Metabolic Regulation of Epigenetic Modifications and Cell Differentiation in Cancer

 , , ,
, , ,  , and
, and 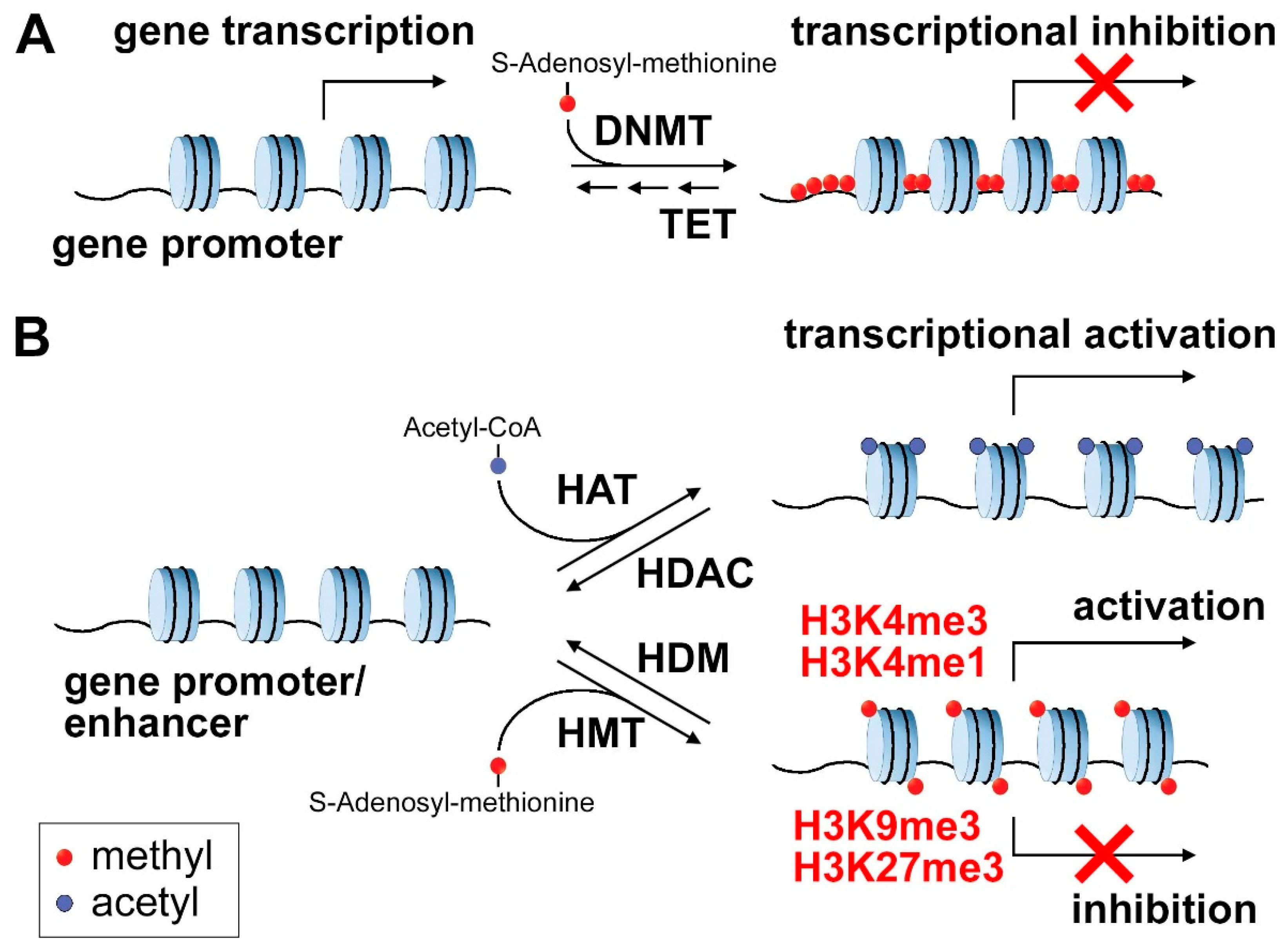{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Epigenetic Regulation in Cancer
2.1. DNA Methylation
2.2. Histone Marks
2.3. Chromatin Remodeling
3. Metabolic Dysregulation as a Driver of Cell Fate in Stem Cells and Cancer
3.1. Mitochondrial Metabolism in the Regulation of Cell Differentiation in Embryonic Stem Cells
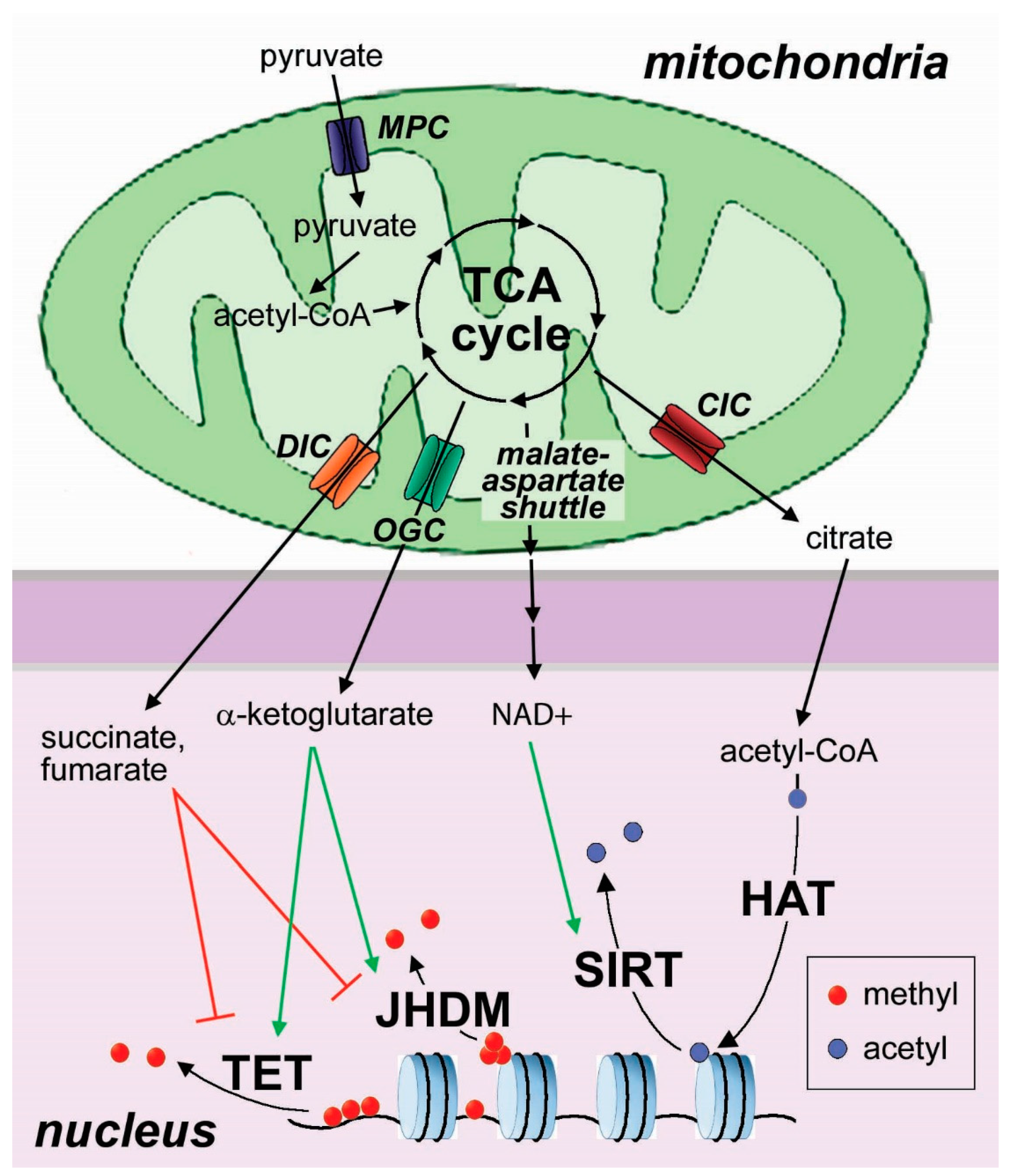
3.2. Mitochondrial Metabolism in the Regulation of Cell Differentiation in Cancer Cells
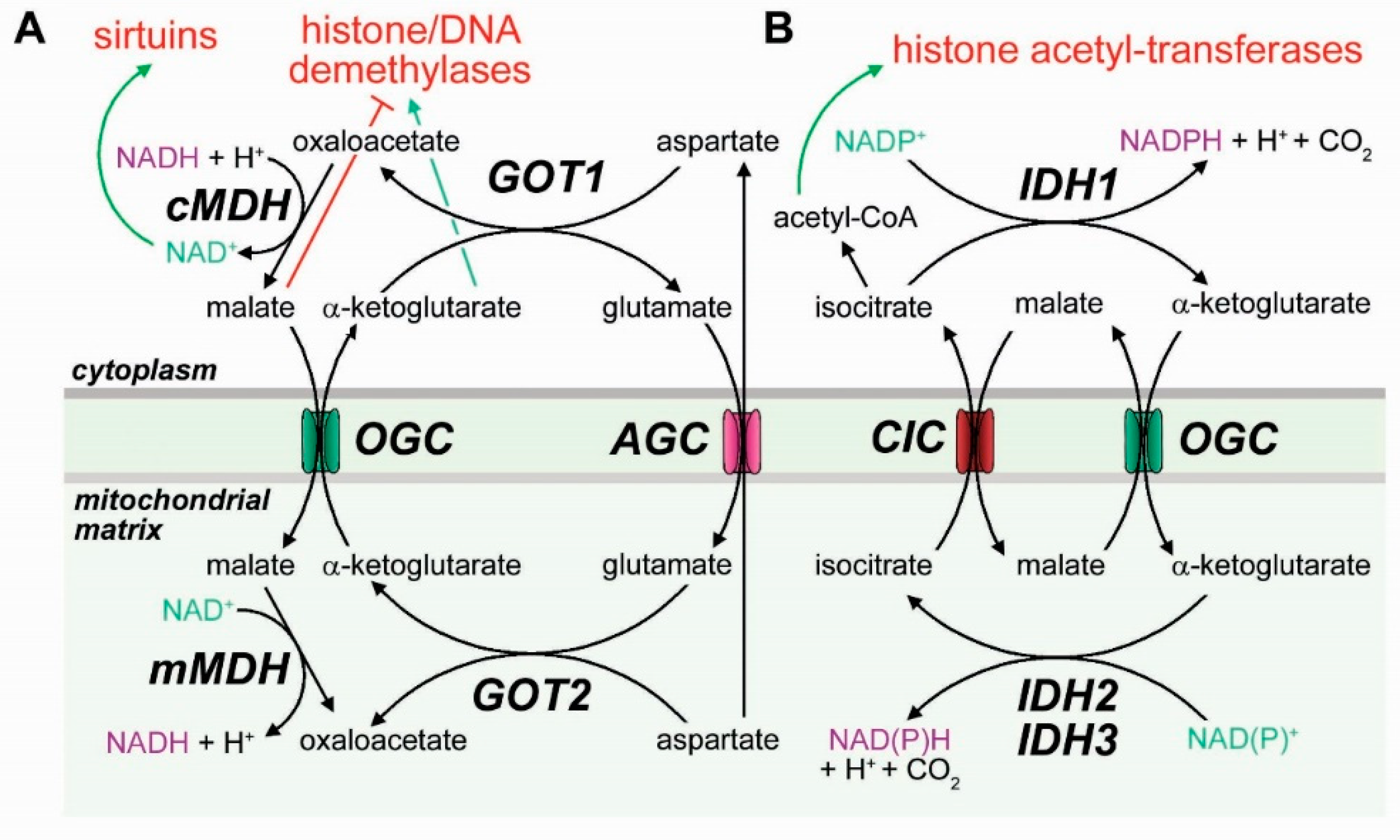
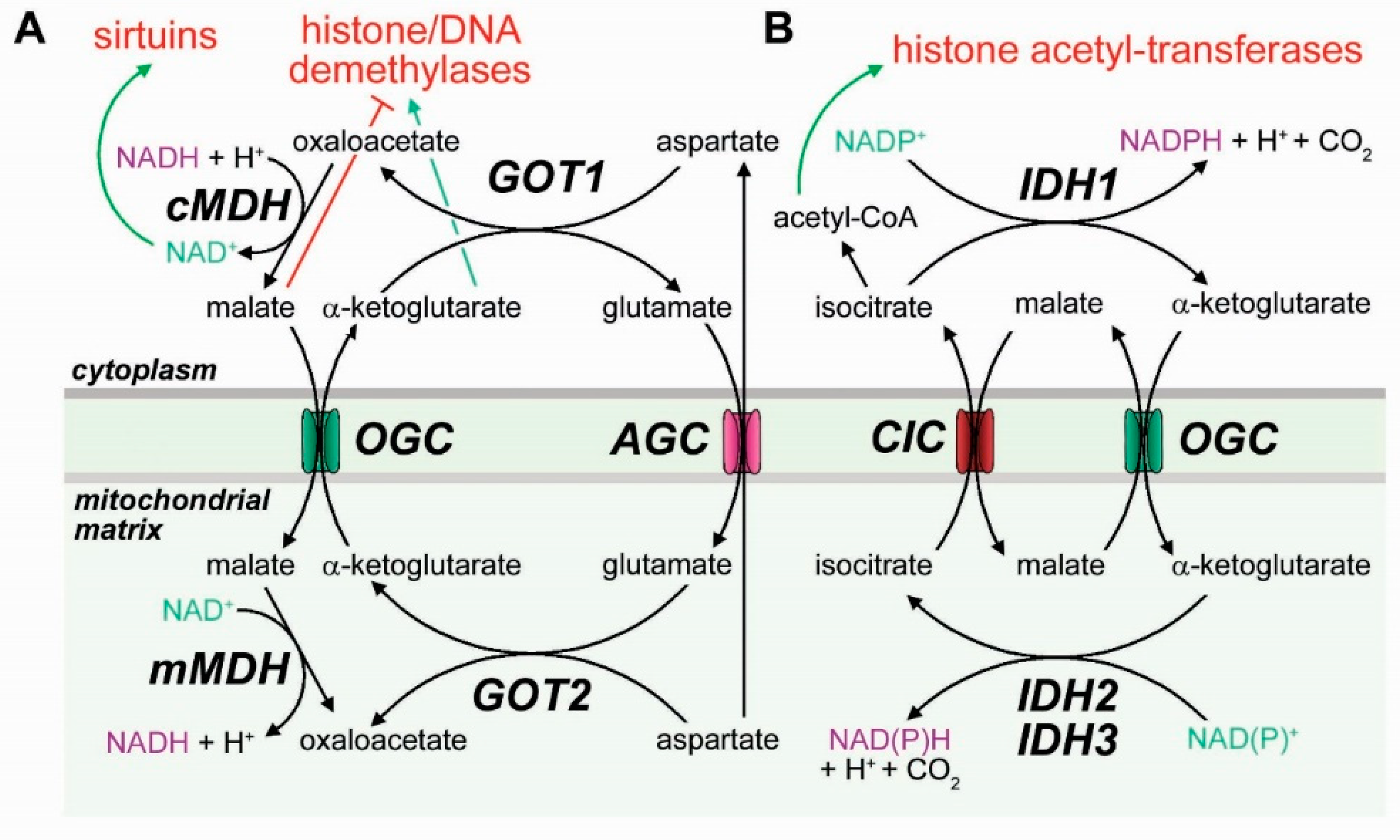
3.3. Mitochondrial Transporters in the Epigenetic Regulation of Cell Differentiation
4. The Role of Hypoxia in Cancer Cell Differentiation
- (1)
- HIF is an α/β heterodimer that binds the hypoxia response elements (HREs) on the promoters of target genes, activating the hypoxic response. Whereas the β subunit is constitutively expressed, HIF-1α and HIF-2α are regulated post-translationally by an O2-dependent mechanism through hydroxylation of proline residues by prolyl hydroxylases (PHDs) and subsequent ubiquitination by pVHL [118,119]. The PHD enzymes are dioxygenases that require for their activity oxygen, as well as iron and αKG [120]. Therefore, HIF-1α/HIF-2α are constitutively degraded during normoxia, whereas during hypoxia they escape ubiquitination and proteasomal degradation and heterodimerize with HIF-1β to regulate transcription of