The Ethanol Fraction of White Rose Petal Extract Abrogates Excitotoxicity-Induced Neuronal Damage In Vivo and In Vitro through Inhibition of Oxidative Stress and Proinflammation


Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation of the WRPE
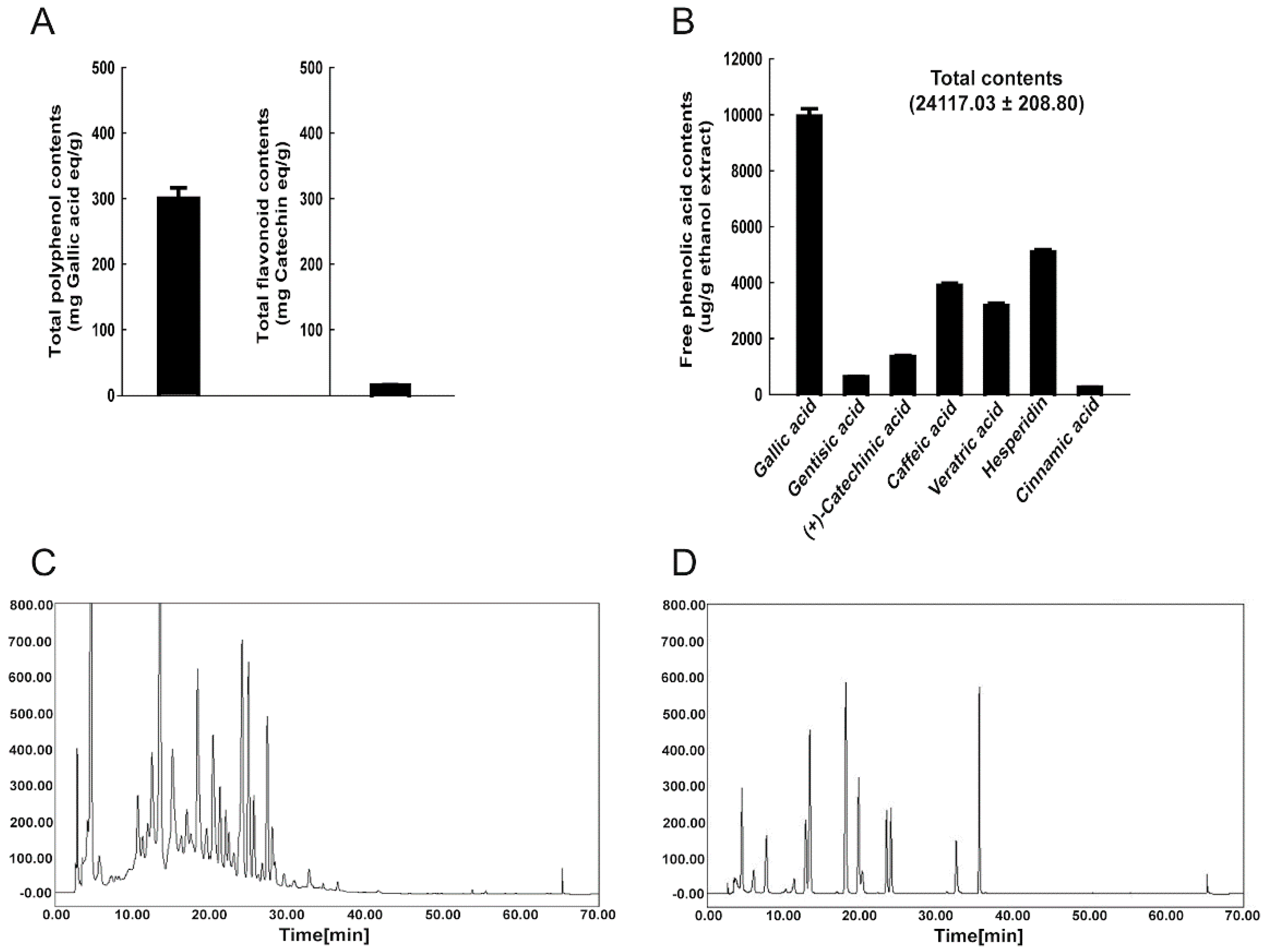
2.2. HPLC Analysis of Free Phenolic Acids
2.3. Determination of Polyphenol and Flavonoid Contents
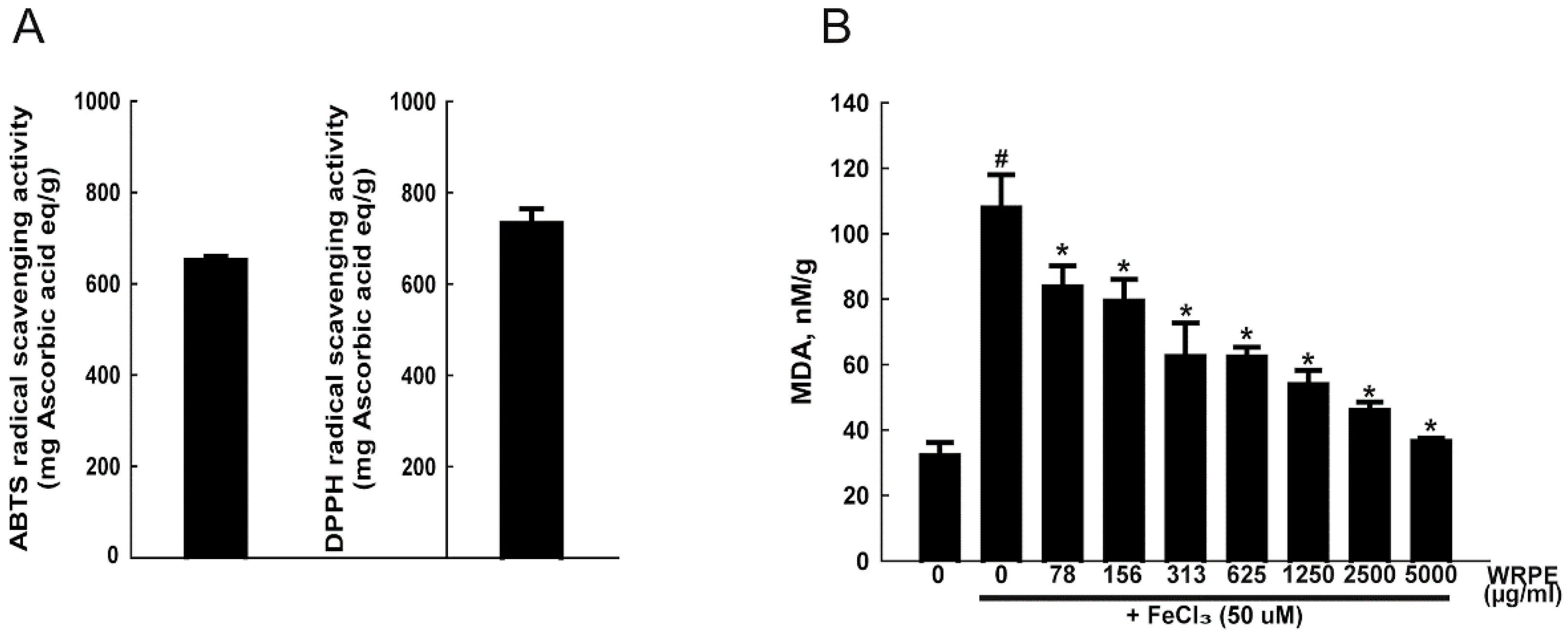
2.4. Measurement of Antioxidant Activity
2.5. Human Neural Stem Cell (NSC) Culture
2.6. Lactate Dehydrogenase (LDH) Assay in NSCs
2.7. Quantitative Real-Time PCR in NSCs
2.8. Western Blot Analysis in NSC
2.9. Animals and Treatment
2.10. Scoring of KA-Induced Seizures
2.11. Analysis of Cytokines and Lipid Peroxidation in the Brain Tissue
2.12. Quantitative Real-Time PCR in the Brain Tissue
2.13. Western Blot Analysis in the Brain Tissue
2.14. Nissl Staining of the Brain Tissue
2.15. Fluoro-Jade C (FJC) Staining of the Brain Tissue
2.16. Glial Fibrillary Acidic Protein (GFAP) Immunostaining of the Brain Tissue
2.17. Statistical Analysis
3. Results
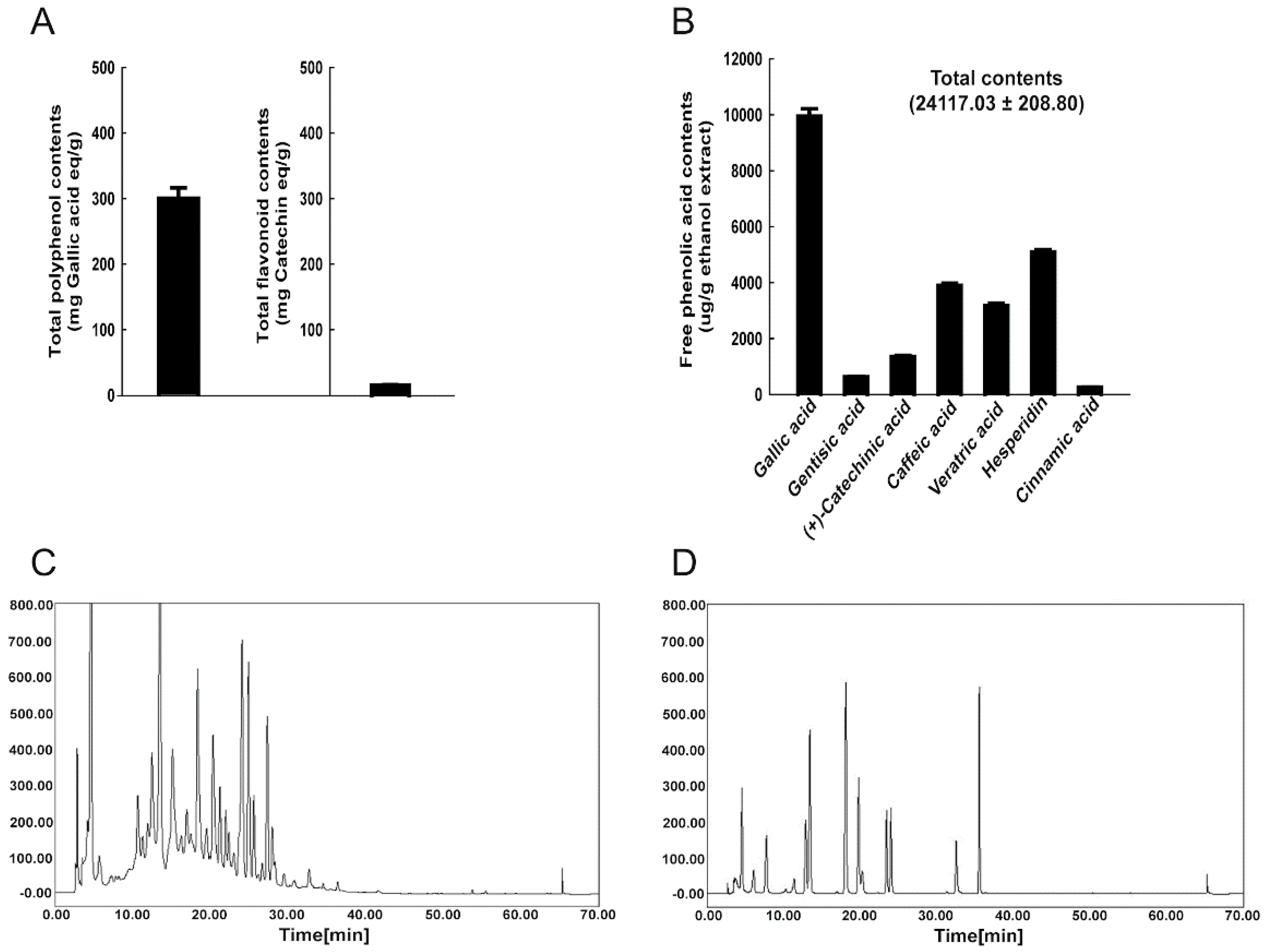
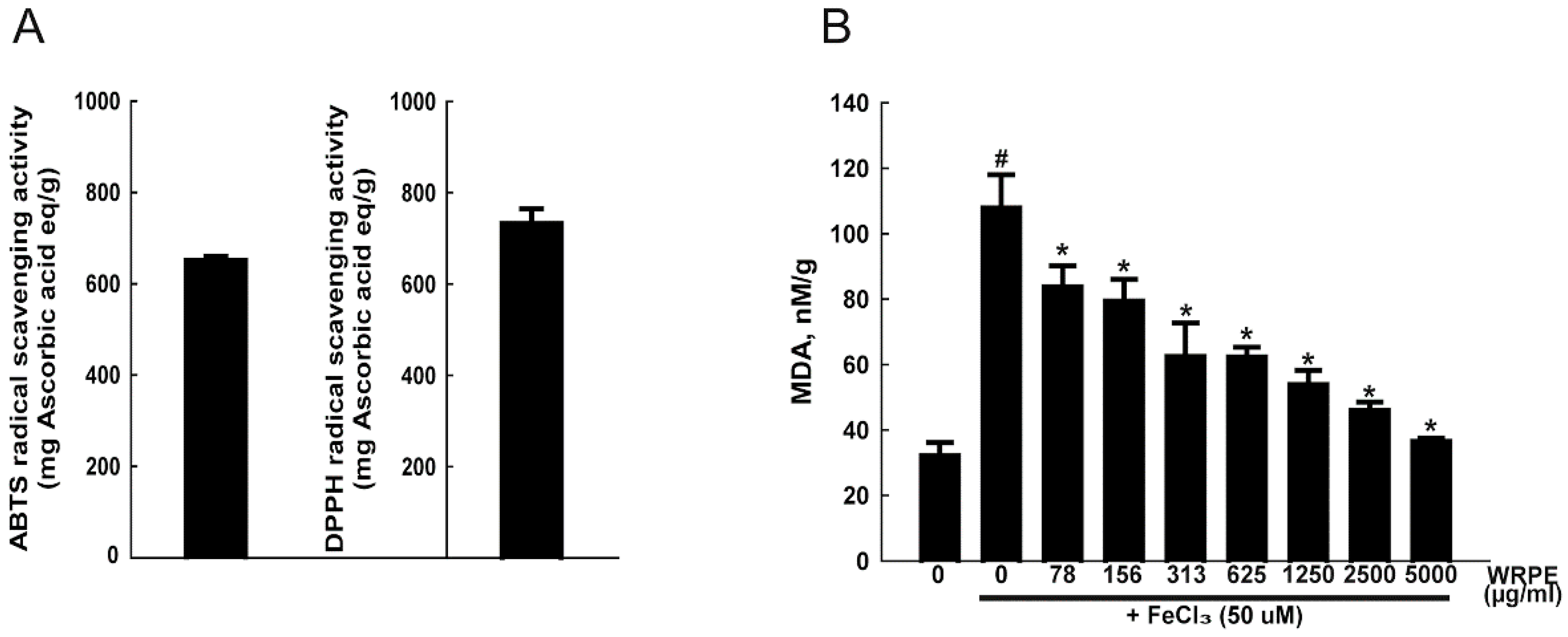
3.1. Phenolic Compounds in WRPE and Antioxidant Activity In Vitro
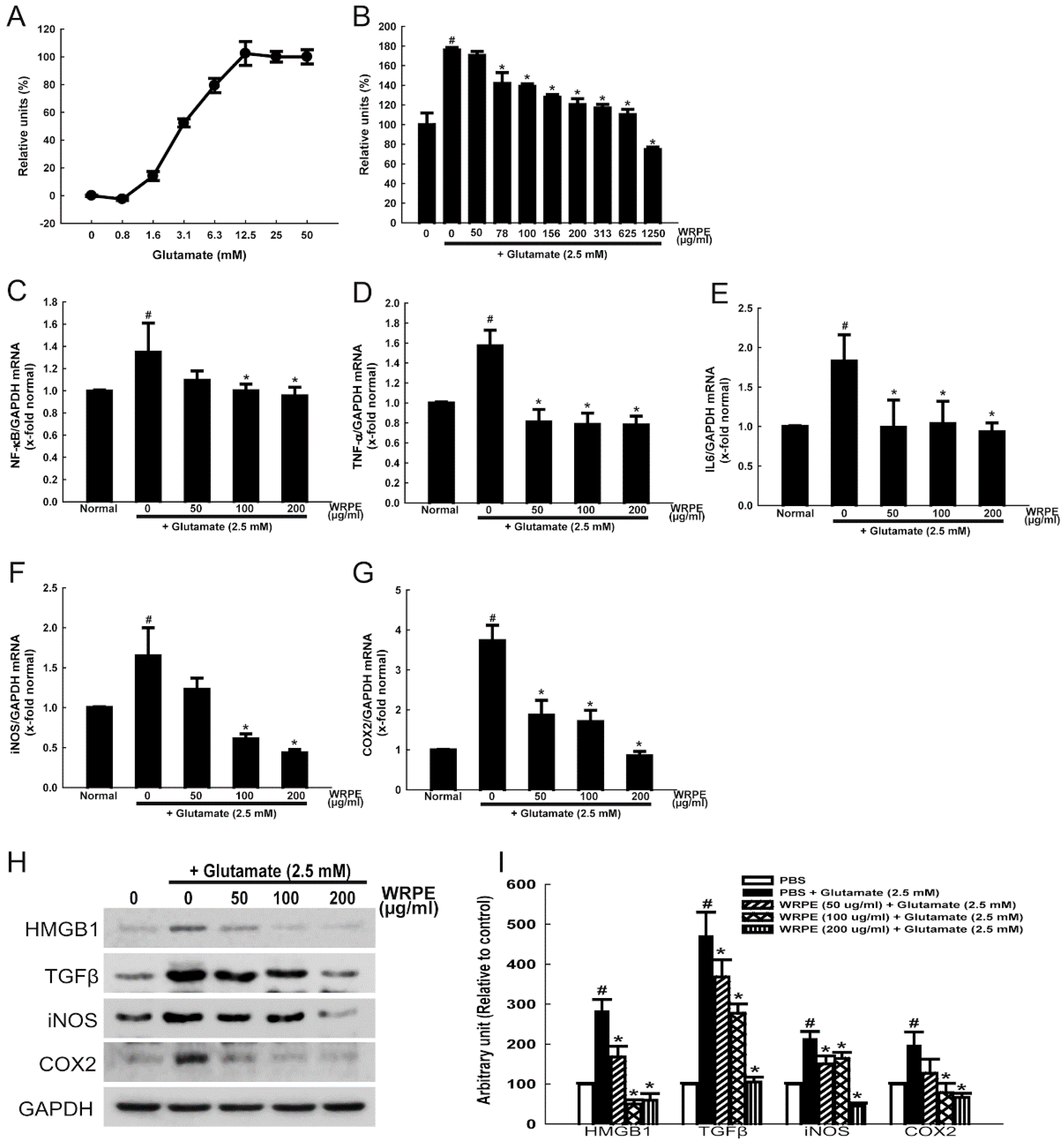
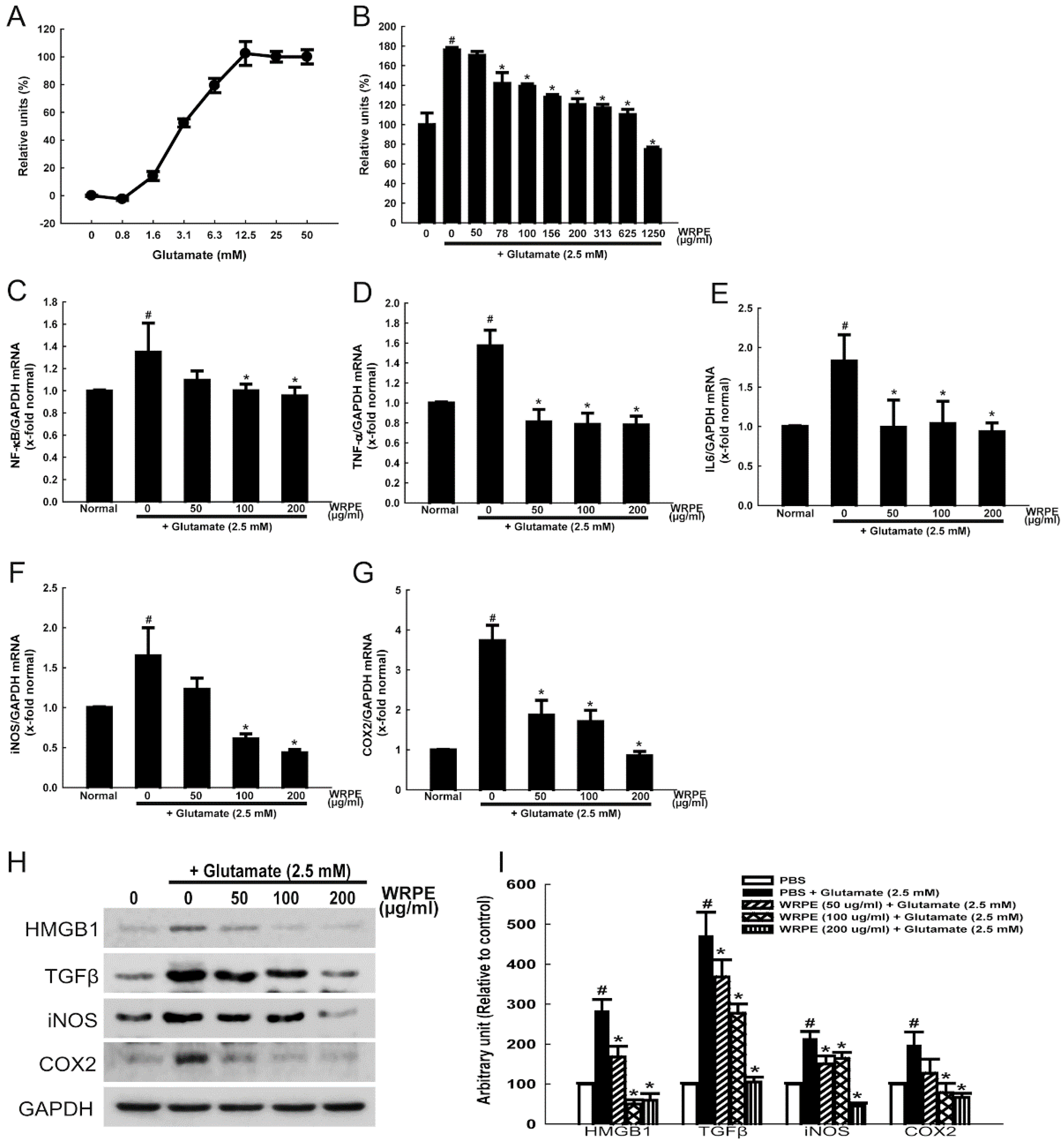
3.2. Neuroprotective and Anti-Inflammatory Activities of WRPE in NSCs
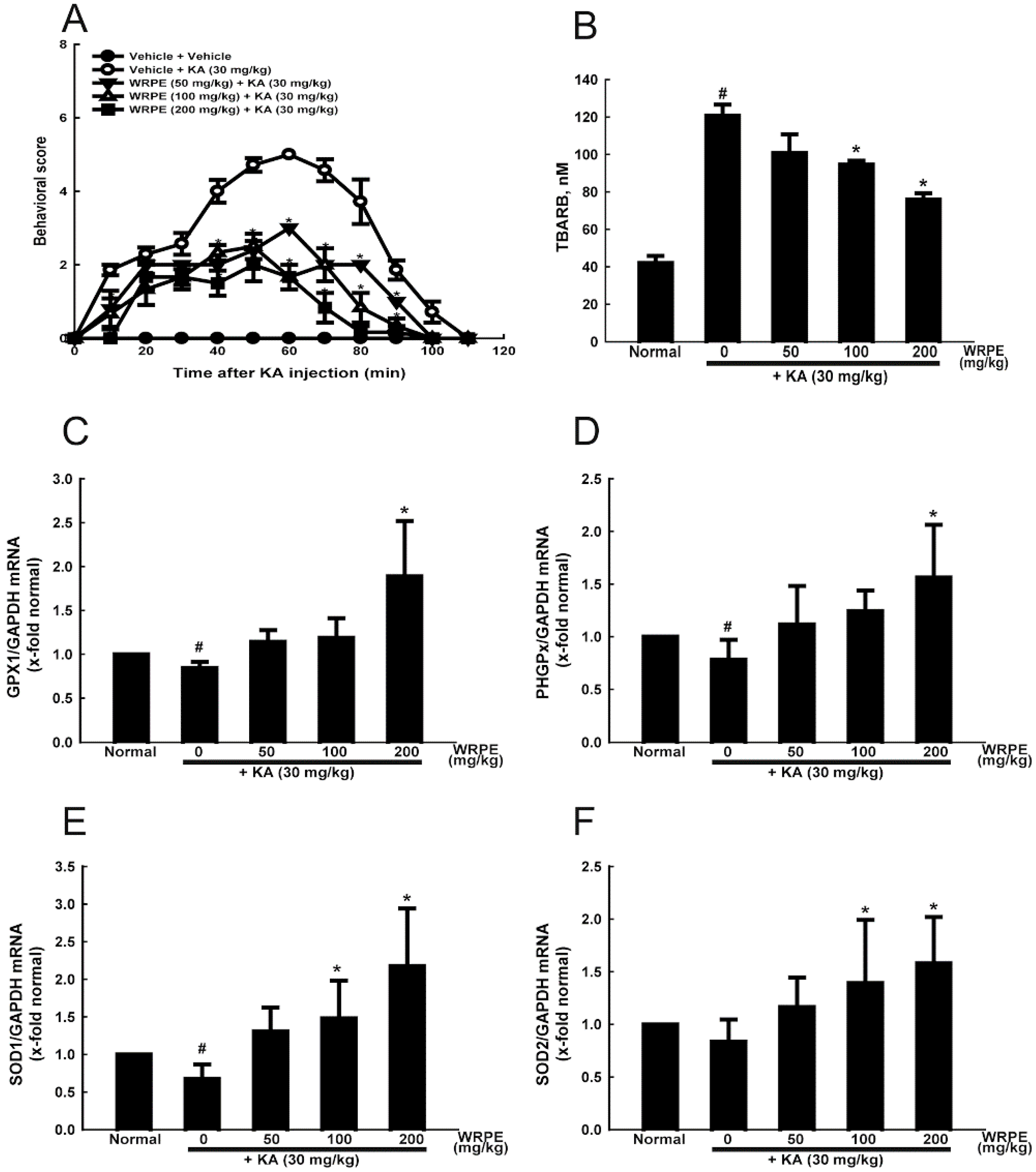
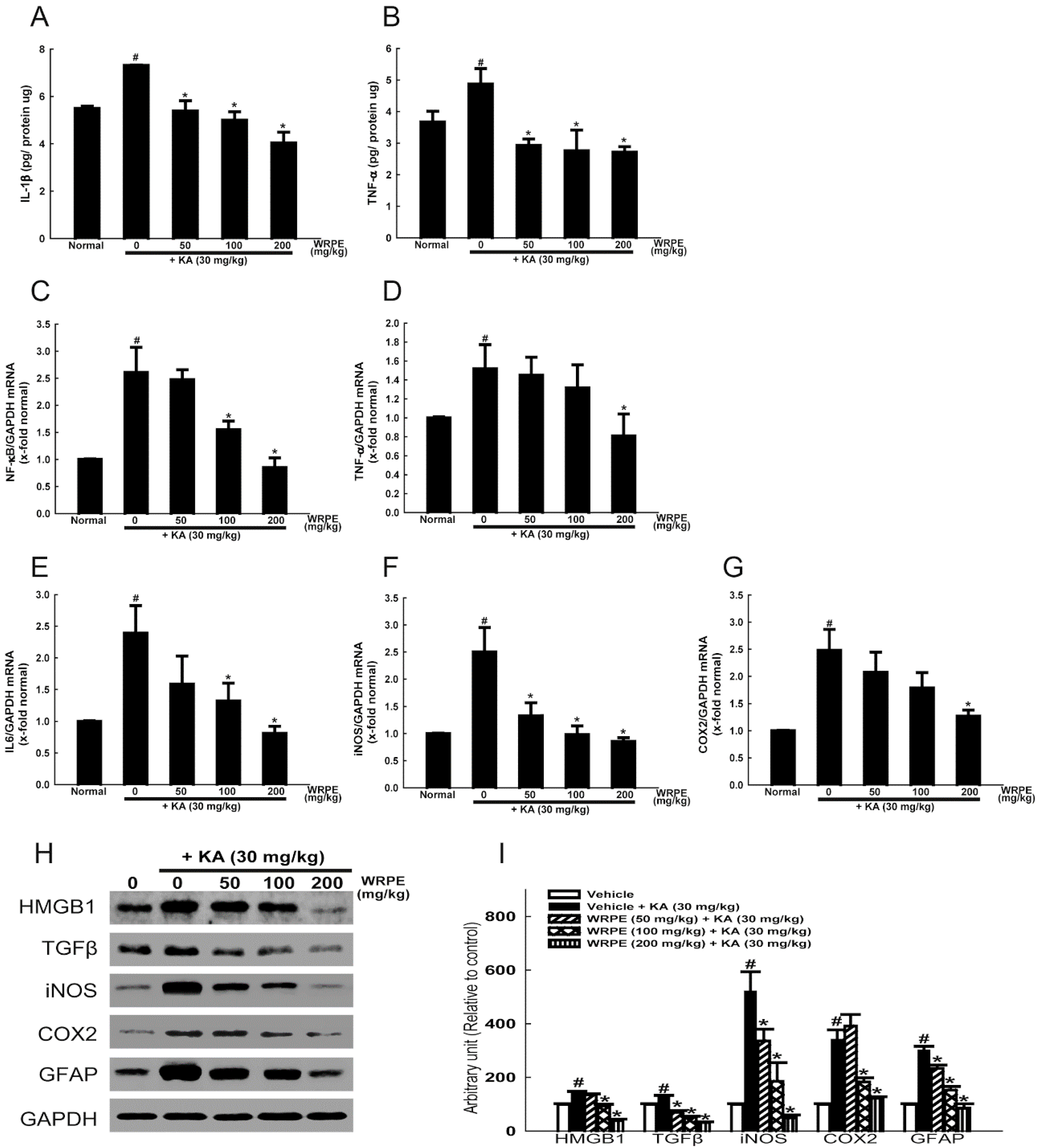
3.3. Anticonvulsant and Antioxidant Activities of WRPE in KA-Challenged Mice
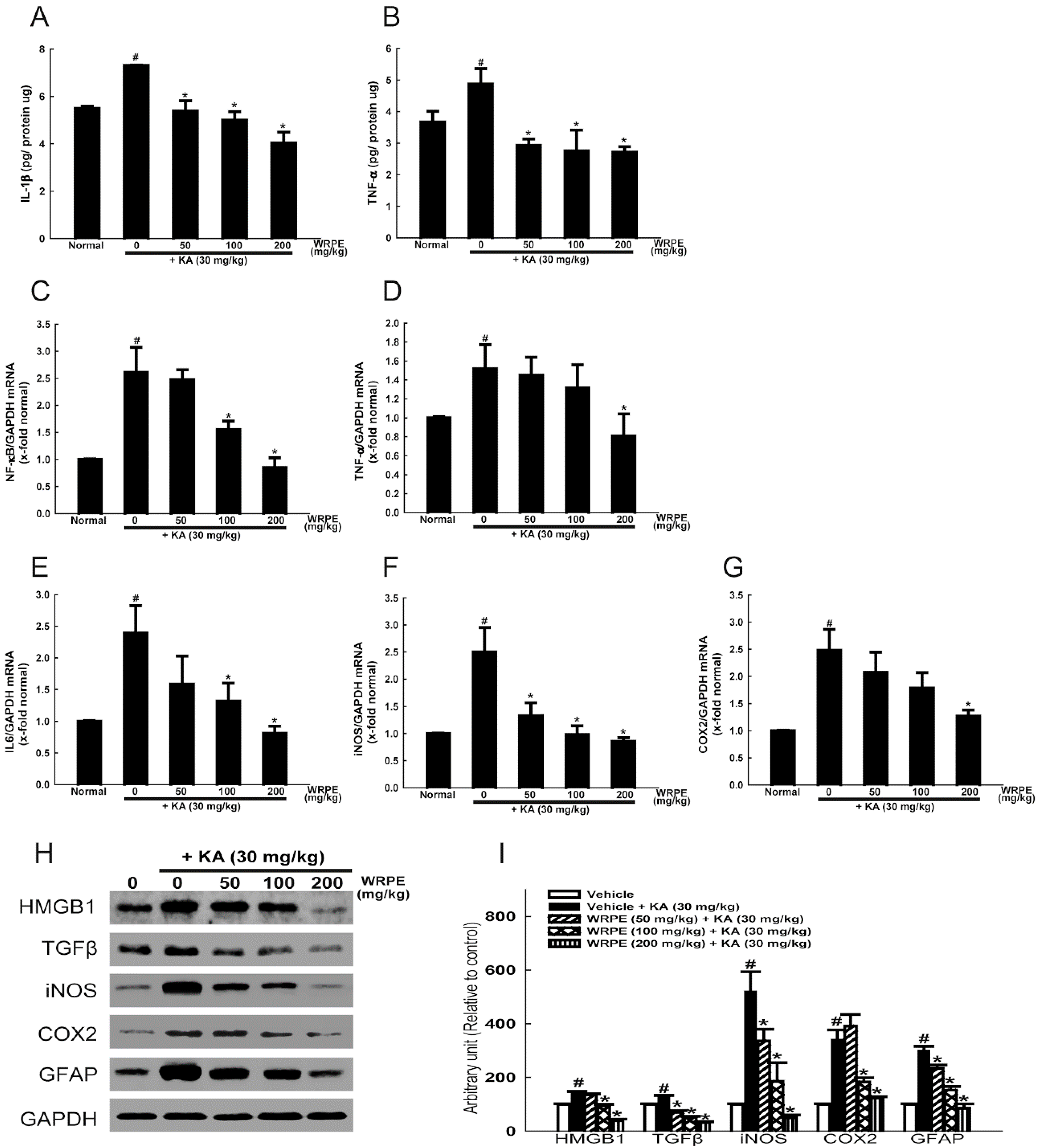
3.4. Anti-Inflammatory Activity of WRPE in KA-Challenged Mice
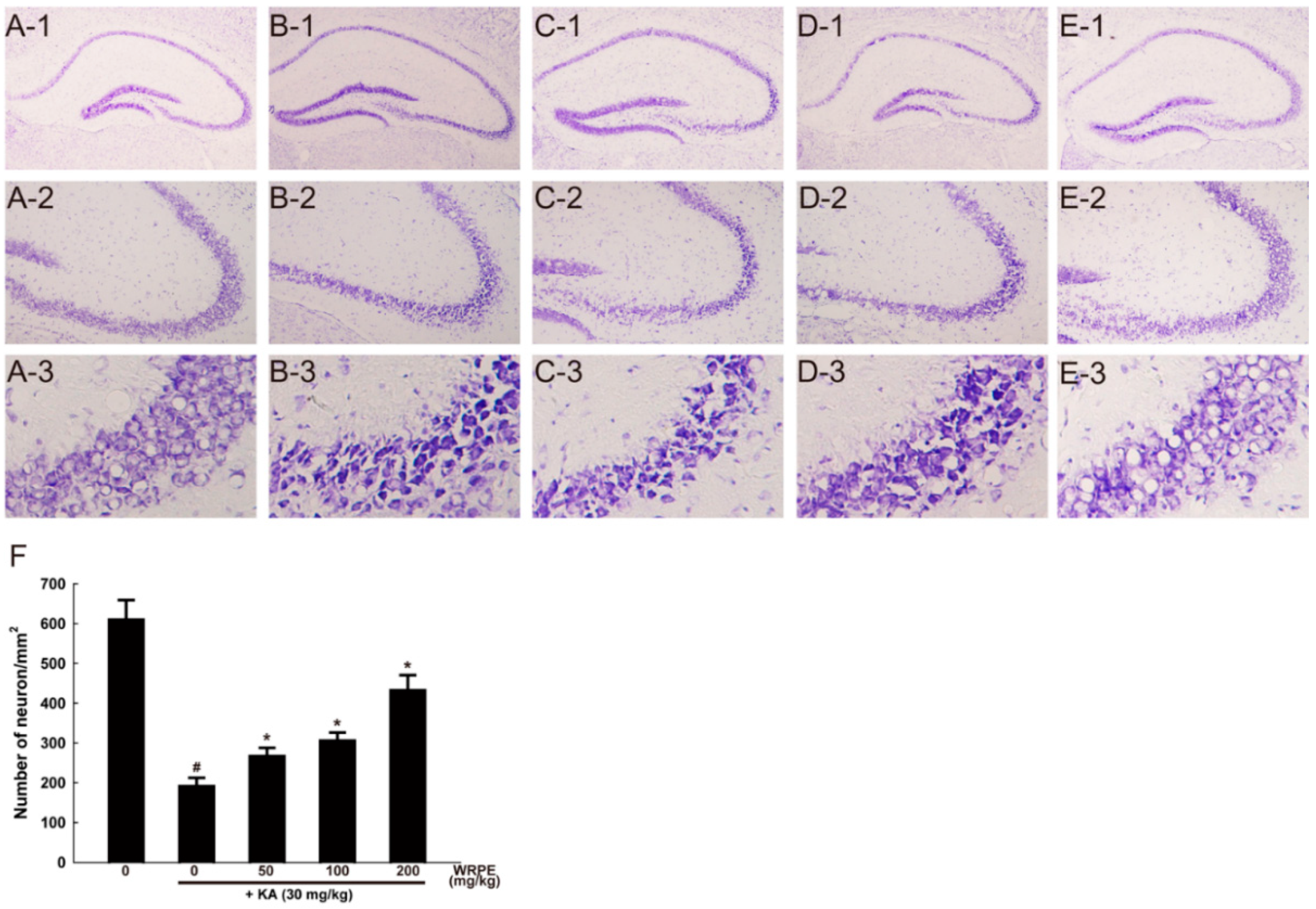
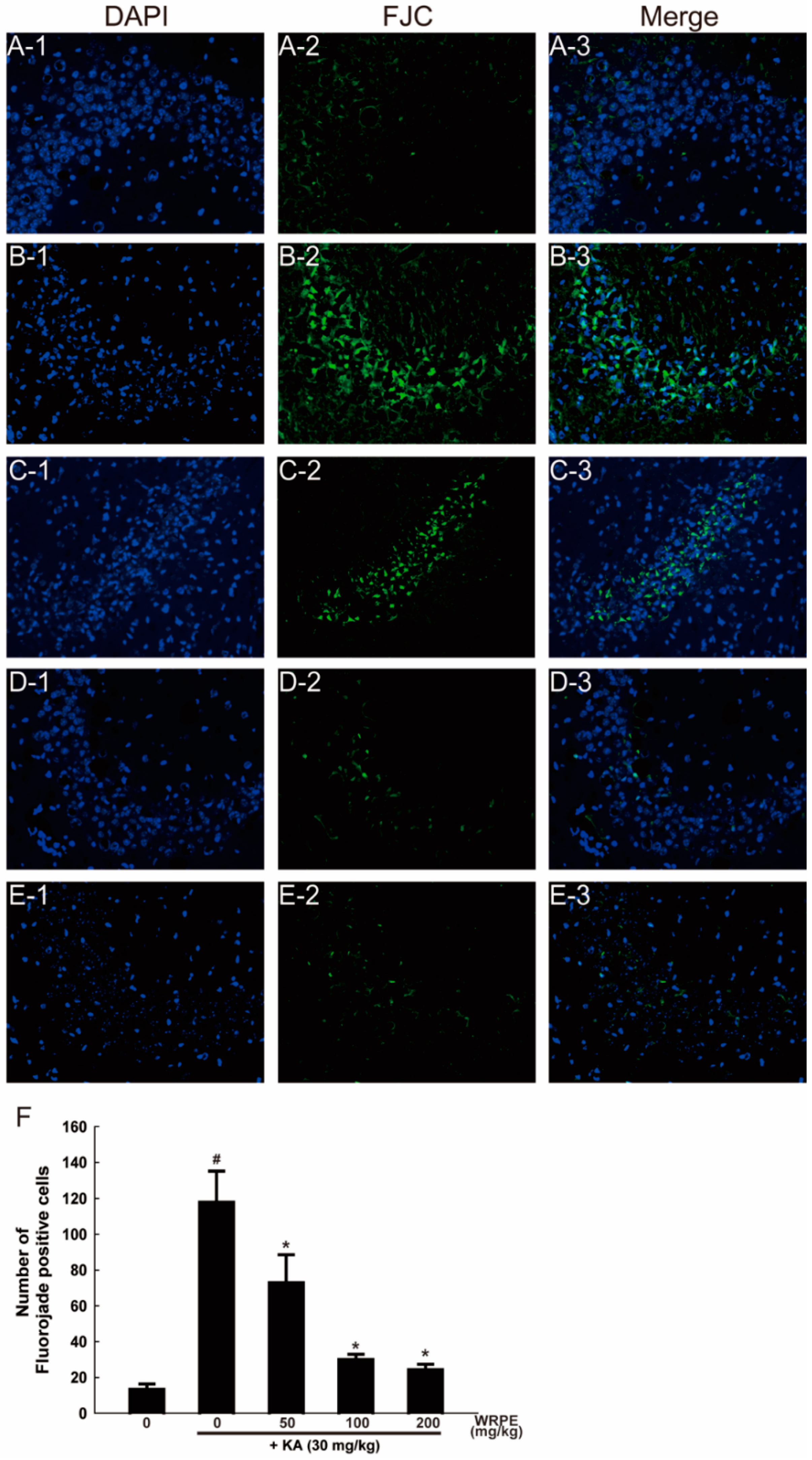
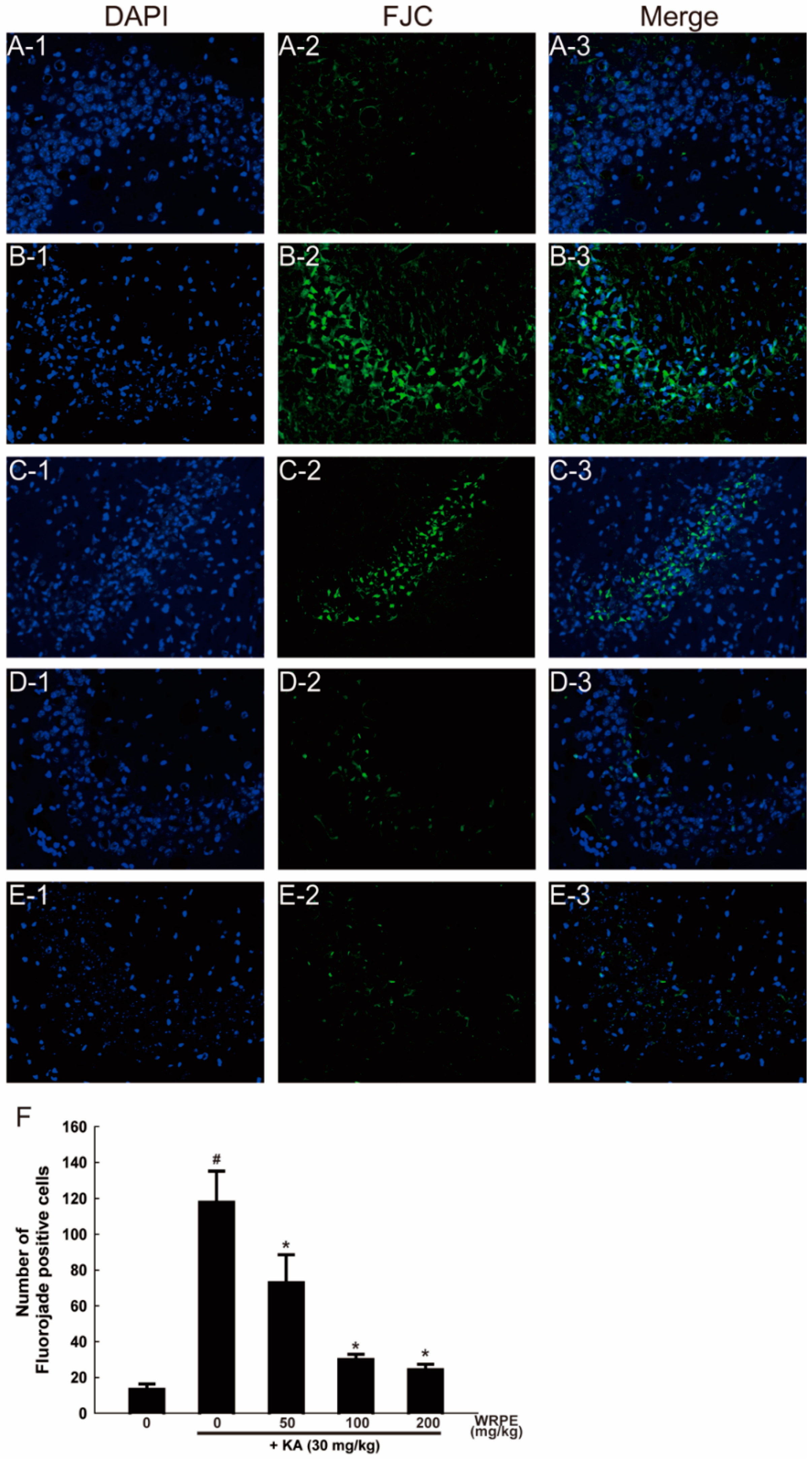
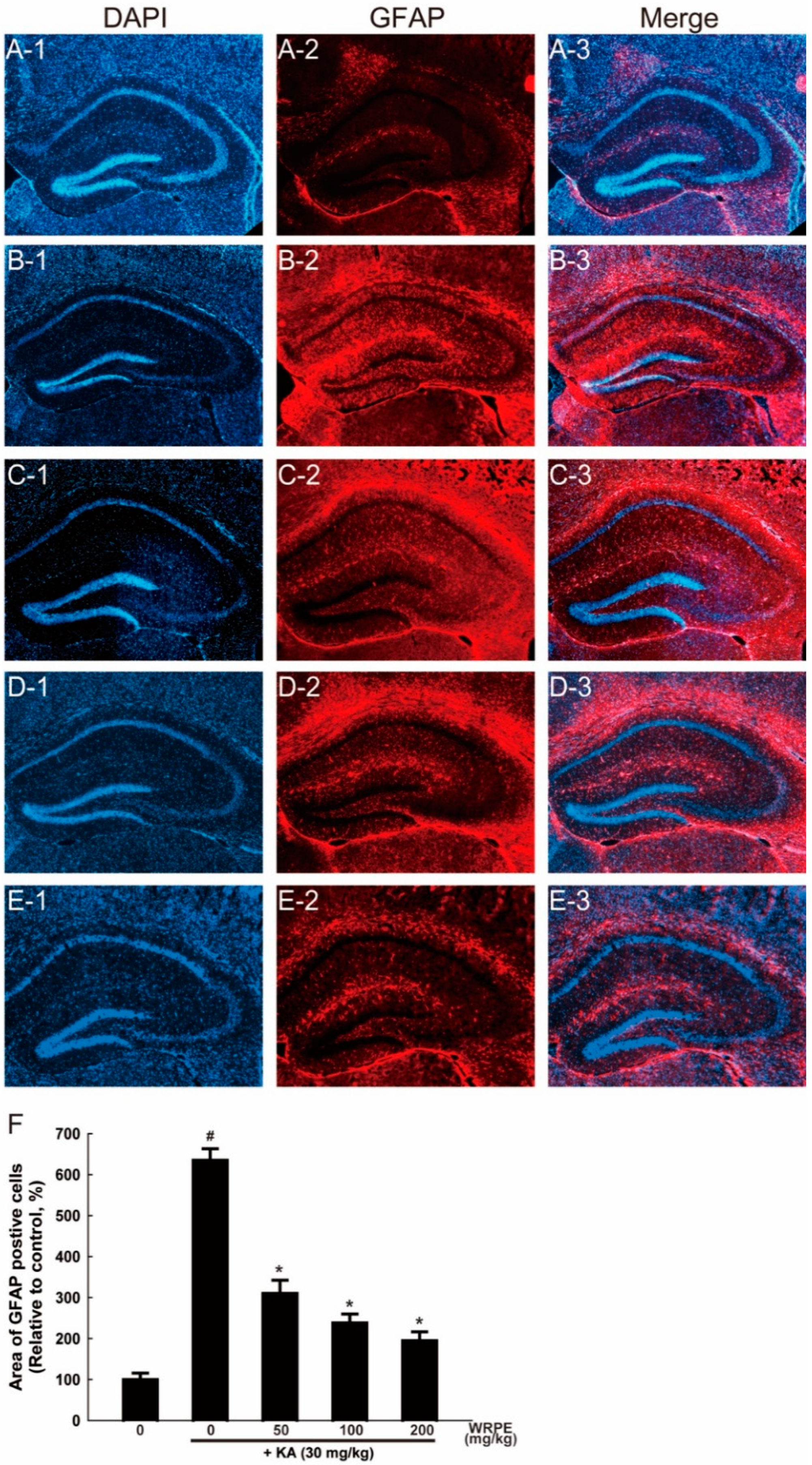
3.5. Neuroprotective Effects of WRPE in KA-Challenged Mice
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Fujikawa, D.G. Prolonged seizures and cellular injury: Understanding the connection. Epilepsy Behav. 2005, 7 (Suppl 3), S3–S11. [Google Scholar] [CrossRef] [PubMed]
