
Targeting CDK9 for the Treatment of Glioblastoma

 , ,
, ,
Abstract
Simple Summary
Abstract
1. Introduction
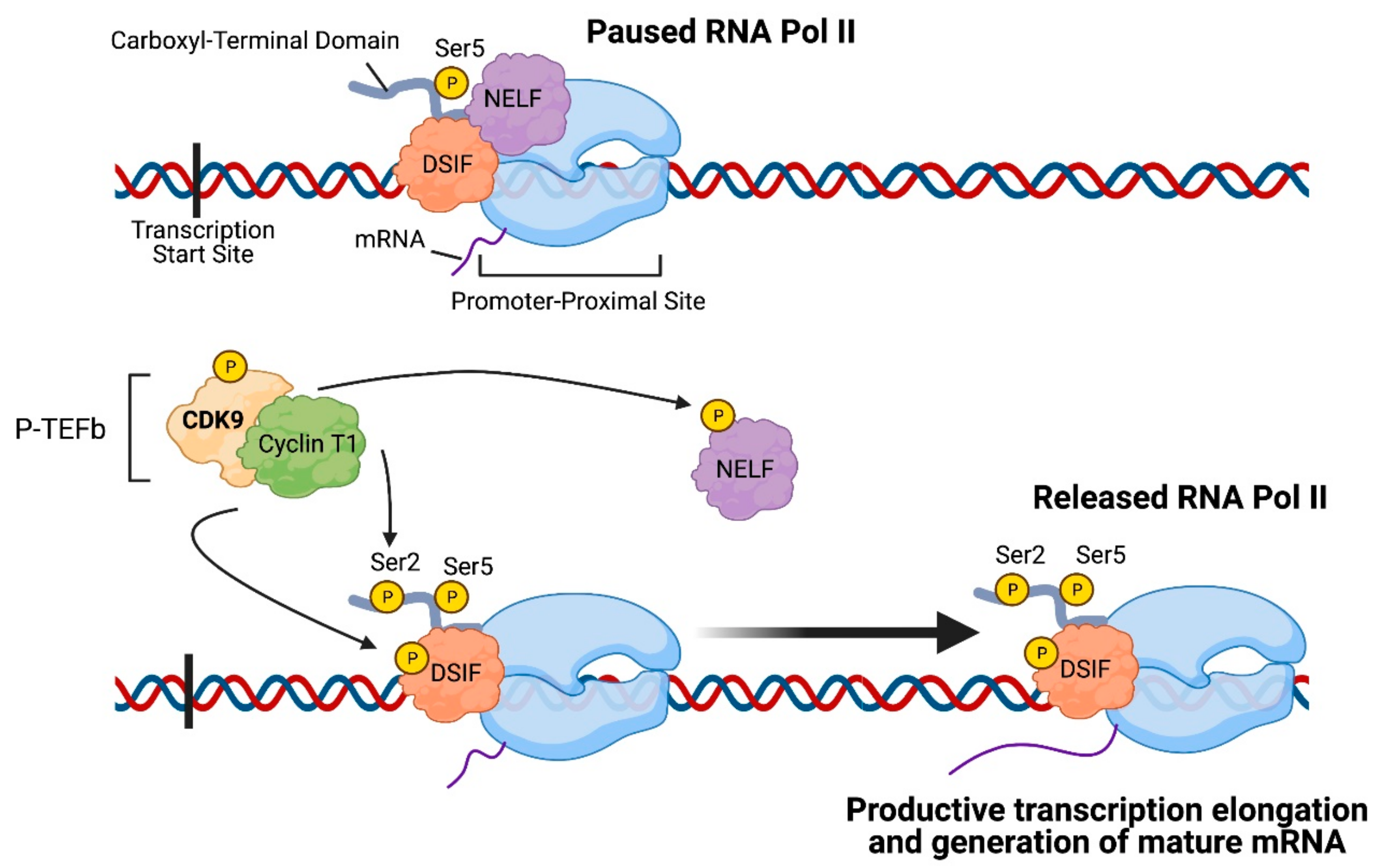
2. CDK9: An Important Regulator of Transcription Elongation
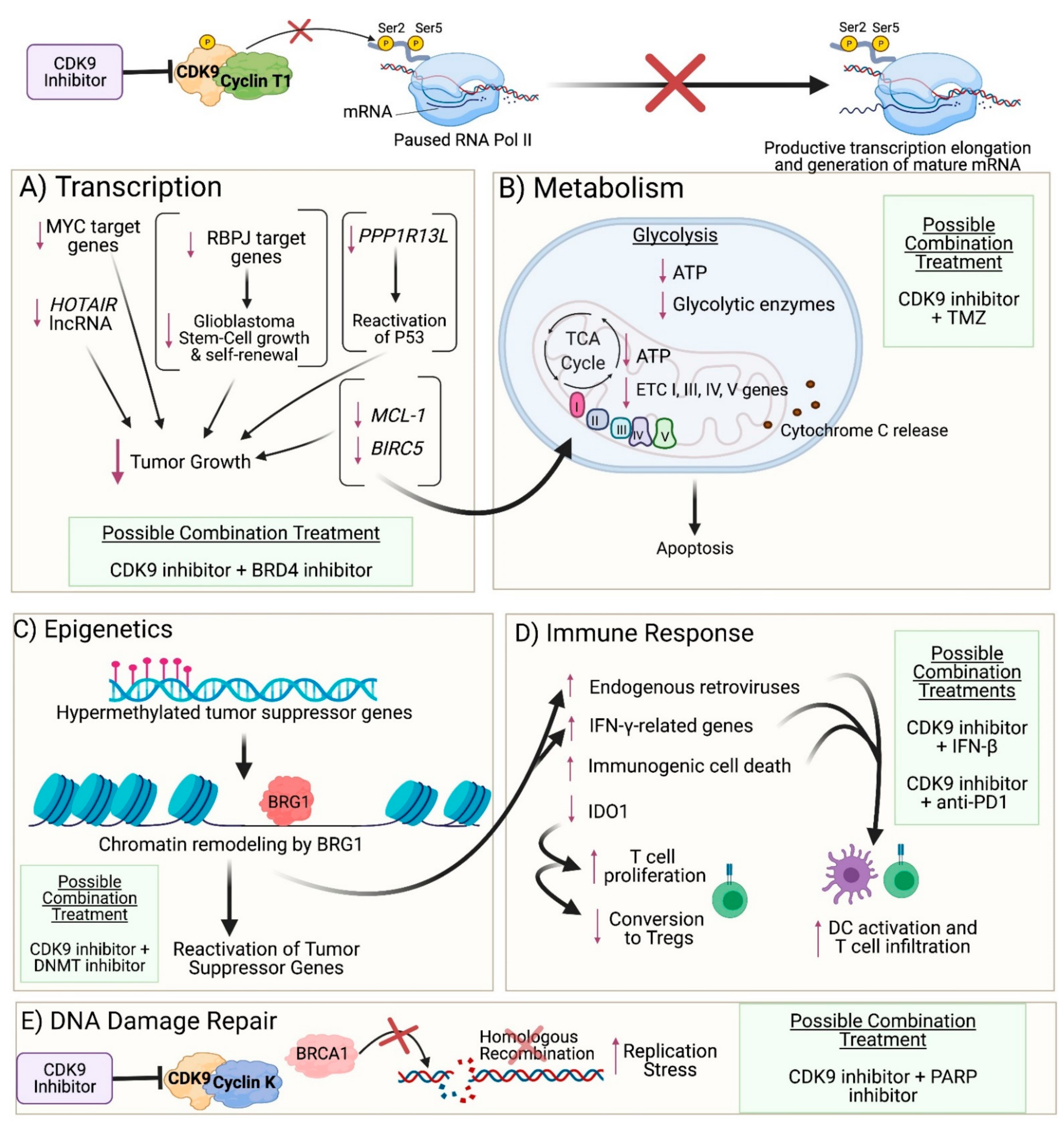
3. Impact of CDK9 Inhibition on Cancer Cells
3.1. Transcription
3.2. Metabolism
3.3. DNA Damage Repair
3.4. Epigenetics
3.5. Immune Response
4. CDK9 Inhibitors in Cancer Clinical Trials
5. Targeting CDK9 in Clinical Trials for Gliomas
5.1. Zotiraciclib as a Promising CDK9 Inhibitor for Treating Glioblastoma
5.2. Clinical Trials Investigating Zotiraciclib in Recurrent and Newly Diagnosed Brain Tumors
5.3. Toxicity Profile of Zotiraciclib
6. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lapointe, S.; Perry, A.; Butowski, N.A. Primary brain tumours in adults. Lancet 2018, 392, 432–446. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Patil, N.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2013–2017. Neuro-Oncology 2020, 22, iv1–iv96. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Juric, V.; Murphy, B. Cyclin-dependent kinase inhibitors in brain cancer: Current state and future directions. Cancer Drug Resist. 2020, 3, 48–62. [Google Scholar] [CrossRef]
- Wong, E.T.; Hess, K.R.; Gleason, M.J.; Jaeckle, K.A.; Kyritsis, A.P.; Prados, M.D.; Levin, V.A.; Yung, W.K.A. Outcomes and Prognostic Factors in Recurrent Glioma Patients Enrolled Onto Phase II Clinical Trials. J. Clin. Oncol. 1999, 17, 2572. [Google Scholar] [CrossRef] [PubMed]
- Fisher, J.P.; Adamson, D.C. Current FDA-Approved Therapies for High-Grade Malignant Gliomas. Biomedicines 2021, 9, 324. [Google Scholar] [CrossRef]
- McLendon, R.; Friedman, A.; Bigner, D.; Van Meir, E.G.; Brat, D.J.; Mastrogianakis, G.M.; Olson, J.J.; Mikkelsen, T.; Lehman, N.; Aldape, K.; et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Aldape, K.; Brindle, K.M.; Chesler, L.; Chopra, R.; Gajjar, A.; Gilbert, M.R.; Gottardo, N.; Gutmann, D.H.; Hargrave, D.; Holland, E.C.; et al. Challenges to curing primary brain tumours. Nat. Rev. Clin. Oncol. 2019, 16, 509–520. [Google Scholar] [CrossRef]
- Prados, M.D.; Byron, S.A.; Tran, N.L.; Phillips, J.J.; Molinaro, A.M.; Ligon, K.L.; Wen, P.Y.; Kuhn, J.G.; Mellinghoff, I.K.; de Groot, J.F.; et al. Toward precision medicine in glioblastoma: The promise and the challenges. Neuro-Oncology 2015, 17, 1051–1063. [Google Scholar] [CrossRef]
