
Botulinum Toxin as a Pain Killer: Players and Actions in Antinociception

Abstract
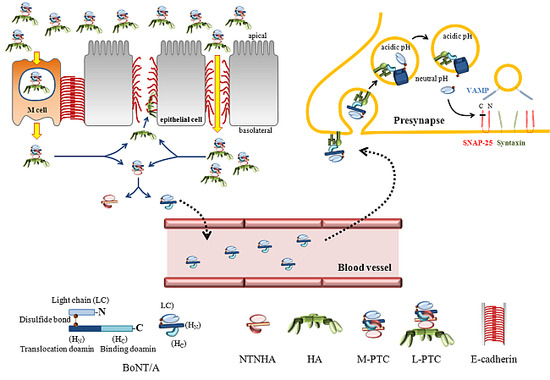
:
1. Introduction
2. BoNTs Complex: Molecular Machines Invading Epithelia and Nerves
2.1. The Components of BoNTs Complex
2.2. The Role of NTNHA
2.3. HA and Gastrointestinal Absorption
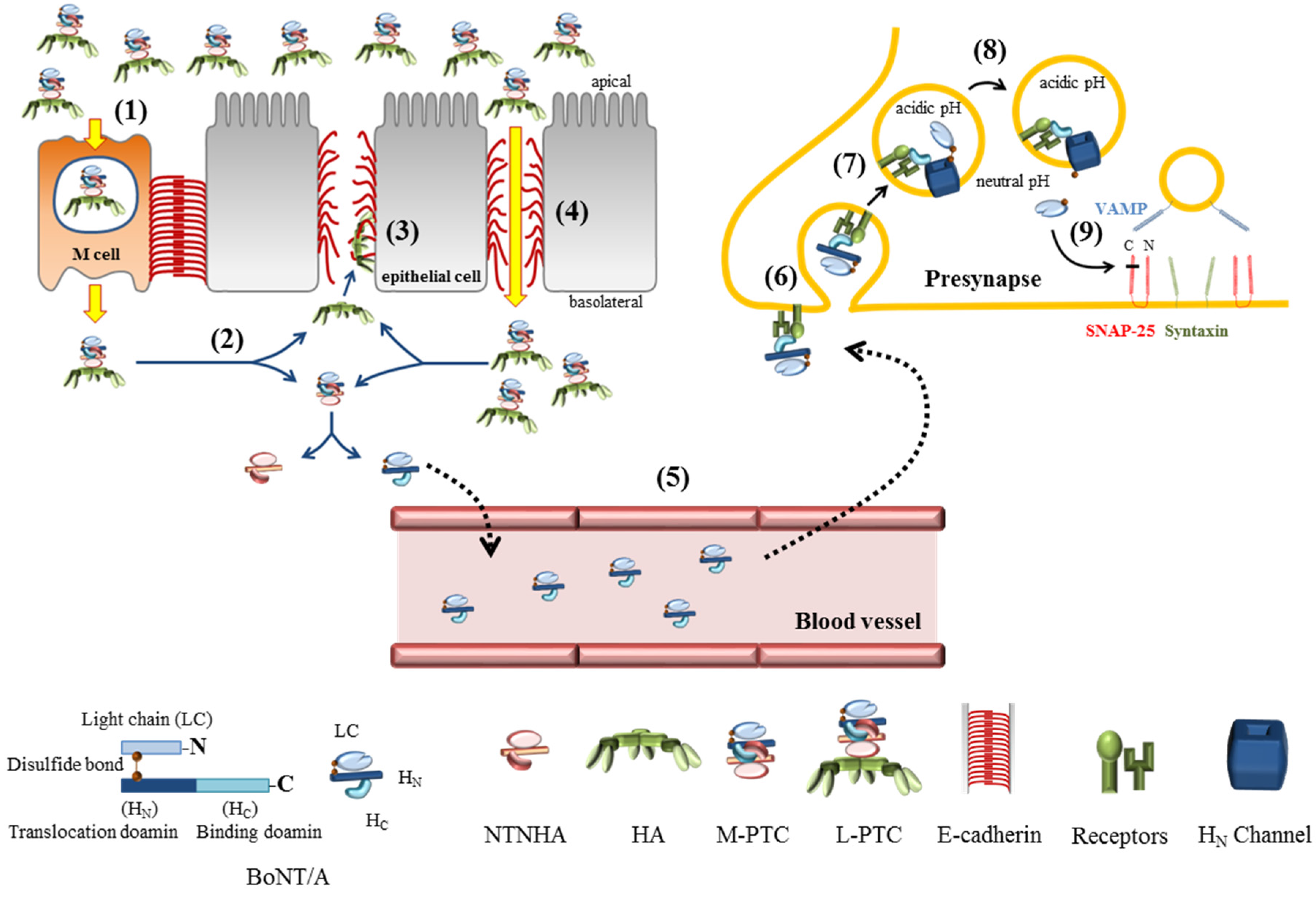
2.4. Uptake at Nerve Endings
2.5. Translocation into the Cytosol to Be an Active Protease
3. BoNTs: A Pain Killer
3.1. Antinociceptive Actions of BoNTs
3.1.1. Glutamate, Substance P (SP) and Cacitonin-Gene Related Peptide (CGRP)
3.1.2. Transient Receptor Potential Vanilloids 1 (TRPV1)
3.1.3. GABAergic and Opioidergic Neurotransmission
3.1.4. Central Effects of BoNTs in Rat Models
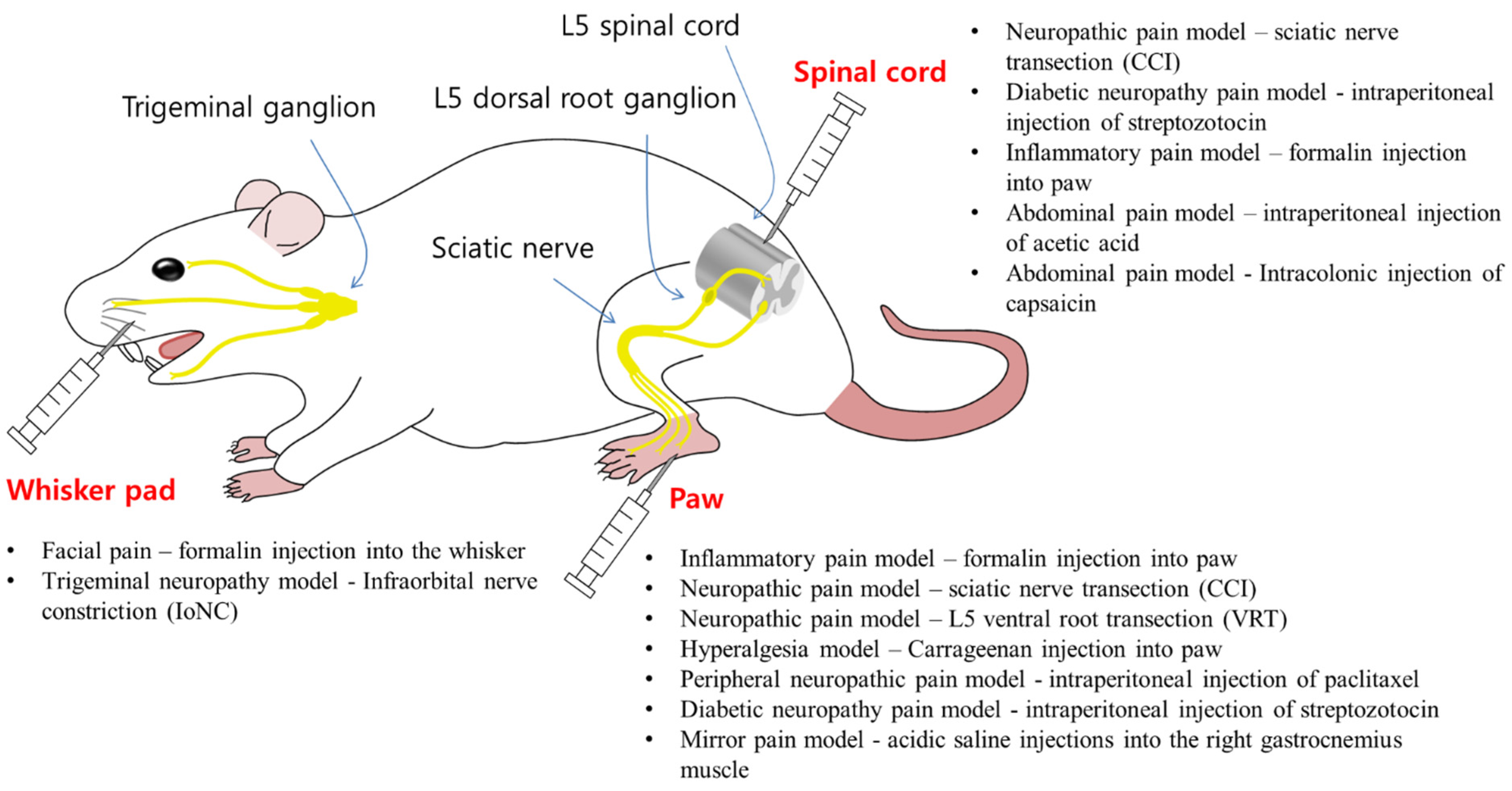
3.1.5. Axonal Transport of BoNTs

{kind=link}
{kind=link}
{kind=link}
| BoNT/A injection | Pain model | Antinociceptive effects |
|---|---|---|
| Paws | Formalin induced inflammatory pain model [73,87,102] | Reduction of enhanced nocifensive behaviors (licking, flinching and shaking) [87,102] |
| Reduction of c-fos early response gene expression [87,102] | ||
| Reduction of enhanced glutamate release in primary afferent terminals [73] | ||
| Sciatic nerve transection (CCI) induced neuropathic model [87,91,102,103,104] | Recovery of paw withdrawal response [87,91,102,103,104] | |
| Cleaved cSNAP-25 detected in paw, sciatic nerve, DRG, and L4/L5 spinal cord (dorsal horn) [103] | ||
| Recovery of thermal hyperalgesia [91,104] | ||
| L5 ventral root transection (VRT) induced neuropathic model [93,94] | Bilateral recovery of decreased paw withdrawal thresholds [93,94] | |
| Reduced expression of TRPV1 and P2X3 in dorsal root ganglion [93,94] | ||
| Carrageenan-induced hyperalgesia [92,105] | Recovery of paw withdrawal response [92,105] | |
| Recovery of thermal hyperalgesia [105] | ||
| Reduction of c-fos early response gene expression in spinal cord [105] | ||
| Paclitaxel-induced peripheral neuropathy model [92] | Bilateral recovery of decreased paw withdrawal thresholds [92] | |
| Diabetic neuropathy pain model [106] | Bilateral recovery of decreased paw withdrawal thresholds [106] | |
| Bilateral recovery of mechanical and thermal hypersensitivity [106] | ||
| Acidic saline induced pain model [90] | Bilateral recovery of decreased paw withdrawal thresholds [90] | |
| Spinal Cord | Sciatic nerve transection (SCI) induced neuropathic model [104] | Reduction of mechanical allodynia and thermal hyperalgesia [104] |
| Diabetic neuropathy pain model [106] | Bilateral recovery of decreased paw withdrawal thresholds [106] | |
| Bilateral recovery of mechanical and thermal hypersensitivity [106] | ||
| Formalin induced inflammatory pain model [107] | Reduction of enhanced nocifensive behaviors (licking, flinching and shaking) [107] | |
| Reduction of CGRP in spinal dorsal horn [107] | ||
| Acetic acid induced abdominal pain [86] | Reduced writhes [86] | |
| Reduction of increased c-fos expression in dorsal horn of the spinal cord (S2/S3segments) [86] | ||
| Reduction of mechanical allodynia [86] | ||
| Face | Formalin-induced facial pain (into the whisker pad) [108] | Reduction of facial rubbing [108] |
| Cleaved cSNAP-25 detected in trigeminal nucleus caudalis (TNC) [108] | ||
| Colchicine-sensitive [108] | ||
| Infraorbital nerve constriction (IoNC) induced trigeminal neuropathy model [95] | Reduction of dural extravasation [95] | |
| Colchicine-sensitive bilateral analgesic effect in trigeminal ganglion [95] |
3.2. Molecular Therapeutics of BoNTs
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cherington, M. Botulism: Update and review. Semin. Neurol. 2004, 24, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, O.; Seveso, M.; Caccin, P.; Schiavo, G.; Montecucco, C. Tetanus and botulinum neurotoxins: Turning bad guys into good by research. Toxicon 2001, 39, 27–41. [Google Scholar] [CrossRef]