- Olney, J.W. Brain lesions, obesity, and other disturbances in mice treated with monosodium glutamate. Science 1969, 164, 719–721. [Google Scholar] [CrossRef] [PubMed]
- Jhamandas, K.H.; Boegman, R.J.; Beninger, R.J. The 1993 upjohn award lecture. Quinolinic acid induced brain neurotransmitter deficits: Modulation by endogenous excitotoxin antagonists. Can. J. Physiol. Pharmacol. 1994, 72, 1473–1482. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Min, H.J.; Shin, J.S. Increased levels of hmgb1 and pro-inflammatory cytokines in children with febrile seizures. J. Neuroinflamm. 2011, 8, 135. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A.; Granata, T. Brain inflammation in epilepsy: Experimental and clinical evidence. Epilepsia 2005, 46, 1724–1743. [Google Scholar] [CrossRef] [PubMed]
- Wheless, J.W.; Clarke, D.F.; Arzimanoglou, A.; Carpenter, D. Treatment of pediatric epilepsy: European expert opinion, 2007. Epileptic Disord. 2007, 9, 353–412. [Google Scholar] [PubMed]
- Peltola, J.; Hurme, M.; Miettinen, A.; Keranen, T. Elevated levels of interleukin-6 may occur in cerebrospinal fluid from patients with recent epileptic seizures. Epilepsy Res. 1998, 31, 129–133. [Google Scholar] [CrossRef]
- Haspolat, S.; Mihci, E.; Coskun, M.; Gumuslu, S.; Ozben, T.; Yegin, O. Interleukin-1beta, tumor necrosis factor-alpha, and nitrite levels in febrile seizures. J. Child Neurol. 2002, 17, 749–751. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A.; Conti, M.; De Luigi, A.; Ravizza, T.; Moneta, D.; Marchesi, F.; De Simoni, M.G. Interleukin-1beta immunoreactivity and microglia are enhanced in the rat hippocampus by focal kainate application: Functional evidence for enhancement of electrographic seizures. J. Neurosci. 1999, 19, 5054–5065. [Google Scholar] [CrossRef] [PubMed]
- Coyle, J.T.; Puttfarcken, P. Oxidative stress, glutamate, and neurodegenerative disorders. Science 1993, 262, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Leite, J.P.; Garcia-Cairasco, N.; Cavalheiro, E.A. New insights from the use of pilocarpine and kainate models. Epilepsy Res. 2002, 50, 93–103. [Google Scholar] [CrossRef]
