Enhancement in Site-Specific Delivery of Carvacrol against Methicillin Resistant Staphylococcus aureus Induced Skin Infections Using Enzyme Responsive Nanoparticles: A Proof of Concept Study

 ,
,  and
and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
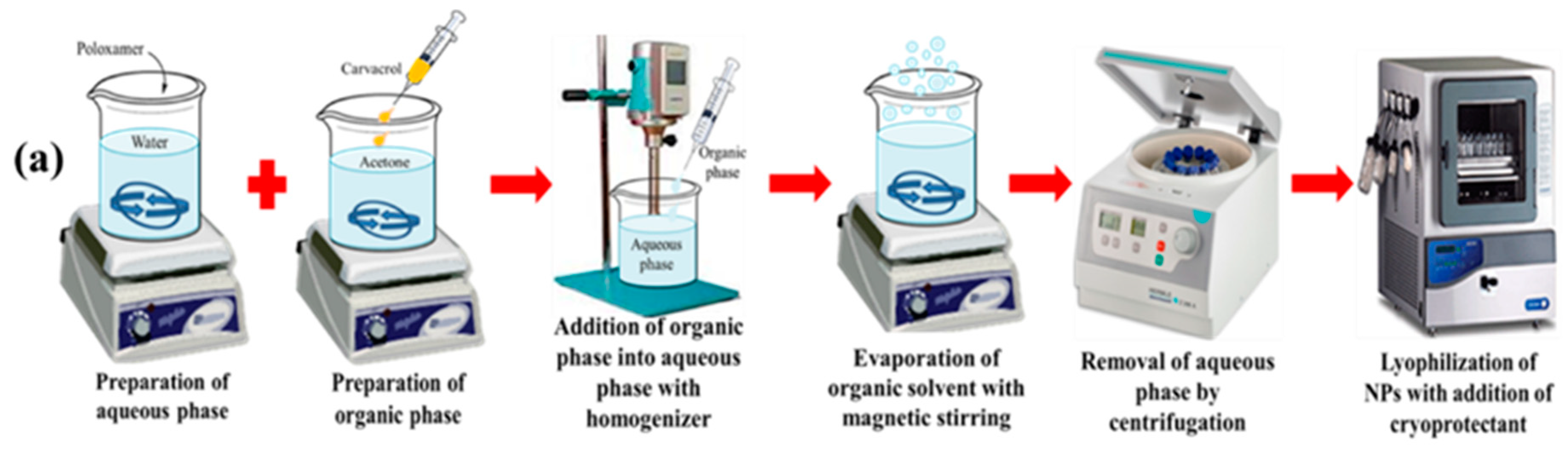
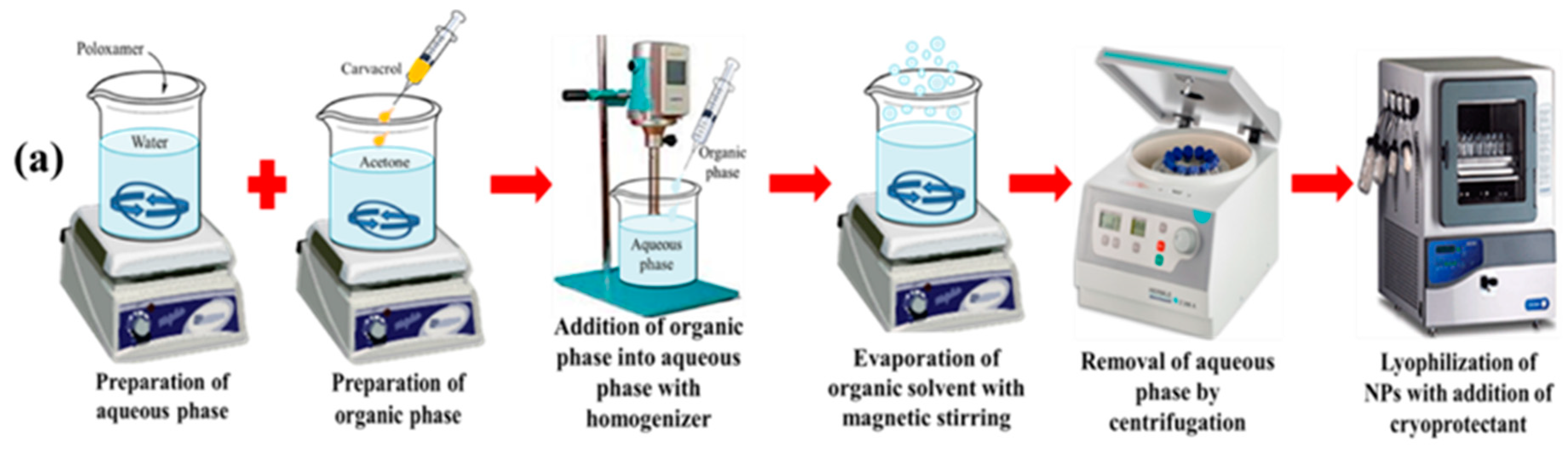
2.2. Preparation of Blank and CAR-PCL NPs
2.3. Optimization of CAR-PCL NPs through Experimental Design
2.4. Physicochemical Characterization of CAR-PCL NPs
2.4.1. Particle Size, Morphology, Polydispersity Index and Zeta Potential
2.4.2. Estimation of Entrapment Efficiency and Percentage Yield
2.5. Fourier Transform-Infrared (FTIR) Spectroscopy
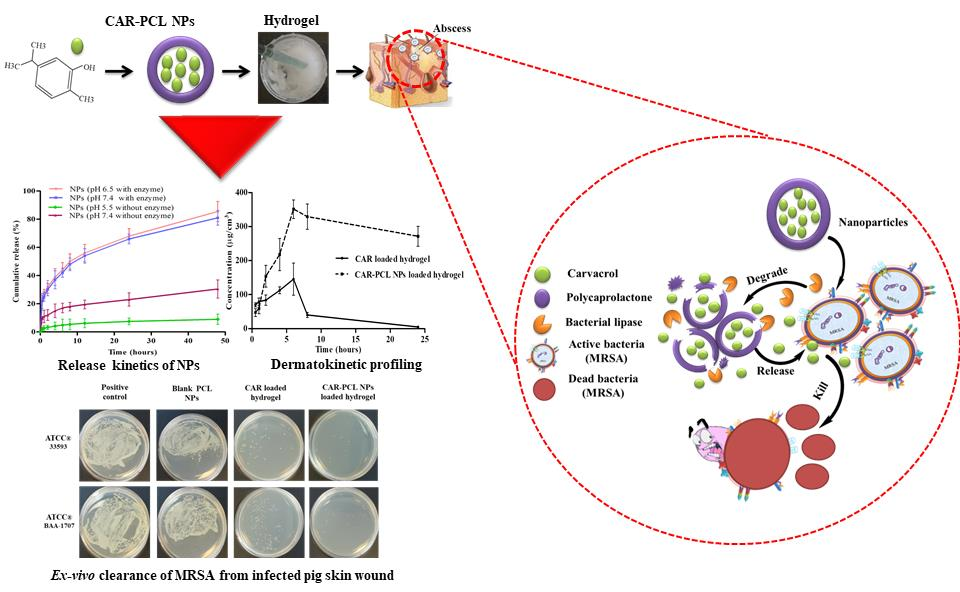
2.6. In-Vitro Release Kinetic Study of CAR-PCL NPs
2.7. In-Vitro Antibacterial Assays
2.7.1. Culture of MRSA Strains
2.7.2. Evaluation of Lipase Producing Activity
2.7.3. Determination of Minimum Inhibitory Concentration and Minimum Bactericidal Concentration
2.7.4. Killing Kinetics of CAR and CAR-PCL NPs
2.8. Preparation of CAR and CAR-PCL NPs Loaded Hydrogel
2.9. Characterization of CAR-PCL NPs Loaded Hydrogel
2.9.1. Physical Appearance, pH, Drug Content and Spreadability of Hydrogel
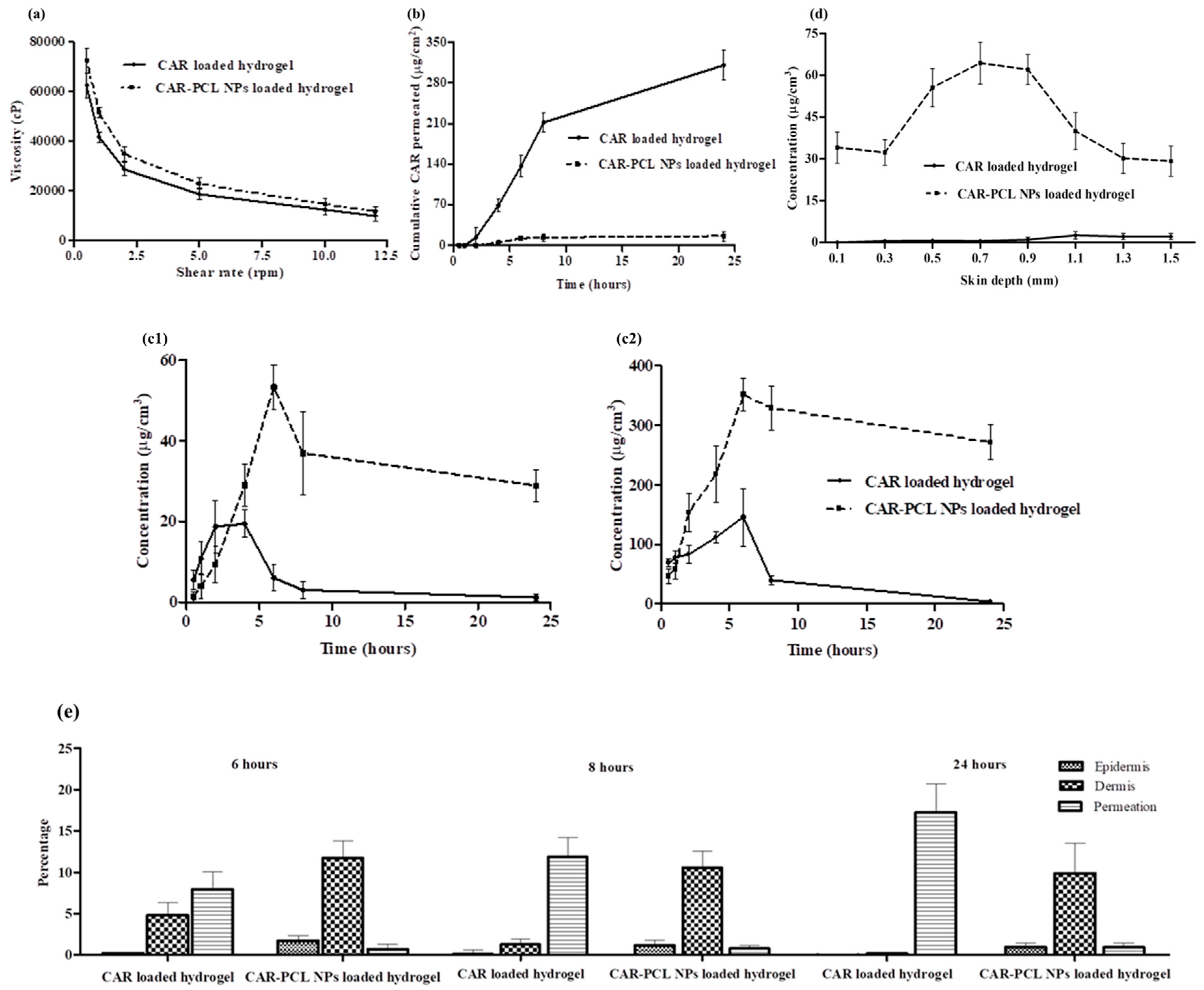
2.9.2. Rheological Evaluation
2.9.3. Extrudability Measurement
2.9.4. Determination of Bioadhesion Time
2.10. Stability Studies of Hydrogel
2.11. Ex-Vivo Dermatokinetic, Skin Deposition, and Distribution Studies
2.11.1. Preparation of Skin Samples
2.11.2. Dermatokinetic and Permeation Studies
2.11.3. Skin Distribution Studies
2.12. Determination of Antibacterial Effectiveness in an Ex-Vivo Pig Skin Wound Model
2.13. Instrumentation and Chromatographic Conditions of Analytical Method
2.14. Statistical Analysis
3. Results and Discussion
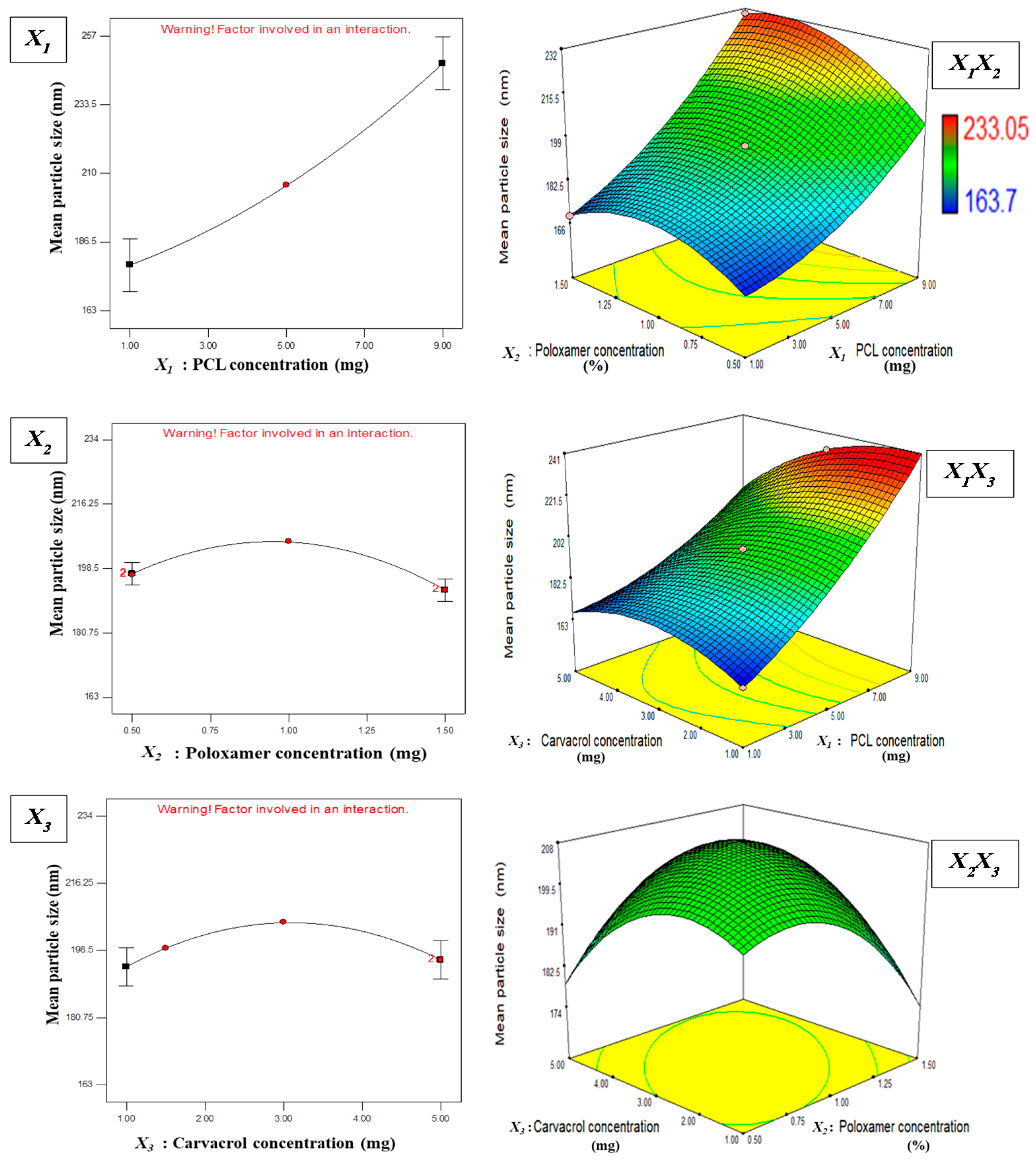
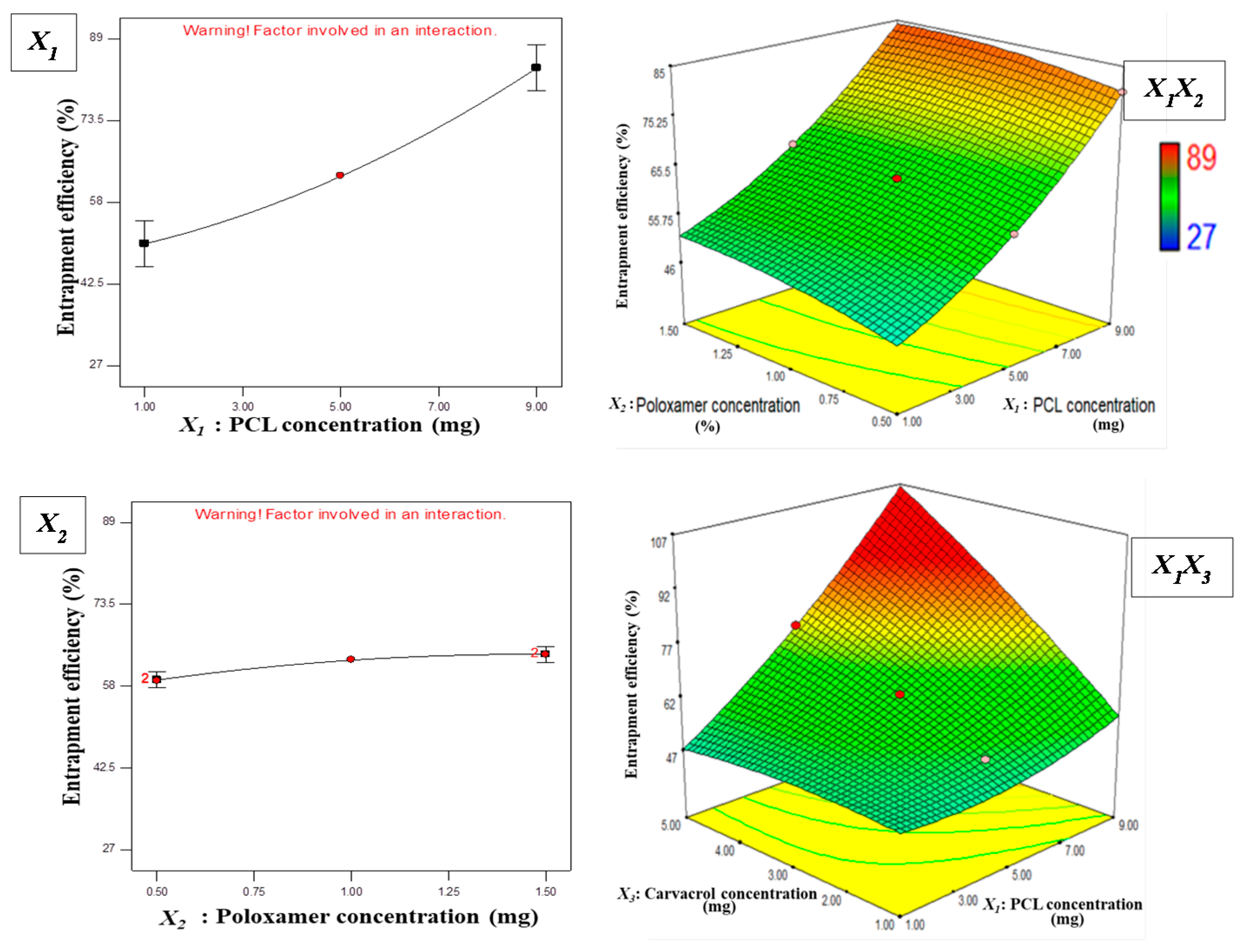
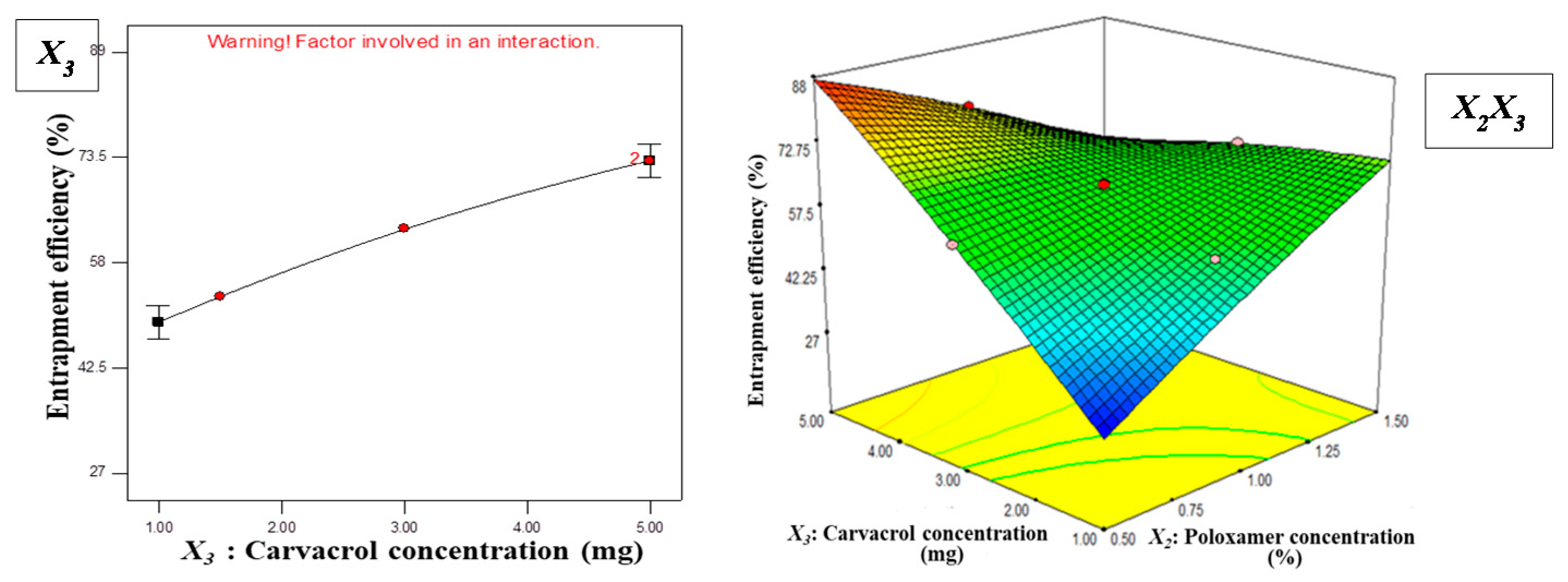
3.1. Statistical Analysis of Experimental Data by Design-Expert Software
3.1.1. Effect of Independent Variables on Particle Size
Y1 = + 205.82 + 34.50X1 − 2.23X2 + 0.91X3
+ 5.83X1X2 − 9.73X1X3 + 9.34X2X3
+ 7.32X12 − 10.9X22 − 10.73X32
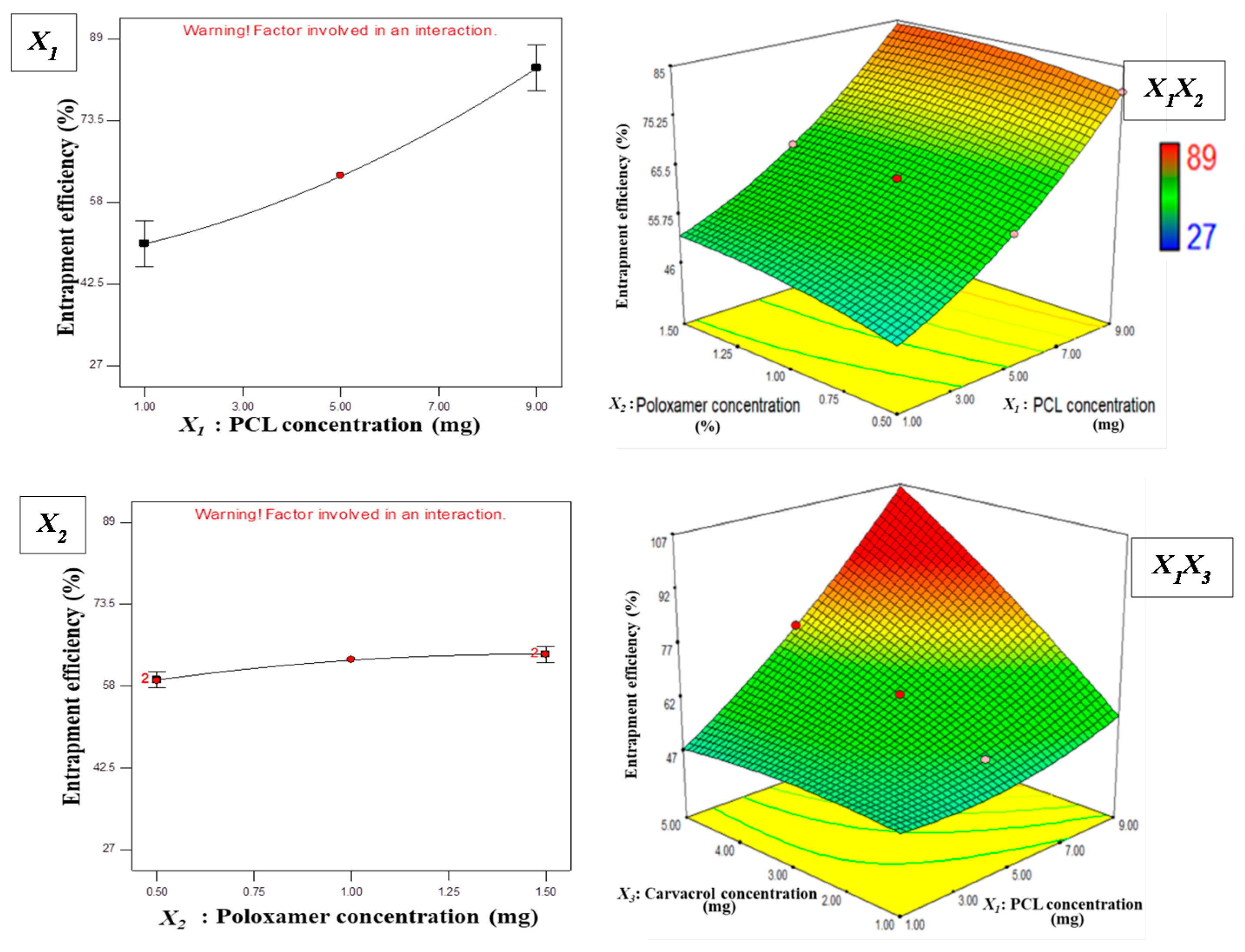
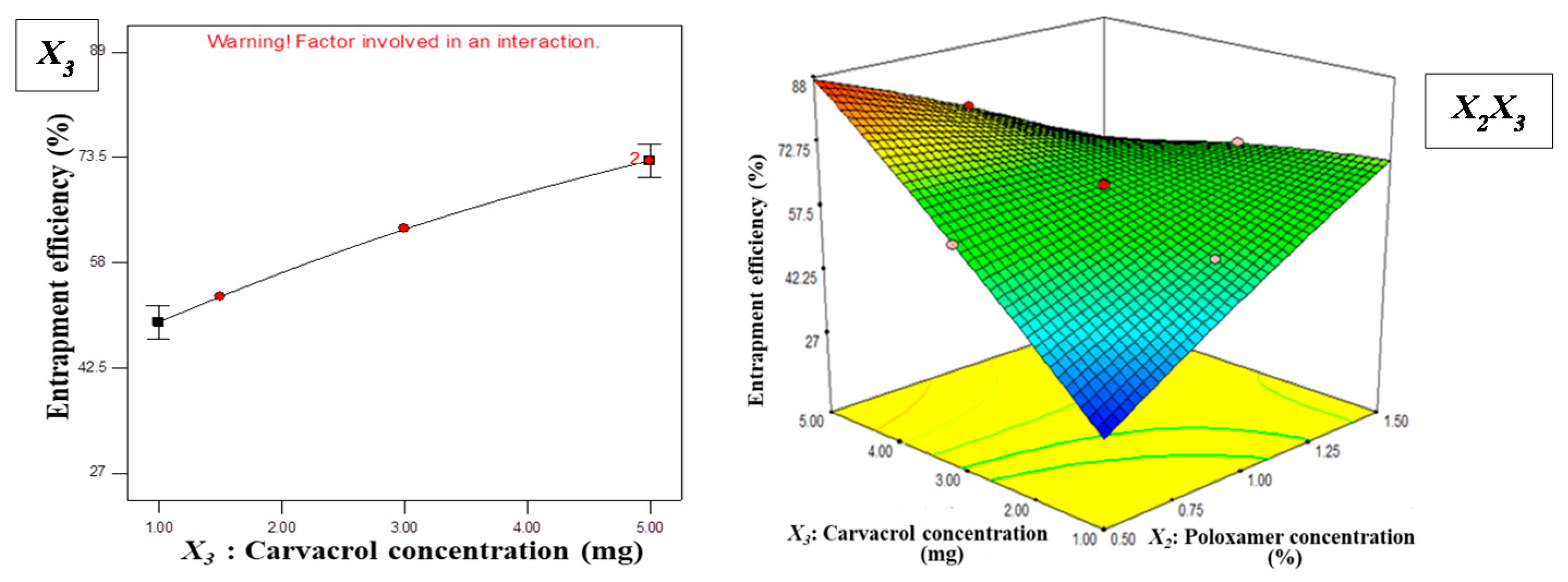
3.1.2. Effect of Independent Variables on EE
Y2 = + 62.91 + 16.69X1 + 2.44X2 + 11.88X3
− 0.29X1X2 + 12.85X1X3 − 18.23X2X3
+ 3.87X12 − 1.32X22 − 1.79X32
3.2. Optimization and Validation
3.3. Physicochemical Characterization of Optimized CAR-PCL NPs
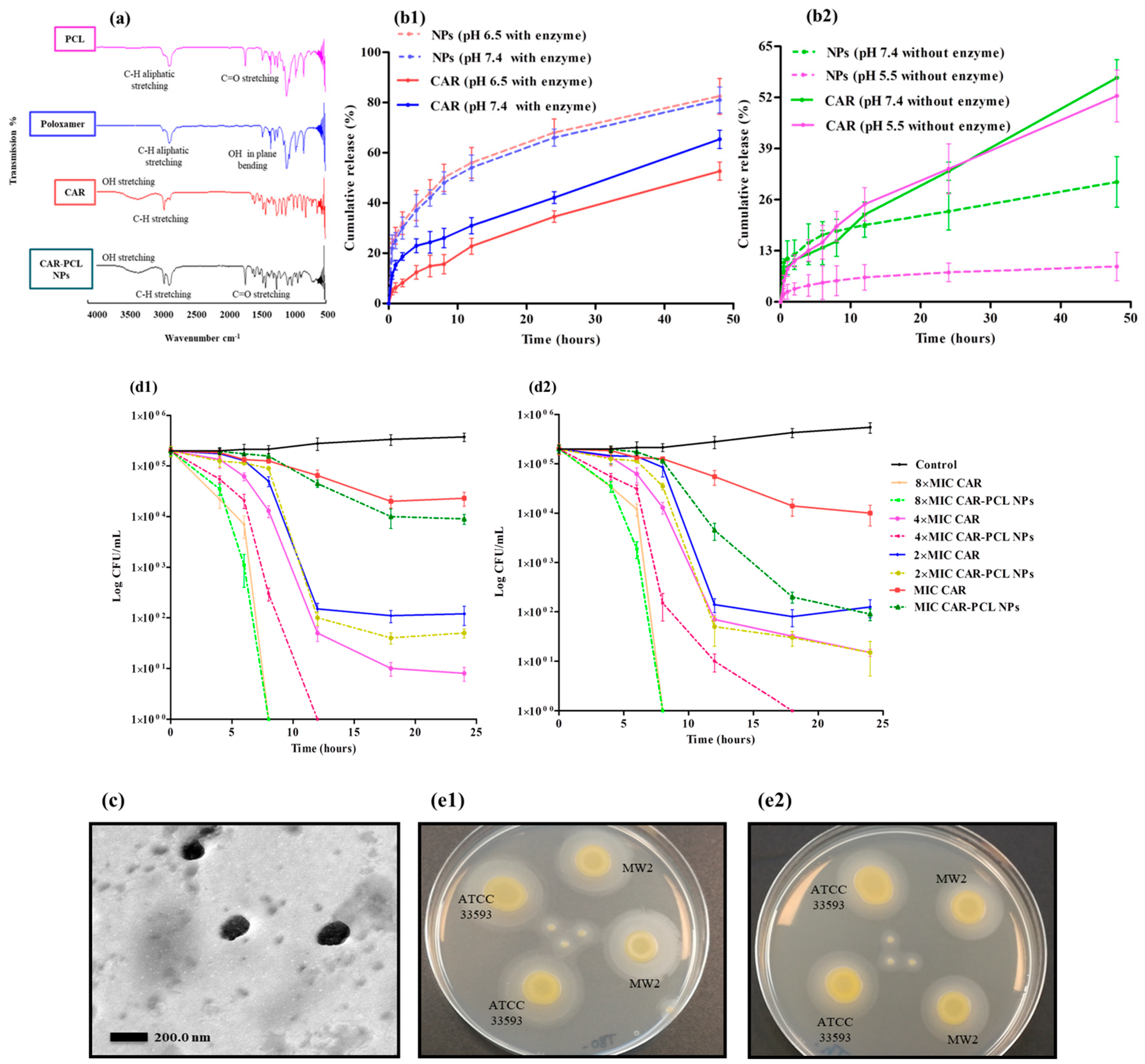
