BRAF Mutated Colorectal Cancer: New Treatment Approaches

 , , ,
, , ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
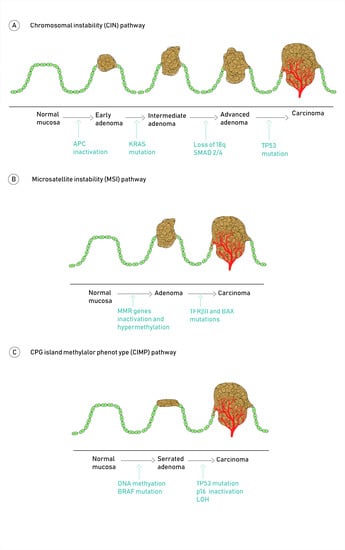
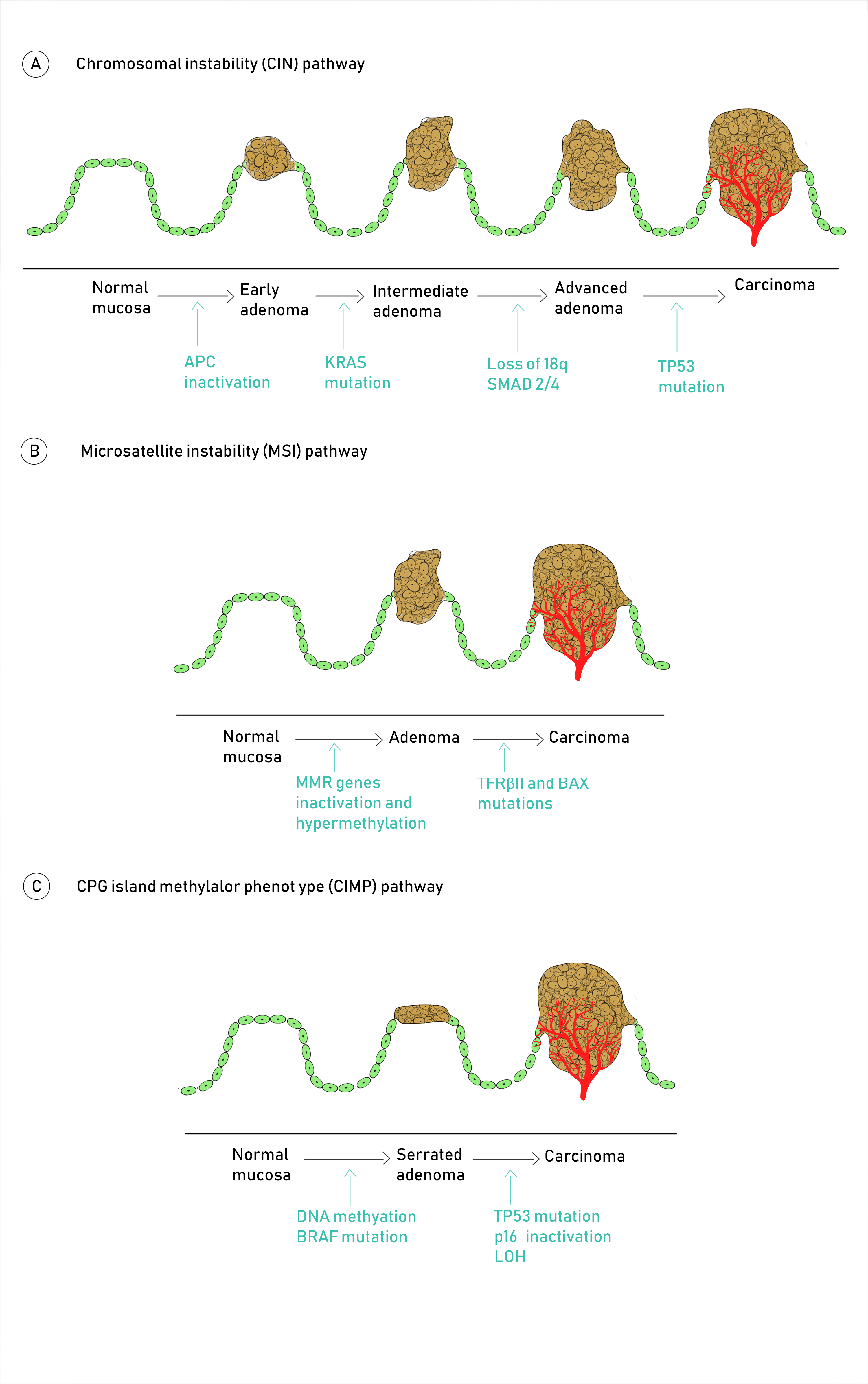
2. Molecular Pathogenesis of Colorectal Cancer
Molecular Pathways Involved in CRC Carcinogenesis
3. Consensus Molecular Subtypes
4. BRAF Mutations in Colorectal Cancers
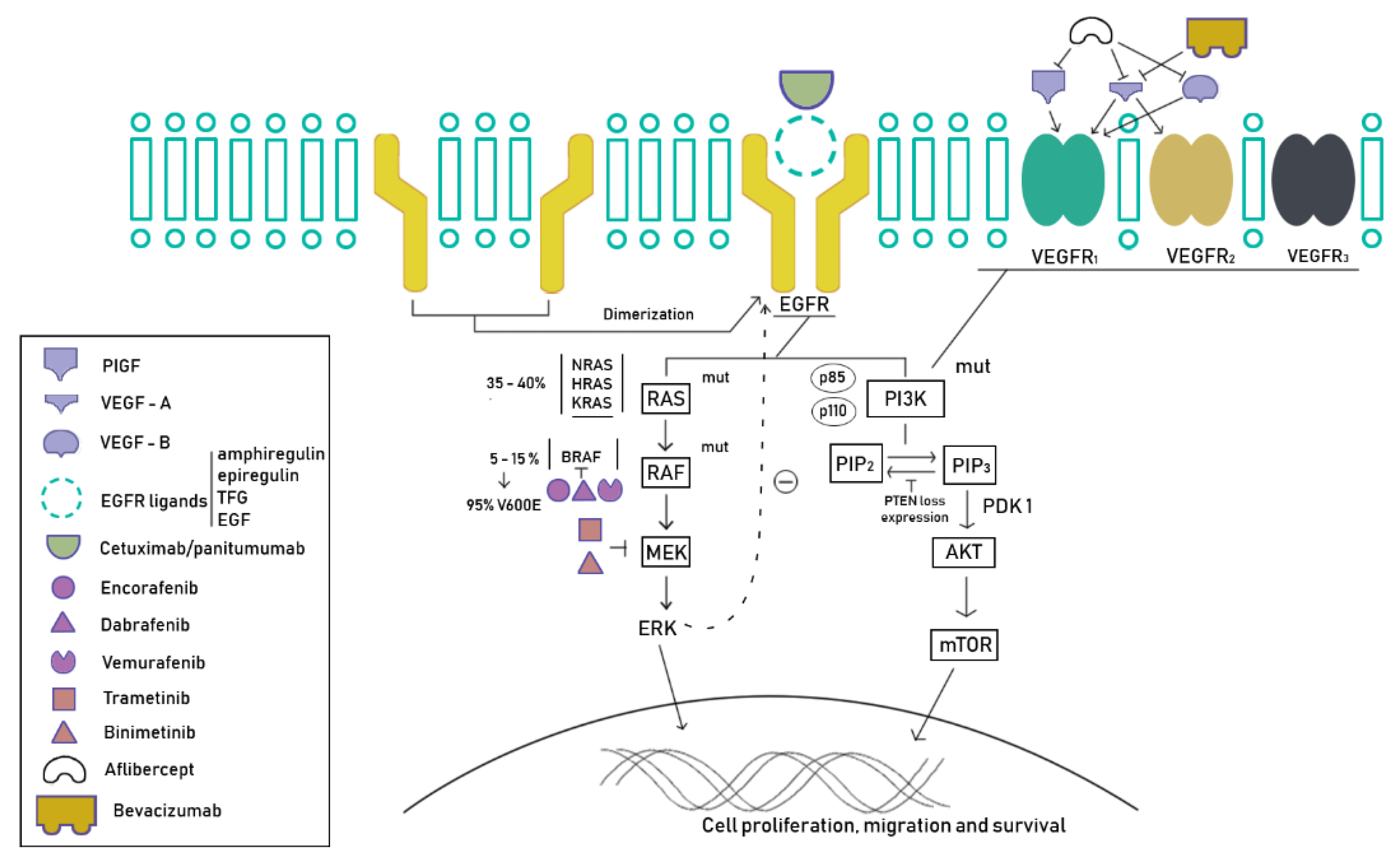
5. New Strategies
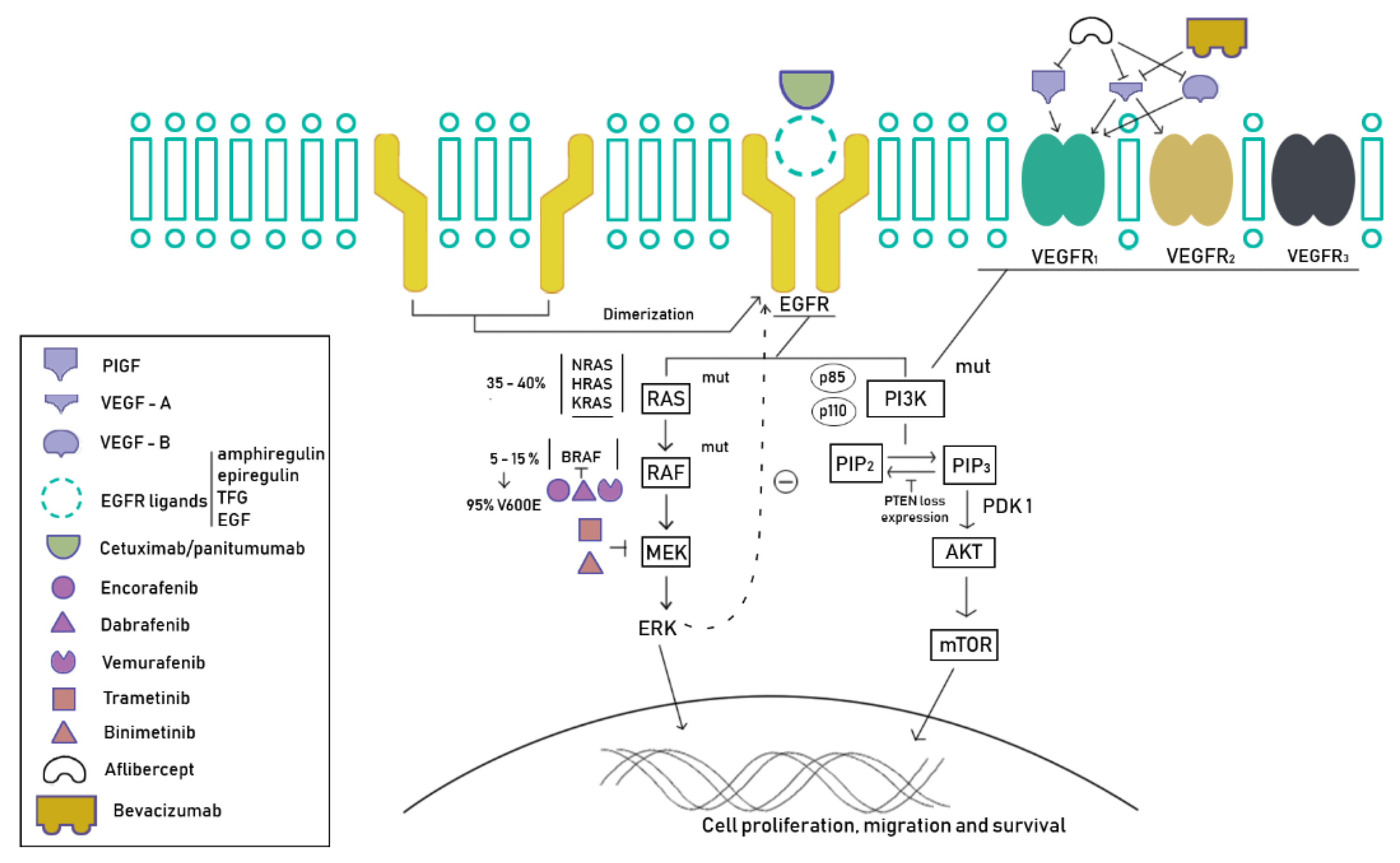
5.1. Tyrosine Kinase Inhibitors
5.2. Immunotherapy
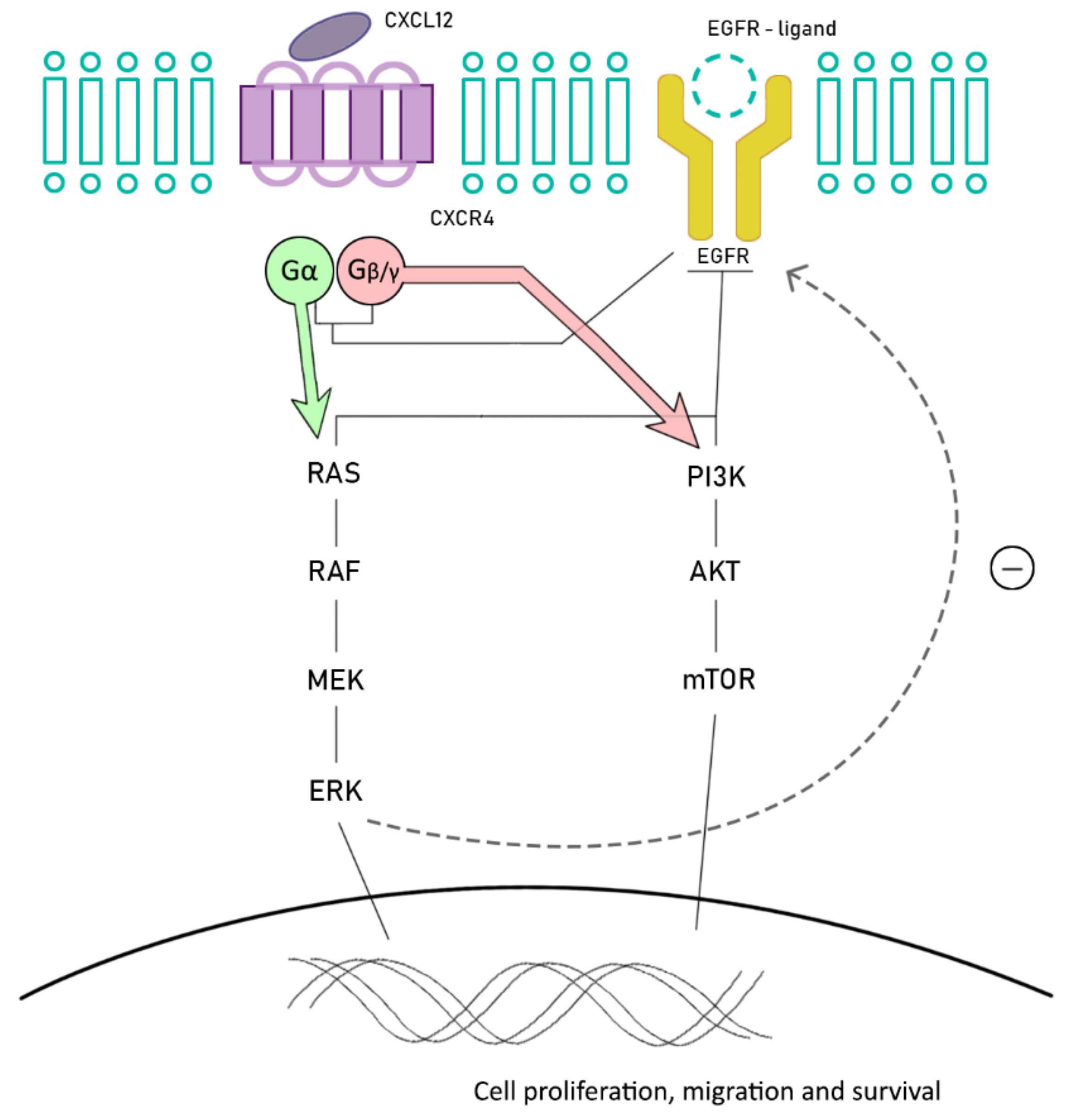
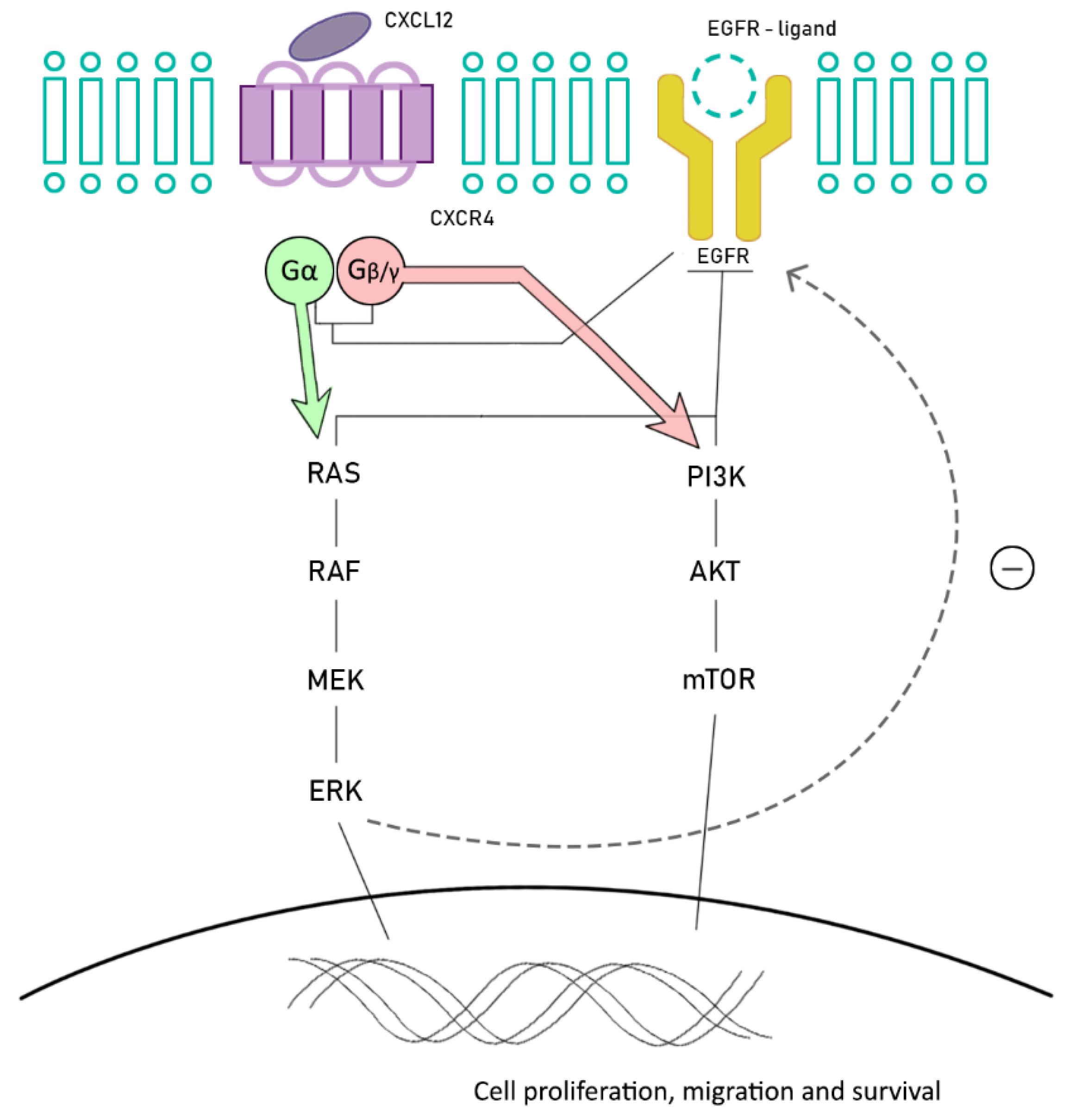
6. Future Promising Strategies: Chemokine Receptors
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rawla, P.; Sunkara, T.; Barsouk, A. Epidemiology of colorectal cancer: Incidence, mortality, survival, and risk factors. Przeglad Gastroenterol. 2019, 14, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Gómez-España, M.A.; Gallego, J.; González-Flores, E.; Maurel, J.; Páez, D.; Sastre, J.; Aparicio, J.; Benavides, M.; Feliu, J.; Vera, R. SEOM clinical guidelines for diagnosis and treatment of metastatic colorectal cancer (2018). Clin. Transl. Oncol. 2019, 21, 46–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linevsky, J.K. Management of Gastrointestinal Diseases; Winawer, S.J., Ed.; Gower Medical Publishing: New York, NY, USA, 1992; p. 1050. [Google Scholar]
- Carini, F.; Mazzola, M.; Rappa, F.; Jurjus, A.; Geagea, A.G.; Al Kattar, S.; Bou-Assi, T.; Jurjus, R.; Damiani, P.; Leone, A.; et al. Colorectal Carcinogenesis: Role of Oxidative Stress and Antioxidants. Anticancer Res. 2017, 37, 4759–4766. [Google Scholar]
- Zauber, A.G.; Winawer, S.J.; O’Brien, M.J.; Lansdorp-Vogelaar, I.; van Ballegooijen, M.; Hankey, B.F.; Shi, W.; Bond, J.H.; Schapiro, M.; Panish, J.F.; et al. Colonoscopic polypectomy and long-term prevention of colorectal-cancer deaths. N. Engl. J. Med. 2012, 366, 687–696. [Google Scholar] [CrossRef]
- De Palma, F.; D’argenio, V.; Pol, J.; Kroemer, G.; Maiuri, M.C.; Salvatore, F. The molecular hallmarks of the serrated pathway in colorectal cancer. Cancers 2019, 11, 1017. [Google Scholar] [CrossRef] [Green Version]
- Barault, L.; Charon-Barra, C.; Jooste, V.; de la Vega, M.F.; Martin, L.; Roignot, P.; Rat, P.; Bouvier, A.M.; Laurent-Puig, P.; Faivre, J.; et al. Hypermethylator phenotype in sporadic colon cancer: Study on a population-based series of 582 cases. Cancer Res. 2008, 68, 8541–8546. [Google Scholar] [CrossRef] [Green Version]