- Ratnam, N.M.; Gilbert, M.R.; Giles, A.J. Immunotherapy in CNS cancers: The role of immune cell trafficking. Neuro-Oncology 2018, 21, 37–46. [Google Scholar] [CrossRef]
- Woroniecka, K.I.; Rhodin, K.E.; Chongsathidkiet, P.; Keith, K.A.; Fecci, P.E. T-cell Dysfunction in Glioblastoma: Applying a New Framework. Clin. Cancer Res. 2018, 24, 3792. [Google Scholar] [CrossRef]
- Prager, B.C.; Bhargava, S.; Mahadev, V.; Hubert, C.G.; Rich, J.N. Glioblastoma Stem Cells: Driving Resilience through Chaos. Trends Cancer 2020, 6, 223–235. [Google Scholar] [CrossRef]
- Romano, G.; Giordano, A. Role of the cyclin-dependent kinase 9-related pathway in mammalian gene expression and human diseases. Cell Cycle 2008, 7, 3664–3668. [Google Scholar] [CrossRef]
- Sobhani, N.; D’Angelo, A.; Pittacolo, M.; Roviello, G.; Miccoli, A.; Corona, S.P.; Bernocchi, O.; Generali, D.; Otto, T. Updates on the CDK4/6 Inhibitory Strategy and Combinations in Breast Cancer. Cells 2019, 8, 321. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.W.; Parikh, M.; Phillips, J.J.; James, C.D.; Molinaro, A.M.; Butowski, N.A.; Clarke, J.L.; Oberheim-Bush, N.A.; Chang, S.M.; Berger, M.S.; et al. Phase-2 trial of palbociclib in adult patients with recurrent RB1-positive glioblastoma. J. Neuro-Oncol. 2018, 140, 477–483. [Google Scholar] [CrossRef]
- Pilot Study of Abemaciclib with Bevacizumab in Recurrent Glioblastoma Patients with Loss of CDKN2A/B or Gain or Amplification of CDK4/6. Available online: https://ClinicalTrials.gov/show/NCT04074785 (accessed on 16 April 2021).
- A Phase 0/II Study of Ribociclib (LEE011) in Combination with Everolimus in Preoperative Recurrent High-Grade Glioma Patients Scheduled for Resection. Available online: https://ClinicalTrials.gov/show/NCT03834740 (accessed on 16 April 2021).
- A Study of Abemaciclib (LY2835219) in Combination with Temozolomide and Irinotecan and Abemaciclib in Combination with Temozolomide in Children and Young Adult Participants with Solid Tumors. Available online: https://ClinicalTrials.gov/show/NCT04238819 (accessed on 16 April 2021).
- Abemaciclib in Children with DIPG or Recurrent/Refractory Solid Tumors. Available online: https://ClinicalTrials.gov/show/NCT02644460 (accessed on 16 April 2021).