- Peng Chen, Z.; Morris, J.G., Jr.; Rodriguez, R.L.; Shukla, A.W.; Tapia-Nunez, J.; Okun, M.S. Emerging opportunities for serotypes of botulinum neurotoxins. Toxins Basel 2012, 4, 1196–1222. [Google Scholar] [CrossRef] [PubMed]
- Masuyer, G.; Chaddock, J.A.; Foster, K.A.; Acharya, K.R. Engineered botulinum neurotoxins as new therapeutics. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 27–51. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.B. Botulinum toxin injection of eye muscles to correct strabismus. Trans. Am. Ophthalmol. Soc. 1981, 79, 734–770. [Google Scholar] [PubMed]
- Chen, S. Clinical uses of botulinum neurotoxins: Current indications, limitations and future developments. Toxins Basel 2012, 4, 913–939. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Jin, R. Assembly and function of the botulinum neurotoxin progenitor complex. Curr. Top. Microbiol. Immunol. 2013, 364, 21–44. [Google Scholar] [PubMed]
- Rigoni, M.; Caccin, P.; Johnson, E.A.; Montecucco, C.; Rossetto, O. Site-directed mutagenesis identifies active-site residues of the light chain of botulinum neurotoxin type A. Biochem. Biophys. Res. Commun. 2001, 288, 1231–1237. [Google Scholar] [CrossRef] [PubMed]
- Schiavo, G.; Matteoli, M.; Montecucco, C. Neurotoxins affecting neuroexocytosis. Physiol. Rev. 2000, 80, 717–766. [Google Scholar] [PubMed]
- Collins, M.D.; East, A.K. Phylogeny and taxonomy of the food-borne pathogen Clostridium botulinum and its neurotoxins. J. Appl. Microbiol. 1998, 84, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Oguma, K.; Inoue, K.; Fujinaga, Y.; Yokota, K.; Watanabe, T.; Ohyama, T.; Takeshi, K.; Inoue, K. Structure and function of Clostridium botulinum progenitor toxin. Toxin Rev. 1999, 18, 17–34. [Google Scholar] [CrossRef]
- Lee, K.; Zhong, X.; Gu, S.; Kruel, A.M.; Dorner, M.B.; Perry, K.; Rummel, A.; Dong, M.; Jin, R. Molecular basis for disruption of E-cadherin adhesion by botulinum neurotoxin A complex. Science 2014, 344, 1405–1410. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Rumpel, S.; Zhou, J.; Strotmeier, J.; Bigalke, H.; Perry, K.; Shoemaker, C.B.; Rummel, A.; Jin, R. Botulinum neurotoxin is shielded by NTNHA in an interlocked complex. Science 2012, 335, 977–981. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T.; Gu, S.; Lam, K.H.; Carter, L.G.; Rummel, A.; Mathews, I.I.; Jin, R. Structural basis of the pH-dependent assembly of a botulinum neurotoxin complex. J. Mol. Biol. 2014, 426, 3773–3782. [Google Scholar] [CrossRef] [PubMed]
- Lam, T.I.; Stanker, L.H.; Lee, K.; Jin, R.; Cheng, L.W. Translocation of botulinum neurotoxin serotype A and associated proteins across the intestinal epithelia. Cell. Microbiol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Fujinaga, Y.; Matsumura, T.; Jin, Y.; Takegahara, Y.; Sugawara, Y. A novel function of botulinum toxin-associated proteins: HA proteins disrupt intestinal epithelial barrier to increase toxin absorption. Toxicon 2009, 54, 583–586. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, T.; Jin, Y.; Kabumoto, Y.; Takegahara, Y.; Oguma, K.; Lencer, W.I.; Fujinaga, Y. The HA proteins of botulinum toxin disrupt intestinal epithelial intercellular junctions to increase toxin absorption. Cell. Microbiol. 2008, 10, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Fujinaga, Y.; Sugawara, Y.; Matsumura, T. Uptake of botulinum neurotoxin in the intestine. Curr. Top. Microbiol. Immunol. 2013, 364, 45–59. [Google Scholar] [PubMed]