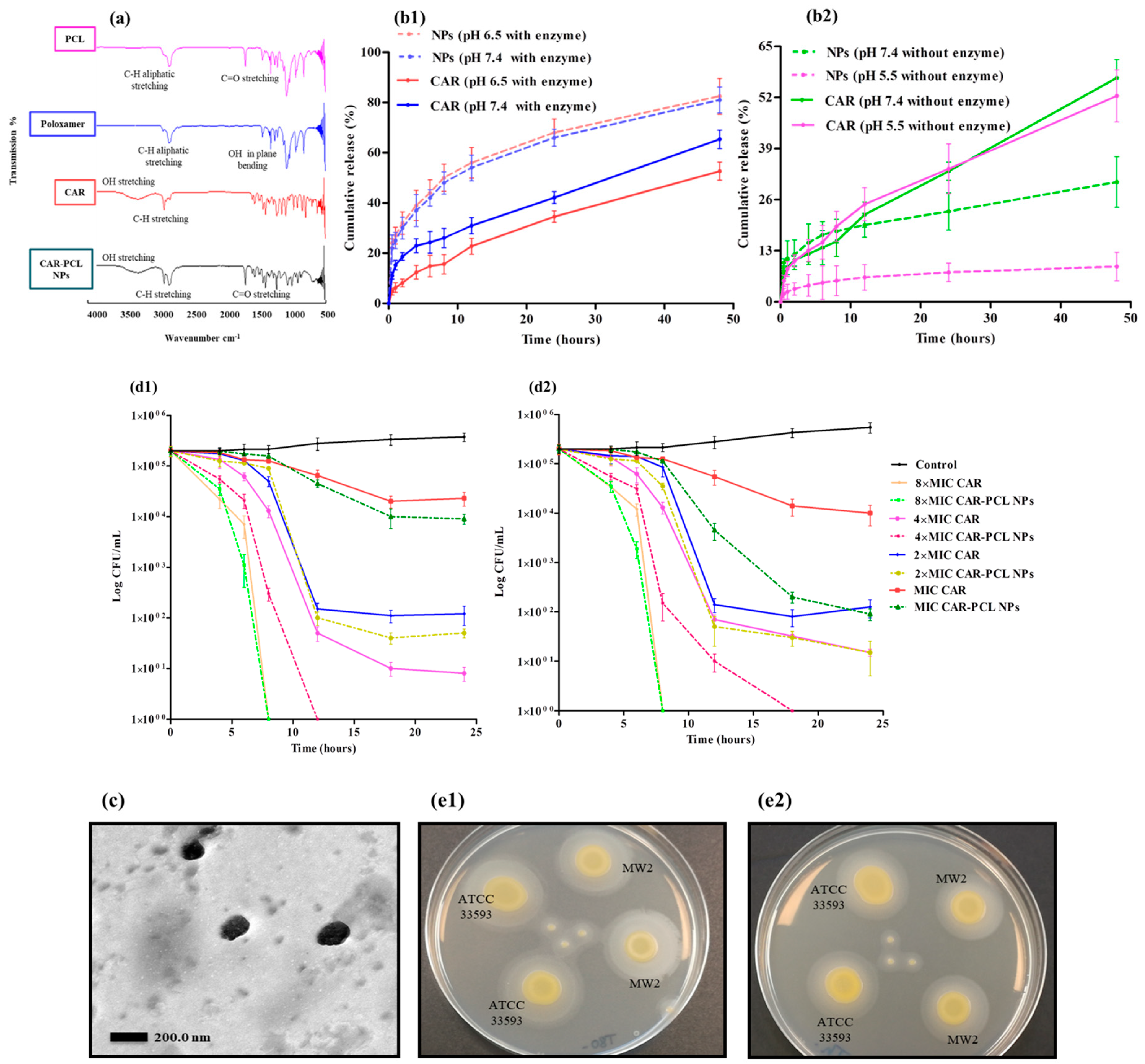
3.4. Fourier Transform-Infrared (FTIR) Spectroscopy
3.5. In-Vitro Release Kinetic Study of CAR-PCL NPs
3.6. In-Vitro Antibacterial Assays
3.6.1. Evaluation of Lipase Producing Activity
3.6.2. Minimum Inhibitory Concentration and Minimum Bactericidal Concentration
3.6.3. Killing Kinetics of CAR and CAR-PCL NPs
3.7. Characterization of CAR-PCL NPs Loaded Hydrogel
3.7.1. Physical Appearance, pH, Drug Content and Spreadability of Hydrogel
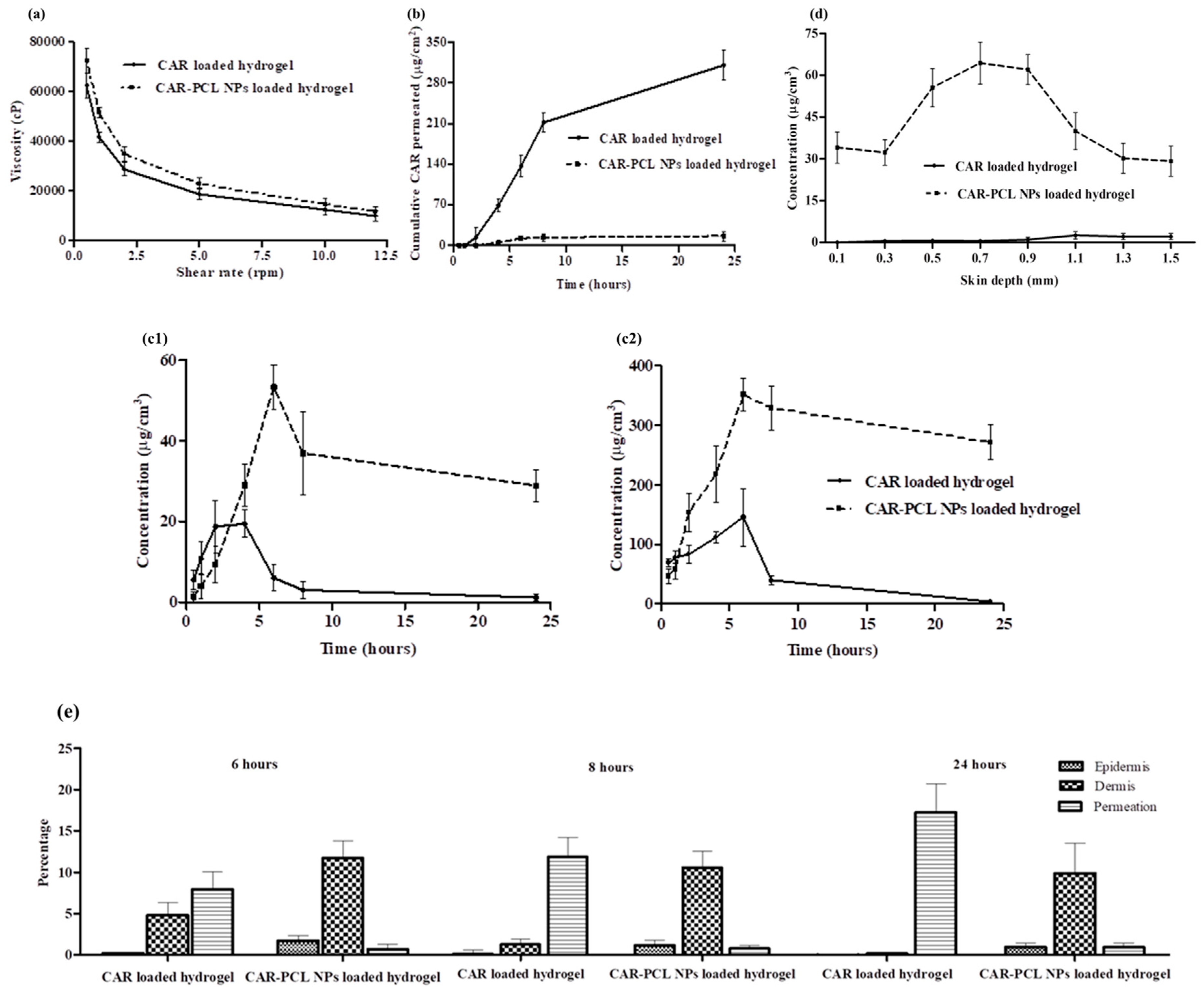
3.7.2. Rheological Evaluation
3.7.3. Extrudability and Bioadhesion Time of Hydrogel
3.8. Storage Stability of Hydrogel
3.9. Ex-Vivo Dermatokinetic, Skin Deposition, and Distribution Studies
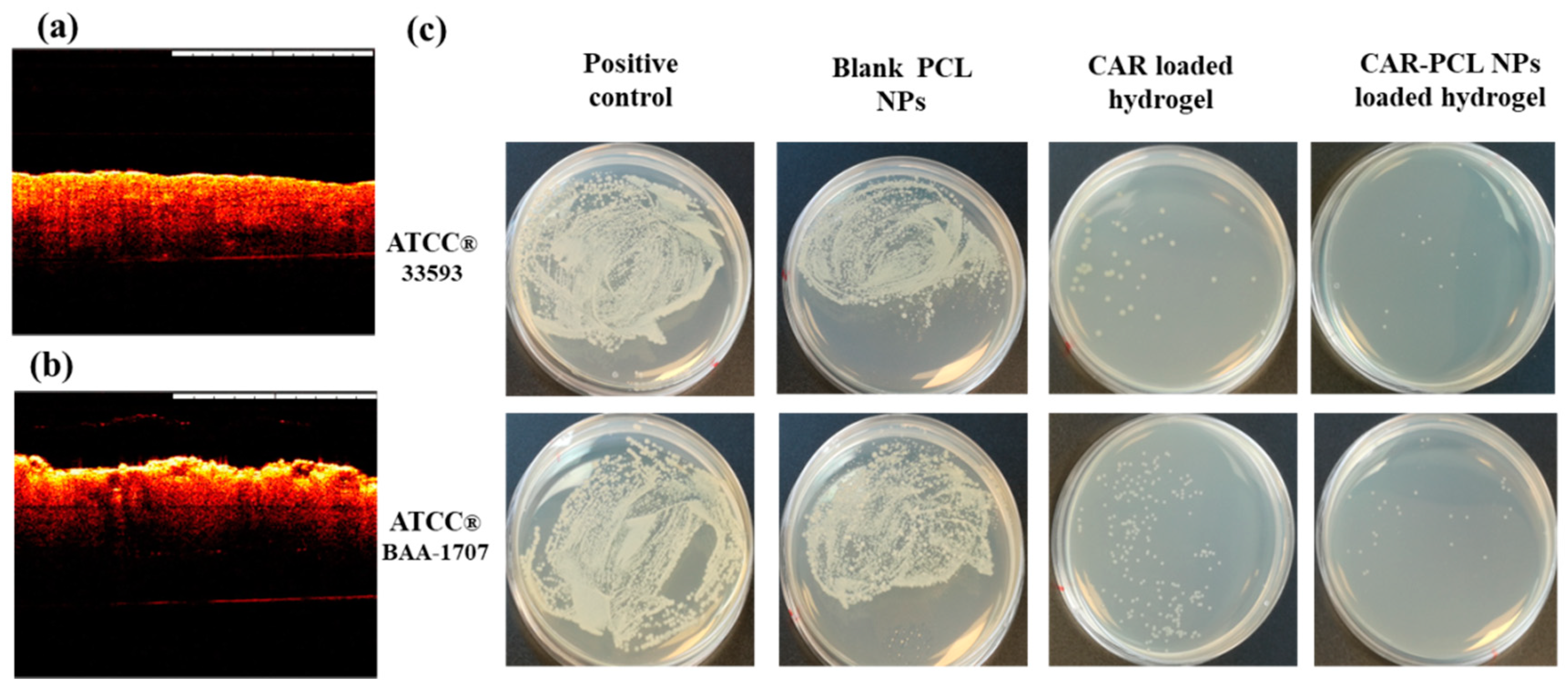
3.10. Determination of Antibacterial Effectiveness in an Ex-Vivo Pig Skin Wound Model
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chokshi, A.; Sifri, Z.; Cennimo, D.; Horng, H. Global contributors to antibiotic resistance. J. Glob. Infect. Dis. 2019, 11, 36–42. [Google Scholar]
- Boswihi, S.S.; Udo, E.E. Methicillin-resistant Staphylococcus aureus: An update on the epidemiology, treatment options and infection control. Curr. Med. Res. Prac. 2018, 8, 18–24. [Google Scholar] [CrossRef]
- Poovelikunnel, T.; Gethin, G.; Humphreys, H. Mupirocin resistance: Clinical implications and potential alternatives for the eradication of MRSA. J. Antimicrob. Chemother. 2015, 70, 2681–2692. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.Y.; Davis, J.S.; Eichenberger, E.; Holland, T.L.; Fowler, V.G. Staphylococcus aureus infections: Epidemiology, pathophysiology, clinical manifestations, and management. Clin. Microbiol. Rev. 2015, 28, 603–661. [Google Scholar] [CrossRef] [PubMed]
- Moran, G.J.; Abrahamian, F.M.; LoVecchio, F.; Talan, D.A. Acute bacterial skin infections: Developments since the 2005 Infectious Diseases Society of America (IDSA) guidelines. J. Emer. Med. 2013, 44, e397–e412. [Google Scholar] [CrossRef] [PubMed]
- Myhrman, E.; Håkansson, J.; Lindgren, K.; Björn, C.; Sjöstrand, V.; Mahlapuu, M. The novel antimicrobial peptide PXL150 in the local treatment of skin and soft tissue infections. Appl. Microbiol. Biotechnol. 2013, 97, 3085–3096. [Google Scholar] [CrossRef] [PubMed]
- Kaur, D.C.; Chate, S.S. Study of antibiotic resistance pattern in methicillin resistant Staphylococcus aureus with special reference to newer antibiotic. J. Glob. Infect. Dis. 2015, 7, 78. [Google Scholar] [CrossRef] [PubMed]