- SJDAWN: St. Jude Children’s Research Hospital Phase 1 Study Evaluating Molecularly-Driven Doublet Therapies for Children and Young Adults with Recurrent Brain Tumors. Available online: https://ClinicalTrials.gov/show/NCT03434262 (accessed on 16 April 2021).
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional Addiction in Cancer. Cell 2017, 168, 629–643. [Google Scholar] [CrossRef]
- Alsfouk, A. Small molecule inhibitors of cyclin-dependent kinase 9 for cancer therapy. J. Enzym. Inhib. Med. Chem. 2021, 36, 693–706. [Google Scholar] [CrossRef]
- Ma, H.; Seebacher, N.A.; Hornicek, F.J.; Duan, Z. Cyclin-dependent kinase 9 (CDK9) is a novel prognostic marker and therapeutic target in osteosarcoma. eBioMedicine 2019, 39, 182–193. [Google Scholar] [CrossRef]
- Li, X.; Seebacher, N.A.; Xiao, T.; Hornicek, F.J.; Duan, Z. Targeting regulation of cyclin dependent kinase 9 as a novel therapeutic strategy in synovial sarcoma. J. Orthop. Res. 2019, 37, 510–521. [Google Scholar] [CrossRef]
- He, S.; Fang, X.; Xia, X.; Hou, T.; Zhang, T. Targeting CDK9: A novel biomarker in the treatment of endometrial cancer. Oncol. Rep. 2020, 44, 1929–1938. [Google Scholar] [CrossRef]
- Goh, K.C.; Novotny-Diermayr, V.; Hart, S.; Ong, L.C.; Loh, Y.K.; Cheong, A.; Tan, Y.C.; Hu, C.; Jayaraman, R.; William, A.D.; et al. TG02, a novel oral multi-kinase inhibitor of CDKs, JAK2 and FLT3 with potent anti-leukemic properties. Leukemia 2012, 26, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Kretz, A.-L.; Schaum, M.; Richter, J.; Kitzig, E.F.; Engler, C.C.; Leithäuser, F.; Henne-Bruns, D.; Knippschild, U.; Lemke, J. CDK9 is a prognostic marker and therapeutic target in pancreatic cancer. Tumor Biol. 2017, 39, 1010428317694304. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Pandey, S.; Travers, M.; Sun, H.; Morton, G.; Madzo, J.; Chung, W.; Khowsathit, J.; Perez-Leal, O.; Barrero, C.A.; et al. Targeting CDK9 Reactivates Epigenetically Silenced Genes in Cancer. Cell 2018, 175, 1244–1258.e1226. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Bhakat, R.; Kling, M.J.; Coulter, D.W.; Chaturvedi, N.K.; Ray, S.; Joshi, S.S. Targeting cyclin-dependent kinase 9 sensitizes medulloblastoma cells to chemotherapy. Biochem. Biophys. Res. Commun. 2019, 520, 250–256. [Google Scholar] [CrossRef]
- Von Achenbach, C.; Le Rhun, E.; Sahm, F.; Wang, S.S.; Sievers, P.; Neidert, M.C.; Rushing, E.J.; Lawhon, T.; Schneider, H.; von Deimling, A.; et al. Sensitivity of human meningioma cells to the cyclin-dependent kinase inhibitor, TG02. Transl. Oncol. 2020, 13, 100852. [Google Scholar] [CrossRef]
- Xie, Q.; Wu, Q.; Kim, L.; Miller, T.E.; Liau, B.B.; Mack, S.C.; Yang, K.; Factor, D.C.; Fang, X.; Huang, Z.; et al. RBPJ maintains brain tumor–initiating cells through CDK9-mediated transcriptional elongation. J. Clin. Investig. 2016, 126, 2757–2772. [Google Scholar] [CrossRef]
- Morales, F.; Giordano, A. Overview of CDK9 as a target in cancer research. Cell Cycle 2016, 15, 519–527. [Google Scholar] [CrossRef]
- Yu, D.S.; Cortez, D. A role for cdk9-cyclin k in maintaining genome integrity. Cell Cycle 2011, 10, 28–32. [Google Scholar] [CrossRef]
- Jonkers, I.; Lis, J.T. Getting up to speed with transcription elongation by RNA polymerase II. Nat. Rev. Mol. Cell Biol. 2015, 16, 167–177. [Google Scholar] [CrossRef]
- Su, Y.-T.; Chen, R.; Wang, H.; Song, H.; Zhang, Q.; Chen, L.-Y.; Lappin, H.; Vasconcelos, G.; Lita, A.; Maric, D.; et al. Novel Targeting of Transcription and Metabolism in Glioblastoma. Clin. Cancer Res. 2018, 24, 1124. [Google Scholar] [CrossRef]
- Liu, W.; Ma, Q.; Wong, K.; Li, W.; Ohgi, K.; Zhang, J.; Aggarwal, A.; Rosenfeld, M.G. Brd4 and JMJD6-associated anti-pause enhancers in regulation of transcriptional pause release. Cell 2013, 155, 1581–1595. [Google Scholar] [CrossRef]
- Vos, S.M.; Farnung, L.; Boehning, M.; Wigge, C.; Linden, A.; Urlaub, H.; Cramer, P. Structure of activated transcription complex Pol II–DSIF–PAF–SPT6. Nature 2018, 560, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.E.; Liau, B.B.; Wallace, L.C.; Morton, A.R.; Xie, Q.; Dixit, D.; Factor, D.C.; Kim, L.J.Y.; Morrow, J.J.; Wu, Q.; et al. Transcription elongation factors represent in vivo cancer dependencies in glioblastoma. Nature 2017, 547, 355–359. [Google Scholar] [CrossRef]
- Bhutada, I.; Chellappan, S.; Padmanabhan, J. Abstract 2310: Targeting transcription-associated CDKs is an effective way to combat glioblastoma and medulloblastoma with minimal effect on primary neurons. Cancer Res. 2018, 78, 2310. [Google Scholar] [CrossRef]
- Guadamillas, M.C.; Cerezo, A.; del Pozo, M.A. Overcoming anoikis—Pathways to anchorage-independent growth in cancer. J. Cell Sci. 2011, 124, 3189. [Google Scholar] [CrossRef]
- Le Rhun, E.; von Achenbach, C.; Lohmann, B.; Silginer, M.; Schneider, H.; Meetze, K.; Szabo, E.; Weller, M. Profound, durable and MGMT-independent sensitivity of glioblastoma cells to cyclin-dependent kinase inhibition. Int. J. Cancer 2019, 145, 242–253. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, J.; Yin, J.; Gan, Y.; Xu, S.; Gu, Y.; Huang, W. Alternative approaches to target Myc for cancer treatment. Signal Transduct. Target. Ther. 2021, 6, 117. [Google Scholar] [CrossRef]
- Tsang, J.; Sung, S.; Gosa, L.; Meetze, K.; Cloughesy, T.; Nathanson, D. EXTH-67. TG02, A brain-penetrant multi-cdk inhibitor, potently suppresses myc-driven glioblastoma. Neuro-Oncology 2017, 19, vi87–vi88. [Google Scholar] [CrossRef]
- Huang, C.-H.; Lujambio, A.; Zuber, J.; Tschaharganeh, D.F.; Doran, M.G.; Evans, M.J.; Kitzing, T.; Zhu, N.; de Stanchina, E.; Sawyers, C.L.; et al. CDK9-mediated transcription elongation is required for MYC addiction in hepatocellular carcinoma. Genes Dev. 2014, 28, 1800–1814. [Google Scholar] [CrossRef]
- A Dose Escalation and Cohort Expansion Study of KB-0742 in Participants with Relapsed or Refractory Solid Tumors or Non-Hodgkin Lymphoma. Available online: https://ClinicalTrials.gov/show/NCT04718675 (accessed on 15 April 2021).