- Lee, K.; Gu, S.; Jin, L.; Le, T.T.; Cheng, L.W.; Strotmeier, J.; Kruel, A.M.; Yao, G.; Perry, K.; Rummel, A.; et al. Structure of a bimodular botulinum neurotoxin complex provides insights into its oral toxicity. PLoS Pathog. 2013, 9, e1003690. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Kotani, M.; Tonozuka, T.; Ide, A.; Oguma, K.; Nishikawa, A. Crystal structure of the HA3 subcomponent of Clostridium botulinum type C progenitor toxin. J. Mol. Biol. 2009, 385, 1193–1206. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Takada, N.; Tonozuka, T.; Sakano, Y.; Oguma, K.; Nishikawa, A. Binding properties of Clostridium botulinum type C progenitor toxin to mucins. Biochim. Biophys. Acta 2007, 1770, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Tonozuka, T.; Ide, A.; Yuzawa, T.; Oguma, K.; Nishikawa, A. Sugar-binding sites of the HA1 subcomponent of Clostridium botulinum type C progenitor toxin. J. Mol. Biol. 2008, 376, 854–867. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Tonozuka, T.; Kotani, M.; Obata, K.; Oguma, K.; Nishikawa, A. Crystallization and preliminary X-ray analysis of the HA3 component of Clostridium botulinum type C progenitor toxin. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2007, 63, 1038–1040. [Google Scholar] [CrossRef] [PubMed]
- Maksymowych, A.B.; Simpson, L.L. Binding and transcytosis of botulinum neurotoxin by polarized human colon carcinoma cells. J. Biol. Chem. 1998, 273, 21950–21957. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, T.; Sugawara, Y.; Yutani, M.; Amatsu, S.; Yagita, H.; Kohda, T.; Fukuoka, S.; Nakamura, Y.; Fukuda, S.; Hase, K.; et al. Botulinum toxin A complex exploits intestinal M cells to enter the host and exert neurotoxicity. Nat. Commun. 2015. [Google Scholar] [CrossRef]
- Amatsu, S.; Sugawara, Y.; Matsumura, T.; Kitadokoro, K.; Fujinaga, Y. Crystal structure of Clostridium botulinum whole hemagglutinin reveals a huge triskelion-shaped molecular complex. J. Biol. Chem. 2013, 288, 35617–35625. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, Y.; Matsumura, T.; Takegahara, Y.; Jin, Y.; Tsukasaki, Y.; Takeichi, M.; Fujinaga, Y. Botulinum hemagglutinin disrupts the intercellular epithelial barrier by directly binding E-cadherin. J. Cell. Biol. 2010, 189, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Couesnon, A.; Pereira, Y.; Popoff, M.R. Receptor-mediated transcytosis of botulinum neurotoxin A through intestinal cell monolayers. Cell. Microbiol. 2008, 10, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Couesnon, A.; Shimizu, T.; Popoff, M.R. Differential entry of botulinum neurotoxin A into neuronal and intestinal cells. Cell. Microbiol. 2009, 11, 289–308. [Google Scholar] [CrossRef] [PubMed]
- Yao, G.; Lee, K.; Gu, S.; Lam, K.H.; Jin, R. Botulinum neurotoxin A complex recognizes host carbohydrates through its hemagglutinin component. Toxins Basel 2014, 6, 624–635. [Google Scholar] [CrossRef] [PubMed]
- Simpson, L. The life history of a botulinum toxin molecule. Toxicon 2013, 68, 40–59. [Google Scholar] [CrossRef] [PubMed]
- Restani, L.; Giribaldi, F.; Manich, M.; Bercsenyi, K.; Menendez, G.; Rossetto, O.; Caleo, M.; Schiavo, G. Botulinum neurotoxins A and E undergo retrograde axonal transport in primary motor neurons. PLoS Pathog. 2012, 8, e1003087. [Google Scholar] [CrossRef] [PubMed]
- Black, J.D.; Dolly, J.O. Interaction of 125I-labeled botulinum neurotoxins with nerve terminals. II. Autoradiographic evidence for its uptake into motor nerves by acceptor-mediated endocytosis. J. Cell Biol. 1986, 103, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Dolly, J.O.; Black, J.; Williams, R.S.; Melling, J. Acceptors for botulinum neurotoxin reside on motor nerve terminals and mediate its internalization. Nature 1984, 307, 457–460. [Google Scholar] [CrossRef] [PubMed]