downstream genes [121]. HIF target genes are involved in the regulation of cell proliferation, angiogenic signalling and altered metabolism [122]. The impact of HIFs on cancer stem cells is mostly attributed to the HIF-dependent activation of stemness factors. Hypoxia signalling can induce HIF-dependent expression of known pluripotent factors, such as KLF4, MYC, OCT4, SOX2, and NANOG, which have an important role in the dedifferentiation process under hypoxic conditions, inducing cancer stemness and repressing cancer cell differentiation [123]. It has been reported that cancer stem-like cells can be induced through de-differentiation under hypoxic conditions in glioma, hepatoma and lung cancer, providing new evidence about tumor development and chemoradiotherapy resistance [124].
- (2)
- Reduced availability of oxygen for the mitochondrial respiratory chain is expected to slow down the oxidation of NADH in the mitochondria, leading to feedback inhibition of the TCA cycle. Hypoxia has been shown to stimulate stemness of breast cancer cells by the production of reactive oxygen species, and this effect is mediated by attenuation of the TCA cycle and accumulation of fumarate [125]. Although the epigenetic consequences of these alterations have not been investigated in detail, they are likely to play a role in cell de-differentiation induced by hypoxia. Prolonged hypoxia can cause reduced activity of nuclear pyruvate dehydrogenase, resulting in reduced nuclear availability of acetyl-CoA and reduced histone acetylation [126].
