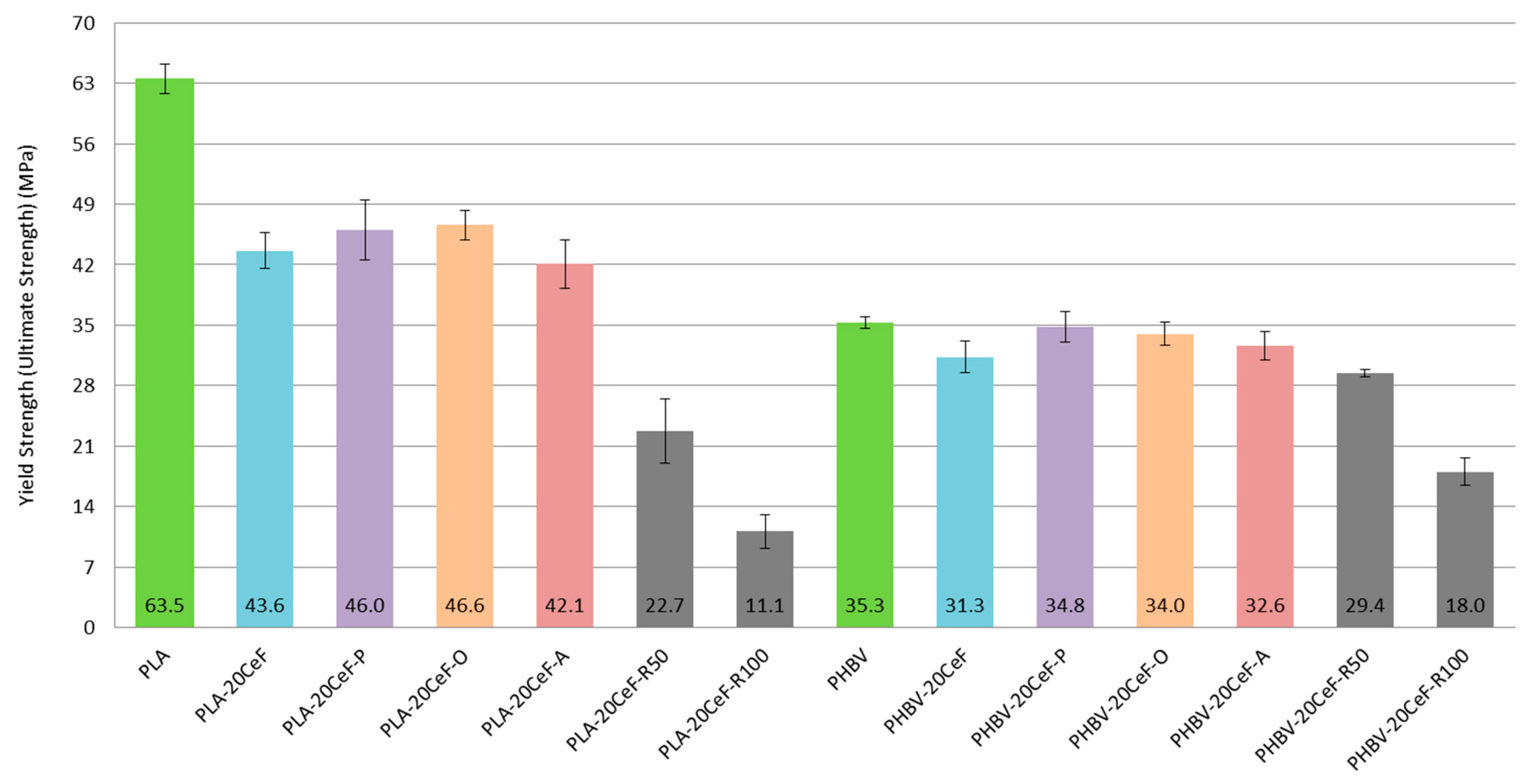
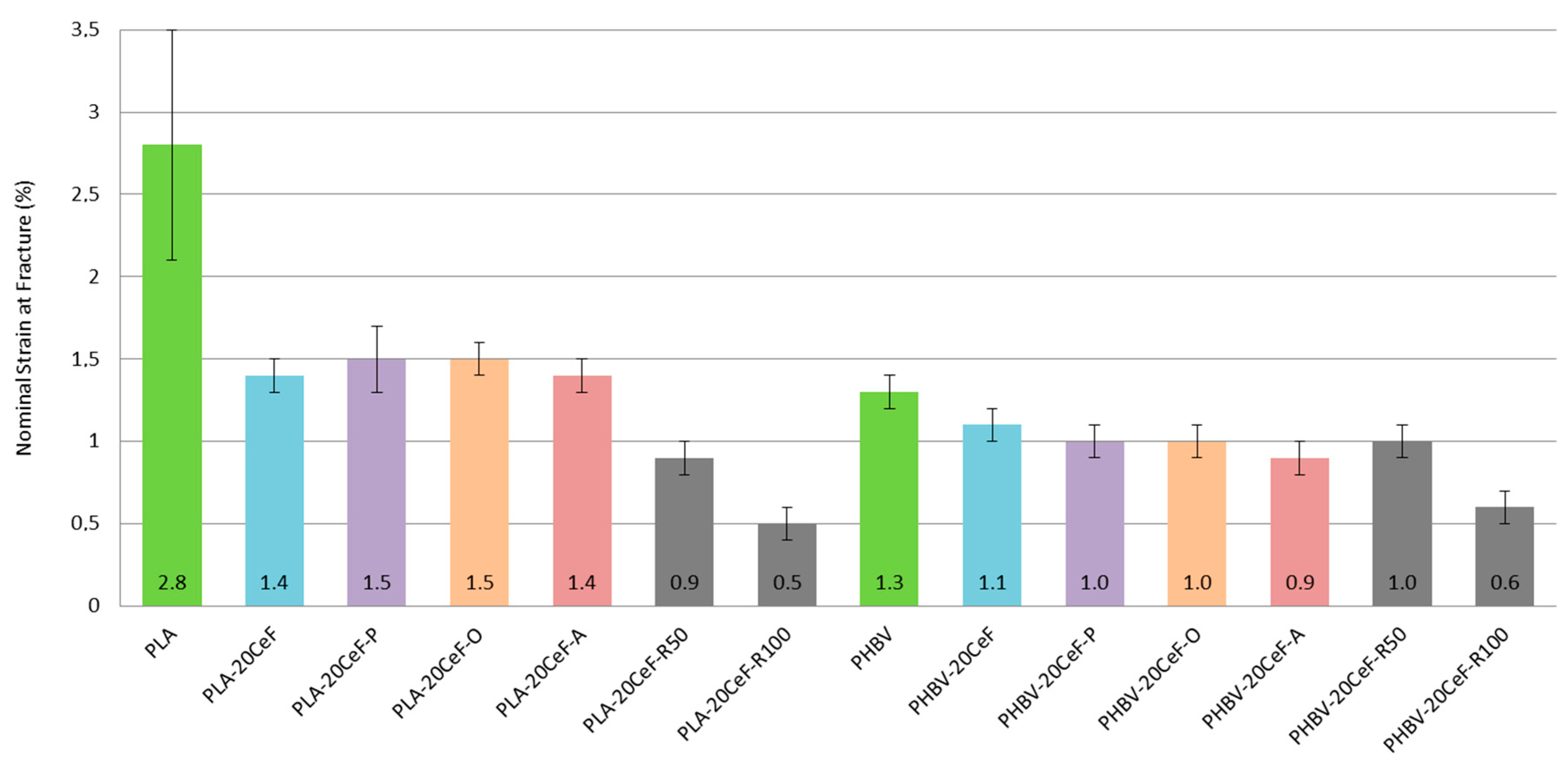
3.1. Tensile Properties
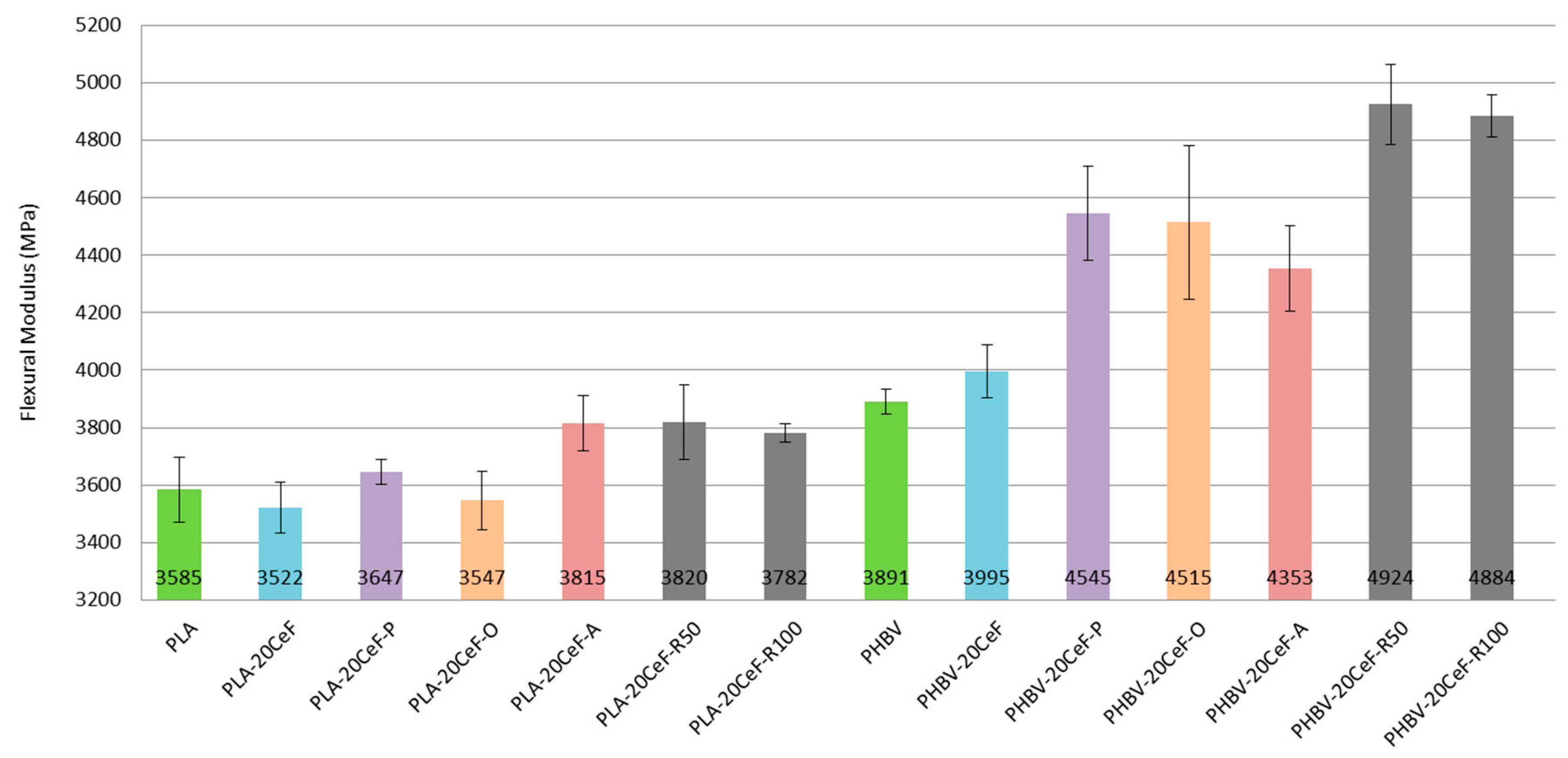
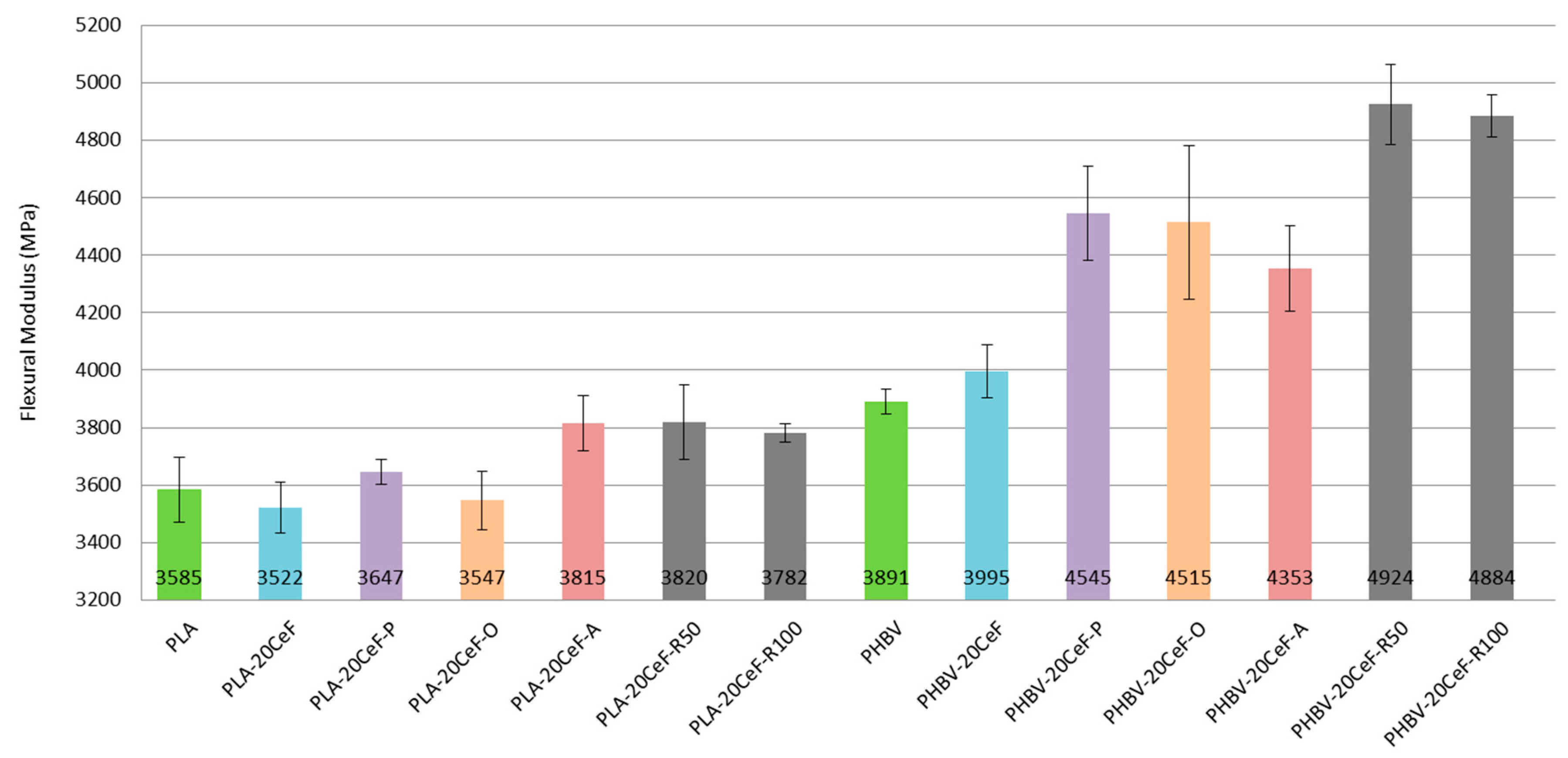
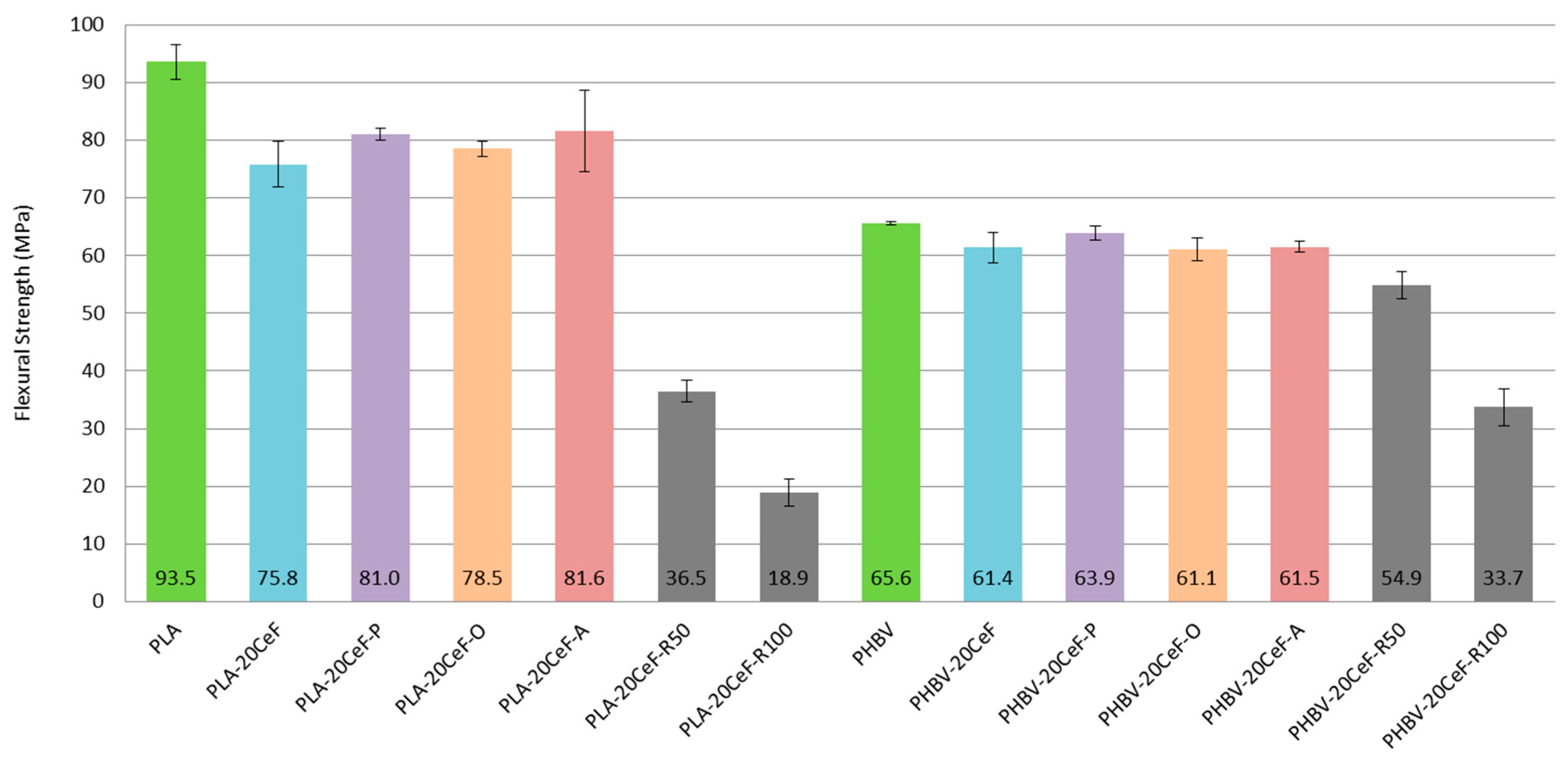
3.2. Flexural Properties
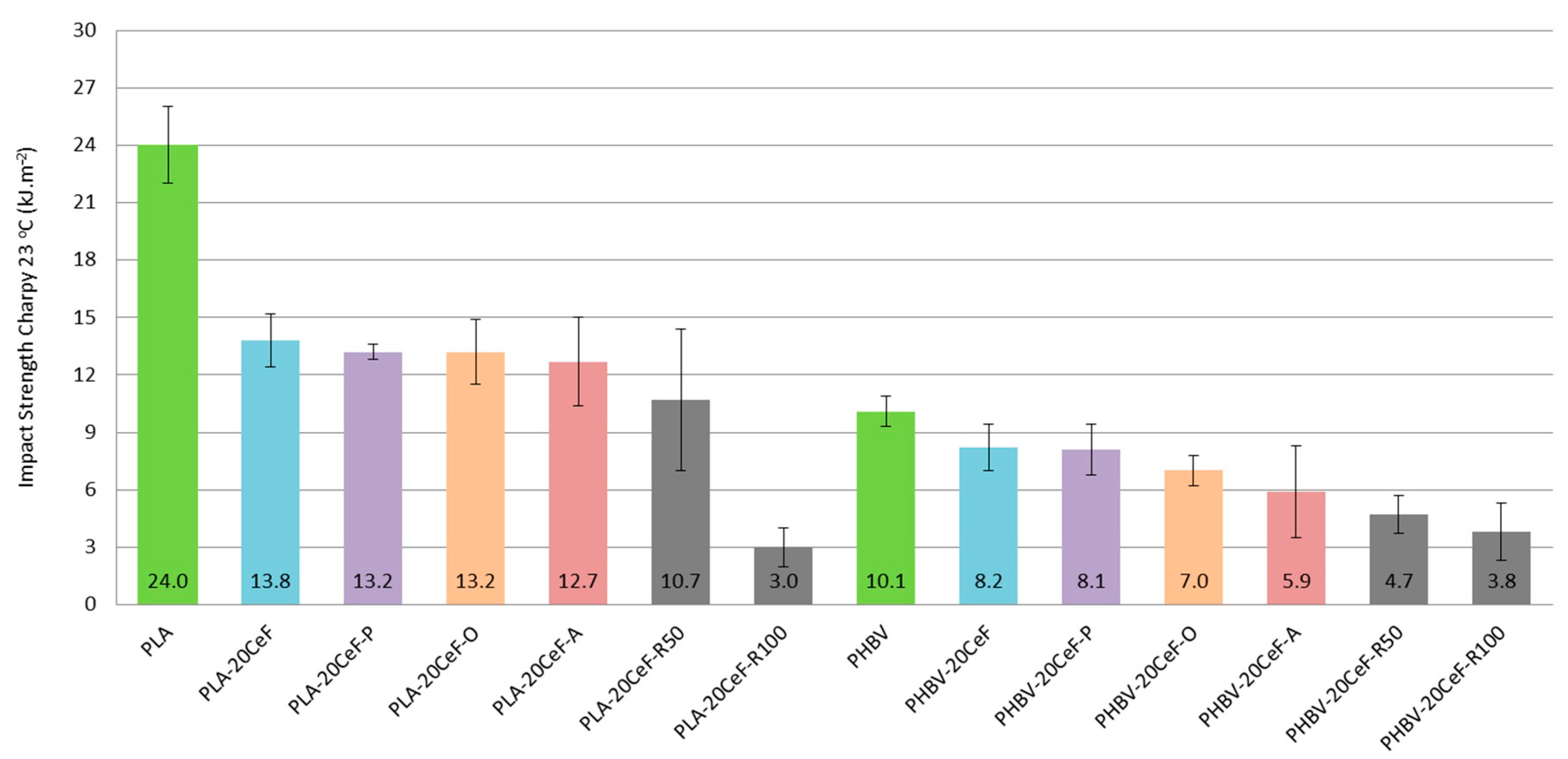
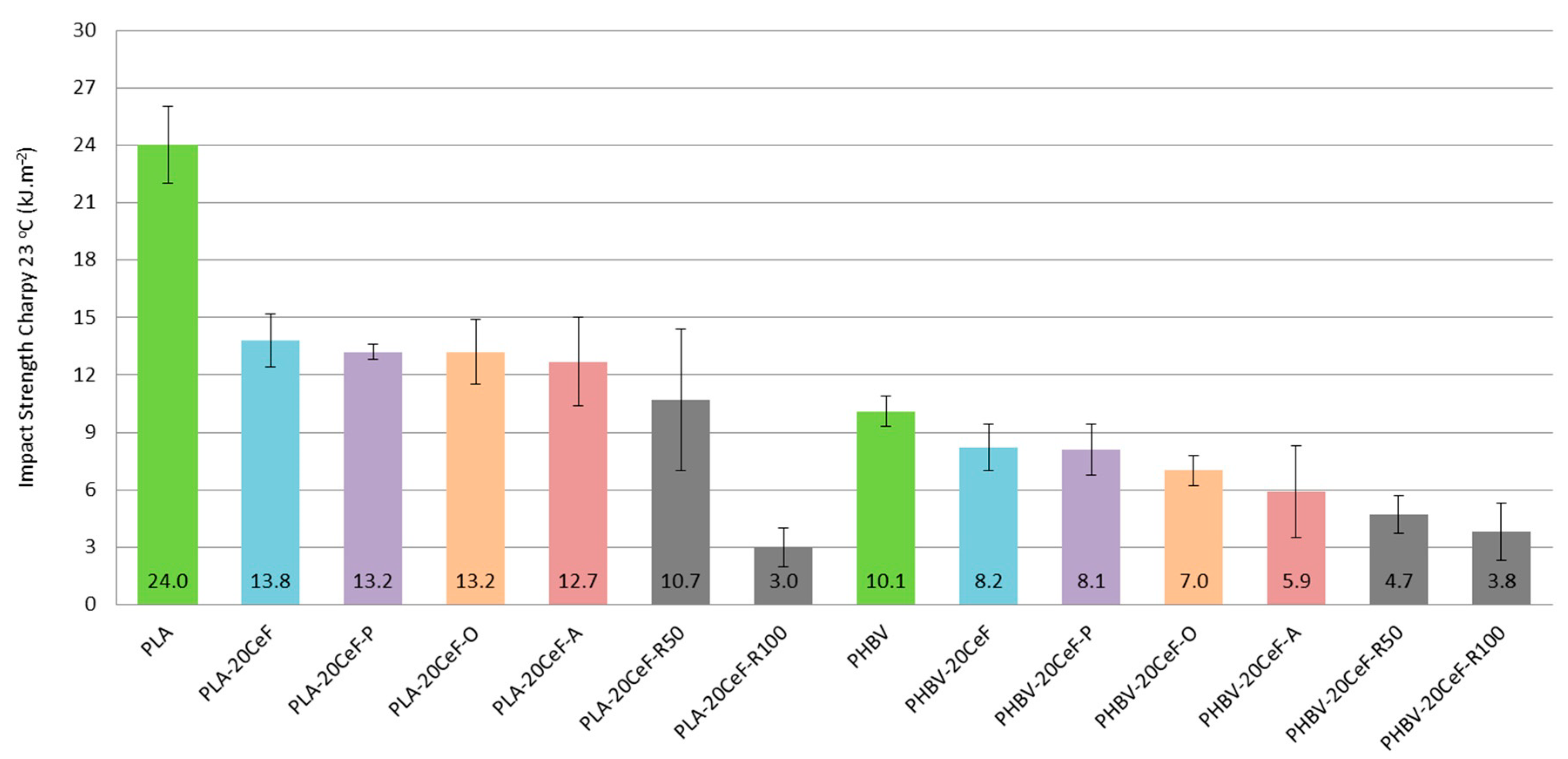
3.3. Charpy Impact Properties
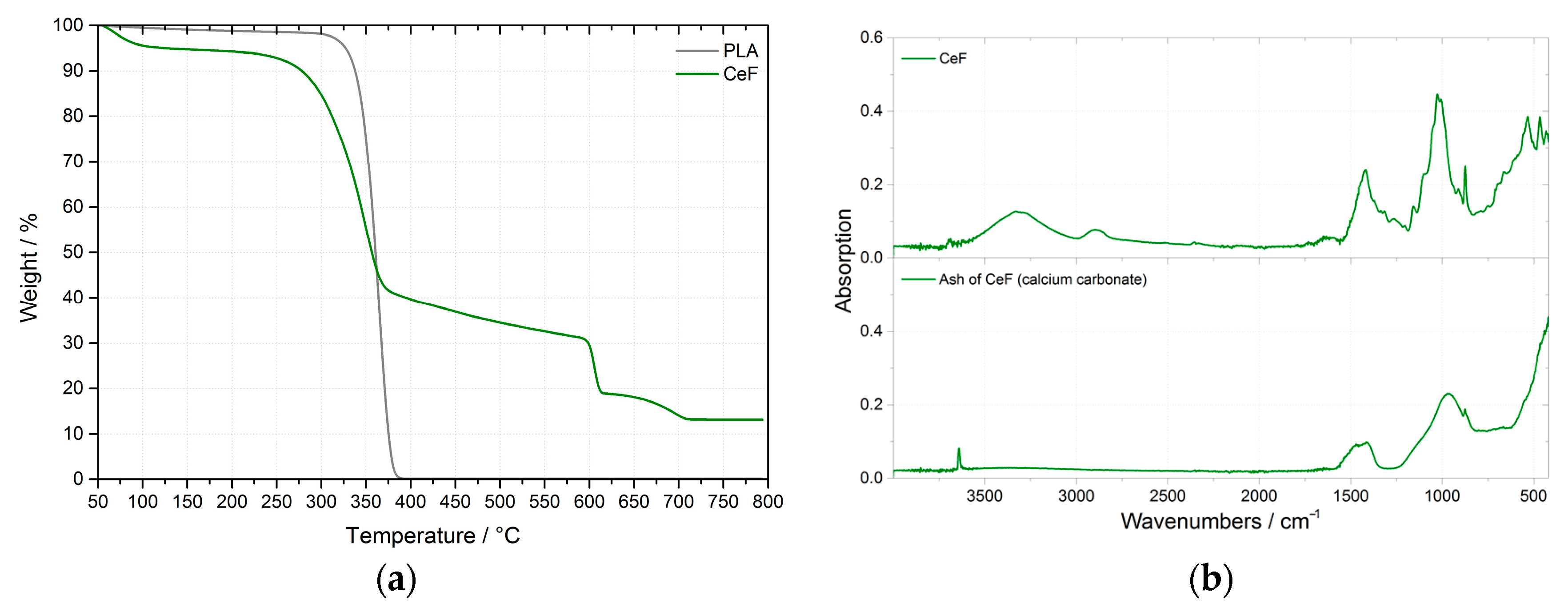
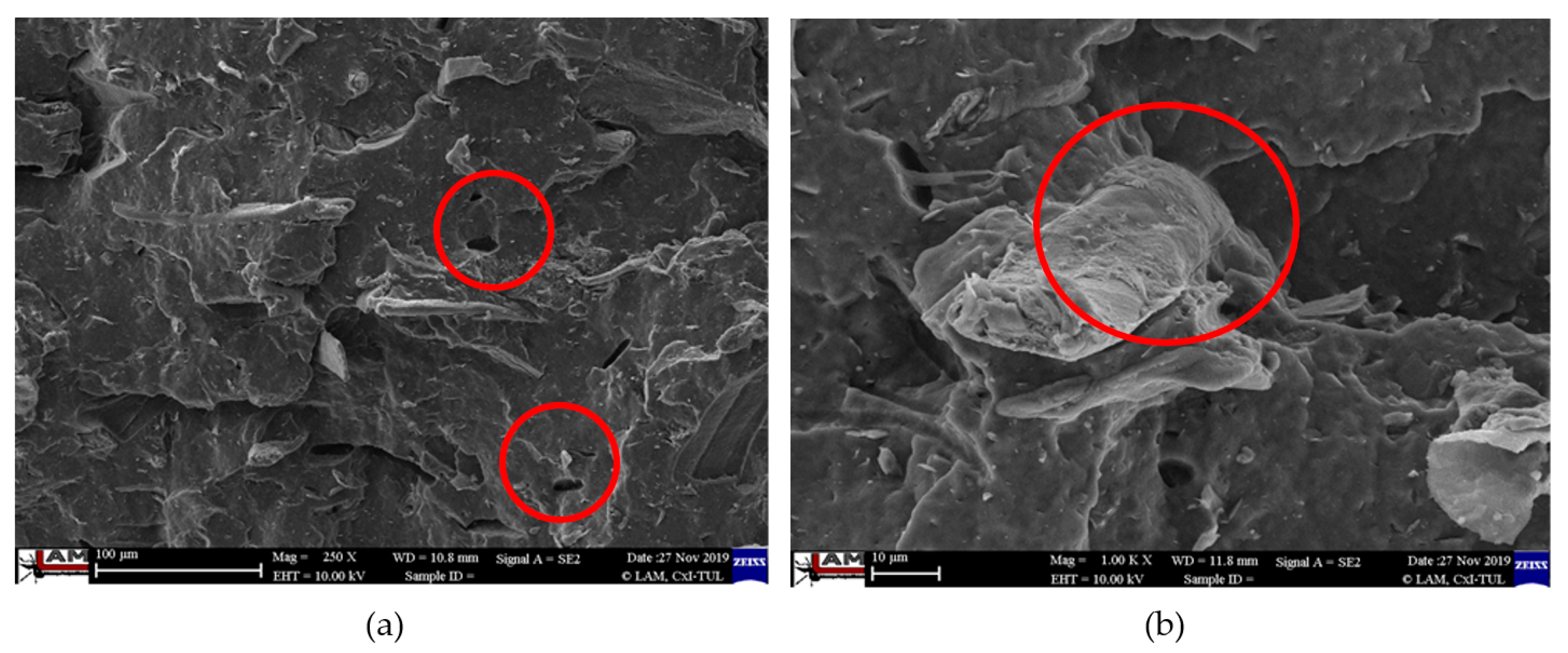
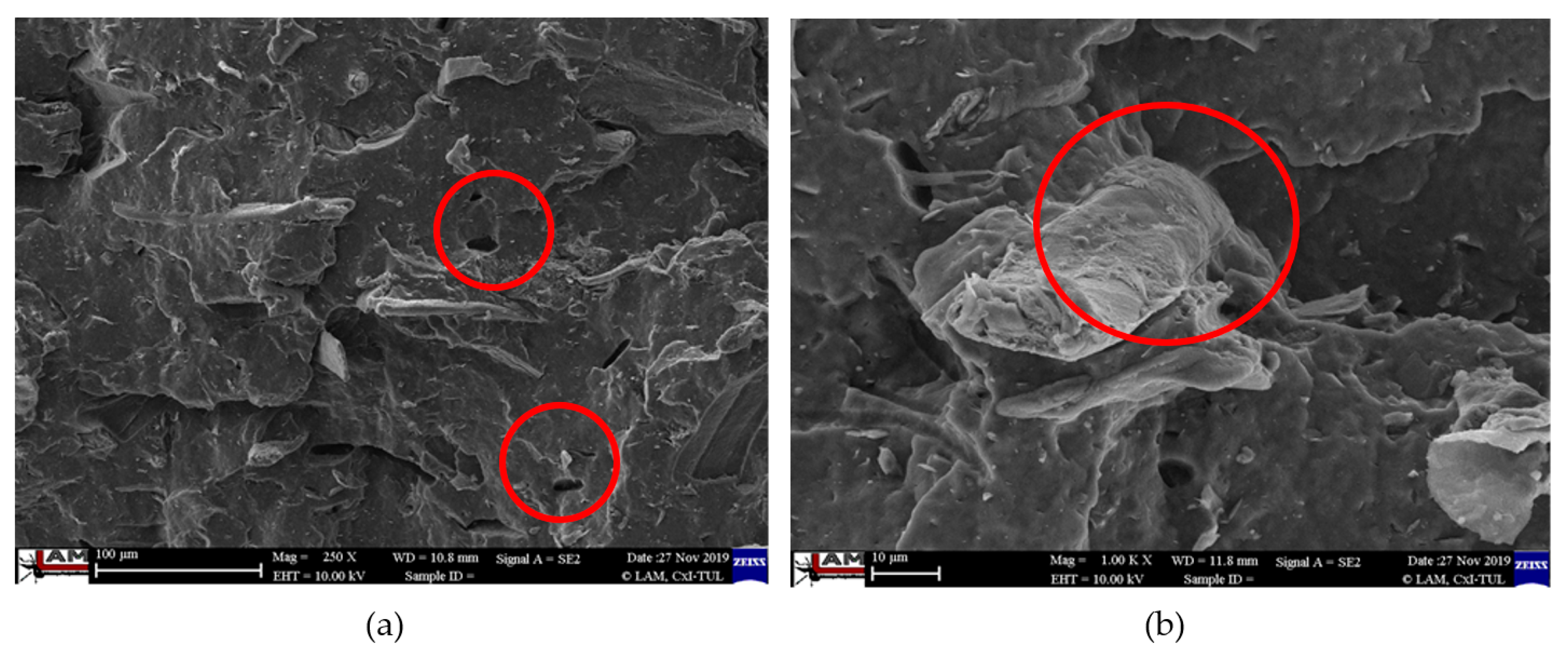
3.4. Characterization of Failure and Microscopy of Fracture Surfaces
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kalia, S.; Dufresne, A.; Cherian, B.M.; Kaith, B.S.; Avérous, L.; Njuguna, J.; Nassiopoulos, E.; Leiza, J.R. Cellulose-Based Bio- and Nanocomposites. Int. J. Polym. Sci. 2011, 2011, 1–35. [Google Scholar] [CrossRef]
- Ganapini, W. Bioplastics: A Case Study of Bioeconomy in Italy; Edizioni Ambiente: Milano, Italy, 2014. [Google Scholar]
- Watkins, E.; Schweitzer, J.-P. Moving Towards a Circular Economy for Plastics in the EU by 2030. Think 2030; Institute for European Environmental Policy (IEEP): Brussels, Belgium, 2018. [Google Scholar]
- La Mantia, F.P.; Morreale, M. Green composites: A brief review. Compos. Part A Appl. Sci. Manuf. 2011, 42, 579–588. [Google Scholar] [CrossRef]
- Marsh, G. Next step for automotive materials. Mater. Today 2003, 4, 36–43. [Google Scholar] [CrossRef]
- Oliveux, G.; Dandy, L.O.; Leeke, G.A. Current status of recycling of fibre reinforced polymers: Review of technologies, reuse and resulting properties. Prog. Mater. Sci. 2015, 72, 61–99. [Google Scholar] [CrossRef] [Green Version]
