
CD8 T Cell–Independent Antitumor Response and Its Potential for Treatment of Malignant Gliomas


{kind=link}
Abstract
:1. Introduction
2. Immunology in the CNS
3. Immunotherapy in Cancer
3.1. Monoclonal Antibodies (mAb)
3.2. Adoptive Cell Transfer (ACT)
3.3. Immune Modulators
3.4. Vaccines
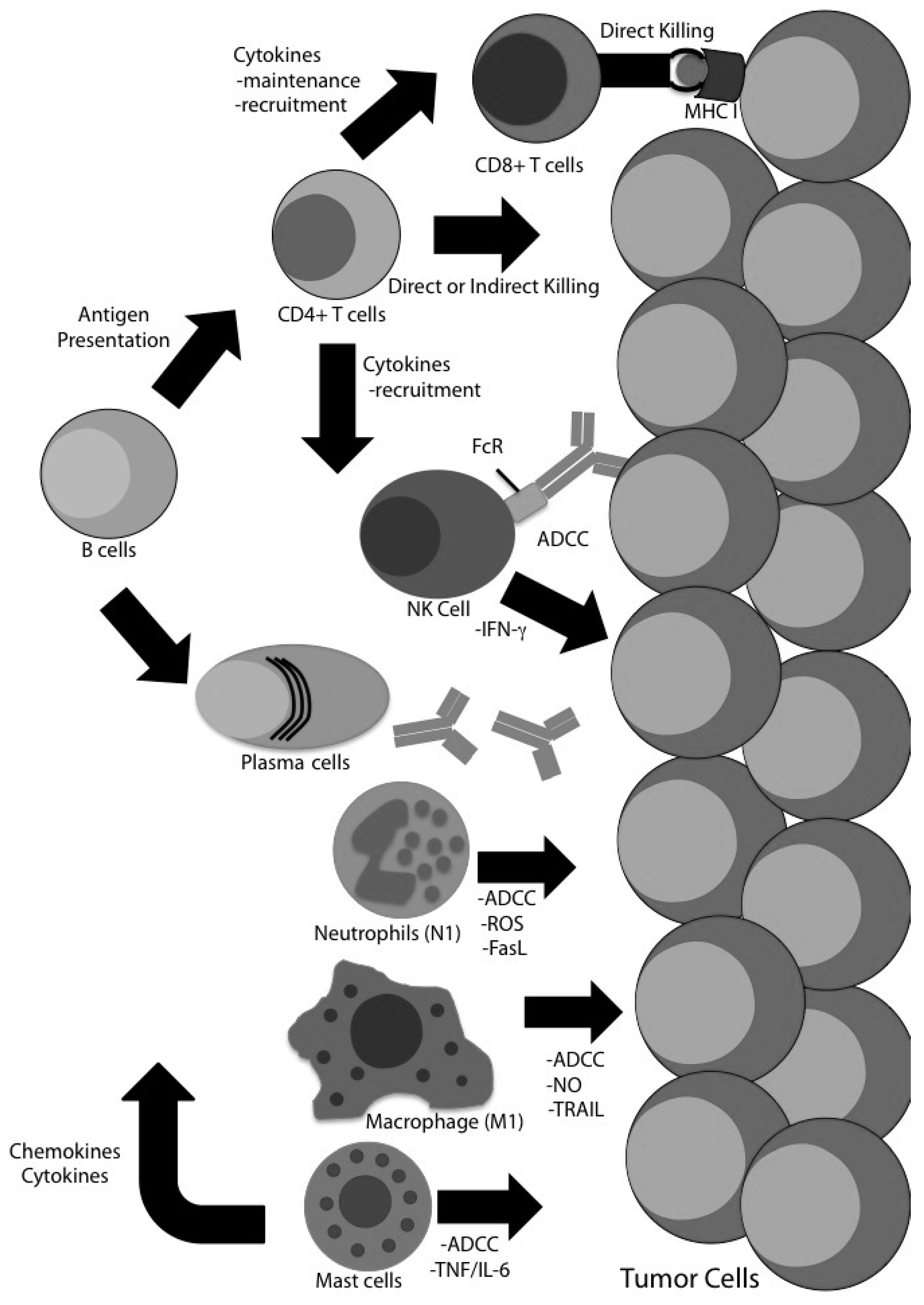
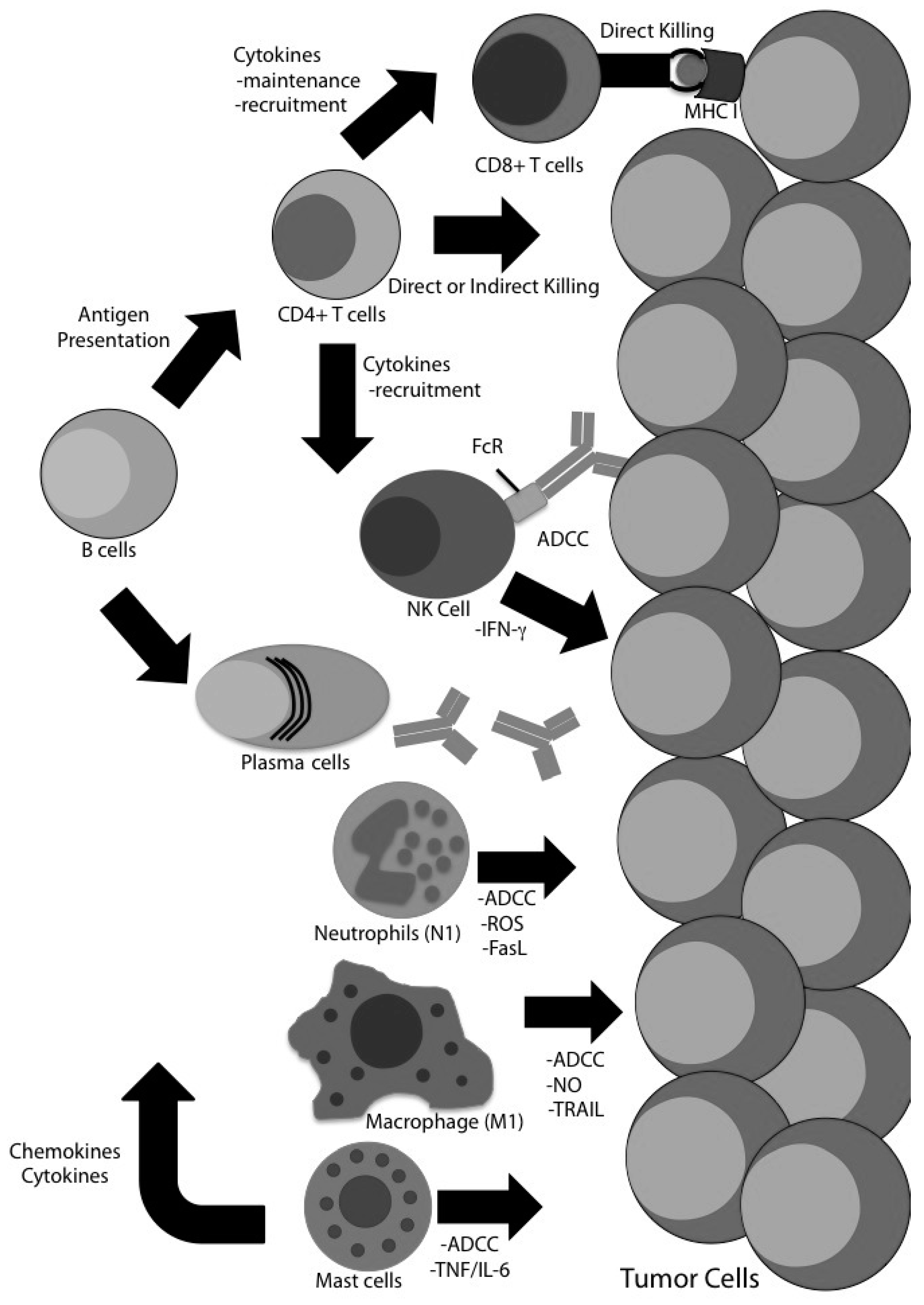
4. Evidence for CD8 T Cell-Independent Tumor Clearance in Glioma
4.1. CD4 T Cells
4.2. Natural Killer (NK) Cells
4.3. B Cells
4.4. Macrophages
4.5. Neutrophils
4.6. Myeloid-Derived Suppressor Cells (MDSC)
4.7. Mast Cells
5. Conclusions: Rethinking the Strategy
Author Contributions
Conflicts of Interest
References
- Wen, P.Y.; Kesari, S. Malignant gliomas in adults. N. Engl. J. Med. 2008, 359, 492–507. [Google Scholar] [CrossRef] [PubMed]
- Gittleman, H.R.; Ostrom, Q.T.; Rouse, C.D.; Dowling, J.A.; de Blank, P.M.; Kruchko, C.A.; Elder, J.B.; Rosenfeld, S.S.; Selman, W.R.; Sloan, A.E.; et al. Trends in central nervous system tumor incidence relative to other common cancers in adults, adolescents, and children in the United States, 2000 to 2010. Cancer 2015, 121, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Gittleman, H.; Liao, P.; Rouse, C.; Chen, Y.; Dowling, J.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2007–2011. Neuro Oncol. 2014, 16. [Google Scholar] [CrossRef] [PubMed]
- Claes, A.; Idema, A.J.; Wesseling, P. Diffuse glioma growth: A guerilla war. Acta Neuropathol. 2007, 114, 443–458. [Google Scholar] [CrossRef] [PubMed]
- Gardner, W.J.; Karnosh, L.J.; McClure, C.C., Jr.; Gardner, A.K. Residual function following hemispherectomy for tumour and for infantile hemiplegia. Brain J. Neurol. 1955, 78, 487–502. [Google Scholar] [CrossRef]
- Friedman, H.S.; Kerby, T.; Calvert, H. Temozolomide and treatment of malignant glioma. Clin. Cancer Res. 2000, 6, 2585–2597. [Google Scholar] [PubMed]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Imperato, J.P.; Paleologos, N.A.; Vick, N.A. Effects of treatment on long-term survivors with malignant astrocytomas. Ann. Neurol. 1990, 28, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Turner, C.D.; Rey-Casserly, C.; Liptak, C.C.; Chordas, C. Late effects of therapy for pediatric brain tumor survivors. J. Child Neurol. 2009, 24, 1455–1463. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.B.; Sturm, E. Conditions determining the transplantability of tissues in the brain. J. Exp. Med. 1923, 38, 183–197. [Google Scholar] [CrossRef] [PubMed]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Medawar, P.B. Immunity to homologous grafted skin: The fate of skin homografts transplanted to the brain, to subcutaneous tissue, and to the anterior chamber of the eye. Br. J. Exp. Pathol. 1948, 29, 58–69. [Google Scholar] [PubMed]
- Scheinberg, L.C.; Kotsilimbas, D.G.; Karpf, R.; Mayer, N. Is the brain “an immunologically privileged site”? 3. Studies based on homologous skin grafts to the brain and subcutaneous tissues. Arch. Neurol. 1966, 15, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Harling-Berg, C.J.; Park, T.J.; Knopf, P.M. Role of the cervical lymphatics in the Th2-type hierarchy of CNS immune regulation. J. Neuroimmunol. 1999, 101, 111–127. [Google Scholar] [CrossRef]