- (3)
- An increasing number of studies have focused on the involvement of hypoxia in regulating multiple components of the epigenetic machinery, including DNA methylation, histone modification, non-coding RNAs (ncRNAs) and chromatin remodelling, affecting the expression of specific genes involved in cancer progression, dedifferentiation, and drug resistance [127,128]. Hypoxia interferes with the activity of oxygen-dependent dioxygenases, including the TET 5-methyl-cytosine dioxygenases and Jumonji-C (JMJC) histone demethylases [129]. As a consequence, hypoxia induces increased trimethylation marks on H3K4 and H3K36 in HeLa cells and in a human fibroblast line, and on H3K9 in several cell lines, including colon carcinoma and epithelial human breast cancer cell lines. These events are driven by the inactivation of the JMJC-containing enzymes, such as lysine demethylase 4A (KDM4A) and 5A (KDM5A). The lack of demethylase activity in hypoxic conditions leads to transcriptional repression of target genes and a phenotype of increased tumor aggressiveness, invasion, migration, and proliferation [130,131]. In addition, hypoxia is able to induce specific changes in the chromatin state associated with EMT. Wu et al. reported that under hypoxia, HDAC3 interacts with hypoxia-induced WDR5, recruits the HMT complex to increase the methylation marks on H3K4, and activates the transcription of mesenchymal genes [132]. Interestingly, the activity of lactate dehydrogenase A, under hypoxic conditions, is able to produce the oncometabolite L-2-hydroxyglutarate, independently of IDH. This event promotes the inhibition of KDM4C and enhanced H3K9 methylation, thus altering the expression of genes involved in cellular differentiation [133,134]. Thienpont et al. have also observed that increased promoter methylation is associated with reduced activity of TET enzymes, which decreases 5-hydroxymethyl-cytosine at gene promoters and enhancers, supporting the hypothesis that hypoxia inhibits TET-mediated DNA demethylation in tumors [135].
5. Therapeutic Implications
6. Concluding Remarks
Funding
Conflicts of Interest
Abbreviations
| 5mC | 5-methyl-cytosine |
| AGC | aspartate-glutamate carrier |
| AML | acute myeloid leukemia |
| ATP | adenosine triphosphate |
| CGI | CpG island |
| CIC | citrate carrier |
| DNA | deoxyribonucleic acid |
| DNMT | DNA methyl-transferase |
| DNMTi | DNA metyl-transferase inhibitors |
| GlcNAc | N-acetylglucosamine |
| GOT1/2 | glutamic-oxaloacetic transaminase |
| H3K27ac | acetylated histone 3 lysine 27 |
| H3K27me | methylated histone 3 lysine 27 |
| H3K4me | methylated histone 3 lysine 4 |
| H3K9me | methylated histone 3 lysine 9 |
| HAT | histone acetyl-transferase |
| HDAC | histone deacetylase |
| HDACi | histone deacetylase inhibitors |
| HDM | histone demethylase |
| HIF | hypoxia inducible factors |
| HMT | histone methyl transferase |
| HMTi | histone methyl-transferase inhibitors |
| IDH1-3 | isocitrate dehydrogenase |
| IGF | insulin-like growth factor |
| JHDM | Jumonji-domain containing histone demethylases |
| cMDH | cytoplasmic malate dehydrogenase |
| mMDH | mitochondrial malate dehydrogenase |
| MDS | myelodysplastic syndromes |
| MLL | mixed-lineage leukemia |
| MPC | mitochondrial pyruvate carrier |
| mtDNA | mitochondrial DNA |
| NAD+ | oxidized nicotinamide adenine dinucleotide |
| NADH | reduced nicotinamide adenine dinucleotide |
| NAMPT | nicotinamide phosphoribosyl-transferase |
| OGC | αKG (oxoglutarate)-glutamate carrier |
| PHD | prolyl hydroxylase |
| PHGDH | phosphoglycerate dehydrogenase |
| RNA | ribonucleic acid |
| TCA | tricarboxylic acid |
| TET | ten-eleven translocation |
| αKG | alpha-ketoglutarate |
References
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Kamphorst, J.J.; Mathew, R.; Chung, M.K.; White, E.; Shlomi, T.; Rabinowitz, J.D. Glutamine-driven oxidative phosphorylation is a major ATP source in transformed mammalian cells in both normoxia and hypoxia. Mol. Syst. Biol. 2013, 9, 712. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeiffer, T.; Schuster, S.; Bonhoeffer, S. Cooperation and competition in the evolution of ATP-producing pathways. Science 2001, 292, 504–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, C.A.; Scafoglio, C. Heterogeneity of Glucose Transport in Lung Cancer. Biomolecules 2020, 10, 868. [Google Scholar] [CrossRef]
- Fendt, S.M.; Bell, E.L.; Keibler, M.A.; Olenchock, B.A.; Mayers, J.R.; Wasylenko, T.M.; Vokes, N.I.; Guarente, L.; Vander Heiden, M.G.; Stephanopoulos, G. Reductive glutamine metabolism is a function of the alpha-ketoglutarate to citrate ratio in cells. Nat. Commun. 2013, 4, 2236. [Google Scholar] [CrossRef] [Green Version]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2011, 481, 380–384. [Google Scholar] [CrossRef] [Green Version]
- Fluegen, G.; Avivar-Valderas, A.; Wang, Y.; Padgen, M.R.; Williams, J.K.; Nobre, A.R.; Calvo, V.; Cheung, J.F.; Bravo-Cordero, J.J.; Entenberg, D.; et al. Phenotypic heterogeneity of disseminated tumour cells is preset by primary tumour hypoxic microenvironments. Nat. Cell Biol. 2017, 19, 120–132. [Google Scholar] [CrossRef] [Green Version]
- Terry, S.; Faouzi Zaarour, R.; Hassan Venkatesh, G.; Francis, A.; El-Sayed, W.; Buart, S.; Bravo, P.; Thiery, J.; Chouaib, S. Role of Hypoxic Stress in Regulating Tumor Immunogenicity, Resistance and Plasticity. Int. J. Mol. Sci. 2018, 19, 3044. [Google Scholar] [CrossRef] [Green Version]
- Quiros, P.M.; Mottis, A.; Auwerx, J. Mitonuclear communication in homeostasis and stress. Nat. Rev. Mol. Cell Biol. 2016, 17, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Campbell, S.L.; Wellen, K.E. Metabolic Signaling to the Nucleus in Cancer. Mol. Cell 2018, 71, 398–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Kanherkar, R.R.; Bhatia-Dey, N.; Csoka, A.B. Epigenetics across the human lifespan. Front. Cell Dev. Biol. 2014, 2, 49. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar] [CrossRef]
- Ushijima, T. Cancer epigenetics: Now harvesting fruit and seeding for common diseases. Biochem. Biophys. Res. Commun. 2014, 455, 1–2. [Google Scholar] [CrossRef]
- Susan, F.; Thea, T.; Joseph, C.; Roadmap, E.C.; Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Roadmap Epigenomics Consortium; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar]
- Beck, S.; Bernstein, B.E.; Campbell, R.M.; Costello, J.F.; Dhanak, D.; Ecker, J.R.; Greally, J.M.; Issa, J.P.; Laird, P.W.; Polyak, K.; et al. A blueprint for an international cancer epigenome consortium. A report from the AACR Cancer Epigenome Task Force. Cancer Res. 2012, 72, 6319–6324. [Google Scholar] [CrossRef] [Green Version]
- The Cancer Genome Atlas Research Network; Weinstein, J.N.; Collisson, E.A.; Mills, C.B.; Mills Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, G.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Adams, D.; Altucci, L.; Antonarakis, S.E.; Ballesteros, J.; Beck, S.; Bird, A.; Bock, C.; Boehm, B.; Campo, E.; Caricasole, A.; et al. BLUEPRINT to decode the epigenetic signature written in blood. Nat. Biotechnol. 2012, 30, 224–226. [Google Scholar] [CrossRef] [Green Version]