- Rybicka, J.; Tiwari, A.; Leeke, G.A. Technology readiness level assessment of composites recycling technologies. J. Clean. Prod. 2016, 112, 1001–1012. [Google Scholar] [CrossRef] [Green Version]
- Mohanty, A.K.; Misra, M.; Hinrichsen, G.I. Biofibres, biodegradable polymers and biocomposites: An overview. Macromol. Mater. Eng. 2000, 276, 1–24. [Google Scholar] [CrossRef]
- Netravali, A.N.; Chabba, S. Composites get greener. Mater. Today 2003, 4, 22–29. [Google Scholar] [CrossRef]
- Jimenez, A.; Peltzer, M.; Ruseckaite, R. Poly (Lactic Acid) Science and Technology: Processing, Properties, Additives and Applications; Royal Society of Chemistry: London, UK, 2014. [Google Scholar]
- Pei, A.; Zhou, Q.; Berglund, L.A. Functionalized cellulose nanocrystals as biobased nucleation agents in poly(l-lactide) (PLLA)—Crystallization and mechanical property effects. Compos. Sci. Technol. 2010, 70, 815–821. [Google Scholar] [CrossRef]
- Nanda, M.R.; Manjusri, M.; Mohanty, A.K. The effects of process engineering on the performance of PLA and PHBV blends. Macromol. Mater. Eng. 2011, 296, 719–728. [Google Scholar] [CrossRef]
- Boufarguine, M.; Guinault, A.; Miquelard-Garnier, G.; Sollogoub, C. PLA/PHBV films with improved mechanical and gas barrier properties. Macromol. Mater. Eng. 2013, 298, 1065–1073. [Google Scholar] [CrossRef] [Green Version]
- Khanna, S.; Srivastava, A.K. Recent advances in microbial polyhydroxyalkanoates. Process Biochem. 2005, 40, 607–619. [Google Scholar] [CrossRef]