- Zheng, F.; Yue, C.; Li, G.; He, B.; Cheng, W.; Wang, X.; Yan, M.; Long, Z.; Qiu, W.; Yuan, Z.; et al. Nuclear AURKA acquires kinase-independent transactivating function to enhance breast cancer stem cell phenotype. Nat. Commun. 2016, 7, 10180. [Google Scholar] [CrossRef]
- Lu, H.; Xue, Y.; Yu, G.K.; Arias, C.; Lin, J.; Fong, S.; Faure, M.; Weisburd, B.; Ji, X.; Mercier, A.; et al. Compensatory induction of MYC expression by sustained CDK9 inhibition via a BRD4-dependent mechanism. eLife 2015, 4, e06535. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Liang, Y.; Tan, Y.; Tang, Y.; Song, H.; Wang, Z.; Li, Y.; Lu, M. CDK9 inhibitors reactivate p53 by downregulating iASPP. Cell Signal. 2020, 67, 109508. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Kang, J.; Liu, F.; Wen, S.; Zeng, X.; Liu, K.; Luo, Y.; Ji, X.; Zhao, S. Overexpression of iASPP-SV in glioma is associated with poor prognosis by promoting cell viability and antagonizing apoptosis. Tumor Biol. 2016, 37, 6323–6330. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.H.; Li, C.H.; He, Q.; Tong, J.H.M.; To, K.-F.; Chen, Y. The Establishment of CDK9/ RNA PolII/H3K4me3/DNA Methylation Feedback Promotes HOTAIR Expression by RNA Elongation Enhancement in Cancer. bioRxiv 2019, 812776. [Google Scholar] [CrossRef]
- Zhou, X.; Ren, Y.; Zhang, J.; Zhang, C.; Zhang, K.; Han, L.; Kong, L.; Wei, J.; Chen, L.; Yang, J.; et al. HOTAIR is a therapeutic target in glioblastoma. Oncotarget 2015, 6, 8353–8365. [Google Scholar] [CrossRef]
- Pastori, C.; Kapranov, P.; Penas, C.; Peschansky, V.; Volmar, C.-H.; Sarkaria, J.N.; Bregy, A.; Komotar, R.; St. Laurent, G.; Ayad, N.G.; et al. The Bromodomain protein BRD4 controls HOTAIR, a long noncoding RNA essential for glioblastoma proliferation. Proc. Natl. Acad. Sci. USA 2015, 112, 8326. [Google Scholar] [CrossRef]
- Wang, J.; Wakeman, T.P.; Lathia, J.D.; Hjelmeland, A.B.; Wang, X.-F.; White, R.R.; Rich, J.N.; Sullenger, B.A. Notch Promotes Radioresistance of Glioma Stem Cells. Stem Cells 2010, 28, 17–28. [Google Scholar] [CrossRef]
- Wolf, A.; Agnihotri, S.; Munoz, D.; Guha, A. Developmental profile and regulation of the glycolytic enzyme hexokinase 2 in normal brain and glioblastoma multiforme. Neurobiol. Dis. 2011, 44, 84–91. [Google Scholar] [CrossRef]
- Yang, W.; Xia, Y.; Ji, H.; Zheng, Y.; Liang, J.; Huang, W.; Gao, X.; Aldape, K.; Lu, Z. Nuclear PKM2 regulates β-catenin transactivation upon EGFR activation. Nature 2011, 480, 118–122. [Google Scholar] [CrossRef]
- Di, H.; Zhang, X.; Guo, Y.; Shi, Y.; Fang, C.; Yuan, Y.; Wang, J.; Shang, C.; Guo, W.; Li, C. Silencing LDHA inhibits proliferation, induces apoptosis and increases chemosensitivity to temozolomide in glioma cells. Oncol. Lett. 2018, 15, 5131–5136. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Liu, H.; Herrmann, C.H.; Chiang, K.; Sung, T.-L.; Moon, S.-H.; Donehower, L.A.; Rice, A.P. 55K isoform of CDK9 associates with Ku70 and is involved in DNA repair. Biochem. Biophys. Res. Commun. 2010, 397, 245–250. [Google Scholar] [CrossRef]
- Yu, D.S.; Zhao, R.; Hsu, E.L.; Cayer, J.; Ye, F.; Guo, Y.; Shyr, Y.; Cortez, D. Cyclin-dependent kinase 9–cyclin K functions in the replication stress response. Embo. Rep. 2010, 11, 876–882. [Google Scholar] [CrossRef]
- Storch, K.; Cordes, N. The impact of CDK9 on radiosensitivity, DNA damage repair and cell cycling of HNSCC cancer cells. Int. J. Oncol. 2016, 48, 191–198. [Google Scholar] [CrossRef]
- Rasmussen, R.D.; Gajjar, M.K.; Tuckova, L.; Jensen, K.E.; Maya-Mendoza, A.; Holst, C.B.; Møllgaard, K.; Rasmussen, J.S.; Brennum, J.; Bartek, J.; et al. BRCA1-regulated RRM2 expression protects glioblastoma cells from endogenous replication stress and promotes tumorigenicity. Nat. Commun. 2016, 7, 13398. [Google Scholar] [CrossRef]
- Nepomuceno, T.C.; Fernandes, V.C.; Gomes, T.T.; Carvalho, R.S.; Suarez-Kurtz, G.; Monteiro, A.N.; Carvalho, M.A. BRCA1 recruitment to damaged DNA sites is dependent on CDK9. Cell Cycle 2017, 16, 665–672. [Google Scholar] [CrossRef]
- Ning, J.-F.; Stanciu, M.; Humphrey, M.R.; Gorham, J.; Wakimoto, H.; Nishihara, R.; Lees, J.; Zou, L.; Martuza, R.L.; Wakimoto, H.; et al. Myc targeted CDK18 promotes ATR and homologous recombination to mediate PARP inhibitor resistance in glioblastoma. Nat. Commun. 2019, 10, 2910. [Google Scholar] [CrossRef]
- Li, J.; Zhi, X.; Chen, S.; Shen, X.; Chen, C.; Yuan, L.; Guo, J.; Meng, D.; Chen, M.; Yao, L. CDK9 inhibitor CDKI-73 is synergetic lethal with PARP inhibitor olaparib in BRCA1 wide-type ovarian cancer. Am. J. Cancer Res. 2020, 10, 1140–1155. [Google Scholar]
- Sizemore, S.T.; Mohammad, R.; Sizemore, G.M.; Nowsheen, S.; Yu, H.; Ostrowski, M.C.; Chakravarti, A.; Xia, F. Synthetic Lethality of PARP Inhibition and Ionizing Radiation is p53-dependent. Mol. Cancer Res. 2018, 16, 1092. [Google Scholar] [CrossRef]
- Carén, H.; Pollard, S.M.; Beck, S. The good, the bad and the ugly: Epigenetic mechanisms in glioblastoma. Mol. Asp. Med. 2013, 34, 849–862. [Google Scholar] [CrossRef]