- Gordon, L.B.; Knopf, P.M.; Cserr, H.F. Ovalbumin is more immunogenic when introduced into brain or cerebrospinal fluid than into extracerebral sites. J. Neuroimmunol. 1992, 40, 81–87. [Google Scholar] [CrossRef]
- Gordon, L.B.; Nolan, S.C.; Cserr, H.F.; Knopf, P.M.; Harling-Berg, C.J. Growth of P511 mastocytoma cells in BALB/c mouse brain elicits CTL response without tumor elimination: A new tumor model for regional central nervous system immunity. J. Immunol. 1997, 159, 2399–2408. [Google Scholar] [PubMed]
- Gordon, L.B.; Nolan, S.C.; Ksander, B.R.; Knopf, P.M.; Harling-Berg, C.J. Normal cerebrospinal fluid suppresses the in vitro development of cytotoxic T cells: Role of the brain microenvironment in CNS immune regulation. J. Neuroimmunol. 1998, 88, 77–84. [Google Scholar] [CrossRef]
- Trandem, K.; Zhao, J.; Fleming, E.; Perlman, S. Highly activated cytotoxic CD8 T cells express protective IL-10 at the peak of coronavirus-induced encephalitis. J. Immunol. 2011, 186, 3642–3652. [Google Scholar] [CrossRef] [PubMed]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. Immune checkpoint targeting in cancer therapy: Toward combination strategies with curative potential. Cell 2015, 161, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Cobleigh, M.A.; Vogel, C.L.; Tripathy, D.; Robert, N.J.; Scholl, S.; Fehrenbacher, L.; Wolter, J.M.; Paton, V.; Shak, S.; Lieberman, G.; et al. Multinational study of the efficacy and safety of humanized anti-HER2 monoclonal antibody in women who have HER2-overexpressing metastatic breast cancer that has progressed after chemotherapy for metastatic disease. J. Clin. Oncol. 1999, 17, 2639–2648. [Google Scholar] [PubMed]
- Sampson, J.H.; Heimberger, A.B.; Archer, G.E.; Aldape, K.D.; Friedman, A.H.; Friedman, H.S.; Gilbert, M.R.; Herndon, J.E., 2nd; McLendon, R.E.; Mitchell, D.A.; et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J. Clin. Oncol. 2010, 28, 4722–4729. [Google Scholar] [CrossRef] [PubMed]
- Sampson, J.H.; Crotty, L.E.; Lee, S.; Archer, G.E.; Ashley, D.M.; Wikstrand, C.J.; Hale, L.P.; Small, C.; Dranoff, G.; Friedman, A.H.; et al. Unarmed, tumor-specific monoclonal antibody effectively treats brain tumors. Proc. Natl. Acad. Sci. USA 2000, 97, 7503–7508. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Restifo, N.P.; Yang, J.C.; Morgan, R.A.; Dudley, M.E. Adoptive cell transfer: A clinical path to effective cancer immunotherapy. Nat. Rev. Cancer 2008, 8, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Vauleon, E.; Avril, T.; Collet, B.; Mosser, J.; Quillien, V. Overview of cellular immunotherapy for patients with glioblastoma. Clin. Dev. Immunol. 2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riddell, S.R.; Jensen, M.C.; June, C.H. Chimeric antigen receptor—Modified T cells: Clinical translation in stem cell transplantation and beyond. Biol. Blood Marrow Transplant. 2013, 19, S2–S5. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Salsman, V.S.; Kew, Y.; Shaffer, D.; Powell, S.; Zhang, Y.J.; Grossman, R.G.; Heslop, H.E.; Gottschalk, S. HER2-specific T cells target primary glioblastoma stem cells and induce regression of autologous experimental tumors. Clin. Cancer Res. 2010, 16, 474–485. [Google Scholar] [CrossRef] [PubMed]