- Mwaikambo, L.Y. Review of the history and application of plant fibre. Afr. J. Sci. Technol. 2006, 7, 120–133. [Google Scholar]
- Mochane, M.J.; Mokhena, T.C.; Sadiku, E.R.; Ray, S.S.; Mofokeng, T.G. Green Polymer Composites Based on Polylactic Acid (PLA) and Fibers. Green Biopolymers and their Nanocomposites; Springer: Singapore, 2019; pp. 29–54. [Google Scholar]
- Mwaikambo, L.Y.; Ansell, M.P. Chemical modification of hemp, sisal, jute, and kapok fibers by alkalization. J. Appl. Polym. Sci. 2002, 84, 2222–2234. [Google Scholar] [CrossRef]
- Kalia, S.; Kaith, B.S.; Kaur, I. Cellulose Fibers: Bio-and Nano-Polymer Composites: Green Chemistry and Technology; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2011. [Google Scholar] [CrossRef]
- Mehta, G.; Drzal, L.T.; Mohanty, A.K.; Misra, M. Effect of fiber surface treatment on the properties of biocomposites from nonwoven industrial hemp fiber mats and unsaturated polyester resin. J. Appl. Polym. Sci. 2006, 99, 1055–1068. [Google Scholar] [CrossRef]
- Tserki, V.; Panayiotou, C.; Zafeiropoulos, N.E. A study of the effect of acetylation and propionylation surface treatments on natural fibres. Compos. Part A Appl. Sci. Manuf. 2005, 36, 1110–1118. [Google Scholar] [CrossRef]
- Mishra, S.; Naik, J.B.; Patil, Y.P. The compatibilising effect of maleic anhydride on swelling and mechanical properties of plant-fiber-reinforced novolac composites. Compos. Sci. Technol. 2000, 60, 1729–1735. [Google Scholar] [CrossRef]
