
Biochemical Characterization of a Recombinant UDP-glucosyltransferase from Rice and Enzymatic Production of Deoxynivalenol-3-O-β-D-glucoside

 , , and
, , and
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Expression and Purification of OsUGT79
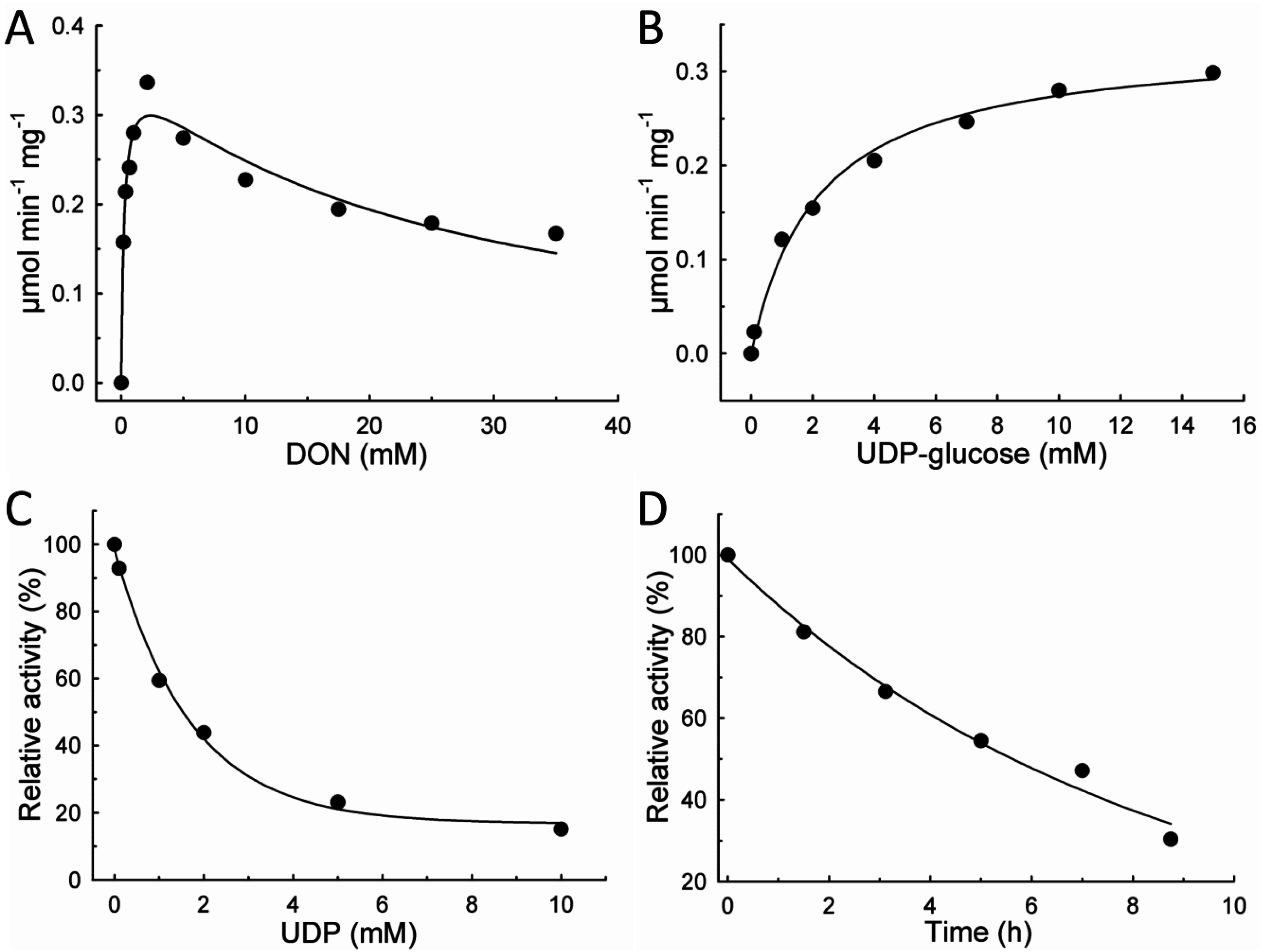
2.2. Kinetic Characteristics of OsUGT79

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Activity (%) |
|---|---|
| Control | 100 |
| EDTA (1 mM) | 98 |
| EDTA (5 mM) | 95 |
| CaCl2 | 108 |
| CuSO4 | ND |
| MgSO4 | 117 |
| MnCl2 | 143 |
| MnSO4 | 139 |
| ZnSO4 | ND |
| FeSO4 | 114 |
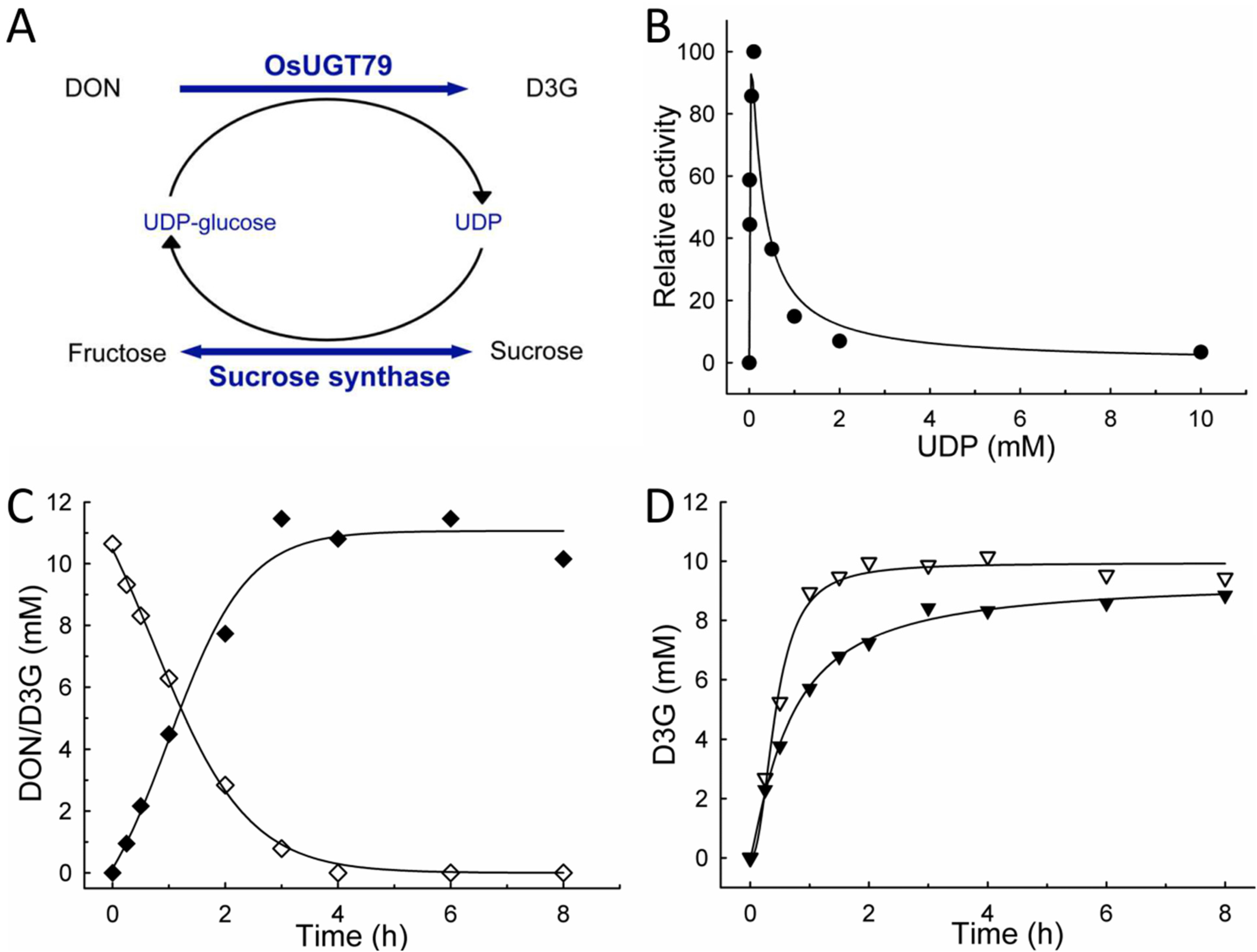
2.3. UDP-Glucose Recycling and DON Production
2.4. Preparative Production and Purification of D3G
3. Experimental Section
3.1. Materials
3.2. Cloning of OsUGT79 and AtSUS1
3.3. Protein Expression and Purification
3.4. Glycosylation Assays
3.5. DON and D3G Determination by HPLC-MS/MS
3.6. Preparative High-Performance Liquid Chromatography
3.7. D3G Purity Analysis by HPLC-UV
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bhat, R.; Rai, R.V.; Karim, A.A. Mycotoxins in food and feed: Present status and future concerns. Compr. Rev. Food Sci. Food Saf. 2010, 9, 57–81. [Google Scholar] [CrossRef]
- Wu, F.; Groopman, J.D.; Pestka, J.J. Public health impacts of foodborne mycotoxins. Annu. Rev. Food Sci. Technol. 2014, 5, 351–372. [Google Scholar] [CrossRef] [PubMed]
- Maresca, M. From the gut to the brain: Journey and pathophysiological effects of the food-associated trichothecene mycotoxin deoxynivalenol. Toxins 2013, 5, 784–820. [Google Scholar] [CrossRef] [PubMed]
- European Commission. Commision Regulation (EC) No 1881/2006 of 19 December 2006 setting maximum levels for certain contaminants in foodstuffs. Off. J. Eur. Union 2006, L364, 5–24. [Google Scholar]
- European Commision. Commission Regulation (EC) No 1126/2006 of 28 September 2008 amending Regulation (EC) No 1881/2006 setting maximum levels for certain contaminants in foodstuffs as regards Fusarium toxins in maize and maize products. Off. J. Eur. Union 2007, L255, 14–17. [Google Scholar]
- Joint FAO/WHO Expert Committee on Food Additives (JECFA). Evaluation of certain contaminants in food. WHO Tech. Rep. Ser. 2011, 959. Available online: http://whqlibdoc.who.int/trs/WHO_TRS_959_eng.pdf (accessed on 17 July 2015).