- Vogelstein, B.; Kinzler, K.W. The multistep nature of cancer. Trends Genet. 1993, 9, 138–141. [Google Scholar] [CrossRef]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/β-Catenin Signaling: Components, Mechanisms, and Diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef] [Green Version]
- Stanczak, A.; Stec, R.; Bodnar, L.; Olszewski, W.; Cichowicz, M.; Kozlowski, W.; Szczylik, C.; Pietrucha, T.; Wieczorek, M.; Lamparska-Przybysz, M. Prognostic significance of Wnt-1, β-catenin and E-cadherin expression in advanced colorectal carcinoma. Pathol. Oncol. Res. 2011, 17, 955–963. [Google Scholar] [CrossRef] [Green Version]
- Al-Sohaily, S.; Biankin, A.; Leong, R.; Kohonen-Corish, M.; Warusavitarne, J. Molecular pathways in colorectal cancer. J. Gastroenterol. Hepatol. 2012, 27, 1423–1431. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, C.C.; Grady, W.M. Colorectal cancer molecular biology moves into clinical practice. Gut 2011, 60, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Kastrinos, F.; Syngal, S. Inherited colorectal cancer syndromes. Cancer J. 2011, 17, 405–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markowitz, S.D.; Bertagnolli, M.M. Molecular origins of cancer: Molecular basis of colorectal cancer. N. Engl. J. Med. 2009, 361, 2449–2460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, A.M.; van Wezel, T.; van den Akker, B.E.; Ventayol García, M.; Ruano, D.; Tops, C.M.; Wagner, A.; Letteboer, T.G.; Gómez-García, E.B.; Devilee, P.; et al. Combined mismatch repair and POLE/POLD1 defects explain unresolved suspected Lynch syndrome cancers. Eur. J. Hum. Genet. 2016, 24, 1089–1092. [Google Scholar] [CrossRef]
- Tran, B.; Kopetz, S.; Tie, J.; Gibbs, P.; Jiang, Z.Q.; Lieu, C.H.; Agarwal, A.; Maru, D.M.; Sieber, O.; Desai, J. Impact of BRAF mutation and microsatellite instability on the pattern of metastatic spread and prognosis in metastatic colorectal cancer. Cancer 2011, 117, 4623–4632. [Google Scholar] [CrossRef] [Green Version]
- Jass, J.R. Serrated route to colorectal cancer: Back street or super highway? J. Pathol. 2001, 193, 283–285. [Google Scholar] [CrossRef]
- Goel, A.; Nagasaka, T.; Arnold, C.N.; Inoue, T.; Hamilton, C.; Niedzwiecki, D.; Compton, C.; Mayer, R.J.; Goldberg, R.; Bertagnolli, M.M.; et al. The CpG Island Methylator Phenotype and Chromosomal Instability Are Inversely Correlated in Sporadic Colorectal Cancer. Gastroenterology 2007, 132, 127–138. [Google Scholar] [CrossRef]
- Jass, J.R. Serrated adenoma of the colorectum and the DNA-methylator phenotype. Nat. Clin. Pract. Oncol. 2005, 2, 398–405. [Google Scholar] [CrossRef]