- Chow, K.K.; Naik, S.; Kakarla, S.; Brawley, V.S.; Shaffer, D.R.; Yi, Z.; Rainusso, N.; Wu, M.F.; Liu, H.; Kew, Y.; et al. T cells redirected to EphA2 for the immunotherapy of glioblastoma. Mol. Ther. 2013, 21, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Kong, S.; Sengupta, S.; Tyler, B.; Bais, A.J.; Ma, Q.; Doucette, S.; Zhou, J.; Sahin, A.; Carter, B.S.; Brem, H.; et al. Suppression of human glioma xenografts with second-generation IL13R-specific chimeric antigen receptor-modified T cells. Clin. Cancer Res. 2012, 18, 5949–5960. [Google Scholar] [CrossRef] [PubMed]
- Miao, H.; Choi, B.D.; Suryadevara, C.M.; Sanchez-Perez, L.; Yang, S.; de Leon, G.; Sayour, E.J.; McLendon, R.; Herndon, J.E., 2nd; Healy, P.; et al. EGFRvIII-specific chimeric antigen receptor T cells migrate to and kill tumor deposits infiltrating the brain parenchyma in an invasive xenograft model of glioblastoma. PLoS ONE 2014, 9, e94281. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Johnson, L.A.; Davis, J.L.; Zheng, Z.; Woolard, K.D.; Reap, E.A.; Feldman, S.A.; Chinnasamy, N.; Kuan, C.T.; Song, H.; et al. Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. Hum. Gene Ther. 2012, 23, 1043–1053. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.A.; Scholler, J.; Ohkuri, T.; Kosaka, A.; Patel, P.R.; McGettigan, S.E.; Nace, A.K.; Dentchev, T.; Thekkat, P.; Loew, A.; et al. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci. Transl. Med. 2015. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.E.; Badie, B.; Barish, M.E.; Weng, L.; Ostberg, J.R.; Chang, W.C.; Naranjo, A.; Starr, R.; Wagner, J.; Wright, C.; et al. Bioactivity and Safety of IL13Ralpha2-Redirected Chimeric Antigen Receptor CD8+ T Cells in Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2015, 21, 4062–4072. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, A.H.; Freeman, G.J. The B7-CD28 superfamily. Nat. Rev. Immunol. 2002, 2, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Bakacs, T.; Mehrishi, J.N.; Moss, R.W. Ipilimumab (Yervoy) and the TGN1412 catastrophe. Immunobiology 2012, 217, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Wolchok, J.D.; Chen, L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 2016. [Google Scholar] [CrossRef] [PubMed]
- Berghoff, A.S.; Kiesel, B.; Widhalm, G.; Rajky, O.; Ricken, G.; Wohrer, A.; Dieckmann, K.; Filipits, M.; Brandstetter, A.; Weller, M.; et al. Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neurooncology 2014, 17, 1064–1075. [Google Scholar] [CrossRef] [PubMed]
- Wainwright, D.A.; Chang, A.L.; Dey, M.; Balyasnikova, I.V.; Kim, C.K.; Tobias, A.; Cheng, Y.; Kim, J.W.; Qiao, J.; Zhang, L.; et al. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clin. Cancer Res. 2014, 20, 5290–5301. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; See, A.P.; Phallen, J.; Jackson, C.M.; Belcaid, Z.; Ruzevick, J.; Durham, N.; Meyer, C.; Harris, T.J.; Albesiano, E.; et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int. J. Radiat. Oncol. Biol. Phys. 2013, 86, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Sharabi, A.B.; Tran, P.T.; Lim, M.; Drake, C.G.; Deweese, T.L. Stereotactic radiation therapy combined with immunotherapy: Augmenting the role of radiation in local and systemic treatment. Oncology 2015, 29, 331–340. [Google Scholar] [PubMed]