- Olney, J.W.; Rhee, V.; Ho, O.L. Kainic acid: A powerful neurotoxic analogue of glutamate. Brain Res. 1974, 77, 507–512. [Google Scholar] [CrossRef]
- Lee, T.H.; Lee, J.G.; Yon, J.M.; Oh, K.W.; Baek, I.J.; Nahm, S.S.; Lee, B.J.; Yun, Y.W.; Nam, S.Y. Capsaicin prevents kainic acid-induced epileptogenesis in mice. Neurochem. Int. 2011, 58, 634–640. [Google Scholar] [CrossRef] [PubMed]
- Floreani, M.; Skaper, S.D.; Facci, L.; Lipartiti, M.; Giusti, P. Melatonin maintains glutathione homeostasis in kainic acid-exposed rat brain tissues. FASEB J. 1997, 11, 1309–1315. [Google Scholar] [CrossRef] [PubMed]
- Saija, A.; Princi, P.; Pisani, A.; Lanza, M.; Scalese, M.; Aramnejad, E.; Ceserani, R.; Costa, G. Protective effect of glutathione on kainic acid-induced neuropathological changes in the rat brain. Gen. Pharmacol. 1994, 25, 97–102. [Google Scholar] [CrossRef]
- Acharya, M.M.; Hattiangady, B.; Shetty, A.K. Progress in neuroprotective strategies for preventing epilepsy. Prog. Neurobiol. 2008, 84, 363–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sok, D.E.; Oh, S.H.; Kim, Y.B.; Kim, M.R. Neuroprotective effect of rough aster butanol fraction against oxidative stress in the brain of mice challenged with kainic acid. J. Agric. Food Chem. 2003, 51, 4570–4575. [Google Scholar] [CrossRef] [PubMed]
- Gupta, Y.K.; Briyal, S. Protective effect of vineatrol against kainic acid induced seizures, oxidative stress and on the expression of heat shock proteins in rats. Eur. Neuropsychopharmacol. 2006, 16, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Gupta, Y.K.; Briyal, S.; Sharma, M. Protective effect of curcumin against kainic acid induced seizures and oxidative stress in rats. Indian J. Physiol. Pharmacol. 2009, 53, 39–46. [Google Scholar] [PubMed]