- Simpson, L.L. The binding fragment from tetanus toxin antagonizes the neuromuscular blocking actions of botulinum toxin. J. Pharmacol. Exp. Ther. 1984, 229, 182–187. [Google Scholar] [PubMed]
- Simpson, L.L. Botulinum toxin and tetanus toxin recognize similar membrane determinants. Brain Res. 1984, 305, 177–180. [Google Scholar] [CrossRef]
- Montecucco, C.; Rossetto, O.; Schiavo, G. Presynaptic receptor arrays for clostridial neurotoxins. Trends Microbiol. 2004, 12, 442–446. [Google Scholar] [CrossRef] [PubMed]
- Montal, M. Botulinum neurotoxin: A marvel of protein design. Annu. Rev. Biochem. 2010, 79, 591–617. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.; Yeh, F.; Tepp, W.H.; Dean, C.; Johnson, E.A.; Janz, R.; Chapman, E.R. SV2 is the protein receptor for botulinum neurotoxin A. Science 2006, 312, 592–596. [Google Scholar] [CrossRef] [PubMed]
- Strotmeier, J.; Mahrhold, S.; Krez, N.; Janzen, C.; Lou, J.; Marks, J.D.; Binz, T.; Rummel, A. Identification of the synaptic vesicle glycoprotein 2 receptor binding site in botulinum neurotoxin A. FEBS Lett. 2014, 588, 1087–1093. [Google Scholar] [CrossRef] [PubMed]
- Benoit, R.M.; Frey, D.; Hilbert, M.; Kevenaar, J.T.; Wieser, M.M.; Stirnimann, C.U.; McMillan, D.; Ceska, T.; Lebon, F.; Jaussi, R.; et al. Structural basis for recognition of synaptic vesicle protein 2C by botulinum neurotoxin A. Nature 2014, 505, 108–111. [Google Scholar] [CrossRef] [PubMed]
- Blasi, J.; Chapman, E.R.; Link, E.; Binz, T.; Yamasaki, S.; de Camilli, P.; Sudhof, T.C.; Niemann, H.; Jahn, R. Botulinum neurotoxin A selectively cleaves the synaptic protein SNAP-25. Nature 1993, 365, 160–163. [Google Scholar] [CrossRef] [PubMed]
- Binz, T.; Rummel, A. Cell entry strategy of clostridial neurotoxins. J. Neurochem. 2009, 109, 1584–1595. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, M.; Takamiya, K.; Aizawa, S.; Furukawa, K.; Furukawa, K. Gangliosides are the binding substances in neural cells for tetanus and botulinum toxins in mice. Biochim. Biophys. Acta 1999, 1441, 1–3. [Google Scholar] [CrossRef]
- Stenmark, P.; Dupuy, J.; Imamura, A.; Kiso, M.; Stevens, R.C. Crystal structure of botulinum neurotoxin type A in complex with the cell surface co-receptor GT1b-insight into the toxin-neuron interaction. PLoS Pathog. 2008, 4, e1000129. [Google Scholar] [CrossRef] [PubMed]
- Colasante, C.; Rossetto, O.; Morbiato, L.; Pirazzini, M.; Molgo, J.; Montecucco, C. Botulinum neurotoxin type A is internalized and translocated from small synaptic vesicles at the neuromuscular junction. Mol. Neurobiol. 2013, 48, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Harper, C.B.; Martin, S.; Nguyen, T.H.; Daniels, S.J.; Lavidis, N.A.; Popoff, M.R.; Hadzic, G.; Mariana, A.; Chau, N.; McCluskey, A.; et al. Dynamin inhibition blocks botulinum neurotoxin type A endocytosis in neurons and delays botulism. J. Biol. Chem. 2011, 286, 35966–35976. [Google Scholar] [CrossRef] [PubMed]
- Takamori, S.; Holt, M.; Stenius, K.; Lemke, E.A.; Gronborg, M.; Riedel, D.; Urlaub, H.; Schenck, S.; Brugger, B.; Ringler, P.; et al. Molecular anatomy of a trafficking organelle. Cell 2006, 127, 831–846. [Google Scholar] [CrossRef] [PubMed]
- Saheki, Y.; de Camilli, P. Synaptic vesicle endocytosis. Cold Spring Harb Perspect. Biol. 2012, 4, a005645. [Google Scholar] [CrossRef] [PubMed]
- Jacky, B.P.; Garay, P.E.; Dupuy, J.; Nelson, J.B.; Cai, B.; Molina, Y.; Wang, J.; Steward, L.E.; Broide, R.S.; Francis, J.; et al. Identification of fibroblast growth factor receptor 3 (FGFR3) as a protein receptor for botulinum neurotoxin serotype A (BoNT/A). PLoS Pathog. 2013, 9, e1003369. [Google Scholar] [CrossRef] [PubMed]