- Ferreira, D.P.; Cruz, J.; Fangueiro, R. Surface modification of natural fibers in polymer composites. In Green Composites for Automotive Applications; Woodhead Publishing: Cambridge, UK, 2019; pp. 3–41. [Google Scholar]
- Kalia, S.; Thakur, K.; Celli, A.; Kiechel, M.A.; Schauer, C.L. Surface modification of plant fibers using environment friendly methods for their application in polymer composites, textile industry and antimicrobial activities: A review. J. Environ. Chem. Eng. 2013, 1, 97–112. [Google Scholar] [CrossRef]
- Sun, D. Surface modification of natural fibers using plasma treatment. In Biodegradable Green Composites; Kalia, S., Ed.; Wiley: Hoboken, NJ, USA, 2016; pp. 18–39. [Google Scholar] [CrossRef]
- Thakur, V.K.; Singha, A.S. Surface Modification of Biopolymers; Wiley: Hoboken, NJ, USA, 2015. [Google Scholar] [CrossRef]
- Oliveira, D.M.; Benini, K.C.C.C.; Monticeli, F.M.; Schukraft, J.P.; Bomfim, A.S.C.; Cioffi, M.O.H.; Voorwald, H.J.C. Permeability of untreated and atmospheric plasma treated coconut fiber mats. Mater. Res. Express 2019, 6, 095323. [Google Scholar] [CrossRef]
- Borůvka, M.; Ngaowthong, C.; Bĕhálek, L.; Habr, J.; Lenfeld, P. Effect of dielectric barrier discharge plasma surface treatment on the properties of pineapple leaf fiber reinforced poly (lactic acid) biocomposites. Mater. Sci. Forum 2016, 862, 156–165. [Google Scholar] [CrossRef]
- Běhálek, L.; Borůvka, M.; Brdlík, P.; Habr, J.; Lenfeld, P.; Kroisová, D.; Veselka, F.; Novák, J. Thermal properties and non-isothermal crystallization kinetics of biocomposites based on poly (lactic acid), rice husks and cellulose fibres. J. Therm. Anal. Calorim. 2020, 142, 629–649. [Google Scholar] [CrossRef]