- Ogino, S.; Kawasaki, T.; Kirkner, G.J.; Ohnishi, M.; Fuchs, C.S. 18q loss of heterozygosity in microsatellite stable colorectal cancer is correlated with CpG island methylator phenotype-negative (CIMP-0) and inversely with CIMP-low and CIMP-high. BMC Cancer 2007, 7, 72. [Google Scholar] [CrossRef] [Green Version]
- Mojarad, E.N.; Kuppen, P.J.; Aghdaei, H.A.; Zali, M.R. The CpG island methylator phenotype (CIMP) in colorectal cancer. Gastroenterol. Hepatol. Bed Bench 2013, 6, 120–128. [Google Scholar]
- Shen, L.; Toyota, M.; Kondo, Y.; Lin, E.; Zhang, L.; Guo, Y.; Hernandez, N.S.; Chen, X.; Ahmed, S.; Konishi, K.; et al. Integrated genetic and epigenetic analysis identifies three different subclasses of colon cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 18654–18659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- French, A.J.; Sargent, D.J.; Burgart, L.J.; Foster, N.R.; Kabat, B.F.; Goldberg, R.; Shepherd, L.; Windschitl, H.E.; Thibodeau, S.N. Prognostic significance of defective mismatch repair and BRAF V600E in patients with colon cancer. Clin. Cancer Res. 2008, 14, 3408–3415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Purcell, R.V.; Visnovska, M.; Biggs, P.J.; Schmeier, S.; Frizelle, F.A. Distinct gut microbiome patterns associate with consensus molecular subtypes of colorectal cancer. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): An open-label, multicentre, phase 2 study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef]
- Lenz, H.J.; Ou, F.S.; Venook, A.P.; Hochster, H.S.; Niedzwiecki, D.; Goldberg, R.M.; Mayer, R.J.; Bertagnolli, M.M.; Blanke, C.D.; Zemla, T.; et al. Impact of consensus molecular subtype on survival in patients with metastatic colorectal cancer: Results from CALGB/SWOG 80405 (Alliance). J. Clin. Oncol. 2019, 37, 1876–1885. [Google Scholar] [CrossRef]
- Song, N.; Pogue-Geile, K.L.; Gavin, P.G.; Yothers, G.; Kim, S.R.; Johnson, N.L.; Lipchik, C.; Allegra, C.J.; Petrelli, N.J.; O’Connell, M.J.; et al. Clinical outcome from oxaliplatin treatment in stage II/III colon cancer according to intrinsic subtypes: Secondary analysis of NSABP C-07/NRG oncology randomized clinical trial. JAMA Oncol. 2016, 2, 1162–1169. [Google Scholar] [CrossRef]
- Prasetyanti, P.R.; van Hooff, S.R.; van Herwaarden, T.; de Vries, N.; Kalloe, K.; Rodermond, H.; van Leersum, R.; de Jong, J.H.; Franitza, M.; Nürnberg, P.; et al. Capturing colorectal cancer inter-tumor heterogeneity in patient-derived xenograft (PDX) models. Int. J. Cancer 2019, 144, 366–371. [Google Scholar] [CrossRef]