- Mohammad, H.P.; Barbash, O.; Creasy, C.L. Targeting epigenetic modifications in cancer therapy: Erasing the roadmap to cancer. Nat. Med. 2019, 25, 403–418. [Google Scholar] [CrossRef] [PubMed]
- You, J.S.; Jones, P.A. Cancer genetics and epigenetics: Two sides of the same coin? Cancer Cell 2012, 22, 9–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miranda-Goncalves, V.; Lameirinhas, A.; Henrique, R.; Jeronimo, C. Metabolism and Epigenetic Interplay in Cancer: Regulation and Putative Therapeutic Targets. Front. Genet. 2018, 9, 427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.; Costello, J. DNA methylation: An epigenetic mark of cellular memory. Exp. Mol. Med. 2017, 49, e322. [Google Scholar] [CrossRef] [Green Version]
- Greenberg, M.V.C.; Bourc’his, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 2019, 20, 590–607. [Google Scholar] [CrossRef]
- Jeziorska, D.M.; Murray, R.J.S.; De Gobbi, M.; Gaentzsch, R.; Garrick, D.; Ayyub, H.; Chen, T.; Li, E.; Telenius, J.; Lynch, M.; et al. DNA methylation of intragenic CpG islands depends on their transcriptional activity during differentiation and disease. Proc. Natl. Acad. Sci. USA 2017, 114, E7526–E7535. [Google Scholar] [CrossRef] [Green Version]
- Du, Q.; Luu, P.L.; Stirzaker, C.; Clark, S.J. Methyl-CpG-binding domain proteins: Readers of the epigenome. Epigenomics 2015, 7, 1051–1073. [Google Scholar] [CrossRef]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [Green Version]
- Feinberg, A.P.; Tycko, B. The history of cancer epigenetics. Nat. Rev. Cancer 2004, 4, 143–153. [Google Scholar] [CrossRef]
- Daskalos, A.; Nikolaidis, G.; Xinarianos, G.; Savvari, P.; Cassidy, A.; Zakopoulou, R.; Kotsinas, A.; Gorgoulis, V.; Field, J.K.; Liloglou, T. Hypomethylation of retrotransposable elements correlates with genomic instability in non-small cell lung cancer. Int. J. Cancer 2009, 124, 81–87. [Google Scholar] [CrossRef]
- Ehrlich, M.; Jackson, K.; Weemaes, C. Immunodeficiency, centromeric region instability, facial anomalies syndrome (ICF). Orphanet J. Rare Dis. 2006, 1, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leick, M.B.; Shoff, C.J.; Wang, E.C.; Congress, J.L.; Gallicano, G.I. Loss of imprinting of IGF2 and the epigenetic progenitor model of cancer. Am. J. Stem Cells 2012, 1, 59–74. [Google Scholar] [PubMed]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome—biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Hatziapostolou, M.; Iliopoulos, D. Epigenetic aberrations during oncogenesis. Cell. Mol. Life Sci 2011, 68, 1681–1702. [Google Scholar] [CrossRef] [PubMed]
- Oki, E.; Oda, S.; Maehara, Y.; Sugimachi, K. Mutated gene-specific phenotypes of dinucleotide repeat instability in human colorectal carcinoma cell lines deficient in DNA mismatch repair. Oncogene 1999, 18, 2143–2147. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.A.; Baylin, S.B. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 2002, 3, 415–428. [Google Scholar] [CrossRef]
- Wong, C.C.; Qian, Y.; Yu, J. Interplay between epigenetics and metabolism in oncogenesis: Mechanisms and therapeutic approaches. Oncogene 2017, 36, 3359–3374. [Google Scholar] [CrossRef]
- Wolf, A.; Agnihotri, S.; Munoz, D.; Guha, A. Developmental profile and regulation of the glycolytic enzyme hexokinase 2 in normal brain and glioblastoma multiforme. Neurobiol. Dis. 2011, 44, 84–91. [Google Scholar] [CrossRef]
- Kim, W.Y.; Sharpless, N.E. The regulation of INK4/ARF in cancer and aging. Cell 2006, 127, 265–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, X.B.; Cai, W.B.; Luo, L.; Liu, L.S.; Shi, H.J.; Chen, M.H. The Prognostic Value of p16 Hypermethylation in Cancer: A Meta-Analysis. PLoS ONE 2013, 8, e66587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintás-Cardama, A.; Kantarjian, H.; Estrov, Z.; Borthakur, G.; Cortes, J.; Verstovsek, S. Therapy with the histone deacetylase inhibitor pracinostat for patients with myelofibrosis. Leuk. Res. 2012, 36, 1124–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanai, Y.; Ushijima, S.; Nakanishi, Y.; Sakamoto, M.; Hirohashi, S. Mutation of the DNA methyltransferase (DNMT) 1 gene in human colorectal cancers. Cancer Lett. 2003, 192, 75–82. [Google Scholar] [CrossRef]
- Ley, T.J.; Ding, L.; Walter, M.J.; McLellan, M.D.; Lamprecht, T.; Larson, D.E.; Kandoth, C.; Payton, J.E.; Baty, J.; Welch, J.; et al. DNMT3A mutations in acute myeloid leukemia. N. Engl. J. Med. 2010, 363, 2424–2433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, X.J.; Xu, J.; Gu, Z.H.; Pan, C.M.; Lu, G.; Shen, Y.; Shi, J.Y.; Zhu, Y.M.; Tang, L.; Zhang, X.W.; et al. Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia. Nat. Genet. 2011, 43, 309–315. [Google Scholar] [CrossRef]
- Tan, A.Y.; Manley, J.L. The TET family of proteins: Functions and roles in disease. J. Mol. Cell Biol. 2009, 1, 82–92. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.R.; Wu, M.J.; Zhang, Y.; Yang, J.Y.; Chang, C.J. TET2 directs mammary luminal cell differentiation and endocrine response. Nat. Commun. 2020, 11, 4642. [Google Scholar] [CrossRef]
- Baylin, S.B.; Jones, P.A. Epigenetic Determinants of Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef] [Green Version]
- Hawkins, R.D.; Hon, G.C.; Yang, C.; Antosiewicz-Bourget, J.E.; Lee, L.K.; Ngo, Q.M.; Klugman, S.; Ching, K.A.; Edsall, L.E.; Ye, Z.; et al. Dynamic chromatin states in human ES cells reveal potential regulatory sequences and genes involved in pluripotency. Cell Res. 2011, 21, 1393–1409. [Google Scholar] [CrossRef] [Green Version]
- Hon, G.C.; Hawkins, R.D.; Ren, B. Predictive chromatin signatures in the mammalian genome. Hum. Mol. Genet. 2009, 18, R195–R201. [Google Scholar] [CrossRef] [Green Version]
- Esteller, M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat. Rev. Genet. 2007, 8, 286–298. [Google Scholar] [CrossRef]
- Iyer, N.G.; Ozdag, H.; Caldas, C. p300/CBP and cancer. Oncogene 2004, 23, 4225–4231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ocker, M.; Schneider-Stock, R. Histone deacetylase inhibitors: Signalling towards p21cip1/waf1. Int. J. Biochem. Cell Biol. 2007, 39, 1367–1374. [Google Scholar] [CrossRef] [PubMed]
- Lakshmikuttyamma, A.; Scott, S.A.; DeCoteau, J.F.; Geyer, C.R. Reexpression of epigenetically silenced AML tumor suppressor genes by SUV39H1 inhibition. Oncogene 2010, 29, 576–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLean, C.M.; Karemaker, I.D.; van Leeuwen, F. The emerging roles of DOT1L in leukemia and normal development. Leukemia 2014, 28, 2131–2138. [Google Scholar] [CrossRef] [PubMed]
- Reynoird, N.; Mazur, P.K.; Stellfeld, T.; Flores, N.M.; Lofgren, S.M.; Carlson, S.M.; Brambilla, E.; Hainaut, P.; Kaznowska, E.B.; Arrowsmith, C.H.; et al. Coordination of stress signals by the lysine methyltransferase SMYD2 promotes pancreatic cancer. Genes Dev. 2016, 30, 772–785. [Google Scholar] [CrossRef]
- Nowak, S.J.; Corces, V.G. Phosphorylation of histone H3: A balancing act between chromosome condensation and transcriptional activation. Trends Genet. 2004, 20, 214–220. [Google Scholar] [CrossRef]