- Maqsood, H.S.; Bashir, U.; Wiener, J.; Puchalski, M.; Sztajnowski, S.; Militky, J. Ozone treatment of jute fibers. Cellulose 2017, 24, 1543–1553. [Google Scholar] [CrossRef]
- Reig, F.B.; Gimeno-Adelantado, J.V.; Moreno Moya, M.C.M. FTIR quantitative analysis of calcium carbonate (calcite) and silica (quartz) mixtures using the constant ratio method. Application to geological samples. Talanta 2002, 58, 811–821. [Google Scholar] [CrossRef]
- Zembouai, I.; Kaci, M.; Bruzaud, S.; Pillin, I. Electron beam radiation effects on properties and ecotoxicity of PHBV/PLA blends in presence of organo-modified montmorillonite. Polym. Degrad. Stab. 2016, 132, 117–126. [Google Scholar] [CrossRef]
- Tiptipakorn, S.; Hemvichain, K.; Pirombua, A. Study of the effect of electron beam on thermal and mechanical properties of poly (lactic acid)/poly (butylene succinate) blends. IOP Conf. Ser. Mater. Sci. Eng. 2019, 526, 012012. [Google Scholar] [CrossRef]
- Luo, S.; Netravali, A.N. Effect of 60Co γ-radiation on the properties of poly (hydroxybutyrate-co-hydroxyvalerate). J. Appl. Polym. Sci. 1999, 73, 1059–1067. [Google Scholar] [CrossRef]
- Oliveira, L.M.; Araujo, E.S.; Guedes, S.M. Gamma irradiation effects on poly (hydroxybutyrate). Polym. Degrad. Stab. 2006, 91, 2157–2162. [Google Scholar] [CrossRef]