- Graziani, F.; Pujol, A.; Nicoletti, C.; Pinton, P.; Armand, L.; di Pasquale, E.; Oswald, I.P.; Perrier, J.; Maresca, M. The Food-Associated Ribotoxin Deoxynivalenol Modulates Inducible NO Synthase in Human Intestinal Cell Model. Toxicol. Sci. 2015, 145, 372–382. [Google Scholar] [CrossRef] [PubMed]
- Pinton, P.; Graziani, F.; Pujol, A.; Nicoletti, C.; Paris, O.; Ernouf, P.; di Pasquale, E.; Perrier, J.; Oswald, I.P.; Maresca, M. Deoxynivalenol inhibits the expression by goblet cells of intestinal mucins through a PKR and MAP kinase dependent repression of the resistin-like molecule β. Mol. Nutr. Food Res. 2015, 59, 1076–1087. [Google Scholar] [CrossRef] [PubMed]
- Gratz, S.W.; Richardson, A.J.; Duncan, G.; Holtrop, G. Annual variation of dietary deoxynivalenol exposure during years of different Fusarium prevalence: A pilot biomonitoring study. Food Addit. Contam. Part A 2014, 31, 1579–1585. [Google Scholar] [CrossRef] [PubMed]
- Šarkanj, B.; Warth, B.; Uhlig, S.; Abia, W.A.; Sulyok, M.; Klapec, T.; Krska, R.; Banjari, I. Urinary analysis reveals high deoxynivalenol exposure in pregnant women from Croatia. Food Chem. Toxicol. 2013, 62, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Warth, B.; Sulyok, M.; Fruhmann, P.; Berthiller, F.; Schuhmacher, R.; Hametner, C.; Adam, G.; Fröhlich, J.; Krska, R. Assessment of human deoxynivalenol exposure using an LC-MS/MS based biomarker method. Toxicol. Lett. 2012, 211, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Berthiller, F.; Crews, C.; Dall’Asta, C.; Saeger, S.D.; Haesaert, G.; Karlovsky, P.; Oswald, I.P.; Seefelder, W.; Speijers, G.; Stroka, J. Masked mycotoxins: A review. Mol. Nutr. Food Res. 2013, 57, 165–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rychlik, M.; Humpf, H.-U.; Marko, D.; Dänicke, S.; Mally, A.; Berthiller, F.; Klaffke, H.; Lorenz, N. Proposal of a comprehensive definition of modified and other forms of mycotoxins including “masked” mycotoxins. Mycotoxin Res. 2014, 30, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Coleman, J.O.D.; Blake-Kalff, M.M.A.; Davies, T.G.E. Detoxification of xenobiotics by plants: Chemical modification and vacuolar compartmentation. Trends Plant Sci. 1997, 2, 144–151. [Google Scholar] [CrossRef]