- Reardon, D.A.; Gokhale, P.C.; Klein, S.R.; Ligon, K.L.; Rodig, S.J.; Ramkissoon, S.H.; Jones, K.L.; Conway, A.S.; Liao, X.; Zhou, J.; et al. Glioblastoma Eradication Following Immune Checkpoint Blockade in an Orthotopic, Immunocompetent Model. Cancer Immunol. Res. 2016, 4, 124–135. [Google Scholar] [CrossRef] [PubMed]
- Pardee, A.D.; Wesa, A.K.; Storkus, W.J. Integrating costimulatory agonists to optimize immune-based cancer therapies. Immunotherapy 2009, 1, 249–264. [Google Scholar] [CrossRef] [PubMed]
- Sugamura, K.; Ishii, N.; Weinberg, A.D. Therapeutic targeting of the effector T-cell co-stimulatory molecule OX40. Nat. Rev. Immunol. 2004, 4, 420–431. [Google Scholar] [CrossRef] [PubMed]
- Okada, H.; Lieberman, F.S.; Walter, K.A.; Lunsford, L.D.; Kondziolka, D.S.; Bejjani, G.K.; Hamilton, R.L.; Torres-Trejo, A.; Kalinski, P.; Cai, Q.; et al. Autologous glioma cell vaccine admixed with interleukin-4 gene transfected fibroblasts in the treatment of patients with malignant gliomas. J. Transl. Med. 2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, A.; Oh, S.; Gharagozlou, S.; Vedi, R.N.; Ericson, K.; Low, W.C.; Chen, W.; Ohlfest, J.R. In vivo vaccination with tumor cell lysate plus CpG oligodeoxynucleotides eradicates murine glioblastoma. J. Immunother. 2007, 30, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Prins, R.M.; Soto, H.; Konkankit, V.; Odesa, S.K.; Eskin, A.; Yong, W.H.; Nelson, S.F.; Liau, L.M. Gene expression profile correlates with T-cell infiltration and relative survival in glioblastoma patients vaccinated with dendritic cell immunotherapy. Clin. Cancer Res. 2011, 17, 1603–1615. [Google Scholar] [CrossRef] [PubMed]
- Ahn, B.J.; Pollack, I.F.; Okada, H. Immune-checkpoint blockade and active immunotherapy for glioma. Cancers 2013, 5, 1379–1412. [Google Scholar] [CrossRef] [PubMed]
- Liau, L.M.; Prins, R.M.; Kiertscher, S.M.; Odesa, S.K.; Kremen, T.J.; Giovannone, A.J.; Lin, J.W.; Chute, D.J.; Mischel, P.S.; Cloughesy, T.F.; et al. Dendritic cell vaccination in glioblastoma patients induces systemic and intracranial T-cell responses modulated by the local central nervous system tumor microenvironment. Clin. Cancer Res. 2005, 11, 5515–5525. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.S.; Wheeler, C.J.; Zeltzer, P.M.; Ying, H.; Finger, D.N.; Lee, P.K.; Yong, W.H.; Incardona, F.; Thompson, R.C.; Riedinger, M.S.; et al. Vaccination of malignant glioma patients with peptide-pulsed dendritic cells elicits systemic cytotoxicity and intracranial T-cell infiltration. Cancer Res. 2001, 61, 842–847. [Google Scholar] [PubMed]
- Xu, L.W.; Chow, K.K.; Lim, M.; Li, G. Current vaccine trials in glioblastoma: A review. J. Immunol. Res. 2014. [Google Scholar] [CrossRef] [PubMed]