- Traczewski, M.M.; Katz, B.D.; Steenbergen, J.N.; Brown, S.D. Inhibitory and bactericidal activities of daptomycin, vancomycin, and teicoplanin against methicillin-resistant Staphylococcus aureus isolates collected from 1985 to 2007. Antimicrob. Agents Chemother. 2009, 53, 1735–1738. [Google Scholar] [CrossRef]
- Krause, K.M.; Renelli, M.; Difuntorum, S.; Wu, T.X.; Debabov, D.V.; Benton, B.M. In-vitro activity of telavancin against resistant gram-positive bacteria. Antimicrob. Agents Chemother. 2008, 52, 2647–2652. [Google Scholar] [CrossRef]
- Gilmer, D.B.; Schmitz, J.E.; Euler, C.W.; Fischetti, V.A. Novel bacteriophage lysin with broad lytic activity protects against mixed infection by Streptococcus pyogenes and methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2013, 57, 2743–2750. [Google Scholar] [CrossRef]
- Prabuseenivasan, S.; Jayakumar, M.; Ignacimuthu, S. In-vitro antibacterial activity of some plant essential oils. BMC Complement. Altern. Med. 2006, 6, 39. [Google Scholar] [CrossRef] [PubMed]
- La Storia, A.; Ercolini, D.; Marinello, F.; Di Pasqua, R.; Villani, F.; Mauriello, G. Atomic force microscopy analysis shows surface structure changes in carvacrol-treated bacterial cells. Res. Microbiol. 2011, 162, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Nostro, A.; Papalia, T. Antimicrobial activity of carvacrol: Current progress and future prospectives. Recent Pat. Anti. Infect. Drug Discov. 2012, 7, 28–35. [Google Scholar] [CrossRef]
- Cristani, M.; D’Arrigo, M.; Mandalari, G.; Castelli, F.; Sarpietro, M.G.; Micieli, D.; Venuti, V.; Bisignano, G.; Saija, A.; Trombetta, D. Interaction of four monoterpenes contained in essential oils with model membranes: Implications for their antibacterial activity. J. Agric. Food Chem. 2007, 55, 6300–6308. [Google Scholar] [CrossRef] [PubMed]
- Marchese, A.; Arciola, C.R.; Coppo, E.; Barbieri, R.; Barreca, D.; Chebaibi, S.; Sobarzo-Sánchez, E.; Nabavi, S.F.; Nabavi, S.M.; Daglia, M. The natural plant compound carvacrol as an antimicrobial and anti-biofilm agent: Mechanisms, synergies and bio-inspired anti-infective materials. Biofouling 2018, 34, 630–656. [Google Scholar] [CrossRef]
- Mouwakeh, A.; Kincses, A.; Nové, M.; Mosolygó, T.; Mohácsi-Farkas, C.; Kiskó, G.; Spengler, G. Nigella sativa essential oil and its bioactive compounds as resistance modifiers against Staphylococcus aureus. Phytother. Res. 2019, 33, 1010–1018. [Google Scholar] [CrossRef]