- Bowles, D.; Lim, E.-K.; Poppenberger, B.; Vaistij, F.E. Glycosyltransferases of lipophilic small molecules. Annu. Rev. Plant Biol. 2006, 57, 567–597. [Google Scholar] [CrossRef] [PubMed]
- Broekaert, N.; Devreese, M.; de Baere, S.; de Backer, P.; Croubels, S. Modified Fusarium mycotoxins unmasked: From occurrence in cereals to animal and human excretion. Food Chem. Toxicol. 2015, 80, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, P.M.; Deleury, E.; Davies, G.J.; Henrissat, B. An evolving hierarchical family classification for glycosyltransferases. J. Mol. Biol. 2003, 328, 307–317. [Google Scholar] [CrossRef]
- Schweiger, W.; Pasquet, J.C.; Nussbaumer, T.; Paris, M.P.K.; Wiesenberger, G.; Macadré, C.; Ametz, C.; Berthiller, F.; Lemmens, M.; Saindrenan, P.; et al. Functional characterization of two clusters of Brachypodium distachyon UDP-glycosyltransferases encoding putative deoxynivalenol detoxification genes. Mol. Plant-Microbe Interact. 2013, 26, 781–792. [Google Scholar] [CrossRef] [PubMed]
- Bowles, D.; Isayenkova, J.; Lim, E.K.; Poppenberger, B. Glycosyltransferases: Managers of small molecules. Curr. Opin. Plant Biol. 2005, 8, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Poppenberger, B.; Berthiller, F.; Lucyshyn, D.; Sieberer, T.; Schuhmacher, R.; Krska, R.; Kuchler, K.; Glössl, J.; Luschnig, C.; Adam, G. Detoxification of the Fusarium mycotoxin deoxynivalenol by a UDP-glucosyltransferase from Arabidopsis thaliana. J. Biol. Chem. 2003, 278, 47905–47914. [Google Scholar] [CrossRef] [PubMed]
- Desmond, O.J.; Manners, J.M.; Schenk, P.M.; Maclean, D.J.; Kazan, K. Gene expression analysis of the wheat response to infection by Fusarium pseudograminearum. Physiol. Mol. Plant Pathol. 2008, 73, 40–47. [Google Scholar] [CrossRef]
- Hill-Ambroz, K.; Webb, C.A.; Matthews, A.R.; Li, W.; Gill, B.S.; Fellers, J.P. Expression analysis and physical mapping of a cDNA library of Fusarium head blight infected wheat spikes. Crop Sci. 2006, 46, S15–S26. [Google Scholar] [CrossRef]
- Ma, L.; Shang, Y.; Cao, A.; Qi, Z.; Xing, L.; Chen, P.; Liu, D.; Wang, X. Molecular cloning and characterization of an up-regulated UDP-glucosyltransferase gene induced by DON from Triticum aestivum L. cv. Wangshuibai. Mol. Biol. Rep. 2010, 37, 785–795. [Google Scholar]