- Mooi, J.K.; Wirapati, P.; Asher, R.; Lee, C.K.; Savas, P.; Price, T.J.; Townsend, A.; Hardingham, J.; Buchanan, D.; Williams, D.; et al. The prognostic impact of consensus molecular subtypes (CMS) and its predictive effects for bevacizumab benefit in metastatic colorectal cancer: Molecular analysis of the AGITG MAX clinical trial. Ann. Oncol. 2018, 29, 2240–2246. [Google Scholar] [CrossRef] [PubMed]
- Thanki, K.; Nicholls, M.E.; Gajjar, A.; Senagore, A.J.; Qiu, S.; Szabo, C.; Hellmich, M.R.; Chao, C. Consensus Molecular Subtypes of Colorectal Cancer and their Clinical Implications. Int. Biol. Biomed. J. 2017, 3, 105–111. [Google Scholar] [PubMed]
- Stintzing, S.; Wirapati, P.; Lenz, H.J.; Neureiter, D.; Fischer von Weikersthal, L.; Decker, T.; Kiani, A.; Kaiser, F.; Al-Batran, S.; Heintges, T.; et al. Consensus molecular subgroups (CMS) of colorectal cancer (CRC) and first-line efficacy of FOLFIRI plus cetuximab or bevacizumab in the FIRE3 (AIO KRK-0306) trial. Ann. Oncol. 2019, 30, 1796–1803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolch, W. Meaningful relationships: The regulation of the Ras/Raf/MEK/ERK pathway by protein interactions. Biochem. J. 2000, 351, 289–305. [Google Scholar] [CrossRef]
- Ducreux, M.; Chamseddine, A.; Laurent-Puig, P.; Smolenschi, C.; Hollebecque, A.; Dartigues, P.; Samallin, E.; Boige, V.; Malka, D.; Gelli, M. Molecular targeted therapy of BRAF-mutant colorectal cancer. Ther. Adv. Med. Oncol. 2019, 11, 1758835919856494. [Google Scholar] [CrossRef] [Green Version]
- Kalady, M. Sessile Serrated Polyps: An Important Route to Colorectal Cancer. J. Natl. Compr. Cancer Netw. 2013, 11, 1585–1594. [Google Scholar] [CrossRef]
- Michaloglou, C.; Vredeveld, L.C.; Mooi, W.J.; Peeper, D.S. BRAF (E600) in benign and malignant human tumours. Oncogene 2008, 27, 877–895. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Loree, J.M.; Yu, C.; Tschautscher, M.; Briggler, A.; Overman, M.; Broaddus, R.; Meric-Bernstam, F.; Jones, J.C.; Balcom, J.; et al. Distinct impacts of KRAS, NRAS and BRAF mutations on survival of patients with metastatic colorectal cancer. J. Clin. Oncol. 2018, 36 (Suppl. 15), 3513. [Google Scholar] [CrossRef]
- Seligmann, J.F.; Fisher, D.; Smith, C.G.; Richman, S.D.; Elliott, F.; Brown, S.; Adams, R.; Maughan, T.; Quirke, P.; Cheadle, J.; et al. Investigating the poor outcomes of BRAF-mutant advanced colorectal cancer: Analysis from 2530 patients in randomised clinical trials. Ann. Oncol. 2017, 28, 562–568. [Google Scholar] [CrossRef]