- Oliveira, L.M.; Araujo, P.L.B.; Araujo, E.S. The effect of gamma radiation on mechanical properties of biodegradable polymers poly (3-hydroxybutyrate) and poly (3-hydroxybutyrate-co-3-hydroxyvalerate). Mater. Res. 2013, 16, 195–203. [Google Scholar] [CrossRef] [Green Version]
- Malinowski, R.; Rytlewski, P.; Żenkiewicz, M. Effects of electron radiation on properties of PLA. Arch. Mater. Sci. 2011, 49, 25–32. [Google Scholar]
- Meihua, L.; Wanxi, Z.; Yuan, Y.; Wei, W.; Chunbai, Z.; Pengyang, D.; Shirley, S. Effects of radiation-induced crosslinking on thermal and mechanical properties of poly (lactic acid) composites reinforced by basalt fiber. Asia-Pac. Symp. Radiat. Chem. 2013, 24 (Suppl. 1), 6. [Google Scholar]
- Dadbin, S.; Naimian, F.; Akhavan, A. Poly (lactic acid)/layered silicate nanocomposite films: Morphology, mechanical properties, and effects of γ-radiation. J. Appl. Polym. Sci. 2011, 122, 142–149. [Google Scholar] [CrossRef]
- Dadbin, S.; Naimian, F.; Akhavan, A.; Hasanpoor, S. Poly(Lactic Acid)/Layered Silicate Nanocomposite Films: Effect of Irradiation; Report of the 2nd RCM on Nanoscale radiation engineering of advanced materials for potential biomedical applications; International Atomic Energy Agency (IAEA): Vienna, Austria, 2010. [Google Scholar]
- Iggui, K.; Kaci, M.; Mahlous, M.; Moigne, N.L.; Bergeret, A. The Effects of Gamma Irradiation on Molecular Weight, Morphology and Physical Properties of PHBV/Cloisite 30B Bionanocomposites. J. Renew. Mater. 2019, 7, 807–820. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Code | Type of Biopolymer | Composition (wt%) | Modification of Fibers | Modification of Composite | |