- Nostro, A.; Scaffaro, R.; Botta, L.; Filocamo, A.; Marino, A.; Bisignano, G. Effect of temperature on the release of carvacrol and cinnamaldehyde incorporated into polymeric systems to control growth and biofilms of Escherichia coli and Staphylococcus aureus. Biofouling 2015, 31, 639–649. [Google Scholar] [CrossRef]
- Scaffaro, R.; Lopresti, F.; D’Arrigo, M.; Marino, A.; Nostro, A. Efficacy of poly (lactic acid)/carvacrol electrospun membranes against Staphylococcus aureus and Candida albicans in single and mixed cultures. Appl. Microbiol. Biotechnol. 2018, 102, 4171–4181. [Google Scholar] [CrossRef]
- Mir, M.; Ahmed, N.; Ur Rehman, A. Recent applications of PLGA based nanostructures in drug delivery. Colloids Surf. B Biointerfaces 2017, 159, 217–231. [Google Scholar] [CrossRef]
- Zhang, Z.-T.; Huang-Fu, M.-Y.; Xu, W.-H.; Han, M. Stimulus-responsive nanoscale delivery systems triggered by the enzymes in the tumor microenvironment. Eur. J. Pharm. Biopharm. 2019, 137, 122–130. [Google Scholar] [CrossRef]
- Chen, H.; Jin, Y.; Wang, J.; Wang, Y.; Jiang, W.; Dai, H.; Pang, S.; Lei, L.; Ji, J.; Wang, B. Design of smart targeted and responsive drug delivery systems with enhanced antibacterial properties. Nanoscale 2018, 10, 20946–20962. [Google Scholar] [CrossRef] [PubMed]
- Xiong, M.-H.; Bao, Y.; Yang, X.-Z.; Wang, Y.-C.; Sun, B.; Wang, J. Lipase-sensitive polymeric triple-layered nanogel for “on-demand” drug delivery. J. Am. Chem. Soc. 2012, 134, 4355–4362. [Google Scholar] [CrossRef] [PubMed]
- Caló, E.; Khutoryanskiy, V.V. Biomedical applications of hydrogels: A review of patents and commercial products. Eur. Polym. J. 2015, 65, 252–267. [Google Scholar] [CrossRef]
- Grip, J.; Engstad, R.E.; Skjævel, I.; Škalko-Basnet, N.; Holsæter, A.M. Sprayable Carbopol hydrogel with soluble beta-1, 3/1, 6-glucan as an active ingredient for wound healing–development and in-vivo evaluation. Eur. J. Pharm. Sci. 2017, 107, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Galindo-Rodríguez, S.A.; Puel, F.; Briançon, S.; Allémann, E.; Doelker, E.; Fessi, H. Comparative scale-up of three methods for producing ibuprofen-loaded nanoparticles. Eur. J. Pharm. Sci. 2005, 25, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Zhang, Y.; Hu, Q.; Zeng, D.; Hua, F.; Meng, W.; Wang, W.; Bao, G.-H. Optimization of paeonol-loaded poly (butyl-2-cyanoacrylate) nanocapsules by central composite design with response surface methodology together with the antibacterial properties. Eur. J. Pharm. Sci. 2017, 101, 189–199. [Google Scholar] [CrossRef]