- Sahin, I.H.; Kazmi, S.M.; Yorio, J.T.; Bhadkamkar, N.A.; Kee, B.K.; Garrett, C. Rare though not mutually exclusive: A report of three cases of concomitant kras and braf mutation and a review of the literature. J. Cancer 2013, 4, 320–322. [Google Scholar] [CrossRef] [Green Version]
- Barras, D.; Missiaglia, E.; Wirapati, P.; Sieber, O.M.; Jorissen, R.N.; Love, C.; Molloy, P.L.; Jones, I.T.; McLaughlin, S.; Gibbs, P.; et al. BRAF V600E mutant colorectal cancer subtypes based on gene expression. Clin. Cancer Res. 2017, 23, 104–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caputo, F.; Santini, C.; Bardasi, C.; Cerma, K.; Casadei-Gardini, A.; Spallanzani, A.; Andrikou, K.; Cascinu, S.; Gelsomino, F. BRAF-mutated colorectal cancer: Clinical and molecular insights. Int. J. Mol. Sci. 2019, 20, 5369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cremolini, C.; Di Bartolomeo, M.; Amatu, A.; Antoniotti, C.; Moretto, R.; Berenato, R.; Perrone, F.; Tamborini, E.; Aprile, G.; Lonardi, S.; et al. BRAF codons 594 and 596 mutations identify a new molecular subtype of metastatic colorectal cancer at favorable prognosis. Ann. Oncol. 2015, 26, 2092–2097. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.C.; Renfro, L.A.; Al-Shamsi, H.O.; Schrock, A.B.; Rankin, A.; Zhang, B.Y.; Kasi, P.M.; Voss, J.S.; Leal, A.D.; Sun, J.; et al. Non-V600BRAF mutations define a clinically distinct molecular subtype of metastatic colorectal cancer. J. Clin. Oncol. 2017, 35, 2624–2630. [Google Scholar] [CrossRef]
- Schirripa, M.; Biason, P.; Cortiula, F.; Pino, M.; Urbano, F.; Antoniotti, C.; Corallo, S.; Gelsomino, F.; Orlandi, A.; Loranrdi, S.; et al. Clinico-pathological and molecular characterization of BRAF mutant metastatic colorectal cancer (mCRC): Are all mutations created equal? Ann. Oncol. 2018, 29 (Suppl. 5), V58. [Google Scholar] [CrossRef]
- Yaeger, R.; Kotani, D.; Mondaca, S.; Parikh, A.R.; Bando, H.; Van Seventer, E.E.; Taniguchi, H.; Zhao, H.; Thant, C.N.; de Stanchina, E.; et al. Response to anti-EGFR therapy in patients with BRAF non-V600–mutant metastatic colorectal cancer. Clin. Cancer Res. 2019, 25, 7089–7097. [Google Scholar] [CrossRef] [Green Version]
- Lochhead, P.; Kuchiba, A.; Imamura, Y.; Liao, X.; Yamauchi, M.; Nishihara, R.; Qian, Z.R.; Morikawa, T.; Shen, J.; Meyerhardt, J.A.; et al. Microsatellite instability and braf mutation testing in colorectal cancer prognostication. J. Natl. Cancer Inst. 2013, 105, 1151–1156. [Google Scholar] [CrossRef] [Green Version]