- Ayyar, B.V.; Aoki, K.R.; Atassi, M.Z. The C-terminal heavy-chain domain of botulinum neurotoxin a is not the only site that binds neurons, as the N-terminal heavy-chain domain also plays a very active role in toxin-cell binding and interactions. Infect. Immun. 2015, 83, 1465–1476. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Sambashivan, S.; Brunger, A.T.; Montal, M. Beltless translocation domain of botulinum neurotoxin A embodies a minimum ion-conductive channel. J. Biol. Chem. 2012, 287, 1657–1661. [Google Scholar] [CrossRef] [PubMed]
- Galloux, M.; Vitrac, H.; Montagner, C.; Raffestin, S.; Popoff, M.R.; Chenal, A.; Forge, V.; Gillet, D. Membrane Interaction of botulinum neurotoxin A translocation (T) domain. The belt region is a regulatory loop for membrane interaction. J. Biol. Chem. 2008, 283, 27668–27676. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Montal, M. Single molecule detection of intermediates during botulinum neurotoxin translocation across membranes. Proc. Natl. Acad. Sci. USA 2007, 104, 10447–10452. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Mushrush, D.J.; Lacy, D.B.; Montal, M. Botulinum neurotoxin devoid of receptor binding domain translocates active protease. PLoS Pathog. 2008, 4, e1000245. [Google Scholar] [CrossRef] [PubMed]
- Koriazova, L.K.; Montal, M. Translocation of botulinum neurotoxin light chain protease through the heavy chain channel. Nat. Struct. Biol. 2003, 10, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Montal, M. Crucial role of the disulfide bridge between botulinum neurotoxin light and heavy chains in protease translocation across membranes. J. Biol. Chem. 2007, 282, 29604–29611. [Google Scholar] [CrossRef] [PubMed]
- Pirazzini, M.; Bordin, F.; Rossetto, O.; Shone, C.C.; Binz, T.; Montecucco, C. The thioredoxin reductase-thioredoxin system is involved in the entry of tetanus and botulinum neurotoxins in the cytosol of nerve terminals. FEBS Lett. 2013, 587, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Suresh, S.; Liu, H.; Tepp, W.H.; Johnson, E.A.; Edwardson, J.M.; Chapman, E.R. Receptor binding enables botulinum neurotoxin B to sense low pH for translocation channel assembly. Cell Host Microbe 2011, 10, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Montal, M. Molecular dissection of botulinum neurotoxin reveals interdomain chaperone function. Toxicon 2013, 75, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A. Synchronized chaperone function of botulinum neurotoxin domains mediates light chain translocation into neurons. Curr. Top. Microbiol. Immunol. 2013, 364, 115–137. [Google Scholar] [PubMed]
- Kalandakanond, S.; Coffield, J.A. Cleavage of SNAP-25 by botulinum toxin type A requires receptor-mediated endocytosis, pH-dependent translocation, and zinc. J. Pharmacol. Exp. Ther. 2001, 296, 980–986. [Google Scholar] [PubMed]
- Binz, T. Clostridial Neurotoxin Light Chains: Devices for SNARE Cleavage Mediated Blockade of Neurotransmission. Curr. Top. Microbiol. Immunol. 2013, 364, 139–157. [Google Scholar] [PubMed]
- Sutton, R.B.; Fasshauer, D.; Jahn, R.; Brunger, A.T. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 A resolution. Nature 1998, 395, 347–353. [Google Scholar] [PubMed]
- Sudhof, T.C. A molecular machine for neurotransmitter release: Synaptotagmin and beyond. Nat. Med. 2013, 19, 1227–1231. [Google Scholar] [CrossRef] [PubMed]
- Matak, I.; Lackovic, Z. Botulinum toxin A, brain and pain. Prog. Neurobiol. 2014, 119–120, 39–59. [Google Scholar] [CrossRef] [PubMed]
- Dressler, D. Botulinum toxin therapy: Its use for neurological disorders of the autonomic nervous system. J. Neurol. 2013, 260, 701–713. [Google Scholar] [CrossRef] [PubMed]
- Mense, S. Neurobiological basis for the use of botulinum toxin in pain therapy. J. Neurol. 2004, 251 (Suppl. 1), I1–I7. [Google Scholar] [CrossRef] [PubMed]