- Wu, Z.; Xu, Q.; Zhang, L.; Kong, D.; Ma, R.; Wang, L. Protective effect of resveratrol against kainate-induced temporal lobe epilepsy in rats. Neurochem. Res. 2009, 34, 1393–1400. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.K.; Guo, H.; Choi, J.K.; Jang, S.K.; Shin, K.; Cha, Y.S.; Choi, Y.; Seo, D.W.; Lee, Y.B.; Joo, S.S.; et al. Extraction conditions of white rose petals for the inhibition of enzymes related to skin aging. Lab. Anim. Res. 2015, 31, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Park, D.; Shin, K.; Choi, Y.; Guo, H.; Cha, Y.; Kim, S.H.; Han, N.S.; Joo, S.S.; Choi, J.K.; Lee, Y.B.; et al. Antimicrobial activities of ethanol and butanol fractions of white rose petal extract. Regul. Toxicol. Pharmacol. 2016, 76, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.H.; Kwon, S.C.; Park, D.; Shin, S.; Jeong, J.H.; Park, S.Y.; Hwang, S.Y.; Kim, Y.B.; Joo, S.S. Anti-allergic effects of white rose petal extract and anti-atopic properties of its hexane fraction. Arch. Pharm. Res. 2009, 32, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Park, D.; Lee, S.H.; Bae, D.K.; Yang, Y.H.; Kyung, J.; Kim, D.; Choi, E.K.; Hong, J.T.; Jeong, H.S.; et al. Neuroprotective effects of a butanol fraction of rosa hybrida petals in a middle cerebral artery occlusion model. Biomol. Ther. (Seoul) 2013, 21, 454–461. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.H.; Ro, H.M.; Kim, S.L.; Kim, H.S.; Chung, I.M. Analysis of isoflavone, phenolic, soyasapogenol, and tocopherol compounds in soybean (glycine max (l.) merrill) germplasms of different seed weights and origins. J. Agric. Food Chem. 2012, 60, 6045–6055. [Google Scholar] [CrossRef] [PubMed]
- Park, D.; Joo, S.S.; Kim, T.K.; Lee, S.H.; Kang, H.; Lee, H.J.; Lim, I.; Matsuo, A.; Tooyama, I.; Kim, Y.B.; et al. Human neural stem cells overexpressing choline acetyltransferase restore cognitive function of kainic acid-induced learning and memory deficit animals. Cell Transplant. 2012, 21, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.G.; Yon, J.M.; Lin, C.; Jung, A.Y.; Jung, K.Y.; Nam, S.Y. Combined treatment with capsaicin and resveratrol enhances neuroprotection against glutamate-induced toxicity in mouse cerebral cortical neurons. Food Chem. Toxicol. 2012, 50, 3877–3885. [Google Scholar] [CrossRef] [PubMed]