- Martinez, R.; Schackert, G. Epigenetic Aberrations in Malignant Gliomas: An Open Door Leading to Better Understanding and Treatment. Epigenetics 2007, 2, 147–150. [Google Scholar] [CrossRef][Green Version]
- Lohmann, B.; Le Rhun, E.; Silginer, M.; Epskamp, M.; Weller, M. Interferon-β sensitizes human glioblastoma cells to the cyclin-dependent kinase inhibitor, TG02. Oncol. Lett. 2020, 19, 2649–2656. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Yuan, Y.; Long Priel, D.A.; Fink, D.; Peer, C.J.; Sissung, T.M.; Su, Y.-T.; Pang, Y.; Yu, G.; Butler, M.K.; et al. Phase I Study of Zotiraciclib in Combination with Temozolomide for Patients with Recurrent High-grade Astrocytomas. Clin. Cancer Res. 2021. [Google Scholar] [CrossRef] [PubMed]
- Attermann, A.S.; Bjerregaard, A.M.; Saini, S.K.; Grønbæk, K.; Hadrup, S.R. Human endogenous retroviruses and their implication for immunotherapeutics of cancer. Ann. Oncol. 2018, 29, 2183–2191. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Zhang, N.; An, Z.; Zheng, W. Abstract B37: Analysis of the differential expression of human endogenous retrovirus in glioblastoma multiforme. Cancer Res. 2020, 80, B37. [Google Scholar] [CrossRef]
- Hossain, D.M.S.; Javaid, S.; Cai, M.; Zhang, C.; Sawant, A.; Hinton, M.; Sathe, M.; Grein, J.; Blumenschein, W.; Pinheiro, E.M.; et al. Dinaciclib induces immunogenic cell death and enhances anti-PD1-mediated tumor suppression. J. Clin. Investig. 2018, 128, 644–654. [Google Scholar] [CrossRef]
- Rajani, K.R.; Carlstrom, L.P.; Parney, I.F.; Johnson, A.J.; Warrington, A.E.; Burns, T.C. Harnessing Radiation Biology to Augment Immunotherapy for Glioblastoma. Front Oncol. 2019, 8, 656. [Google Scholar] [CrossRef]
- Riess, C.; Schneider, B.; Kehnscherper, H.; Gesche, J.; Irmscher, N.; Shokraie, F.; Classen, C.F.; Wirthgen, E.; Domanska, G.; Zimpfer, A.; et al. Activation of the Kynurenine Pathway in Human Malignancies Can Be Suppressed by the Cyclin-Dependent Kinase Inhibitor Dinaciclib. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Hellvard, A.; Zeitlmann, L.; Heiser, U.; Kehlen, A.; Niestroj, A.; Demuth, H.-U.; Koziel, J.; Delaleu, N.; Jan, P.; Mydel, P. Inhibition of CDK9 as a therapeutic strategy for inflammatory arthritis. Sci. Rep. 2016, 6, 31441. [Google Scholar] [CrossRef]
- Chen, R.; Tsai, J.; Thompson, P.A.; Chen, Y.; Xiong, P.; Liu, C.; Burrows, F.; Sivina, M.; Burger, J.A.; Keating, M.J.; et al. The multi-kinase inhibitor TG02 induces apoptosis and blocks B-cell receptor signaling in chronic lymphocytic leukemia through dual mechanisms of action. Blood Cancer J. 2021, 11, 57. [Google Scholar] [CrossRef]
- Chen, E.W.; Tay, N.Q.; Brzostek, J.; Gascoigne, N.R.J.; Rybakin, V. A Dual Inhibitor of Cdc7/Cdk9 Potently Suppresses T Cell Activation. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Wu, T.; Qin, Z.; Tian, Y.; Wang, J.; Xu, C.; Li, Z.; Bian, J. Recent Developments in the Biology and Medicinal Chemistry of CDK9 Inhibitors: An Update. J. Med. Chem. 2020, 63, 13228–13257. [Google Scholar] [CrossRef]
- Romano, G. Deregulations in the cyclin-dependent kinase-9-related pathway in cancer: Implications for drug discovery and development. ISRN Oncol. 2013, 2013, 305371. [Google Scholar] [CrossRef]
- Scott, E.N.; Thomas, A.L.; Molife, L.R.; Ahmed, S.; Blagden, S.; Fong, P.C.; Kowal, K.; McCoy, C.; Wiesinger, H.; Steward, W.; et al. A phase I dose escalation study of the pharmacokinetics and tolerability of ZK 304709, an oral multi-targeted growth inhibitor (MTGI™), in patients with advanced solid tumours. Cancer Chemother. Pharmacol. 2009, 64, 425–429. [Google Scholar] [CrossRef]
- Heath, E.I.; Bible, K.; Martell, R.E.; Adelman, D.C.; LoRusso, P.M. A phase 1 study of SNS-032 (formerly BMS-387032), a potent inhibitor of cyclin-dependent kinases 2, 7 and 9 administered as a single oral dose and weekly infusion in patients with metastatic refractory solid tumors. Investig. New Drugs 2008, 26, 59–65. [Google Scholar] [CrossRef]
- Richters, A.; Doyle, S.K.; Freeman, D.B.; Lee, C.; Leifer, B.S.; Jagannathan, S.; Kabinger, F.; Koren, J.V.; Struntz, N.B.; Urgiles, J.; et al. Modulating Androgen Receptor-Driven Transcription in Prostate Cancer with Selective CDK9 Inhibitors. Cell Chem. Biol. 2021, 28, 134–147.e114. [Google Scholar] [CrossRef]
- Colotta, F.; Moll, J.; Valsasina, B.; Vanotti, E.; Rainoldi, S.; Sola, F.; Marchesi, V.; Menichincheri, M.; Ciavolella, A.; Patton, V.; et al. Abstract DD01-01: NMS-1116354: More than an inhibitor of Cdc 7 kinase in S-phase. Cancer Res. 2010, 70, DD01-01. [Google Scholar] [CrossRef]
- Van der Biessen, D.A.J.; Burger, H.; de Bruijn, P.; Lamers, C.H.J.; Naus, N.; Loferer, H.; Wiemer, E.A.C.; Mathijssen, R.H.J.; de Jonge, M.J.A. Phase I Study of RGB-286638, A Novel, Multitargeted Cyclin-Dependent Kinase Inhibitor in Patients with Solid Tumors. Clin. Cancer Res. 2014, 20, 4776. [Google Scholar] [CrossRef]