- Kenechukwu, F.C.; Attama, A.A.; Ibezim, E.C.; Nnamani, P.O.; Umeyor, C.E.; Uronnachi, E.M.; Gugu, T.H.; Momoh, M.A.; Ofokansi, K.C.; Akpa, P.A. Surface-modified mucoadhesive microgels as a controlled release system for miconazole nitrate to improve localized treatment of vulvovaginal candidiasis. Eur. J. Pharm. Sci. 2018, 111, 358–375. [Google Scholar] [CrossRef]
- Abdel Messih, H.A.; Ishak, R.A.; Geneidi, A.S.; Mansour, S. Nanoethosomes for transdermal delivery of tropisetron HCl: Multi-factorial predictive modeling, characterization, and ex-vivo skin permeation. Drug Dev. Ind. Pharm. 2017, 43, 958–971. [Google Scholar] [CrossRef]
- Seong, J.S.; Yun, M.E.; Park, S.N. Surfactant-stable and pH-sensitive liposomes coated with N-succinyl-chitosan and chitooligosaccharide for delivery of quercetin. Carbohydr. Polym. 2018, 181, 659–667. [Google Scholar] [CrossRef]
- Radaelli, M.; Silva, B.P.d.; Weidlich, L.; Hoehne, L.; Flach, A.; Costa, L.A.M.A.d.; Ethur, E.M. Antimicrobial activities of six essential oils commonly used as condiments in Brazil against Clostridium perfringens. Braz. J. Microbiol. 2016, 47, 424–430. [Google Scholar] [CrossRef]
- Khan, I.; Bahuguna, A.; Kumar, P.; Bajpai, V.K.; Kang, S.C. Antimicrobial potential of carvacrol against uropathogenic Escherichia coli via membrane disruption, depolarization, and reactive oxygen species generation. Front. Microbiol. 2017, 8, 2421. [Google Scholar] [CrossRef] [PubMed]
- Helal, D.A.; El-Rhman, D.A.; Abdel-Halim, S.A.; El-Nabarawi, M.A. Formulation and evaluation of fluconazole topical gel. Int. J. Pharm. Pharm. Sci. 2012, 4, 176–183. [Google Scholar]
- Zeb, A.; Qureshi, O.S.; Yu, C.-H.; Akram, M.; Kim, H.-S.; Kim, M.-S.; Kang, J.-H.; Majid, A.; Chang, S.-Y.; Bae, O.-N. Enhanced anti-rheumatic activity of methotrexate-entrapped ultradeformable liposomal gel in adjuvant-induced arthritis rat model. Int. J. Pharm. 2017, 525, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Gade, R.; Rao, C.B.; Ayanampudi, A.; Vegendla, M.R.; Bhai, V.A.; Nama, S. Formulation and evaluation of mephenesin topical gel. World J. Pharm. Pharm. Sci. 2013, 2, 1475–1489. [Google Scholar]
- Naz, K.; Shahnaz, G.; Ahmed, N.; Qureshi, N.A.; Sarwar, H.S.; Imran, M.; Khan, G.M. Formulation and in-vitro characterization of thiolated buccoadhesive film of fluconazole. AAPS PharmSciTech 2017, 18, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Permana, A.D.; McCrudden, M.T.; Donnelly, R.F. Enhanced Intradermal Delivery of Nanosuspensions of Antifilariasis Drugs Using Dissolving Microneedles: A Proof of Concept Study. Pharmaceutics 2019, 11, 346. [Google Scholar] [CrossRef] [Green Version]