- Phuphanich, S.; Wheeler, C.J.; Rudnick, J.D.; Mazer, M.; Wang, H.; Nuno, M.A.; Richardson, J.E.; Fan, X.; Ji, J.; Chu, R.M.; et al. Phase I trial of a multi-epitope-pulsed dendritic cell vaccine for patients with newly diagnosed glioblastoma. Cancer Immunol. Immunother. 2013, 62, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Okada, H.; Kalinski, P.; Ueda, R.; Hoji, A.; Kohanbash, G.; Donegan, T.E.; Mintz, A.H.; Engh, J.A.; Bartlett, D.L.; Brown, C.K.; et al. Induction of CD8+ T-cell responses against novel glioma-associated antigen peptides and clinical activity by vaccinations with {alpha}-type 1 polarized dendritic cells and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in patients with recurrent malignant glioma. J. Clin. Oncol. 2011, 29, 330–336. [Google Scholar] [PubMed]
- Dutoit, V.; Herold-Mende, C.; Hilf, N.; Schoor, O.; Beckhove, P.; Bucher, J.; Dorsch, K.; Flohr, S.; Fritsche, J.; Lewandrowski, P.; et al. Exploiting the glioblastoma peptidome to discover novel tumour-associated antigens for immunotherapy. Brain J. Neurol. 2012, 135, 1042–1054. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.A.; Lechner, M.G.; Popescu, F.E.; Bedi, J.; Decker, S.A.; Hu, P.; Erickson, J.R.; O’Sullivan, M.G.; Swier, L.; Salazar, A.M.; et al. An in vivo immunotherapy screen of costimulatory molecules identifies Fc-OX40L as a potent reagent for the treatment of established murine gliomas. Clin. Cancer Res. 2012, 18, 4657–4668. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.A.; Erickson, J.R.; Johnson, C.S.; Seiler, C.E.; Bedi, J.; Hu, P.; Pluhar, G.E.; Epstein, A.L.; Ohlfest, J.R. CD8+ T cell-independent tumor regression induced by Fc-OX40L and therapeutic vaccination in a mouse model of glioma. J. Immunol. 2014, 192, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Zagzag, D.; Salnikow, K.; Chiriboga, L.; Yee, H.; Lan, L.; Ali, M.A.; Garcia, R.; Demaria, S.; Newcomb, E.W. Downregulation of major histocompatibility complex antigens in invading glioma cells: Stealth invasion of the brain. Lab. Investig. 2005, 85, 328–341. [Google Scholar] [CrossRef] [PubMed]
- Kane, A.; Yang, I. Interferon-gamma in brain tumor immunotherapy. Neurosurg. Clin. N. Am. 2010, 21, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.F. The role of MHC class II-restricted tumor antigens and CD4+ T cells in antitumor immunity. Trends Immunol. 2001, 22, 269–276. [Google Scholar] [CrossRef]
- Hung, K.; Hayashi, R.; Lafond-Walker, A.; Lowenstein, C.; Pardoll, D.; Levitsky, H. The central role of CD4(+) T cells in the antitumor immune response. J. Exp. Med. 1998, 188, 2357–2368. [Google Scholar] [CrossRef] [PubMed]
- Perez-Diez, A.; Joncker, N.T.; Choi, K.; Chan, W.F.; Anderson, C.C.; Lantz, O.; Matzinger, P. CD4 cells can be more efficient at tumor rejection than CD8 cells. Blood 2007, 109, 5346–5354. [Google Scholar] [CrossRef] [PubMed]
- Soghoian, D.Z.; Streeck, H. Cytolytic CD4(+) T cells in viral immunity. Expert Rev. Vaccines 2010, 9, 1453–1463. [Google Scholar] [CrossRef] [PubMed]
- Robbins, P.F.; El-Gamil, M.; Li, Y.F.; Zeng, G.; Dudley, M.; Rosenberg, S.A. Multiple HLA class II-restricted melanocyte differentiation antigens are recognized by tumor-infiltrating lymphocytes from a patient with melanoma. J. Immunol. 2002, 169, 6036–6047. [Google Scholar] [CrossRef] [PubMed]