- Steiner, B.; Kurz, H.; Lemmens, M.; Buerstmayr, H. Differential gene expression of related wheat lines with contrasting levels of head blight resistance after Fusarium graminearum inoculation. Theor. Appl. Genet. 2009, 118, 753–764. [Google Scholar] [CrossRef] [PubMed]
- Lemmens, M.; Scholz, U.; Berthiller, F.; Dall’Asta, C.; Koutnik, A.; Schuhmacher, R.; Adam, G.; Buerstmayr, H.; Mesterházy, Á.; Krska, R.; et al. The ability to detoxify the mycotoxin deoxynivalenol colocalizes with a major quantitative trait locus for Fusarium head blight resistance in wheat. Mol. Plant-Microbe Interact. 2005, 18, 1318–1324. [Google Scholar] [CrossRef] [PubMed]
- Horevaj, P.; Gale, L.R.; Milus, E.A. Resistance in winter wheat lines to initial infection and spread within spikes by deoxynivalenol and nivalenol chemotypes of Fusarium graminearum. Plant Dis. 2011, 95, 31–37. [Google Scholar] [CrossRef]
- Gunnaiah, R.; Kushalappa, A.C.; Duggavathi, R.; Fox, S.; Somers, D.J. Integrated metabolo-proteomic approach to decipher the mechanisms by which wheat QTL (Fhb1) contributes to resistance against Fusarium graminearum. PLoS ONE 2012, 7, e40695. [Google Scholar] [CrossRef] [PubMed]
- Pasquet, J.-C. Détoxication des Mycotoxines par les Plantes : Analyse de l’Interaction entre Brachypodium distachyon et Fusarium graminearum. Doctoral Thesis, Ecole doctorale Sciences du Végétal, Orsay, France, November 2014. [Google Scholar]
- Li, X.; Shin, S.; Heinen, S.; Dill-Macky, R.; Berthiller, F.; Clemente, T.; McCormick, S.; Muehlbauer, G.J. Transgenic wheat expressing a barley UDP-glucosyltransferase detoxifies deoxynivalenol and provides high levels of resistance to Fusarium graminearum. Mol. Plant-Microbe Interact. 2015. submitted. [Google Scholar]
- Berthiller, F.; Dall’asta, C.; Corradini, R.; Marchelli, R.; Sulyok, M.; Krska, R.; Adam, G.; Schuhmacher, R. Occurrence of deoxynivalenol and its 3-β-d-glucoside in wheat and maize. Food Addit. Contam. Part A 2009, 26, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Desmarchelier, A.; Seefelder, W. Survey of deoxynivalenol and deoxynivalenol-3-glucoside in cereal-based products by liquid chromatography electrospray ionization tandem mass spectrometry. World Mycotoxin J. 2011, 4, 29–35. [Google Scholar] [CrossRef]