- Gonsalves, W.I.; Mahoney, M.R.; Sargent, D.J.; Nelson, G.D.; Alberts, S.R.; Sinicrope, F.A.; Goldberg, R.M.; Limburg, P.J.; Thibodeau, S.N.; Grothey, A.; et al. Patient and tumor characteristics and BRAF and KRAS mutations in colon cancer, NCCTG/Alliance N0147. J. Natl. Cancer Inst. 2014, 106, dju106. [Google Scholar] [CrossRef]
- Manthravadi, S.; Sun, W.; Saeed, A. Prognostic impact of BRAF V600E mutation in patients with non-metastatic colorectal cancer with microsatellite instability: A systematic review and meta-analysis. J. Clin. Oncol. 2018, 36, 3597. [Google Scholar] [CrossRef]
- Rowland, A.; Dias, M.M.; Wiese, M.D.; Kichenadasse, G.; McKinnon, R.A.; Karapetis, C.S.; Sorich, M.J. Meta-analysis of BRAF mutation as a predictive biomarker of benefit from anti-EGFR monoclonal antibody therapy for RAS wild-type metastatic colorectal cancer. Br. J. Cancer 2015, 112, 1888–1894. [Google Scholar] [CrossRef] [Green Version]
- Pietrantonio, F.; Petrelli, F.; Coinu, A.; Di Bartolomeo, M.; Borgonovo, K.; Maggi, C.; Cabiddu, M.; Iacovelli, R.; Bossi, I.; Lonati, V.; et al. Predictive role of BRAF mutations in patients with advanced colorectal cancer receiving cetuximab and panitumumab: A meta-analysis. Eur. J. Cancer 2015, 51, 587–594. [Google Scholar] [CrossRef] [PubMed]
- De Roock, W.; Claes, B.; Bernasconi, D.; De Schutter, J.; Biesmans, B.; Fountzilas, G.; Kalogeras, K.T.; Kotoula, V.; Papamichael, D.; Laurent-Puig, P.; et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: A retrospective consortium analysis. Lancet Oncol. 2010, 11, 753–762. [Google Scholar] [CrossRef]
- Di Nicolantonio, F.; Martini, M.; Molinari, F.; Sartore-Bianchi, A.; Arena, S.; Saletti, P.; De Dosso, S.; Mazzucchelli, L.; Frattini, M.; Siena, S.; et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J. Clin. Oncol. 2008, 26, 5705–5712. [Google Scholar] [CrossRef]
- Geissler, M.; Riera-Knorrenschild, J.; Tannapfel, A.; Greeve, J.; Florschütz, A.; Wessendorf, S.; Ettrich, T.; Kanzler, S.; Nörenberg, D.; Ricke, J.; et al. mFOLFOXIRI + panitumumab versus FOLFOXIRI as first-line treatment in patients with RAS wild- type metastatic colorectal cancer m(CRC): A randomized phase II VOLFI trial of the AIO (AIO-KRK0109). J. Clin. Oncol. 2019, 37, 3401–3411. [Google Scholar] [CrossRef]
- NCCN Clinical Practice Guidelines in Oncology. Colon Cancer, version 3.2020. Available online: https://www.nccn.org/professionals/physician_gls/pdf/colon.pdf (accessed on 22 May 2020).