- Hafner, M.; Mills, C.E.; Subramanian, K.; Chen, C.; Chung, M.; Boswell, S.A.; Everley, R.A.; Liu, C.; Walmsley, C.S.; Juric, D.; et al. Multiomics Profiling Establishes the Polypharmacology of FDA-Approved CDK4/6 Inhibitors and the Potential for Differential Clinical Activity. Cell Chem. Biol. 2019, 26, 1067–1080.e1068. [Google Scholar] [CrossRef]
- Braal, C.L.; Jongbloed, E.M.; Wilting, S.M.; Mathijssen, R.H.J.; Koolen, S.L.W.; Jager, A. Inhibiting CDK4/6 in Breast Cancer with Palbociclib, Ribociclib, and Abemaciclib: Similarities and Differences. Drugs 2021, 81, 317–331. [Google Scholar] [CrossRef]
- Portman, N.; Alexandrou, S.; Carson, E.; Wang, S.; Lim, E.; Caldon, C.E. Overcoming CDK4/6 inhibitor resistance in ER-positive breast cancer. Endocr. Relat. Cancer 2019, 26, R15–R30. [Google Scholar] [CrossRef]
- Zotiraciclib (TG02) Plus Dose-Dense or Metronomic Temozolomide Followed by Randomized Phase II Trial of Zotiraciclib (TG02) Plus Temozolomide Versus Temozolomide Alone in Adults with Recurrent Anaplastic Astrocytoma and Glioblastoma. Available online: https://ClinicalTrials.gov/show/NCT02942264 (accessed on 5 April 2021).
- Phase I Clinical Study of Oral TG02 Capsule in the Treatment of Recurrent Progressive High-grade Glioma Patients. Available online: https://ClinicalTrials.gov/show/NCT03904628 (accessed on 5 April 2021).
- Study of TG02 in Elderly Newly Diagnosed or Adult Relapsed Patients with Anaplastic Astrocytoma or Glioblastoma. Available online: https://ClinicalTrials.gov/show/NCT03224104 (accessed on 5 April 2021).



{kind=link}
{kind=link}
| No. | Inhibitor Name | Targets (Including CDK9) | Investigated in Clinical Trial for Gliomas | Clinical Trial (with NCT Identifier from ClinicalTrials.gov accessed on 16 April 2021), If Applicable) | Phase | Trial Status | Cancer Type |
|---|---|---|---|---|---|---|---|
| 1 | AT7519 | CDK1, CDK2, CDK4, CDK5, CDK6, CDK9 [78] | N/A | NCT01652144: A Phase II Study of AT7519M, a CDK Inhibitor, in Patients with Relapsed Mantle Cell Lymphoma | II | Completed | Mantle Cell Lymphoma |
| NCT01627054: A Phase II Study of AT7519M, a CDK Inhibitor, in Patients with Relapsed and/or Refractory Chronic Lymphocytic Leukemia | II | Completed | Refractory Chronic Lymphocytic Leukemia | ||||
| NCT01183949: Effect of AT7519M Alone and AT7519M Plus Bortezomib in Patients with Previously Treated Multiple Myeloma | I/II | Completed | Multiple Myeloma | ||||
| NCT02503709: Onalespib and CDKI AT7519 in Treating Patients with Solid Tumors That Are Metastatic or Cannot Be Removed by Surgery | I | Active, not recruiting | Advanced Malignant Solid Neoplasms, Metastatic Malignant Solid Neoplasms, and Unresectable Solid Neoplasms | ||||
| NCT00390117: AT7519M in Treating Patients with Advanced or Metastatic Solid Tumors or Refractory Non-Hodgkin’s Lymphoma | I | Completed | Advanced or Metastatic Solid Tumors and Refractory Non-Hodgkin’s Lymphoma | ||||
| 2 | Atuveciclib/BAY-1143572 | CDK2 and CDK9 [78] | N/A | NCT02345382: Phase I Dose Escalation of BAY1143572 in Subjects with Acute Leukemia | I | Completed | Acute Leukemia |
| NCT01938638: Open Label Phase I Dose Escalation Study with BAY1143572 in Patients with Advanced Cancer | I | Completed | Advanced Cancers | ||||
| 3 | AZD-4573 | CDK1 and CDK9 [78] | N/A | NCT03263637: Study to Assess Safety, Tolerability, Pharmacokinetics and Antitumor Activity of AZD4573 in Relapsed/Refractory Haematological Malignancies | I | Recruiting | Relapsed/Refractory Haematological Malignancies |
| 4 | BAY-1251152 | CDK9 [78] | N/A | NCT02745743: Phase I Trial of BAY1251152 for Advanced Blood Cancers | I | Completed | Advanced Blood Cancers |
| NCT02635672: Phase I Dose Escalation Study for BAY 1251152 in Patients with Advanced Cancer | I | Active, not recruiting | Advanced Cancers | ||||
| 5 | BTX-A51 | CDK7 and CDK9 [78] | N/A | NCT04243785: A Study of BTX-A51 in People with Relapsed or Refractory Acute Myeloid Leukemia or High-Risk Myelodysplastic Syndrome | I | Recruiting | Acute Myeloid Leukemiad and Myelodysplastic Syndrome |
| 6 | CYC065/Fadraciclib | CDK2, CDK5, CDK7, CDK9 [78] | N/A | NCT03739554: CYC065 CDK Inhibitor and Venetoclax Study in Relapsed/Refractory CLL | I | Recruiting | Relapsed or Refractory Chronic Lymphocytic Leukemia |
| NCT04017546: CYC065 CDK Inhibitor and Venetoclax Study in Relapsed/Refractory AML or MDS | I | Recruiting | Acute Myeloid Leukemia and Myelodysplastic Syndromes | ||||
| 7 | Dinaciclib/SCH-727965 | CDK1, CDK2, CDK5, CDK9 [78] | N/A | NCT00732810: SCH-727965 in Patients with Advanced Breast and Lung Cancers | II | Completed | Breast Neoplasms and Non-Small-Cell Lung Cancer |
| NCT00798213: SCH-727965 in Patients with Acute Myelogenous Leukemia and Acute Lymphoblastic Leukemia | II | Terminated | Acute Myelogenous Leukemia and Acute Lymphoblastic Leukemia | ||||
| NCT00871663: Phase 1 Weekly Dosing of SCH 727965 in Patients with Advanced Cancer | II | Completed | Solid Tumors, Non-Hodgkin Lymphoma, Multiple Myeloma, and Chronic Lymphocytic Leukemia | ||||