- Kostelanska, M.; Hajslova, J.; Zachariasova, M.; Malachova, A.; Kalachova, K.; Poustka, J.; Fiala, J.; Scott, P.M.; Berthiller, F.; Krska, R. Occurrence of deoxynivalenol and its major conjugate, deoxynivalenol-3-glucoside, in beer and some brewing intermediates. J. Agric. Food Chem. 2009, 57, 3187–3194. [Google Scholar] [CrossRef] [PubMed]
- De Nijs, M.; van den Top, H.; Portier, L.; Oegema, G.; Kramer, E.; van Egmond, H.; Hoogenboom, L. Digestibility and absorption of deoxynivalenol-3-β-glucoside in in vitro models. World Mycotoxin J. 2012, 5, 319–324. [Google Scholar] [CrossRef]
- Malachova, A.; Stockova, L.; Wakker, A.; Varga, E.; Krska, R.; Michlmayr, H.; Adam, G.; Berthiller, F. Critical Evaluation of indirect methods for the determination of modified deoxynivalenol in cereals. Anal. Bioanal. Chem. 2015, in press. [Google Scholar] [CrossRef] [PubMed]
- Berthiller, F.; Krska, R.; Domig, K.J.; Kneifel, W.; Juge, N.; Schuhmacher, R.; Adam, G. Hydrolytic fate of deoxynivalenol-3-glucoside during digestion. Toxicol. Lett. 2011, 206, 264–267. [Google Scholar] [CrossRef] [PubMed]
- Dall’Erta, A.; Cirlini, M.; Dall’Asta, M.; del Rio, D.; Galaverna, G.; Dall’Asta, C. Masked mycotoxins are efficiently hydrolyzed by human colonic microbiota releasing their aglycones. Chem. Res. Toxicol. 2013, 26, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Nagl, V.; Schwartz, H.; Krska, R.; Moll, W.-D.; Knasmüller, S.; Ritzmann, M.; Adam, G.; Berthiller, F. Metabolism of the masked mycotoxin deoxynivalenol-3-glucoside in rats. Toxicol. Lett. 2012, 213, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Nagl, V.; Woechtl, B.; Schwartz-Zimmermann, H.E.; Hennig-Pauka, I.; Moll, W.-D.; Adam, G.; Berthiller, F. Metabolism of the masked mycotoxin deoxynivalenol-3-glucoside in pigs. Toxicol. Lett. 2014, 229, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Gratz, S.W.; Duncan, G.; Richardson, A.J. The human fecal microbiota metabolizes deoxynivalenol and deoxynivalenol-3-glucoside and may be responsible for urinary deepoxy-deoxynivalenol. Appl. Environ. Microbiol. 2013, 79, 1821–1825. [Google Scholar] [CrossRef] [PubMed]
- European Food Safety Authority (EFSA). Scientific opinion on the risks for human and animal health related to the presence of modified forms of certain mycotoxins in food and feed. EFSA J. 2014, 12, 3916. [Google Scholar]