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ince, W.L.; Jubb, A.M.; Holden, S.N.; Holmgren, E.B.; Tobin, P.; Sridhar, M.; Hurwitz, H.I.; Kabbinavar, F.; Novotny, W.F.; Hillan, K.J.; et al. Association of k-ras, b-raf, and p53 status with the treatment effect of bevacizumab. J. Natl. Cancer Inst. 2005, 97, 981–989. [Google Scholar] [CrossRef]
- Innocenti, F.; Ou, F.S.; Qu, X.; Zemla, T.J.; Niedzwiecki, D.; Tam, R.; Mahajan, S.; Goldberg, R.M.; Bertagnolli, M.M.; Blanke, C.D.; et al. Mutational Analysis of Patients with Colorectal Cancer in CALGB/SWOG 80405 Identifies New Roles of Microsatellite Instability and Tumor Mutational Burden for Patient Outcome. J. Clin. Oncol. 2019, 37, 1217–1227. [Google Scholar] [CrossRef]
- Masi, G.; Loupakis, F.; Salvatore, L.; Fornaro, L.; Cremolini, C.; Cupini, S.; Ciarlo, A.; Del Monte, F.; Cortesi, E.; Amoroso, D.; et al. Bevacizumab with FOLFOXIRI (irinotecan, oxaliplatin, fluorouracil, and folinate) as first-line treatment for metastatic colorectal cancer: A phase 2 trial. Lancet Oncol. 2010, 11, 845–852. [Google Scholar] [CrossRef]
- Loupakis, F.; Cremolini, C.; Salvatore, L.; Masi, G.; Sensi, E.; Schirripa, M.; Michelucci, A.; Pfanner, E.; Brunetti, I.; Lupi, C.; et al. FOLFOXIRI plus bevacizumab as first-line treatment in BRAF mutant metastatic colorectal cancer. Eur. J. Cancer 2014, 50, 57–63. [Google Scholar] [CrossRef]
- Cremolini, C.; Loupakis, F.; Antoniotti, C.; Lupi, C.; Sensi, E.; Lonardi, S.; Mezi, S.; Tomasello, G.; Ronzoni, M.; Zaniboni, A.; et al. FOLFOXIRI plus bevacizumab versus FOLFIRI plus bevacizumab as first-line treatment of patients with metastatic colorectal cancer: Updated overall survival and molecular subgroup analyses of the open-label, phase 3 TRIBE study. Lancet Oncol. 2015, 16, 1306–1315. [Google Scholar] [CrossRef]
- Corcoran, R.B.; Atreya, C.E.; Falchook, G.S.; Kwak, E.L.; Ryan, D.P.; Bendell, J.C.; Hamid, O.; Messersmith, W.A.; Daud, A.; Kurzrock, R.; et al. Combined BRAF and MEK inhibition with dabrafenib and trametinib in BRAF V600-Mutant colorectal cancer. J. Clin. Oncol. 2015, 33, 4023–4031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prahallad, A.; Sun, C.; Huang, S.; Di Nicolantonio, F.; Salazar, R.; Zecchin, D.; Beijersbergen, R.L.; Bardelli, A.; Bernards, R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012, 483, 100–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyman, D.M.; Puzanov, I.; Subbiah, V.; Faris, J.E.; Chau, I.; Blay, J.Y.; Wolf, J.; Raje, N.S.; Diamond, E.L.; Hollebecque, A.; et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N. Engl. J. Med. 2015, 373, 726–736. [Google Scholar] [CrossRef] [PubMed]
- Yaeger, R.; Cercek, A.; O’Reilly, E.M.; Reidy, D.L.; Kemeny, N.; Wolinsky, T.; Capanu, M.; Gollub, M.J.; Rosen, N.; Berger, M.F.; et al. Pilot trial of combined BRAF and EGFR inhibition in BRAF-mutant metastatic colorectal cancer patients. Clin. Cancer Res. 2015, 21, 1313–1320. [Google Scholar] [CrossRef] [Green Version]
- Yaeger, R.; Yao, Z.; Hyman, D.M.; Hechtman, J.F.; Vakiani, E.; Zhao, H.; Su, W.; Wang, L.; Joelson, A.; Cercek, A.; et al. Mechanisms of acquired resistance to BRAF V600E inhibition in colon cancers converge on RAF dimerization and are sensitive to its inhibition. Cancer Res. 2017, 77, 6513–6523. [Google Scholar] [CrossRef] [Green Version]
- Van Cutsem, E.; Cuyle, P.; Huijberts, S.; Schellens, J.; Elez, E.; Yaeger, R.; Fakih, M.; Montagut, C.; Peeters, M.; Desai, J.; et al. BEACON CRC study safety lead-in: Assessment of the BRAF inhibitor encorafenib + MEK inhibitor binimetinib + anti-epidermal growth factor receptor antibody cetuximab for BRAFV600E metastatic colorectal cancer. Ann. Oncol. 2018, 29 (Suppl. 5), V109. [Google Scholar] [CrossRef]
- Kopetz, S.; Grothey, A.; Yaeger, R.; Van Cutsem, E.; Desai, J.; Yoshino, T.; Wasan, H.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib, Binimetinib, and Cetuximab in BRAFV600E-Mutated Colorectal Cancer. N. Engl. J. Med. 2019, 381, 1632–1643. [Google Scholar] [CrossRef] [Green Version]
- Mao, M.; Tian, F.; Mariadason, J.M.; Tsao, C.C.; Lemos, R.; Dayyani, F., Jr.; Gopal, Y.N.; Jiang, Z.Q.; Wistuba, I.I.; Tang, X.M.; et al. Resistance to BRAF inhibition in BRAF-mutant colon cancer can be overcome with PI3K inhibition or demethylating agents. Clin. Cancer Res. 2013, 19, 657–667. [Google Scholar] [CrossRef] [Green Version]