- Kumar, R.; Horikoshi, N.; Singh, M.; Gupta, A.; Misra, H.S.; Albuquerque, K.; Hunt, C.R.; Pandita, T.K. Chromatin modifications and the DNA damage response to ionizing radiation. Front. Oncol. 2012, 2, 214. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Thompson, C.B. Metabolic regulation of epigenetics. Cell Metab. 2012, 16, 9–17. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro-Silva, C.; Vermeulen, W.; Lans, H. SWI/SNF: Complex complexes in genome stability and cancer. DNA Repair 2019, 77, 87–95. [Google Scholar] [CrossRef]
- Holoch, D.; Moazed, D. RNA-mediated epigenetic regulation of gene expression. Nat. Rev. Genet. 2015, 16, 71–84. [Google Scholar] [CrossRef]
- Böhmdorfer, G.; Wierzbicki, A.T. Control of Chromatin Structure by Long Noncoding RNA. Trends Cell Biol. 2015, 25, 623–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Y.; Tang, S.; Li, X. Sirtuins in Metabolic and Epigenetic Regulation of Stem Cells. Trends Endocrinol. Metab. 2019, 30, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Tohyama, S.; Fujita, J.; Hishiki, T.; Matsuura, T.; Hattori, F.; Ohno, R.; Kanazawa, H.; Seki, T.; Nakajima, K.; Kishino, Y.; et al. Glutamine Oxidation Is Indispensable for Survival of Human Pluripotent Stem Cells. Cell Metab. 2016, 23, 663–674. [Google Scholar] [CrossRef] [Green Version]
- Carey, B.W.; Finley, L.W.; Cross, J.R.; Allis, C.D.; Thompson, C.B. Intracellular alpha-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 2015, 518, 413–416. [Google Scholar] [CrossRef] [Green Version]
- Agathocleous, M.; Meacham, C.E.; Burgess, R.J.; Piskounova, E.; Zhao, Z.; Crane, G.M.; Cowin, B.L.; Bruner, E.; Murphy, M.M.; Chen, W.; et al. Ascorbate regulates haematopoietic stem cell function and leukaemogenesis. Nature 2017, 549, 476–481. [Google Scholar] [CrossRef]
- Losman, J.A.; Kaelin, W.G., Jr. What a difference a hydroxyl makes: Mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev. 2013, 27, 836–852. [Google Scholar] [CrossRef] [Green Version]
- Ye, C.; Tu, B.P. Sink into the Epigenome: Histones as Repositories That Influence Cellular Metabolism. Trends Endocrinol. Metab. 2018, 29, 626–637. [Google Scholar] [CrossRef]
- Zhao, W.; Qi, X.; Liu, L.; Ma, S.; Liu, J.; Wu, J. Epigenetic Regulation of m(6)A Modifications in Human Cancer. Mol. Ther. Nucleic Acids 2020, 19, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Reid, M.A.; Dai, Z.; Locasale, J.W. The impact of cellular metabolism on chromatin dynamics and epigenetics. Nat. Cell Biol. 2017, 19, 1298–1306. [Google Scholar] [CrossRef] [PubMed]
- Hwang, I.Y.; Kwak, S.; Lee, S.; Kim, H.; Lee, S.E.; Kim, J.H.; Kim, Y.A.; Jeon, Y.K.; Chung, D.H.; Jin, X.; et al. Psat1-Dependent Fluctuations in alpha-Ketoglutarate Affect the Timing of ESC Differentiation. Cell Metab. 2016, 24, 494–501. [Google Scholar] [CrossRef] [Green Version]
- Finley, L.W.S.; Vardhana, S.A.; Carey, B.W.; Alonso-Curbelo, D.; Koche, R.; Chen, Y.; Wen, D.; King, B.; Radler, M.R.; Rafii, S.; et al. Pluripotency transcription factors and Tet1/2 maintain Brd4-independent stem cell identity. Nat. Cell Biol. 2018, 20, 565–574. [Google Scholar] [CrossRef]
- Chung, T.L.; Brena, R.M.; Kolle, G.; Grimmond, S.M.; Berman, B.P.; Laird, P.W.; Pera, M.F.; Wolvetang, E.J. Vitamin C promotes widespread yet specific DNA demethylation of the epigenome in human embryonic stem cells. Stem Cells 2010, 28, 1848–1855. [Google Scholar] [CrossRef] [PubMed]
- Boyer, L.A.; Plath, K.; Zeitlinger, J.; Brambrink, T.; Medeiros, L.A.; Lee, T.I.; Levine, S.S.; Wernig, M.; Tajonar, A.; Ray, M.K.; et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 2006, 441, 349–353. [Google Scholar] [CrossRef]
- Collinson, A.; Collier, A.J.; Morgan, N.P.; Sienerth, A.R.; Chandra, T.; Andrews, S.; Rugg-Gunn, P.J. Deletion of the Polycomb-Group Protein EZH2 Leads to Compromised Self-Renewal and Differentiation Defects in Human Embryonic Stem Cells. Cell Rep. 2016, 17, 2700–2714. [Google Scholar] [CrossRef]
- TeSlaa, T.; Chaikovsky, A.C.; Lipchina, I.; Escobar, S.L.; Hochedlinger, K.; Huang, J.; Graeber, T.G.; Braas, D.; Teitell, M.A. Alpha-Ketoglutarate Accelerates the Initial Differentiation of Primed Human Pluripotent Stem Cells. Cell Metab. 2016, 24, 485–493. [Google Scholar] [CrossRef] [Green Version]
- Calvanese, V.; Lara, E.; Suarez-Alvarez, B.; Abu Dawud, R.; Vazquez-Chantada, M.; Martinez-Chantar, M.L.; Embade, N.; Lopez-Nieva, P.; Horrillo, A.; Hmadcha, A.; et al. Sirtuin 1 regulation of developmental genes during differentiation of stem cells. Proc. Natl. Acad. Sci. USA 2010, 107, 13736–13741. [Google Scholar] [CrossRef] [Green Version]
- Heo, J.; Lim, J.; Lee, S.; Jeong, J.; Kang, H.; Kim, Y.; Kang, J.W.; Yu, H.Y.; Jeong, E.M.; Kim, K.; et al. Sirt1 Regulates DNA Methylation and Differentiation Potential of Embryonic Stem Cells by Antagonizing Dnmt3l. Cell Rep. 2017, 18, 1930–1945. [Google Scholar] [CrossRef] [Green Version]
- Tang, S.; Fang, Y.; Huang, G.; Xu, X.; Padilla-Banks, E.; Fan, W.; Xu, Q.; Sanderson, S.M.; Foley, J.F.; Dowdy, S.; et al. Methionine metabolism is essential for SIRT1-regulated mouse embryonic stem cell maintenance and embryonic development. EMBO J. 2017, 36, 3175–3193. [Google Scholar] [CrossRef] [PubMed]
- Etchegaray, J.P.; Chavez, L.; Huang, Y.; Ross, K.N.; Choi, J.; Martinez-Pastor, B.; Walsh, R.M.; Sommer, C.A.; Lienhard, M.; Gladden, A.; et al. The histone deacetylase SIRT6 controls embryonic stem cell fate via TET-mediated production of 5-hydroxymethylcytosine. Nat. Cell Biol. 2015, 17, 545–557. [Google Scholar] [CrossRef] [PubMed]
- Cha, Y.; Han, M.J.; Cha, H.J.; Zoldan, J.; Burkart, A.; Jung, J.H.; Jang, Y.; Kim, C.H.; Jeong, H.C.; Kim, B.G.; et al. Metabolic control of primed human pluripotent stem cell fate and function by the miR-200c-SIRT2 axis. Nat. Cell Biol. 2017, 19, 445–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Liau, B.B.; Sievers, C.; Donohue, L.K.; Gillespie, S.M.; Flavahan, W.A.; Miller, T.E.; Venteicher, A.S.; Hebert, C.H.; Carey, C.D.; Rodig, S.J.; et al. Adaptive Chromatin Remodeling Drives Glioblastoma Stem Cell Plasticity and Drug Tolerance. Cell Stem Cell 2017, 20, 233–246.e237. [Google Scholar] [CrossRef] [Green Version]
- Oshimori, N.; Oristian, D.; Fuchs, E. TGF-beta promotes heterogeneity and drug resistance in squamous cell carcinoma. Cell 2015, 160, 963–976. [Google Scholar] [CrossRef] [Green Version]
- Balanis, N.G.; Sheu, K.M.; Esedebe, F.N.; Patel, S.J.; Smith, B.A.; Park, J.W.; Alhani, S.; Gomperts, B.N.; Huang, J.; Witte, O.N.; et al. Pan-cancer Convergence to a Small-Cell Neuroendocrine Phenotype that Shares Susceptibilities with Hematological Malignancies. Cancer Cell 2019, 36, 17–34.e17. [Google Scholar] [CrossRef]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef] [Green Version]
- Turcan, S.; Rohle, D.; Goenka, A.; Walsh, L.A.; Fang, F.; Yilmaz, E.; Campos, C.; Fabius, A.W.; Lu, C.; Ward, P.S.; et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012, 483, 479–483. [Google Scholar] [CrossRef]
- Xiao, M.; Yang, H.; Xu, W.; Ma, S.; Lin, H.; Zhu, H.; Liu, L.; Liu, Y.; Yang, C.; Xu, Y.; et al. Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012, 26, 1326–1338. [Google Scholar] [CrossRef] [Green Version]