- Racine, R.J. Modification of seizure activity by electrical stimulation. Ii. Motor seizure. Electroencephalogr. Clin. Neurophysiol. 1972, 32, 281–294. [Google Scholar] [CrossRef]
- Heinemann, U.; Draguhn, A.; Ficker, E.; Stabel, J.; Zhang, C.L. Strategies for the development of drugs for pharmacoresistant epilepsies. Epilepsia 1994, 35 (Suppl. 5), S10–S21. [Google Scholar] [CrossRef] [PubMed]
- Penkowa, M.; Florit, S.; Giralt, M.; Quintana, A.; Molinero, A.; Carrasco, J.; Hidalgo, J. Metallothionein reduces central nervous system inflammation, neurodegeneration, and cell death following kainic acid-induced epileptic seizures. J. Neurosci. Res. 2005, 79, 522–534. [Google Scholar] [CrossRef] [PubMed]
- Park, D.; Jeon, J.H.; Kwon, S.C.; Shin, S.; Jang, J.Y.; Jeong, H.S.; Lee, D.I.; Kim, Y.B.; Joo, S.S. Antioxidative activities of white rose flower extract and pharmaceutical advantages of its hexane fraction via free radical scavenging effects. Biochem. Cell Biol. 2009, 87, 943–952. [Google Scholar] [CrossRef] [PubMed]
- Heim, K.E.; Tagliaferro, A.R.; Bobilya, D.J. Flavonoid antioxidants: Chemistry, metabolism and structure-activity relationships. J. Nutr. Biochem. 2002, 13, 572–584. [Google Scholar] [CrossRef]
- Zhang, A.; Fang, Y.; Wang, H.; Li, H.; Zhang, Z. Free-radical scavenging properties and reducing power of grape cane extracts from 11 selected grape cultivars widely grown in china. Molecules 2011, 16, 10104–10122. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Pandey, A.K. Chemistry and biological activities of flavonoids: An overview. Sci. World J. 2013, 2013, 162750. [Google Scholar] [CrossRef] [PubMed]
- Kiasalari, Z.; Roghani, M.; Khalili, M.; Rahmati, B.; Baluchnejadmojarad, T. Antiepileptogenic effect of curcumin on kainate-induced model of temporal lobe epilepsy. Pharm. Biol. 2013, 51, 1572–1578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vezzani, A.; French, J.; Bartfai, T.; Baram, T.Z. The role of inflammation in epilepsy. Nat. Rev. Neurol. 2011, 7, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Plata-Salaman, C.R.; Ilyin, S.E.; Turrin, N.P.; Gayle, D.; Flynn, M.C.; Romanovitch, A.E.; Kelly, M.E.; Bureau, Y.; Anisman, H.; McIntyre, D.C. Kindling modulates the IL-1beta system, TNF-alpha, TGF-beta1, and neuropeptide mrnas in specific brain regions. Brain Res. Mol. Brain Res. 2000, 75, 248–258. [Google Scholar] [CrossRef]