- Freund, B.; Schwartz, M. Temporal relationship of muscle weakness and pain reduction in subjects treated with botulinum toxin A. J. Pain 2003, 4, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Argoff, C.E. A focused review on the use of botulinum toxins for neuropathic pain. Clin. J. Pain 2002, 18, S177–S181. [Google Scholar] [CrossRef] [PubMed]
- Silberstein, S.; Mathew, N.; Saper, J.; Jenkins, S. Botulinum toxin type A as a migraine preventive treatment. For the BOTOX Migraine Clinical Research Group. Headache 2000, 40, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, A.; Smith, H.S. Botulinum toxins: Mechanisms of action, antinociception and clinical applications. Toxicology 2013, 306, 124–146. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.; Khanijou, S.; Rubino, J.; Aoki, K.R. Subcutaneous administration of botulinum toxin A reduces formalin-induced pain. Pain 2004, 107, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Bittencourt da Silva, L.; Karshenas, A.; Bach, F.W.; Rasmussen, S.; Arendt-Nielsen, L.; Gazerani, P. Blockade of glutamate release by botulinum neurotoxin type A in humans: A dermal microdialysis study. Pain Res. Manag. 2014, 19, 126–132. [Google Scholar] [PubMed]
- Rapp, D.E.; Turk, K.W.; Bales, G.T.; Cook, S.P. Botulinum toxin type a inhibits calcitonin gene-related peptide release from isolated rat bladder. J. Urol. 2006, 175, 1138–1142. [Google Scholar] [CrossRef]
- Lucioni, A.; Bales, G.T.; Lotan, T.L.; McGehee, D.S.; Cook, S.P.; Rapp, D.E. Botulinum toxin type A inhibits sensory neuropeptide release in rat bladder models of acute injury and chronic inflammation. BJU Int. 2008, 101, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Durham, P.L.; Cady, R.; Cady, R. Regulation of calcitonin gene-related peptide secretion from trigeminal nerve cells by botulinum toxin type A: Implications for migraine therapy. Headache 2004, 44, 35–42, discussion 42–33. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Ovsepian, S.V.; Wang, J.; Pickering, M.; Sasse, A.; Aoki, K.R.; Lawrence, G.W.; Dolly, J.O. Activation of TRPV1 mediates calcitonin gene-related peptide release, which excites trigeminal sensory neurons and is attenuated by a retargeted botulinum toxin with anti-nociceptive potential. J. Neurosci. 2009, 29, 4981–4992. [Google Scholar] [CrossRef] [PubMed]
- Caterina, M.J.; Julius, D. The vanilloid receptor: A molecular gateway to the pain pathway. Annu. Rev. Neurosci. 2001, 24, 487–517. [Google Scholar] [CrossRef] [PubMed]
- Adcock, J.J. TRPV1 receptors in sensitisation of cough and pain reflexes. Pulm. Pharmacol. Ther. 2009, 22, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Min, J.W.; Liu, W.H.; He, X.H.; Peng, B.W. Different types of toxins targeting TRPV1 in pain. Toxicon 2013, 71, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Apostolidis, A.; Popat, R.; Yiangou, Y.; Cockayne, D.; Ford, A.P.; Davis, J.B.; Dasgupta, P.; Fowler, C.J.; Anand, P. Decreased sensory receptors P2X3 and TRPV1 in suburothelial nerve fibers following intradetrusor injections of botulinum toxin for human detrusor overactivity. J. Urol. 2005, 174, 977–982, discussion 982–973. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Shibata, M.; Toriumi, H.; Iwashita, T.; Funakubo, M.; Sato, H.; Kuroi, T.; Ebine, T.; Koizumi, K.; Suzuki, N. Reduction of TRPV1 expression in the trigeminal system by botulinum neurotoxin type-A. Neurobiol. Dis. 2012, 48, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Morenilla-Palao, C.; Planells-Cases, R.; Garcia-Sanz, N.; Ferrer-Montiel, A. Regulated exocytosis contributes to protein kinase C potentiation of vanilloid receptor activity. J. Biol. Chem. 2004, 279, 25665–25672. [Google Scholar] [CrossRef] [PubMed]