- Mir, M.; Ishtiaq, S.; Rabia, S.; Khatoon, M.; Zeb, A.; Khan, G.M.; Ur Rehman, A.; Ud Din, F. Nanotechnology: From in-vivo imaging system to controlled drug delivery. Nanoscale Res. Lett. 2017, 12, 500. [Google Scholar] [CrossRef]
- Verma, D.D.; Verma, S.; Blume, G.; Fahr, A. Particle size of liposomes influences dermal delivery of substances into skin. Int. J. Pharm. 2003, 258, 141–151. [Google Scholar] [CrossRef]
- Ahmadi, M.; Vahabzadeh, F.; Bonakdarpour, B.; Mofarrah, E.; Mehranian, M. Application of the central composite design and response surface methodology to the advanced treatment of olive oil processing wastewater using Fenton’s peroxidation. J. Hazard. Mater. 2005, 123, 187–195. [Google Scholar] [CrossRef]
- Budhian, A.; Siegel, S.J.; Winey, K.I. Haloperidol-loaded PLGA nanoparticles: Systematic study of particle size and drug content. Int. J. Pharm. 2007, 336, 367–375. [Google Scholar] [CrossRef]
- Dubey, N.; Varshney, R.; Shukla, J.; Ganeshpurkar, A.; Hazari, P.P.; Bandopadhaya, G.P.; Mishra, A.K.; Trivedi, P. Synthesis and evaluation of biodegradable PCL/PEG nanoparticles for neuroendocrine tumor targeted delivery of somatostatin analog. Drug Dev. 2012, 19, 132–142. [Google Scholar] [CrossRef] [Green Version]
- Shalaby, K.S.; Soliman, M.E.; Casettari, L.; Bonacucina, G.; Cespi, M.; Palmieri, G.F.; Sammour, O.A.; El Shamy, A.A. Determination of factors controlling the particle size and entrapment efficiency of noscapine in PEG/PLA nanoparticles using artificial neural networks. Int. J. Nanomed. 2014, 9, 4953–4964. [Google Scholar]
- Keawchaoon, L.; Yoksan, R. Preparation, characterization and in-vitro release study of carvacrol-loaded chitosan nanoparticles. Colloids Surf. B Biointerfaces 2011, 84, 163–171. [Google Scholar] [CrossRef]
- Snehalatha, M.; Venugopal, K.; Saha, R.N. Etoposide-loaded PLGA and PCL nanoparticles I: Preparation and effect of formulation variables. Drug Deliv. 2008, 15, 267–275. [Google Scholar] [CrossRef]
- Kaasalainen, M.; Mäkilä, E.; Riikonen, J.; Kovalainen, M.; Järvinen, K.; Herzig, K.-H.; Lehto, V.-P.; Salonen, J. Effect of isotonic solutions and peptide adsorption on zeta potential of porous silicon nanoparticle drug delivery formulations. Int. J. Pharm. 2012, 431, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Kohli, A.; Alpar, H. Potential use of nanoparticles for transcutaneous vaccine delivery: Effect of particle size and charge. Int. J. Pharm. 2004, 275, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, D.B.; Amiji, M.M. Poly (ethylene oxide)-modified poly (ɛ-caprolactone) nanoparticles for targeted delivery of tamoxifen in breast cancer. Int. J. Pharm. 2005, 293, 261–270. [Google Scholar] [CrossRef]