| NCT01096342: Dinaciclib in Treating Patients with Relapsed or Refractory Multiple Myeloma | II | Completed | Refractory Multiple Myeloma | ||||
| NCT01650727: A Study of Dinaciclib in Combination with Rituximab in Participants with Chronic Lymphocytic Leukemia and Small Lymphocytic Lymphoma | I | Completed | Chronic Lymphocytic Leukemia and Small Lymphocytic Lymphoma | ||||
| 8 | Flavopiridol/Alvocidib | CDK1, CDK2, CDK4, CDK5, CDK6, CDK7, CDK9, GSK3β [32,77,78] | N/A | NCT00083122: Cisplatin and Flavopiridol in Treating Patients with Advanced Ovarian Epithelial Cancer or Primary Peritoneal Cancer | II | Completed | Ovarian Epithelial Cancer and Primary Peritoneal Cancer |
| NCT00407966: Alvocidib, Cytarabine, and Mitoxantrone in Treating Patients with Newly Diagnosed Acute Myeloid Leukemia | II | Completed | Acute Myeloid Leukemia | ||||
| NCT00464633: Alvocidib in Patients with Previously Treated Chronic Lymphocytic Leukemia or Prolymphocytic Leukemia Arising From Chronic Lymphocytic Leukemia (CLL) | II | Completed | Chronic Lymphocytic Leukemia, Prolymphocytic Leukemia arising from Chronic Lymphocytic Leukemia | ||||
| NCT03593915: Study of Alvocidib Plus Decitabine or Azacitidine in Patients with MDS | Ib/II | Active, not recruiting | Myelodysplastic Syndromes | ||||
| NCT00112723: Flavopiridol in Treating Patients with Relapsed or Refractory Lymphoma or Multiple Myeloma | I/II | Terminated | Lymphoma and Multiple Myeloma | ||||
| NCT00112684: Alvocidib in Treating Patients with Locally Advanced or Metastatic Solid Tumors | I | Terminated | Advanced or Metastatic Solid Tumors | ||||
| NCT00082784: Bortezomib and Flavopiridol in Treating Patients with Recurrent or Refractory Indolent B-Cell Neoplasms | I | Completed | B-Cell Neoplasms | ||||
| NCT00470197: Flavopiridol, Cytarabine, and Mitoxantrone in Treating Patients with Relapsed or Refractory Acute Leukemia | I | Completed | Relapsed or Refractory Acute Leukemia | ||||
| 9 | KB-0742 | CDK9 [82] | N/A | NCT04718675: A Dose Escalation and Cohort Expansion Study of KB-0742 in Participants with Relapsed or Refractory Solid Tumors or Non-Hodgkin’s Lymphoma | I | Recruiting | Relapsed or Refractory Solid Tumors and Non-Hodgkin’s Lymphoma |
| 10 | NMS-1116354 | CDK9, CDC7 [77,83] | N/A | NCT01016327: Study of NMS-1116354 in Solid Tumors | I | Terminated (Discontinuation of clinical investigation of drug) | Advanced Solid Tumors |
| NCT01092052: Study of NMS-1116354 in Advanced/Metastatic Solid Tumors | I | Terminated (Discontinuation of clinical investigation of drug) | Advanced/Metastatic Solid Tumors | ||||
| 11 | RGB-286638 | CDK1, CDK2, CDK3, CDK4, CDK5, and CDK9 (less active against CDK6 and CDK7) [84] | N/A | NCT01168882: Safety and Tolerability of RGB-286638 in Patients with Selected, Relapsed or Refractory Hematological Malignancies | I | withdrawn | Hematological Malignancies |
| 12 | Riviciclib/P-276-00 | CDK1, CDK2, CDK4, CDK6, CDK9 [78] | N/A | NCT00824343: A Phase II Clinical Trial to Study the Efficacy and Safety of a New Drug P276-00 in Treatment of Recurrent and/or Locally Advanced Head and Neck Cancer (MONARCH) | II | Completed | Advanced Head and Neck Cancer |
| NCT00843050: A Phase II Study to Evaluate Efficacy and Safety of P276-00 in Relapsed and/or Refractory Mantle Cell Lymphoma | II | Terminated (based on interim results; no major safety or tolerability concerns) | Mantle Cell Lymphoma | ||||
| NCT00898287: Safety and Efficacy Study of P276-00 in Combination with Gemcitabine in Patients with Advanced Pancreatic Cancer (SAVIOR) | I/II | Completed | Pancreatic Cancer | ||||
| NCT00899054: Safety and Efficacy Study of P276-00 in Combination with Radiation in Subjects with Advanced Head and Neck Cancer (SPARK) | I/II | Completed | Squamous Cell Carcinoma of Head and Neck | ||||
| NCT00882063: Study To Evaluate Safety and Efficacy of P276-00 in Subjects with Refractory Multiple Myeloma | I/II | Completed | Refractory Multiple Myeloma | ||||
| NCT01333137: A Clinical Trial Comparing Gemcitabine and Carboplatin with and without P276-00 in Subjects with Metastatic Triple Negative Breast Cancer, with a Run-in of Escalating Dose of P276-00 Added to Gemcitabine and Carboplatin | I | Terminated | Metastatic Triple Negative Breast Cancer | ||||
| 13 | Roniciclib/BAY-1000394 | CDK1, CDK2, CDK4, CDK5, CDK7, CDK9 [78] | N/A | NCT02656849: BAY 1000394 for MCL-1-, MYC-, and CCNE1-Amplified Tumors | II | withdrawn (Development of BAY1000394 has been terminated by Bayer) | Solid Tumors |
| NCT02161419: RONICICLIB/Placebo in Combination with Chemotherapy in Small Cell Lung Cancer (CONCEPT-SCLC) | II | Terminated | Small Cell Lung Carcinoma | ||||
| NCT02522910: An Open-label Phase Ib/II Study of BAY 1000394 (Roniciclib) in Combination with Docetaxel in Second- or Third-line Treatment of Patients with Advanced Non-small Cell Lung Cancer (NSCLC) | Ib/II | withdrawn | Non-Small Cell Lung Cancer | ||||