|---|---|---|---|---|---|
| Biopolymer | Cellulose Fibers | ||||
| PLA | PLA | 100 | 0 | No | No |
| PLA-20CeF | PLA | 80 | 20 | No | No |
| PLA-20CeF-P | PLA | 80 | 20 | Plasma | No |
| PLA-20CeF-O | PLA | 80 | 20 | Ozone | No |
| PLA-20CeF-A | PLA | 80 | 20 | Acetylation | No |
| PLA-20CeF-R50 | PLA | 80 | 20 | No | Radiation |
| PLA-20CeF-R100 | PLA | 80 | 20 | No | Radiation |
| PHBV | PHBV | 100 | 0 | No | No |
| PHBV-20CeF | PHBV | 80 | 20 | No | No |
| PHBV-20CeF-P | PHBV | 80 | 20 | Plasma | No |
| PHBV-20CeF-O | PHBV | 80 | 20 | Ozone | No |
| PHBV-20CeF-A | PHBV | 80 | 20 | Acetylation | No |
| PHBV-20CeF-R50 | PHBV | 80 | 20 | No | Radiation |
| PHBV-20CeF-R100 | PHBV | 80 | 20 | No | Radiation |
| Sample Code (First Heating) | Temperature of Cold Crystallization (°C) Tp,cc | Enthalpy of Cold Crystallization (J/g) ΔHcc | Temperature of Premelt Crystallization (°C) Tp,pc | Enthalpy of Premelt Crystallization (J/g) ΔHpc | Melting Temperature (°C) Tp,m | Enthalpy of Melting (J/g) ΔHm | Degree of Crystallinity (%) Xc |
|---|---|---|---|---|---|---|---|
| PLA | 103.2 | −30.30 | 156.7 | −0.81 | 171.7 | 40.74 | 10.3 |
| PLA-20CeF | 97.4 | −27.52 | 155.7 | −2.54 | 171.7 | 41.74 | 15.7 |
| PLA-20CeF-P | 97.7 | −25.60 | 155.9 | −2.44 | 171.3 | 41.70 | 18.3 |
| PLA-20CeF-O | 97.5 | −28.16 | 155.9 | −2.56 | 171.7 | 40.67 | 13.4 |
| PLA-20CeF-A | 96.8 | −26.43 | 154.2 | −2.41 | 169.1 | 39.09 | 13.8 |
| PLA-20CeF-R50 | 98.6 | −28.58 | 154.3 | −1.25 | 167.6 | 42.92 | 17.6 |
| PLA-20CeF-R100 | 98.9 | −32.10 | 165.9 | 43.67 | 15.5 |