- Jankowsky, J.L.; Patterson, P.H. The role of cytokines and growth factors in seizures and their sequelae. Prog. Neurobiol. 2001, 63, 125–149. [Google Scholar] [CrossRef]
- Turrin, N.P.; Rivest, S. Innate immune reaction in response to seizures: Implications for the neuropathology associated with epilepsy. Neurobiol. Dis. 2004, 16, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Ravizza, T.; Gagliardi, B.; Noe, F.; Boer, K.; Aronica, E.; Vezzani, A. Innate and adaptive immunity during epileptogenesis and spontaneous seizures: Evidence from experimental models and human temporal lobe epilepsy. Neurobiol. Dis. 2008, 29, 142–160. [Google Scholar] [CrossRef] [PubMed]
- Allan, S.M.; Tyrrell, P.J.; Rothwell, N.J. Interleukin-1 and neuronal injury. Nat. Rev. Immunol. 2005, 5, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Doble, A. The role of excitotoxicity in neurodegenerative disease: Implications for therapy. Pharmacol. Ther. 1999, 81, 163–221. [Google Scholar] [CrossRef]
- Maroso, M.; Balosso, S.; Ravizza, T.; Liu, J.; Aronica, E.; Iyer, A.M.; Rossetti, C.; Molteni, M.; Casalgrandi, M.; Manfredi, A.A.; et al. Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. Nat. Med. 2010, 16, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, M.E.; Manfredi, A.A. High-mobility group box 1 (HMGB1) protein at the crossroads between innate and adaptive immunity. Immunol. Rev. 2007, 220, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Puttachary, S.; Sharma, S.; Stark, S.; Thippeswamy, T. Seizure-induced oxidative stress in temporal lobe epilepsy. BioMed Res. Int. 2015, 2015, 745613. [Google Scholar] [CrossRef] [PubMed]
- Rana, A.; Musto, A.E. The role of inflammation in the development of epilepsy. J. Neuroinflamm. 2018, 15, 144. [Google Scholar] [CrossRef] [PubMed]