- Bardoni, R.; Takazawa, T.; Tong, C.K.; Choudhury, P.; Scherrer, G.; Macdermott, A.B. Pre- and postsynaptic inhibitory control in the spinal cord dorsal horn. Ann. N. Y. Acad. Sci. 2013, 1279, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Drinovac, V.; Bach-Rojecky, L.; Babic, A.; Lackovic, Z. Antinociceptive effect of botulinum toxin type A on experimental abdominal pain. Eur. J. Pharmacol. 2014, 745, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Drinovac, V.; Bach-Rojecky, L.; Matak, I.; Lackovic, Z. Involvement of mu-opioid receptors in antinociceptive action of botulinum toxin type A. Neuropharmacology 2013, 70, 331–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vacca, V.; Marinelli, S.; Eleuteri, C.; Luvisetto, S.; Pavone, F. Botulinum neurotoxin A enhances the analgesic effects on inflammatory pain and antagonizes tolerance induced by morphine in mice. Brain Behav. Immun. 2012, 26, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Vacca, V.; Marinelli, S.; Luvisetto, S.; Pavone, F. Botulinum toxin A increases analgesic effects of morphine, counters development of morphine tolerance and modulates glia activation and mu opioid receptor expression in neuropathic mice. Brain Behav. Immun. 2013, 32, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Bach-Rojecky, L.; Lackovic, Z. Central origin of the antinociceptive action of botulinum toxin type A. Pharmacol. Biochem. Behav. 2009, 94, 234–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bach-Rojecky, L.; Relja, M.; Lackovic, Z. Botulinum toxin type A in experimental neuropathic pain. J. Neural. Transm. 2005, 112, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Favre-Guilmard, C.; Auguet, M.; Chabrier, P.E. Different antinociceptive effects of botulinum toxin type A in inflammatory and peripheral polyneuropathic rat models. Eur. J. Pharmacol. 2009, 617, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Cheng, J.; Dai, J.; Zhang, D. Botulinum toxin decreases hyperalgesia and inhibits P2X3 receptor over-expression in sensory neurons induced by ventral root transection in rats. Pain Med. 2011, 12, 1385–1394. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Cheng, J.; Zhuang, Y.; Qu, W.; Muir, J.; Liang, H.; Zhang, D. Botulinum toxin type A reduces hyperalgesia and TRPV1 expression in rats with neuropathic pain. Pain Med. 2013, 14, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Filipovic, B.; Matak, I.; Bach-Rojecky, L.; Lackovic, Z. Central action of peripherally applied botulinum toxin type A on pain and dural protein extravasation in rat model of trigeminal neuropathy. PLoS ONE 2012, 7, e29803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habermann, E. 125I-labeled neurotoxin from Clostridium botulinum A: Preparation, binding to synaptosomes and ascent to the spinal cord. Naunyn Schmiedebergs Arch. Pharmacol. 1974, 281, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Wiegand, H.; Erdmann, G.; Wellhoner, H.H. 125I-labelled botulinum A neurotoxin: Pharmacokinetics in cats after intramuscular injection. Naunyn Schmiedebergs Arch. Pharmacol. 1976, 292, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Antonucci, F.; Rossi, C.; Gianfranceschi, L.; Rossetto, O.; Caleo, M. Long-distance retrograde effects of botulinum neurotoxin A. J. Neurosci. 2008, 28, 3689–3696. [Google Scholar] [CrossRef] [PubMed]