- Spagnolo, F.; Ghiorzo, P.; Queirolo, P. Overcoming resistance to BRAF inhibition in BRAF-mutated metastatic melanoma. Oncotarget 2014, 5, 10206–10221. [Google Scholar] [CrossRef] [Green Version]
- Notarangelo, T.; Sisinni, L.; Condelli, V.; Landriscina, M. Dual EGFR and BRAF blockade overcomes resistance to vemurafenib in BRAF mutated thyroid carcinoma cells. Cancer Cell Int. 2017, 17, 86. [Google Scholar] [CrossRef]
- Poulikakos, P.I.; Persaud, Y.; Janakiraman, M.; Kong, X.; Ng, C.; Moriceau, G.; Shi, H.; Atefi, M.; Titz, B.; Gabay, M.T.; et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature 2011, 480, 387–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vido, M.J.; Le, K.; Hartsough, E.J.; Aplin, A.E. BRAF Splice Variant Resistance to RAF Inhibitor Requires Enhanced MEK Association. Cell Rep. 2018, 25, 1501–1510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stagni, C.; Zamuner, C.; Elefanti, L.; Zanin, T.; Bianco, P.D.; Sommariva, A.; Fabozzi, A.; Pigozzo, J.; Mocellin, S.; Montesco, M.C.; et al. BRAF Gene Copy Number and Mutant Allele Frequency Correlate With Time to Progression in Metastatic Melanoma Patients Treated With MAPK Inhibitors. Mol. Cancer Ther. 2018, 17, 1332–1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sale, M.J.; Balmanno, K.; Saxena, J.; Ozono, E.; Wojdyla, K.; McIntyre, R.E.; Gilley, R.; Woroniuk, A.; Howarth, K.D.; Hughes, G.; et al. MEK1/2 Inhibitor Withdrawal Reverses Acquired Resistance Driven by BRAF V600E Amplification Whereas KRAS G13D Amplification Promotes EMT-chemoresistance. Nat. Commun. 2019, 10, 2030. [Google Scholar] [CrossRef] [Green Version]
- Overman, M.J.; Lonardi, S.; Wong, K.Y.; Lenz, H.J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Van Cutsem, E.; McDermott, R.; Hill, A.; et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J. Clin. Oncol. 2018, 36, 773–779. [Google Scholar] [CrossRef]
- Xu, C.; Zheng, L.; Li, D.; Chen, G.; Gu, J.; Chen, J.; Yao, Q. CXCR4 overexpression is correlated with poor prognosis in colorectal cancer. Life Sci. 2018, 208, 333–340. [Google Scholar] [CrossRef]
- Yu, X.; Wang, D.; Wang, X.; Sun, S.; Zhang, Y.; Wang, S.; Miao, R.; Xu, X.; Qu, X. CXCL12/CXCR4 promotes inflammation-driven colorectal cancer progression through activation of RhoA signaling by sponging miR-133a-3p. J. Exp. Clin. Cancer Res. 2019, 38, 32. [Google Scholar] [CrossRef]
- Gala, M.K.; Austin, T.; Ogino, S.; Chan, A.T. TFF2-CXCR4 axis is associated with BRAF V600E colon cancer. Cancer Prev. Res. 2015, 8, 614–619. [Google Scholar] [CrossRef] [Green Version]
- Ottaiano, A.; Scala, S.; Normanno, N.; Botti, G.; Tatangelo, F.; Di Mauro, A.; Capozzi, M.; Facchini, S.; Tafuto, S.; Nasti, G. Prognostic and Predictive Role of CXC Chemokine Receptor 4 in Metastatic Colorectal Cancer Patients. Appl. Immunohistochem. Mol. Morphol. 2020. [Google Scholar] [CrossRef]
- Kim, J.; Takeuchi, H.; Lam, S.T.; Turner, R.R.; Wang, H.J.; Kuo, C.; Foshag, L.; Bilchik, A.J.; Hoon, D.S. Chemokine receptor CXCR4 expression in colorectal cancer patients increases the risk for recurrence and for poor survival. J. Clin. Oncol. 2005, 23, 2744–2753. [Google Scholar] [CrossRef]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Molina-Cerrillo, J.; San Román, M.; Pozas, J.; Alonso-Gordoa, T.; Pozas, M.; Conde, E.; Rosas, M.; Grande, E.; García-Bermejo, M.L.; Carrato, A. BRAF Mutated Colorectal Cancer: New Treatment Approaches. Cancers 2020, 12, 1571. https://doi.org/10.3390/cancers12061571
Molina-Cerrillo J, San Román M, Pozas J, Alonso-Gordoa T, Pozas M, Conde E, Rosas M, Grande E, García-Bermejo ML, Carrato A. BRAF Mutated Colorectal Cancer: New Treatment Approaches. Cancers. 2020; 12(6):1571. https://doi.org/10.3390/cancers12061571
Chicago/Turabian StyleMolina-Cerrillo, Javier, María San Román, Javier Pozas, Teresa Alonso-Gordoa, Miguel Pozas, Elisa Conde, Marta Rosas, Enrique Grande, María Laura García-Bermejo, and Alfredo Carrato. 2020. "BRAF Mutated Colorectal Cancer: New Treatment Approaches" Cancers 12, no. 6: 1571. https://doi.org/10.3390/cancers12061571