- Morris, J.P.T.; Yashinskie, J.J.; Koche, R.; Chandwani, R.; Tian, S.; Chen, C.C.; Baslan, T.; Marinkovic, Z.S.; Sanchez-Rivera, F.J.; Leach, S.D.; et al. Alpha-Ketoglutarate links p53 to cell fate during tumour suppression. Nature 2019, 573, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Guerra, F.; Arbini, A.A.; Moro, L. Mitochondria and cancer chemoresistance. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 686–699. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Ma, J.; Lu, W. The Significance of Mitochondrial Dysfunction in Cancer. Int. J. Mol. Sci. 2020, 21, 5598. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.Q.; Li, Q.; Wang, G.H.; Sun, F.F.; Huang, G.J.; Bian, X.W.; Yu, S.C.; Qian, G.S. Mitochondrial and energy metabolism-related properties as novel indicators of lung cancer stem cells. Int. J. Cancer 2011, 129, 820–831. [Google Scholar] [CrossRef]
- Facucho-Oliveira, J.M.; Alderson, J.; Spikings, E.C.; Egginton, S.; St John, J.C. Mitochondrial DNA replication during differentiation of murine embryonic stem cells. J. Cell Sci. 2007, 120, 4025–4034. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; St John, J.C. Modulation of mitochondrial DNA copy number in a model of glioblastoma induces changes to DNA methylation and gene expression of the nuclear genome in tumours. Epigenetics Chromatin 2018, 11, 53. [Google Scholar] [CrossRef] [Green Version]
- Pan, M.; Reid, M.A.; Lowman, X.H.; Kulkarni, R.P.; Tran, T.Q.; Liu, X.; Yang, Y.; Hernandez-Davies, J.E.; Rosales, K.K.; Li, H.; et al. Regional glutamine deficiency in tumours promotes dedifferentiation through inhibition of histone demethylation. Nat. Cell Biol. 2016, 18, 1090–1101. [Google Scholar] [CrossRef]
- Margueron, R.; Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef] [Green Version]
- Tran, T.Q.; Hanse, E.A.; Habowski, A.N.; Li, H.; Gabra, M.B.I.; Yang, Y.; Lowman, X.H.; Ooi, A.M.; Liao, S.Y.; Edwards, R.A.; et al. Alpha-Ketoglutarate attenuates Wnt signaling and drives differentiation in colorectal cancer. Nat. Cancer 2020, 1, 345–358. [Google Scholar] [CrossRef]
- Liu, L.; Liu, C.; Zhang, Q.; Shen, J.; Zhang, H.; Shan, J.; Duan, G.; Guo, D.; Chen, X.; Cheng, J.; et al. SIRT1-mediated transcriptional regulation of SOX2 is important for self-renewal of liver cancer stem cells. Hepatology 2016, 64, 814–827. [Google Scholar] [CrossRef] [Green Version]
- Pruitt, K.; Zinn, R.L.; Ohm, J.E.; McGarvey, K.M.; Kang, S.H.; Watkins, D.N.; Herman, J.G.; Baylin, S.B. Inhibition of SIRT1 reactivates silenced cancer genes without loss of promoter DNA hypermethylation. PLoS Genet. 2006, 2, e40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucena-Cacace, A.; Otero-Albiol, D.; Jimenez-Garcia, M.P.; Munoz-Galvan, S.; Carnero, A. NAMPT Is a Potent Oncogene in Colon Cancer Progression that Modulates Cancer Stem Cell Properties and Resistance to Therapy through Sirt1 and PARP. Clin. Cancer Res. 2018, 24, 1202–1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gujar, A.D.; Le, S.; Mao, D.D.; Dadey, D.Y.; Turski, A.; Sasaki, Y.; Aum, D.; Luo, J.; Dahiya, S.; Yuan, L.; et al. An NAD+-dependent transcriptional program governs self-renewal and radiation resistance in glioblastoma. Proc. Natl. Acad. Sci. USA 2016, 113, E8247–E8256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCommis, K.S.; Finck, B.N. Mitochondrial pyruvate transport: A historical perspective and future research directions. Biochem. J. 2015, 466, 443–454. [Google Scholar] [CrossRef] [Green Version]
- Bender, T.; Martinou, J.C. The mitochondrial pyruvate carrier in health and disease: To carry or not to carry? Biochim. Biophys. Acta 2016, 1863, 2436–2442. [Google Scholar] [CrossRef]
- Schell, J.C.; Wisidagama, D.R.; Bensard, C.; Zhao, H.; Wei, P.; Tanner, J.; Flores, A.; Mohlman, J.; Sorensen, L.K.; Earl, C.S.; et al. Control of intestinal stem cell function and proliferation by mitochondrial pyruvate metabolism. Nat. Cell Biol. 2017, 19, 1027–1036. [Google Scholar] [CrossRef]
- Bensard, C.L.; Wisidagama, D.R.; Olson, K.A.; Berg, J.A.; Krah, N.M.; Schell, J.C.; Nowinski, S.M.; Fogarty, S.; Bott, A.J.; Wei, P.; et al. Regulation of Tumor Initiation by the Mitochondrial Pyruvate Carrier. Cell Metab. 2020, 31, 284–300.e287. [Google Scholar] [CrossRef]
- Tompkins, S.C.; Sheldon, R.D.; Rauckhorst, A.J.; Noterman, M.F.; Solst, S.R.; Buchanan, J.L.; Mapuskar, K.A.; Pewa, A.D.; Gray, L.R.; Oonthonpan, L.; et al. Disrupting Mitochondrial Pyruvate Uptake Directs Glutamine into the TCA Cycle away from Glutathione Synthesis and Impairs Hepatocellular Tumorigenesis. Cell Rep. 2019, 28, 2608–2619.e2606. [Google Scholar] [CrossRef]
- Bader, D.A.; Hartig, S.M.; Putluri, V.; Foley, C.; Hamilton, M.P.; Smith, E.A.; Saha, P.K.; Panigrahi, A.; Walker, C.; Zong, L.; et al. Mitochondrial pyruvate import is a metabolic vulnerability in androgen receptor-driven prostate cancer. Nat. Metab. 2019, 1, 70–85. [Google Scholar] [CrossRef]
- Buffet, A.; Morin, A.; Castro-Vega, L.J.; Habarou, F.; Lussey-Lepoutre, C.; Letouze, E.; Lefebvre, H.; Guilhem, I.; Haissaguerre, M.; Raingeard, I.; et al. Germline Mutations in the Mitochondrial 2-Oxoglutarate/Malate Carrier SLC25A11 Gene Confer a Predisposition to Metastatic Paragangliomas. Cancer Res. 2018, 78, 1914–1922. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.S.; Lee, H.; Lee, S.; Kang, J.H.; Lee, S.H.; Kim, S.G.; Cho, E.S.; Kim, N.H.; Yook, J.I.; Kim, S.Y. Loss of SLC25A11 causes suppression of NSCLC and melanoma tumor formation. EBioMedicine 2019, 40, 184–197. [Google Scholar] [CrossRef] [Green Version]
- Pike, L.S.; Smift, A.L.; Croteau, N.J.; Ferrick, D.A.; Wu, M. Inhibition of fatty acid oxidation by etomoxir impairs NADPH production and increases reactive oxygen species resulting in ATP depletion and cell death in human glioblastoma cells. Biochim. Biophys. Acta 2011, 1807, 726–734. [Google Scholar] [CrossRef] [Green Version]
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer metabolism: Fatty acid oxidation in the limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factor 1: Oxygen homeostasis and disease pathophysiology. Trends Mol. Med. 2001, 7, 345–350. [Google Scholar] [CrossRef]
- Kung, A.L.; Wang, S.; Klco, J.M.; Kaelin, W.G.; Livingston, D.M. Suppression of tumor growth through disruption of hypoxia-inducible transcription. Nat. Med. 2000, 6, 1335–1340. [Google Scholar] [CrossRef]
- Aebersold, D.M.; Burri, P.; Beer, K.T.; Laissue, J.; Djonov, V.; Greiner, R.H.; Semenza, G.L. Expression of hypoxia-inducible factor-1α: A novel predictive and prognostic parameter in the radiotherapy of oropharyngeal cancer. Cancer Res. 2001, 61, 2911–2916. [Google Scholar]
- Koukourakis, M.I.; Giatromanolaki, A.; Skarlatos, J.; Corti, L.; Blandamura, S.; Piazza, M.; Gatter, K.C.; Harris, A.L. Hypoxia inducible factor (HIF-1a and HIF-2a) expression in early esophageal cancer and response to photodynamic therapy and radiotherapy. Cancer Res. 2001, 61, 1830–1832. [Google Scholar]
- Semenza, G.L. Hypoxia-inducible factor 1: Master regulator of O2 homeostasis. Curr. Opin. Genet. Dev. 1998, 8, 588–594. [Google Scholar] [CrossRef]