- Schweiger, W.; Boddu, J.; Shin, S.; Poppenberger, B.; Berthiller, F.; Lemmens, M.; Muehlbauer, G.J.; Adam, G. Validation of a candidate deoxynivalenol-inactivating UDP-glucosyltransferase from barley by heterologous expression in yeast. Mol. Plant-Microbe Interact. 2010, 23, 977–986. [Google Scholar] [CrossRef] [PubMed]
- Son, M.H.; Kim, B.-G.; Kim, D.H.; Jin, M.; Kim, K.; Ahn, J.-H. Production of flavonoid O-glucoside using sucrose synthase and flavonoid O-glucosyltransferase fusion protein. J. Microbiol. Biotechnol. 2009, 19, 709–712. [Google Scholar] [PubMed]
- Masada, S.; Kawase, Y.; Nagatoshi, M.; Oguchi, Y.; Terasaka, K.; Mizukami, H. An efficient chemoenzymatic production of small molecule glucosides with in situ UDP-glucose recycling. FEBS Lett. 2007, 581, 2562–2566. [Google Scholar] [CrossRef] [PubMed]
- Rocco, C.; Dennison, K.; Klenchin, V.A.; Rayment, I.; Escalante-Semerena, J. Construction and use of new cloning vectors for the rapid isolation of recombinant proteins from Escherichia coli. Plasmid 2008, 59, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Lairson, L.; Henrissat, B.; Davies, G.; Withers, S. Glycosyltransferases: Structures, functions, and mechanisms. Biochemistry 2008, 77, 521. [Google Scholar] [CrossRef] [PubMed]
- Moréra, S.; Larivière, L.; Kurzeck, J.; Aschke-Sonnenborn, U.; Freemont, P.S.; Janin, J.; Rüger, W. High resolution crystal structures of T4 phage β-glucosyltransferase: Induced fit and effect of substrate and metal binding. J. Mol. Biol. 2001, 311, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Larivière, L.; Gueguen-Chaignon, V.; Moréra, S. Crystal structures of the T4 phage β-glucosyltransferase and the D100A mutant in complex with UDP-glucose: Glucose binding and identification of the catalytic base for a direct displacement mechanism. J. Mol. Biol. 2003, 330, 1077–1086. [Google Scholar] [CrossRef]
- Ford, C.M.; Boss, P.K.; Høj, P.B. Cloning and characterization of vitis vinifera UDP-Glucose: Flavonoid 3-O-glucosyltransferase, a homologue of the enzyme encoded by the maize bronze-1 locus that may primarily serve to glucosylate anthocyanidins in vivo. J. Biol. Chem. 1998, 273, 9224–9233. [Google Scholar] [CrossRef] [PubMed]
- Almagro, G.; Baroja-Fernández, E.; Muñoz, F.J.; Bahaji, A.; Etxeberria, E.; Li, J.; Montero, M.; Hidalgo, M.; Sesma, M.T.; Pozueta-Romero, J. No evidence for the occurrence of substrate inhibition of Arabidopsis thaliana sucrose synthase-1 (AtSUS1) by fructose and UDP-glucose. Plant Signal. Behav. 2012, 7, 799–802. [Google Scholar] [CrossRef] [PubMed]