- Dudek, F.E. Mechanisms of seizure-induced inflammation of the brain: Many possible roles for neuronal cox-2. Epilepsy Curr. 2012, 12, 115–117. [Google Scholar] [CrossRef] [PubMed]
- Kim, G. Exercise-Induced Adult Neruogenesis and the Seizure Threshold: The Role of Cox-2. B.S. Thesis, Syracuse University, New York, NY, USA, 2015. [Google Scholar]
- Salvadori, M.G.; Bandero, C.R.; Jesse, A.C.; Gomes, A.T.; Rambo, L.M.; Bueno, L.M.; Bortoluzzi, V.T.; Oliveira, M.S.; Mello, C.F. Prostaglandin E(2) potentiates methylmalonate-induced seizures. Epilepsia 2012, 53, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A.; Friedman, A.; Dingledine, R.J. The role of inflammation in epileptogenesis. Neuropharmacology 2013, 69, 16–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, D.; Joo, S.S.; Lee, H.J.; Choi, K.C.; Kim, S.U.; Kim, Y.B. Microtubule-associated protein 2, an early blood marker of ischemic brain injury. J. Neurosci. Res. 2012, 90, 461–467. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Instruments | HPLC (ACME 9000 system, Younglin, Anyang, Korea) |
| Column | Mightysil RP-18GP column (5 μm, 4.6 × 250 mm) |
| Mobile phase | A (0.1% (v/v) acetic acid in acetonitrile), B (0.1% (v/v) acetic acid in water) 0 → 2 min (A:B = 92:8 → A:B = 90:10) 2 → 27 min (A:B = 90:10 → A:B = 70:30) 27 → 50 min (A:B = 70:30 → A:B = 10:90) 50 → 51 min (A:B = 10:90 → A:B = 0:100) 51 → 60 min (A:B = 0:100) 60 → 70 min (A:B = 0:100 → A:B = 92:8) |
| Detector | UV detector (280 nm) |
| Flow rate | 1.0 mL/min |
| Injection volume | 20 μL |
| Gene Name | Accession No. | Human Primer | |
| Forward (5′-3′) | Reverse (5′-3′) | ||
| COX2 | NM_000963 | AAGTGCGATTGTACCCGGAC | GTGCACTGTGTTTGGAGTGG |
| GPx1 | NM_000581 | ACACCCAGATGAACGAGCTG | CAAACTGGTTGCACGGGAAG |
| IL6 | NM_001318095 | AGTGAGGAACAAGCCAGAGC | ATTTGTGGTTGGGTCAGGGG |
| iNOS | XM_011524860.2 | ACAACAAATTCAGGTACGCTGTG | TCTGATCAATGTCATGAGCAAAGG |
| NF-kB | NM_001165412 | GTGGTGCGGCTCATGTTTAC | CGTCTGATACCACGGGTTCC |
| PHGPx | NM_002085 | CAGTGAGGCAAGACCGAAGT | CCGAACTGGTTACACGGGAA |
| SOD1 | NM_000454 | ATGACTTGGGCAAAGGTGGA | GGGCGATCCCAATTACACCA |