- Dudley, M.E.; Wunderlich, J.R.; Robbins, P.F.; Yang, J.C.; Hwu, P.; Schwartzentruber, D.J.; Topalian, S.L.; Sherry, R.; Restifo, N.P.; Hubicki, A.M.; et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 2002, 298, 850–854. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.; Bunse, L.; Pusch, S.; Sahm, F.; Wiestler, B.; Quandt, J.; Menn, O.; Osswald, M.; Oezen, I.; Ott, M.; et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature 2014, 512, 324–327. [Google Scholar] [CrossRef] [PubMed]
- Sayour, E.J.; McLendon, P.; McLendon, R.; De Leon, G.; Reynolds, R.; Kresak, J.; Sampson, J.H.; Mitchell, D.A. Increased proportion of FoxP3+ regulatory T cells in tumor infiltrating lymphocytes is associated with tumor recurrence and reduced survival in patients with glioblastoma. Cancer Immunol. Immunother. 2015, 64, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Pere, H.; Tanchot, C.; Bayry, J.; Terme, M.; Taieb, J.; Badoual, C.; Adotevi, O.; Merillon, N.; Marcheteau, E.; Quillien, V.R.; et al. Comprehensive analysis of current approaches to inhibit regulatory T cells in cancer. Oncoimmunology 2012, 1, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Wainwright, D.A.; Dey, M.; Chang, A.; Lesniak, M.S. Targeting Tregs in Malignant Brain Cancer: Overcoming IDO. Front. Immunol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Sevko, A.; Sade-Feldman, M.; Kanterman, J.; Michels, T.; Falk, C.S.; Umansky, L.; Ramacher, M.; Kato, M.; Schadendorf, D.; Baniyash, M.; et al. Cyclophosphamide promotes chronic inflammation-dependent immunosuppression and prevents antitumor response in melanoma. J. Investig. Dermatol. 2013, 133, 1610–1619. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, J.; Yamazaki, S.; Takahashi, T.; Ishida, Y.; Sakaguchi, S. Stimulation of CD25(+)CD4(+) regulatory T cells through GITR breaks immunological self-tolerance. Nat. Immunol. 2002, 3, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Yang, I.; Han, S.J.; Sughrue, M.E.; Tihan, T.; Parsa, A.T. Immune cell infiltrate differences in pilocytic astrocytoma and glioblastoma: Evidence of distinct immunological microenvironments that reflect tumor biology. J. Neurosurg. 2011, 115, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, N.; Murata, S.; Ueki, T.; Mekata, E.; Reilly, R.T.; Jaffee, E.M.; Tani, T. OX40 costimulation can abrogate Foxp3+ regulatory T cell-mediated suppression of antitumor immunity. Int. J. Cancer 2009, 125, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.; Chen, Y.; Xiao, W.; Sun, R.; Tian, Z. NK cell-based immunotherapy for malignant diseases. Cell. Mol. Immunol. 2013, 10, 230–252. [Google Scholar] [CrossRef] [PubMed]
- Prins, R.M.; Vo, D.D.; Khan-Farooqi, H.; Yang, M.Y.; Soto, H.; Economou, J.S.; Liau, L.M.; Ribas, A. NK and CD4 cells collaborate to protect against melanoma tumor formation in the brain. J. Immunol. 2006, 177, 8448–8455. [Google Scholar] [CrossRef] [PubMed]
- Crawford, A.; Macleod, M.; Schumacher, T.; Corlett, L.; Gray, D. Primary T cell expansion and differentiation in vivo requires antigen presentation by B cells. J. Immunol. 2006, 176, 3498–3506. [Google Scholar] [CrossRef] [PubMed]