- Mika, J.; Rojewska, E.; Makuch, W.; Korostynski, M.; Luvisetto, S.; Marinelli, S.; Przewlocka, B.; Pavone, F. The effect of botulinum neurotoxin A on sciatic nerve injury-induced neuroimmunological changes in rat dorsal root ganglia and spinal cord. Neuroscience 2011, 175, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Restani, L.; Antonucci, F.; Gianfranceschi, L.; Rossi, C.; Rossetto, O.; Caleo, M. Evidence for anterograde transport and transcytosis of botulinum neurotoxin A (BoNT/A). J. Neurosci. 2011, 31, 15650–15659. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, G.W.; Ovsepian, S.V.; Wang, J.; Aoki, K.R.; Dolly, J.O. Extravesicular intraneuronal migration of internalized botulinum neurotoxins without detectable inhibition of distal neurotransmission. Biochem. J. 2012, 441, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Drinovac, V.; Bach-Rojecky, L.; Lackovic, Z. Association of antinociceptive action of botulinum toxin type A with GABA-A receptor. J. Neural Transm. 2014, 121, 665–669. [Google Scholar] [CrossRef] [PubMed]
- Marinelli, S.; Vacca, V.; Ricordy, R.; Uggenti, C.; Tata, A.M.; Luvisetto, S.; Pavone, F. The analgesic effect on neuropathic pain of retrogradely transported botulinum neurotoxin A involves Schwann cells and astrocytes. PLoS ONE 2012, 7, e47977. [Google Scholar] [CrossRef] [PubMed]
- Marinelli, S.; Luvisetto, S.; Cobianchi, S.; Makuch, W.; Obara, I.; Mezzaroma, E.; Caruso, M.; Straface, E.; Przewlocka, B.; Pavone, F. Botulinum neurotoxin type A counteracts neuropathic pain and facilitates functional recovery after peripheral nerve injury in animal models. Neuroscience 2010, 171, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Shin, M.C.; Yukihira, T.; Ito, Y.; Akaike, N. Antinociceptive effects of A1 and A2 type botulinum toxins on carrageenan-induced hyperalgesia in rat. Toxicon 2013, 64, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Bach-Rojecky, L.; Salkovic-Petrisic, M.; Lackovic, Z. Botulinum toxin type A reduces pain supersensitivity in experimental diabetic neuropathy: Bilateral effect after unilateral injection. Eur. J. Pharmacol. 2010, 633, 10–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, W.H.; Shin, T.J.; Kim, H.J.; Lee, J.K.; Suh, H.W.; Lee, S.C.; Seo, K. Intrathecal administration of botulinum neurotoxin type A attenuates formalin-induced nociceptive responses in mice. Anesth. Analg. 2011, 112, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Matak, I.; Bach-Rojecky, L.; Filipovic, B.; Lackovic, Z. Behavioral and immunohistochemical evidence for central antinociceptive activity of botulinum toxin A. Neuroscience 2011, 186, 201–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mustafa, G.; Anderson, E.M.; Bokrand-Donatelli, Y.; Neubert, J.K.; Caudle, R.M. Anti-nociceptive effect of a conjugate of substance P and light chain of botulinum neurotoxin type A. Pain 2013, 154, 2547–2553. [Google Scholar] [CrossRef] [PubMed]
- North, R.A. Molecular physiology of P2X receptors. Physiol. Rev. 2002, 82, 1013–1067. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Meng, J.; Wang, J.; Hearty, S.; Dolly, J.O.; O’Kennedy, R. Targeted delivery of a SNARE protease to sensory neurons using a single chain antibody (scFv) against the extracellular domain of P2X(3) inhibits the release of a pain mediator. Biochem. J. 2014, 462, 247–256. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, D.-W.; Lee, S.-K.; Ahnn, J. Botulinum Toxin as a Pain Killer: Players and Actions in Antinociception. Toxins 2015, 7, 2435-2453. https://doi.org/10.3390/toxins7072435
Kim D-W, Lee S-K, Ahnn J. Botulinum Toxin as a Pain Killer: Players and Actions in Antinociception. Toxins. 2015; 7(7):2435-2453. https://doi.org/10.3390/toxins7072435
Chicago/Turabian StyleKim, Dong-Wan, Sun-Kyung Lee, and Joohong Ahnn. 2015. "Botulinum Toxin as a Pain Killer: Players and Actions in Antinociception" Toxins 7, no. 7: 2435-2453. https://doi.org/10.3390/toxins7072435
APA StyleKim, D.-W., Lee, S.-K., & Ahnn, J. (2015). Botulinum Toxin as a Pain Killer: Players and Actions in Antinociception. Toxins, 7(7), 2435-2453. https://doi.org/10.3390/toxins7072435