| SOD2 | NM_000636 | GGAGAACCCAAAGGGGAGTTG | GCCGTCAGCTTCTCCTTAAAC |
| TNF-α | NM_000594 | GCTGCACTTTGGAGTGATCG | TCACTCGGGGTTCGAGAAGA |
| GAPDH | NM_001289746 | AAGAAGGTGGTGAAGCAG | GTCAAAGGTGGAGGAGTG |
| Gene Name | Accession No. | Mouse Primer | |
| Forward (5′-3′) | Reverse (5′-3′) | ||
| COX2 | NM_011198 | GAACCTGCAGTTTGCTGTGG | ACTCTGTTGTGCTCCCGAAG |
| IL-6 | NM_031168.1 | TCCAGTTGCCTTCTTGGGAC | AGTCTCCTCTCCGGACTTGT |
| iNOS | NM_010927.3 | CTATGGCCGCTTTGATGTGC | TTGGGATGCTCCATGGTCAC |
| NF-kB | NM_008689 | CACTGCTCAGGTCCACTGTC | CTGTCACTATCCCGGAGTTCA |
| SOD2 | NM_013671 | GGAGCAAGGTCGCTTACAGA | GTGCTCCCACACGTCAATC |
| TNF-α | NM_013693 | TACCTTGTTGCCTCCTCTT | GTCACCAAATCAGCGTTATTAAG |
| GAPDH | NM_008084 | CGTGCCGCCTGGAGAAACC | TGGAAGAGTGGGAGTTGCTGTTG |
| Epitope | Company | Cat. Number | Dilution | 2° Ab (IgG) |
|---|---|---|---|---|
| HMGB1 | Cell Signaling | #3935 | 1:1000 | anti-rabbit |
| TGFβ | Santa Cruz | sc-146 | 1:200 | anti-rabbit |
| iNOS | Santa Cruz | sc-651 | 1:200 | anti-rabbit |
| COX2 | Santa Cruz | sc-1746 | 1:500 | anti-goat |
| GFAP | Santa Cruz | sc-51908 | 1:200 | anti-mouse |
| GAPDH | Cell Signaling | #2118 | 1:1000 | anti-rabbit |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yon, J.-M.; Kim, Y.-B.; Park, D. The Ethanol Fraction of White Rose Petal Extract Abrogates Excitotoxicity-Induced Neuronal Damage In Vivo and In Vitro through Inhibition of Oxidative Stress and Proinflammation. Nutrients 2018, 10, 1375. https://doi.org/10.3390/nu10101375
Yon J-M, Kim Y-B, Park D. The Ethanol Fraction of White Rose Petal Extract Abrogates Excitotoxicity-Induced Neuronal Damage In Vivo and In Vitro through Inhibition of Oxidative Stress and Proinflammation. Nutrients. 2018; 10(10):1375. https://doi.org/10.3390/nu10101375
Chicago/Turabian StyleYon, Jung-Min, Yun-Bae Kim, and Dongsun Park. 2018. "The Ethanol Fraction of White Rose Petal Extract Abrogates Excitotoxicity-Induced Neuronal Damage In Vivo and In Vitro through Inhibition of Oxidative Stress and Proinflammation" Nutrients 10, no. 10: 1375. https://doi.org/10.3390/nu10101375