- Zheng, D.; Li, X.; Xu, H.; Lu, X.; Hu, Y.; Fan, W. Study on docetaxel-loaded nanoparticles with high antitumor efficacy against malignant melanoma. Acta Biochim. Biophys. Sin. 2009, 41, 578–587. [Google Scholar] [CrossRef] [Green Version]
- Kalita, S.; Devi, B.; Kandimalla, R.; Sharma, K.K.; Sharma, A.; Kalita, K.; Kataki, A.C.; Kotoky, J. Chloramphenicol encapsulated in poly-ε-caprolactone–pluronic composite: Nanoparticles for treatment of MRSA-infected burn wounds. Int. J. Nanomed. 2015, 10, 2971–2984. [Google Scholar]
- Bruin, S.; Jongen, T.R. Food process engineering: The last 25 years and challenges ahead. Compr. Rev. Food Sci. Food Saf. 2003, 2, 42–81. [Google Scholar] [CrossRef]
- Chawla, J.S.; Amiji, M.M. Biodegradable poly (ε-caprolactone) nanoparticles for tumor-targeted delivery of tamoxifen. Int. J. Pharm. 2002, 249, 127–138. [Google Scholar] [CrossRef]
- Papadimitriou, S.; Bikiaris, D. Novel self-assembled core–shell nanoparticles based on crystalline amorphous moieties of aliphatic copolyesters for efficient controlled drug release. J. Control. Release 2009, 138, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Jim, T.; Gan, Z.; Zhao, Y.; Wang, S. A heterogeneous catalytic kinetics for enzymatic biodegradation of poly (ϵ-caprolactone) nanoparticles in aqueous solution. Polymer 2000, 41, 3593–3597. [Google Scholar] [CrossRef]
- Gul, R.; Ahmed, N.; Ullah, N.; Khan, M.I.; Elaissari, A. Biodegradable Ingredient-Based Emulgel Loaded with Ketoprofen Nanoparticles. AAPS PharmSciTech 2018, 19, 1869–1881. [Google Scholar] [CrossRef]
- Ramnath, L.; Sithole, B.; Govinden, R. Identification of lipolytic enzymes isolated from bacteria indigenous to Eucalyptus wood species for application in the pulping industry. Biotechnol. Rep. 2017, 15, 114–124. [Google Scholar] [CrossRef]
- Keepers, T.R.; Gomez, M.; Celeri, C.; Nichols, W.W.; Krause, K.M. Bactericidal activity, absence of serum effect, and time-kill kinetics of ceftazidime-avibactam against β-lactamase-producing Enterobacteriaceae and Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2014, 58, 5297–5305. [Google Scholar] [CrossRef] [Green Version]
- Kamimura, J.A.; Santos, E.H.; Hill, L.E.; Gomes, C.L. Antimicrobial and antioxidant activities of carvacrol microencapsulated in hydroxypropyl-beta-cyclodextrin. LWT Food Sci. Technol. 2014, 57, 701–709. [Google Scholar] [CrossRef]