| Sample Code (First Heating) | Melting Temperature (°C) Tp,m1 | Melting Temperature (°C) Tp,m2 | Enthalpy of Melting (J/g) ΔHm | Enthalpy of Melting (J/g) ΔHm1 | Enthalpy of Melting (J/g) ΔHm2 | Degree of Crystallinity (%) Xc |
|---|---|---|---|---|---|---|
| PHBV | 177.4 | 177.4 | 93.45 | 63.7 | ||
| PHBV-20CeF | 171.7 | 174.3 | 82.48 | 59.88 | 22.60 | 70.3 |
| PHBV-20CeF-P | 171.9 | 174.9 | 82.85 | 69.66 | 13.19 | 70.6 |
| PHBV-20CeF-O | 171.0 | 174.7 | 82.00 | 69.88 | 12.12 | 69.9 |
| PHBV-20CeF-A | 172.7 | 177.0 | 84.95 | 71.52 | 13.43 | 72.4 |
| PHBV-20CeF-R50 | 169.9 | 176.1 | 86.65 | 71.36 | 15.29 | 73.9 |
| PHBV-20CeF-R100 | 169.3 | 174.7 | 82.71 | 71.51 | 11.20 | 70.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lenfeld, P.; Brdlík, P.; Borůvka, M.; Běhálek, L.; Habr, J. Effect of Radiation Crosslinking and Surface Modification of Cellulose Fibers on Properties and Characterization of Biopolymer Composites. Polymers 2020, 12, 3006. https://doi.org/10.3390/polym12123006
Lenfeld P, Brdlík P, Borůvka M, Běhálek L, Habr J. Effect of Radiation Crosslinking and Surface Modification of Cellulose Fibers on Properties and Characterization of Biopolymer Composites. Polymers. 2020; 12(12):3006. https://doi.org/10.3390/polym12123006
Chicago/Turabian StyleLenfeld, Petr, Pavel Brdlík, Martin Borůvka, Luboš Běhálek, and Jiří Habr. 2020. "Effect of Radiation Crosslinking and Surface Modification of Cellulose Fibers on Properties and Characterization of Biopolymer Composites" Polymers 12, no. 12: 3006. https://doi.org/10.3390/polym12123006