- Deeken, J.F.; Loscher, W. The blood-brain barrier and cancer: Transporters, treatment, and Trojan horses. Clin. Cancer Res. 2007, 13, 1663–1674. [Google Scholar] [CrossRef] [PubMed]
- Pallasch, C.P.; Struss, A.K.; Munnia, A.; Konig, J.; Steudel, W.I.; Fischer, U.; Meese, E. Autoantibodies against GLEA2 and PHF3 in glioblastoma: Tumor-associated autoantibodies correlated with prolonged survival. Int. J. Cancer 2005, 117, 456–459. [Google Scholar] [CrossRef] [PubMed]
- Candolfi, M.; Curtin, J.F.; Yagiz, K.; Assi, H.; Wibowo, M.K.; Alzadeh, G.E.; Foulad, D.; Muhammad, A.K.; Salehi, S.; Keech, N.; et al. B cells are critical to T-cell-mediated antitumor immunity induced by a combined immune-stimulatory/conditionally cytotoxic therapy for glioblastoma. Neoplasia 2011, 13, 947–960. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P. The interaction of anticancer therapies with tumor-associated macrophages. J. Exp. Med. 2015, 212, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Komohara, Y.; Ohnishi, K.; Kuratsu, J.; Takeya, M. Possible involvement of the M2 anti-inflammatory macrophage phenotype in growth of human gliomas. J. Pathol. 2008, 216, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; Chiorean, E.G.; Fishman, M.P.; Saboury, B.; Teitelbaum, U.R.; Sun, W.; Huhn, R.D.; Song, W.; Li, D.; Sharp, L.L.; et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 2011, 331, 1612–1616. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.F.; Yang, D.; Suki, D.; Aldape, K.; Grimm, E.; Heimberger, A.B. The role of human glioma-infiltrating microglia/macrophages in mediating antitumor immune responses. Neurooncology 2006, 8, 261–279. [Google Scholar] [CrossRef] [PubMed]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef] [PubMed]
- MacMicking, J.; Xie, Q.W.; Nathan, C. Nitric oxide and macrophage function. Annu. Rev. Immunol. 1997, 15, 323–350. [Google Scholar] [CrossRef] [PubMed]
- De Palma, M.; Lewis, C.E. Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell 2013, 23, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Griffith, T.S.; Wiley, S.R.; Kubin, M.Z.; Sedger, L.M.; Maliszewski, C.R.; Fanger, N.A. Monocyte-mediated tumoricidal activity via the tumor necrosis factor-related cytokine, TRAIL. J. Exp. Med. 1999, 189, 1343–1354. [Google Scholar] [CrossRef] [PubMed]
- Fossati, G.; Ricevuti, G.; Edwards, S.W.; Walker, C.; Dalton, A.; Rossi, M.L. Neutrophil infiltration into human gliomas. Acta Neuropathol. 1999, 98, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Piao, Y.; Holmes, L.; Fuller, G.N.; Henry, V.; Tiao, N.; de Groot, J.F. Neutrophils promote the malignant glioma phenotype through S100A4. Clin. Cancer Res. 2014, 20, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Gregory, A.D.; Houghton, A.M. Tumor-associated neutrophils: New targets for cancer therapy. Cancer Res. 2011, 71, 2411–2416. [Google Scholar] [CrossRef] [PubMed]
- Fridlender, Z.G.; Albelda, S.M. Tumor-associated neutrophils: Friend or foe? Carcinogenesis 2012, 33, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef] [PubMed]