- Kaelin, W.G. Proline hydroxylation and gene expression. Annu. Rev. Biochem. 2005, 74, 115–128. [Google Scholar] [CrossRef]
- Epstein, A.C.; Gleadle, J.M.; McNeill, L.A.; Hewitson, K.S.; O’Rourke, J.; Mole, D.R.; Mukherji, M.; Metzen, E.; Wilson, M.I.; Dhanda, A.; et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 2001, 107, 43–54. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, K.; Frew, I.J.; Hagensen, M.; Skals, M.; Habelhah, H.; Bhoumik, A.; Kadoya, T.; Erdjument-Bromage, H.; Tempst, P.; Frappell, P.B.; et al. Siah2 regulates stability of prolyl-hydroxylases, controls HIF1alpha abundance, and modulates physiological responses to hypoxia. Cell 2004, 117, 941–952. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, R.; Hardy, A.; Schofield, C.J. The human oxygen sensing machinery and its manipulation. Chem. Soc. Rev. 2008, 37, 1308–1319. [Google Scholar] [CrossRef]
- Mathieu, J.; Zhang, Z.; Zhou, W.; Wang, A.J.; Heddleston, J.M.; Pinna, C.M.; Hubaud, A.; Stadler, B.; Choi, M.; Bar, M.; et al. HIF induces human embryonic stem cell markers in cancer cells. Cancer Res. 2011, 71, 4640–4652. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Wan, W.W.; Xiong, S.L.; Feng, H.; Wu, N. Cancer stem-like cells can be induced through dedifferentiation under hypoxic conditions in glioma, hepatoma and lung cancer. Cell Death Discov. 2017, 3, 16105. [Google Scholar] [CrossRef]
- Tang, K.; Yu, Y.; Zhu, L.; Xu, P.; Chen, J.; Ma, J.; Zhang, H.; Fang, H.; Sun, W.; Zhou, L.; et al. Hypoxia-reprogrammed tricarboxylic acid cycle promotes the growth of human breast tumorigenic cells. Oncogene 2019, 38, 6970–6984. [Google Scholar] [CrossRef] [Green Version]
- Eguchi, K.; Nakayama, K. Prolonged hypoxia decreases nuclear pyruvate dehydrogenase complex and regulates the gene expression. Biochem. Biophys. Res. Commun. 2019, 520, 128–135. [Google Scholar] [CrossRef]
- Yang, Y.; Yang, X.; Yang, Y.; Zhu, H.; Chen, X.; Zhang, H.; Wang, F.; Qin, Q.; Cheng, H.; Sun, X. Exosomes: A Promising Factor Involved in Cancer Hypoxic Microenvironments. Curr. Med. Chem. 2015, 22, 4189–4195. [Google Scholar] [CrossRef]
- Axelson, H.; Fredlund, E.; Ovenberger, M.; Landberg, G.; Pahlman, S. Hypoxia-induced dedifferentiation of tumor cells--a mechanism behind heterogeneity and aggressiveness of solid tumors. Semin. Cell Dev. Biol. 2005, 16, 554–563. [Google Scholar] [CrossRef]
- Batie, M.; Rocha, S. JmjC histone demethylases act as chromatin oxygen sensors. Mol. Cell Oncol. 2019, 6, 1608501. [Google Scholar] [CrossRef] [Green Version]
- Batie, M.; Frost, J.; Frost, M.; Wilson, J.W.; Schofield, P.; Rocha, S. Hypoxia induces rapid changes to histone methylation and reprograms chromatin. Science 2019, 363, 1222–1226. [Google Scholar] [CrossRef]
- Dobrynin, G.; McAllister, T.E.; Leszczynska, K.B.; Ramachandran, S.; Krieg, A.J.; Kawamura, A.; Hammond, E.M. KDM4A regulates HIF-1 levels through H3K9me3. Sci. Rep. 2017, 7, 11094. [Google Scholar] [CrossRef]
- Wu, M.Z.; Tsai, Y.P.; Yang, M.H.; Huang, C.H.; Chang, S.Y.; Chang, C.C.; Teng, S.C.; Wu, K.J. Interplay between HDAC3 and WDR5 is essential for hypoxia-induced epithelial-mesenchymal transition. Mol. Cell 2011, 43, 811–822. [Google Scholar] [CrossRef]
- Intlekofer, A.M.; Dematteo, R.G.; Venneti, S.; Finley, L.W.; Lu, C.; Judkins, A.R.; Rustenburg, A.S.; Grinaway, P.B.; Chodera, J.D.; Cross, J.R.; et al. Hypoxia Induces Production of L-2-Hydroxyglutarate. Cell Metab. 2015, 22, 304–311. [Google Scholar] [CrossRef] [Green Version]
- Oldham, W.M.; Clish, C.B.; Yang, Y.; Loscalzo, J. Hypoxia-Mediated Increases in L-2-hydroxyglutarate Coordinate the Metabolic Response to Reductive Stress. Cell Metab. 2015, 22, 291–303. [Google Scholar] [CrossRef] [Green Version]
- Thienpont, B.; Steinbacher, J.; Zhao, H.; D’Anna, F.; Kuchnio, A.; Ploumakis, A.; Ghesquiere, B.; Van Dyck, L.; Boeckx, B.; Schoonjans, L.; et al. Tumour hypoxia causes DNA hypermethylation by reducing TET activity. Nature 2016, 537, 63–68. [Google Scholar] [CrossRef]
- Sia, J.; Szmyd, R.; Hau, E.; Gee, H.E. Molecular Mechanisms of Radiation-Induced Cancer Cell Death: A Primer. Front. Cell Dev. Biol. 2020, 8, 41. [Google Scholar] [CrossRef]
- Vander Linden, C.; Corbet, C. Therapeutic Targeting of Cancer Stem Cells: Integrating and Exploiting the Acidic Niche. Front. Oncol. 2019, 9, 159. [Google Scholar] [CrossRef] [Green Version]
- Najafi, M.; Farhood, B.; Mortezaee, K.; Kharazinejad, E.; Majidpoor, J.; Ahadi, R. Hypoxia in solid tumors: A key promoter of cancer stem cell (CSC) resistance. J. Cancer Res. Clin. Oncol. 2020, 146, 19–31. [Google Scholar] [CrossRef]
- Macedo-Silva, C.; Miranda-Goncalves, V.; Henrique, R.; Jeronimo, C.; Bravo, I. The Critical Role of Hypoxic Microenvironment and Epigenetic Deregulation in Esophageal Cancer Radioresistance. Genes 2019, 10, 927. [Google Scholar] [CrossRef] [Green Version]
- Huijskens, M.J.; Wodzig, W.K.; Walczak, M.; Germeraad, W.T.; Bos, G.M. Ascorbic acid serum levels are reduced in patients with hematological malignancies. Results Immunol. 2016, 6, 8–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cimmino, L.; Neel, B.G.; Aifantis, I. Vitamin C in Stem Cell Reprogramming and Cancer. Trends Cell Biol. 2018, 28, 698–708. [Google Scholar] [CrossRef]
- Scafoglio, C.; Hirayama, B.A.; Kepe, V.; Liu, J.; Ghezzi, C.; Satyamurthy, N.; Moatamed, N.A.; Huang, J.; Koepsell, H.; Barrio, J.R.; et al. Functional expression of sodium-glucose transporters in cancer. Proc. Natl. Acad. Sci. USA 2015, 112, E4111–E4119. [Google Scholar] [CrossRef] [Green Version]
- Scafoglio, C.R.; Villegas, B.; Abdelhady, G.; Bailey, S.T.; Liu, J.; Shirali, A.S.; Wallace, W.D.; Magyar, C.E.; Grogan, T.R.; Elashoff, D.; et al. Sodium-glucose transporter 2 is a diagnostic and therapeutic target for early-stage lung adenocarcinoma. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef]
- Hsieh, M.H.; Choe, J.H.; Gadhvi, J.; Kim, Y.J.; Arguez, M.A.; Palmer, M.; Gerold, H.; Nowak, C.; Do, H.; Mazambani, S.; et al. p63 and SOX2 Dictate Glucose Reliance and Metabolic Vulnerabilities in Squamous Cell Carcinomas. Cell Rep. 2019, 28, 1860–1878.e1869. [Google Scholar] [CrossRef] [Green Version]
- Momcilovic, M.; Bailey, S.T.; Lee, J.T.; Fishbein, M.C.; Braas, D.; Go, J.; Graeber, T.G.; Parlati, F.; Demo, S.; Li, R.; et al. The GSK3 Signaling Axis Regulates Adaptive Glutamine Metabolism in Lung Squamous Cell Carcinoma. Cancer Cell 2018, 33, 905–921.e905. [Google Scholar] [CrossRef] [Green Version]
- Pajak, B.; Siwiak, E.; Soltyka, M.; Priebe, A.; Zielinski, R.; Fokt, I.; Ziemniak, M.; Jaskiewicz, A.; Borowski, R.; Domoradzki, T.; et al. 2-Deoxy-d-Glucose and Its Analogs: From Diagnostic to Therapeutic Agents. Int. J. Mol. Sci. 2019, 21, 234. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Herschman, H.R. A Tumor Agnostic Therapeutic Strategy for Hexokinase 1-Null/Hexokinase 2-Positive Cancers. Cancer Res. 2019, 79, 5907–5914. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Qu, C.; Liu, T.; Wang, C. PFKFB3 inhibitors as potential anticancer agents: Mechanisms of action, current developments, and structure-activity relationships. Eur. J. Med. Chem. 2020, 203, 112612. [Google Scholar] [CrossRef]