- Altpeter, F.; Posselt, U. Production of high quantities of 3-acetyldeoxynivalenol and deoxynivalenol. Appl. Microbiol. Biotechnol. 1994, 41, 384–387. [Google Scholar] [CrossRef]
- Chen, G.; Qiu, N.; Karrer, C.; Caspers, P.; Page, M. Restriction site-free insertion of PCR products directionally into vectors. BioTechniques 2000, 28, 498–500, 504–505. [Google Scholar] [PubMed]
- Van den Ent, F.; Löwe, J. RF cloning: A restriction-free method for inserting target genes into plasmids. J. Biochem. Biophys. Methods 2006, 67, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Paris, M.P.K.; Schweiger, W.; Hametner, C.; Stückler, R.; Muehlbauer, G.J.; Varga, E.; Krska, R.; Berthiller, F.; Adam, G. Zearalenone-16-O-glucoside: A new masked mycotoxin. J. Agric. Food Chem. 2014, 62, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- Krenn, P.; Berthiller, F.; Schweiger, W.; Hametner, C.; Ludwig, R.; Adam, G.; Krska, R.; Schuhmacher, R. Production of zearalenone-4-glucoside, a-zearalenol-4-glucoside and β-zearalenol-4-glucoside. Mycotoxin Res. 2007, 23, 180–184. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Michlmayr, H.; Malachová, A.; Varga, E.; Kleinová, J.; Lemmens, M.; Newmister, S.; Rayment, I.; Berthiller, F.; Adam, G. Biochemical Characterization of a Recombinant UDP-glucosyltransferase from Rice and Enzymatic Production of Deoxynivalenol-3-O-β-D-glucoside. Toxins 2015, 7, 2685-2700. https://doi.org/10.3390/toxins7072685
Michlmayr H, Malachová A, Varga E, Kleinová J, Lemmens M, Newmister S, Rayment I, Berthiller F, Adam G. Biochemical Characterization of a Recombinant UDP-glucosyltransferase from Rice and Enzymatic Production of Deoxynivalenol-3-O-β-D-glucoside. Toxins. 2015; 7(7):2685-2700. https://doi.org/10.3390/toxins7072685
Chicago/Turabian StyleMichlmayr, Herbert, Alexandra Malachová, Elisabeth Varga, Jana Kleinová, Marc Lemmens, Sean Newmister, Ivan Rayment, Franz Berthiller, and Gerhard Adam. 2015. "Biochemical Characterization of a Recombinant UDP-glucosyltransferase from Rice and Enzymatic Production of Deoxynivalenol-3-O-β-D-glucoside" Toxins 7, no. 7: 2685-2700. https://doi.org/10.3390/toxins7072685