| NCT01573338: Clinical Study to Evaluate the Maximum Tolerated Dose of BAY1000394 When Given Together with Chemotherapy and the Effectiveness of This Combination Treatment in Shrinking a Specific Type of Lung Tumors (Small Cell Lung Cancer) | I/II | Terminated | Small Cell Lung Cancer | ||||
| NCT01188252: Clinical Study to Evaluate the Maximum Tolerated Dose of BAY1000394 Given in a 3 Days on/4 Days Off Schedule in Subjects with Advanced Malignancies | I | Completed | Advanced Malignancies | ||||
| 14 | Roscovitine/Seliciclib/CYC202 | CDK2, CDK7, CDK9, DIRK1A, ERK1 [32,78] | N/A | NCT00372073: Efficacy Study of Oral Seliciclib to Treat Non-Small Cell Lung Cancer | II | Terminated | Non-Small Cell Lung Cancer |
| NCT01333423: Maximum Tolerated Dose (MTD) of Liposomal Doxorubicin in Combination with Seliciclib for Patients with Metastatic Triple Negative Breast Cancer (TNBC) | I | withdrawn | Metastatic Triple Negative Breast Cancer | ||||
| 15 | SNS-032 | CDK1, CDK2, CDK5, CDK7, CDK9 [78] | N/A | NCT00292864: Safety Assessment of One-hour Infusions of SNS-032 for the Treatment of Select Advanced Solid Tumors | I | Terminated (based on report published by study investigators) [81] | Metastatic Refractory Solid Tumors |
| NCT00446342: Study of Intravenously Administered SNS-032 in Patients with Advanced B-lymphoid Malignancies | I | Completed | B-lymphoid Malignancies, Chronic Lymphocytic Leukemia and Mantle Cell Lymphoma, and Multiple Myeloma | ||||
| 16 | TP-1287 | CDK1, CDK2, CDK4, CDK6, CDK7, CDK9 [78] | N/A | NCT03604783: Phase I, First-in-human Study of Oral TP-1287 in Patients with Advanced Solid Tumors | I | Recruiting | Advanced Solid Tumors |
| 17 | Voruciclib/P-1446 | CDK1, CDK2, CDK4, CDK5, CDK6, CDK8, CDK9 [78] | N/A | NCT03547115: A Phase 1 Study of Voruciclib in Subjects with B-Cell Malignancies or AML | I | Recruiting | Follicular Lymphoma, Mantle Cell Lymphoma, Marginal Zone Lymphoma, Small Lymphocytic Lymphoma, Chronic Lymphocytic Leukemia, and Diffuse Large B-cell Lymphoma, Acute Myeloid Leukemia |
| 18 | ZK-304709 | CDK1, CDK2, CDK4, CDK7, CDK9, VEGFR1, VEGFR2, VEGFR3, PDGFRβ, FLT3 [78,79] | N/A | (NCT Indicator N/A): A Phase I Dose Escalation Study of the Pharmacokinetics and Tolerability of ZK-304709, an Oral Multi-Targeted Growth Inhibitor (MTGI™), in Patients with Advanced Solid Tumors [80] | I | Terminated | Advanced Solid Tumors |
| 19 | Zotiraciclib/TG02/SB1317 | CDK1, CDK2, CDK5, CDK7, CDK9, JAK2, FLT3 [26] | Yes | NCT02942264: Zotiraciclib (TG02) Plus Dose-Dense or Metronomic Temozolomide Followed by Randomized Phase II Trial of Zotiraciclib (TG02) Plus Temozolomide Versus Temozolomide Alone in Adults with Recurrent Anaplastic Astrocytoma and Glioblastoma | I/II | Phase I Completed | Recurrent Anaplastic Astrocytoma and Glioblastoma |
| NCT03224104: Study of TG02 in Elderly Newly Diagnosed or Adult Relapsed Patients with Anaplastic Astrocytoma or Glioblastoma (STEAM) | I | Recruiting | Anaplastic Astrocytoma and Glioblastoma | ||||
| NCT01204164: Phase I Clinical Study of Oral TG02 Capsule in the Treatment of Recurrent/Progressive High-grade Glioma Patients | I | Recruiting | Recurrent/Progressive High-Grade Glioma | ||||
| NCT03738111: Study of TG02 Citrate in Patients with Advanced Hepatocellular Carcinoma | I | withdrawn | Advanced Hepatocellular Carcinoma | ||||
| NCT02933944: Exploratory Study of TG02-treatment as Monotherapy or in Combination with Pembrolizumab to Assess Safety and Immune Activation in Patients with Locally Advanced Primary and Recurrent Oncogenic RAS Exon 2 Mutant Colorectal Cancer | I | Terminated | Colorectal Cancer | ||||
| NCT01204164: Phase 1 Study of TG02 Citrate in Patients with Advanced Hematological Malignancies (TG02-101) | I | Completed | Advanced Hematological Malignancies | ||||
| NCT01699152: Phase 1 Study of TG02 Citrate in Patients with Chronic Lymphocytic Leukemia and Small Lymphocytic Lymphoma | I | Completed | Chronic Lymphocytic Leukemia and Small Lymphocytic Lymphoma |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ranjan, A.; Pang, Y.; Butler, M.; Merchant, M.; Kim, O.; Yu, G.; Su, Y.-T.; Gilbert, M.R.; Levens, D.; Wu, J. Targeting CDK9 for the Treatment of Glioblastoma. Cancers 2021, 13, 3039. https://doi.org/10.3390/cancers13123039
Ranjan A, Pang Y, Butler M, Merchant M, Kim O, Yu G, Su Y-T, Gilbert MR, Levens D, Wu J. Targeting CDK9 for the Treatment of Glioblastoma. Cancers. 2021; 13(12):3039. https://doi.org/10.3390/cancers13123039
Chicago/Turabian StyleRanjan, Alice, Ying Pang, Madison Butler, Mythili Merchant, Olga Kim, Guangyang Yu, Yu-Ting Su, Mark R. Gilbert, David Levens, and Jing Wu. 2021. "Targeting CDK9 for the Treatment of Glioblastoma" Cancers 13, no. 12: 3039. https://doi.org/10.3390/cancers13123039
APA StyleRanjan, A., Pang, Y., Butler, M., Merchant, M., Kim, O., Yu, G., Su, Y.-T., Gilbert, M. R., Levens, D., & Wu, J. (2021). Targeting CDK9 for the Treatment of Glioblastoma. Cancers, 13(12), 3039. https://doi.org/10.3390/cancers13123039