- James, B.R.; Anderson, K.G.; Brincks, E.L.; Kucaba, T.A.; Norian, L.A.; Masopust, D.; Griffith, T.S. CpG-mediated modulation of MDSC contributes to the efficacy of Ad5-TRAIL therapy against renal cell carcinoma. Cancer Immunol. Immunother. 2014, 63, 1213–1227. [Google Scholar] [CrossRef] [PubMed]
- Shirota, Y.; Shirota, H.; Klinman, D.M. Intratumoral injection of CpG oligonucleotides induces the differentiation and reduces the immunosuppressive activity of myeloid-derived suppressor cells. J. Immunol. 2012, 188, 1592–1599. [Google Scholar] [CrossRef] [PubMed]
- Zoglmeier, C.; Bauer, H.; Norenberg, D.; Wedekind, G.; Bittner, P.; Sandholzer, N.; Rapp, M.; Anz, D.; Endres, S.; Bourquin, C. CpG blocks immunosuppression by myeloid-derived suppressor cells in tumor-bearing mice. Clin. Cancer Res. 2011, 17, 1765–1775. [Google Scholar] [CrossRef] [PubMed]
- Lechner, M.G.; Liebertz, D.J.; Epstein, A.L. Characterization of cytokine-induced myeloid-derived suppressor cells from normal human peripheral blood mononuclear cells. J. Immunol. 2010, 185, 2273–2284. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Purwar, R.; Schlapbach, C.; Xiao, S.; Kang, H.S.; Elyaman, W.; Jiang, X.; Jetten, A.M.; Khoury, S.J.; Fuhlbrigge, R.C.; Kuchroo, V.K.; et al. Robust tumor immunity to melanoma mediated by interleukin-9-producing T cells. Nat. Med. 2012, 18, 1248–1253. [Google Scholar] [CrossRef] [PubMed]
- Oldford, S.A.; Haidl, I.D.; Howatt, M.A.; Leiva, C.A.; Johnston, B.; Marshall, J.S. A critical role for mast cells and mast cell-derived IL-6 in TLR2-mediated inhibition of tumor growth. J. Immunol. 2010, 185, 7067–7076. [Google Scholar] [CrossRef] [PubMed]
- Oldford, S.A.; Marshall, J.S. Mast cells as targets for immunotherapy of solid tumors. Mol. Immunol. 2015, 63, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Polajeva, J.; Sjosten, A.M.; Lager, N.; Kastemar, M.; Waern, I.; Alafuzoff, I.; Smits, A.; Westermark, B.; Pejler, G.; Uhrbom, L.; et al. Mast cell accumulation in glioblastoma with a potential role for stem cell factor and chemokine CXCL12. PLoS ONE 2011, 6, e25222. [Google Scholar] [CrossRef] [PubMed]
- Valzasina, B.; Guiducci, C.; Dislich, H.; Killeen, N.; Weinberg, A.D.; Colombo, M.P. Triggering of OX40 (CD134) on CD4(+)CD25+ T cells blocks their inhibitory activity: A novel regulatory role for OX40 and its comparison with GITR. Blood 2005, 105, 2845–2851. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murphy, K.A.; Griffith, T.S. CD8 T Cell–Independent Antitumor Response and Its Potential for Treatment of Malignant Gliomas. Cancers 2016, 8, 71. https://doi.org/10.3390/cancers8080071
Murphy KA, Griffith TS. CD8 T Cell–Independent Antitumor Response and Its Potential for Treatment of Malignant Gliomas. Cancers. 2016; 8(8):71. https://doi.org/10.3390/cancers8080071
Chicago/Turabian StyleMurphy, Katherine A., and Thomas S. Griffith. 2016. "CD8 T Cell–Independent Antitumor Response and Its Potential for Treatment of Malignant Gliomas" Cancers 8, no. 8: 71. https://doi.org/10.3390/cancers8080071
APA StyleMurphy, K. A., & Griffith, T. S. (2016). CD8 T Cell–Independent Antitumor Response and Its Potential for Treatment of Malignant Gliomas. Cancers, 8(8), 71. https://doi.org/10.3390/cancers8080071