- Joshi, M.; Patravale, V. Formulation and evaluation of nanostructured lipid carrier (NLC)–based gel of Valdecoxib. Drug Dev. Ind. Pharm. 2006, 32, 911–918. [Google Scholar] [CrossRef]
- Dar, M.J.; Din, F.U.; Khan, G.M. Sodium stibogluconate loaded nano-deformable liposomes for topical treatment of leishmaniasis: Macrophage as a target cell. Drug Deliv. 2018, 25, 1595–1606. [Google Scholar] [CrossRef]
- Aiyalu, R.; Govindarjan, A.; Ramasamy, A. Formulation and evaluation of topical herbal gel for the treatment of arthritis in animal model. Braz. J. Pharm. Sci. 2016, 52, 493–507. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Run | Independent Variables | Dependent Variables | |||
|---|---|---|---|---|---|
| X1 (mg/mL) | X2 (%) | X3 (mg/mL) | Y1 (nm ± SD) | Y2 (% ± SD) | |
| F1 | 5.00 | 1.00 | 3.00 | 206 ± 2.61 | 63 ± 0.71 |
| F2 | 5.00 | 1.50 | 3.00 | 192.5 ± 0.21 | 64 ± 2.13 |
| F3 | 5.00 | 1.00 | 5.00 | 196 ± 3.60 | 73 ± 1.94 |
| F4 | 9.00 | 1.50 | 1.00 | 229 ± 5.79 | 76 ± 3.21 |
| F5 | 1.00 | 1.50 | 5.00 | 168.8 ± 1.76 | 30.5 ± 5.47 |
| F6 | 5.00 | 1.00 | 1.50 | 199.1 ± 0.98 | 53 ± 2.91 |
| F7 | 5.00 | 0.30 | 3.00 | 187.6 ± 0.56 | 57 ± 1.98 |
| F8 | 9.00 | 0.50 | 3.00 | 233.05 ± 1.20 | 80 ± 3.16 |
| F9 | 9.00 | 1.50 | 5.00 | 230.0 ± 0.21 | 89 ± 1.82 |
| F10 | 1.00 | 0.50 | 1.00 | 163.7 ± 1.13 | 27 ± 4.37 |
| F11 | 5.00 | 0.50 | 3.00 | 196.7 ± 2.89 | 59 ± 2.63 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mir, M.; Ahmed, N.; Permana, A.D.; Rodgers, A.M.; Donnelly, R.F.; Rehman, A.u. Enhancement in Site-Specific Delivery of Carvacrol against Methicillin Resistant Staphylococcus aureus Induced Skin Infections Using Enzyme Responsive Nanoparticles: A Proof of Concept Study. Pharmaceutics 2019, 11, 606. https://doi.org/10.3390/pharmaceutics11110606
Mir M, Ahmed N, Permana AD, Rodgers AM, Donnelly RF, Rehman Au. Enhancement in Site-Specific Delivery of Carvacrol against Methicillin Resistant Staphylococcus aureus Induced Skin Infections Using Enzyme Responsive Nanoparticles: A Proof of Concept Study. Pharmaceutics. 2019; 11(11):606. https://doi.org/10.3390/pharmaceutics11110606
Chicago/Turabian StyleMir, Maria, Naveed Ahmed, Andi Dian Permana, Aoife Maria Rodgers, Ryan F. Donnelly, and Asim.ur. Rehman. 2019. "Enhancement in Site-Specific Delivery of Carvacrol against Methicillin Resistant Staphylococcus aureus Induced Skin Infections Using Enzyme Responsive Nanoparticles: A Proof of Concept Study" Pharmaceutics 11, no. 11: 606. https://doi.org/10.3390/pharmaceutics11110606