- Shackelford, D.B.; Abt, E.; Gerken, L.; Vasquez, D.S.; Seki, A.; Leblanc, M.; Wei, L.; Fishbein, M.C.; Czernin, J.; Mischel, P.S.; et al. LKB1 inactivation dictates therapeutic response of non-small cell lung cancer to the metabolism drug phenformin. Cancer Cell 2013, 23, 143–158. [Google Scholar] [CrossRef] [Green Version]
- Vasan, K.; Werner, M.; Chandel, N.S. Mitochondrial Metabolism as a Target for Cancer Therapy. Cell Metab. 2020, 32, 341–352. [Google Scholar] [CrossRef]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef]
- Wang, P.; Wang, Z.; Liu, J. Role of HDACs in normal and malignant hematopoiesis. Mol. Cancer 2020, 19, 5. [Google Scholar] [CrossRef] [Green Version]
- Debeb, B.G.; Lacerda, L.; Xu, W.; Larson, R.; Solley, T.; Atkinson, R.; Sulman, E.P.; Ueno, N.T.; Krishnamurthy, S.; Reuben, J.M.; et al. Histone deacetylase inhibitors stimulate dedifferentiation of human breast cancer cells through WNT/beta-catenin signaling. Stem Cells 2012, 30, 2366–2377. [Google Scholar] [CrossRef] [Green Version]
- Laugesen, A.; Helin, K. Chromatin repressive complexes in stem cells, development, and cancer. Cell Stem Cell 2014, 14, 735–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rugo, H.S.; Jacobs, I.; Sharma, S.; Scappaticci, F.; Paul, T.A.; Jensen-Pergakes, K.; Malouf, G.G. The Promise for Histone Methyltransferase Inhibitors for Epigenetic Therapy in Clinical Oncology: A Narrative Review. Adv. Ther. 2020, 37, 3059–3082. [Google Scholar] [CrossRef]
- McCabe, M.T.; Graves, A.P.; Ganji, G.; Diaz, E.; Halsey, W.S.; Jiang, Y.; Smitheman, K.N.; Ott, H.M.; Pappalardi, M.B.; Allen, K.E.; et al. Mutation of A677 in histone methyltransferase EZH2 in human B-cell lymphoma promotes hypertrimethylation of histone H3 on lysine 27 (H3K27). Proc. Natl. Acad. Sci. USA 2012, 109, 2989–2994. [Google Scholar] [CrossRef] [Green Version]
- Varambally, S.; Dhanasekaran, S.M.; Zhou, M.; Barrette, T.R.; Kumar-Sinha, C.; Sanda, M.G.; Ghosh, D.; Pienta, K.J.; Sewalt, R.G.; Otte, A.P.; et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002, 419, 624–629. [Google Scholar] [CrossRef]
- Mohammad, F.; Weissmann, S.; Leblanc, B.; Pandey, D.P.; Hojfeldt, J.W.; Comet, I.; Zheng, C.; Johansen, J.V.; Rapin, N.; Porse, B.T.; et al. EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat. Med. 2017, 23, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Roberts, C.W. Targeting EZH2 in cancer. Nat. Med. 2016, 22, 128–134. [Google Scholar] [CrossRef]
- Adelaiye-Ogala, R.; Budka, J.; Damayanti, N.P.; Arrington, J.; Ferris, M.; Hsu, C.C.; Chintala, S.; Orillion, A.; Miles, K.M.; Shen, L.; et al. EZH2 Modifies Sunitinib Resistance in Renal Cell Carcinoma by Kinome Reprogramming. Cancer Res. 2017, 77, 6651–6666. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.W.; Hua, K.T.; Li, K.C.; Kao, H.F.; Hong, R.L.; Ko, J.Y.; Hsiao, M.; Kuo, M.L.; Tan, C.T. Histone Methyltransferase G9a Drives Chemotherapy Resistance by Regulating the Glutamate-Cysteine Ligase Catalytic Subunit in Head and Neck Squamous Cell Carcinoma. Mol. Cancer Ther. 2017, 16, 1421–1434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Dong, X.; Ren, Y.; Luo, J.; Liu, P.; Su, D.; Yang, X. Targeting EHMT2 reverses EGFR-TKI resistance in NSCLC by epigenetically regulating the PTEN/AKT signaling pathway. Cell Death Dis. 2018, 9, 129. [Google Scholar] [CrossRef] [Green Version]
- Nassa, G.; Salvati, A.; Tarallo, R.; Gigantino, V.; Alexandrova, E.; Memoli, D.; Sellitto, A.; Rizzo, F.; Malanga, D.; Mirante, T.; et al. Inhibition of histone methyltransferase DOT1L silences ERalpha gene and blocks proliferation of antiestrogen-resistant breast cancer cells. Sci. Adv. 2019, 5, eaav5590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lochmann, T.L.; Powell, K.M.; Ham, J.; Floros, K.V.; Heisey, D.A.R.; Kurupi, R.I.J.; Calbert, M.L.; Ghotra, M.S.; Greninger, P.; Dozmorov, M.; et al. Targeted inhibition of histone H3K27 demethylation is effective in high-risk neuroblastoma. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Ravasio, R.; Ceccacci, E.; Nicosia, L.; Hosseini, A.; Rossi, P.L.; Barozzi, I.; Fornasari, L.; Zuffo, R.D.; Valente, S.; Fioravanti, R.; et al. Targeting the scaffolding role of LSD1 (KDM1A) poises acute myeloid leukemia cells for retinoic acid-induced differentiation. Sci. Adv. 2020, 6, eaax2746. [Google Scholar] [CrossRef] [Green Version]
- Heuser, M.; Yun, H.; Thol, F. Epigenetics in myelodysplastic syndromes. Semin. Cancer Biol. 2018, 51, 170–179. [Google Scholar] [CrossRef]
- Wu, Q.; Yang, Z.; Xia, L.; Nie, Y.; Wu, K.; Shi, Y.; Fan, D. Methylation of miR-129-5p CpG island modulates multi-drug resistance in gastric cancer by targeting ABC transporters. Oncotarget 2014, 5, 11552–11563. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Wang, R.; Song, H.; Huang, G.; Chen, L. Secreted frizzled related protein 1 modulates taxane resistance of human lung adenocarcinoma. Mol. Med. 2014, 20, 164–178. [Google Scholar] [CrossRef]
- De La Rosa, J.; Urdiciain, A.; Zazpe, I.; Zelaya, M.V.; Melendez, B.; Rey, J.A.; Idoate, M.A.; Castresana, J.S. The synergistic effect of DZNEP, panobinostat and temozolomide reduces clonogenicity and induces apoptosis in glioblastoma cells. Int. J. Oncol. 2020, 56, 283–300. [Google Scholar] [CrossRef]
- Dimopoulos, K.; Sogaard Helbo, A.; Fibiger Munch-Petersen, H.; Sjo, L.; Christensen, J.; Sommer Kristensen, L.; Asmar, F.; Hermansen, N.E.U.; O’Connel, C.; Gimsing, P.; et al. Dual inhibition of DNMTs and EZH2 can overcome both intrinsic and acquired resistance of myeloma cells to IMiDs in a cereblon-independent manner. Mol. Oncol. 2018, 12, 180–195. [Google Scholar] [CrossRef]
- Klaus, C.R.; Iwanowicz, D.; Johnston, D.; Campbell, C.A.; Smith, J.J.; Moyer, M.P.; Copeland, R.A.; Olhava, E.J.; Scott, M.P.; Pollock, R.M.; et al. DOT1L inhibitor EPZ-5676 displays synergistic antiproliferative activity in combination with standard of care drugs and hypomethylating agents in MLL-rearranged leukemia cells. J. Pharmacol. Exp. Ther. 2014, 350, 646–656. [Google Scholar] [CrossRef] [Green Version]
- Steele, N.; Finn, P.; Brown, R.; Plumb, J.A. Combined inhibition of DNA methylation and histone acetylation enhances gene re-expression and drug sensitivity in vivo. Br. J. Cancer 2009, 100, 758–763. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saggese, P.; Sellitto, A.; Martinez, C.A.; Giurato, G.; Nassa, G.; Rizzo, F.; Tarallo, R.; Scafoglio, C. Metabolic Regulation of Epigenetic Modifications and Cell Differentiation in Cancer. Cancers 2020, 12, 3788. https://doi.org/10.3390/cancers12123788
Saggese P, Sellitto A, Martinez CA, Giurato G, Nassa G, Rizzo F, Tarallo R, Scafoglio C. Metabolic Regulation of Epigenetic Modifications and Cell Differentiation in Cancer. Cancers. 2020; 12(12):3788. https://doi.org/10.3390/cancers12123788
Chicago/Turabian StyleSaggese, Pasquale, Assunta Sellitto, Cesar A. Martinez, Giorgio Giurato, Giovanni Nassa, Francesca Rizzo, Roberta Tarallo, and Claudio Scafoglio. 2020. "Metabolic Regulation of Epigenetic Modifications and Cell Differentiation in Cancer" Cancers 12, no. 12: 3788. https://doi.org/10.3390/cancers12123788