
NXT1, a Novel Influenza A NP Binding Protein, Promotes the Nuclear Export of NP via a CRM1-Dependent Pathway

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells, Viruses, and Transfections
2.2. Plasmid Construction
2.3. Pull-down and Immunoprecipitation Assays
2.4. Western Blot Analysis
2.5. NXT1 Knockdown
2.6. Cell Viability
2.7. Virus Replication
2.8. Immunofluorescence Staining
2.9. Fluorescence In Situ Hybridization (FISH)
3. Results
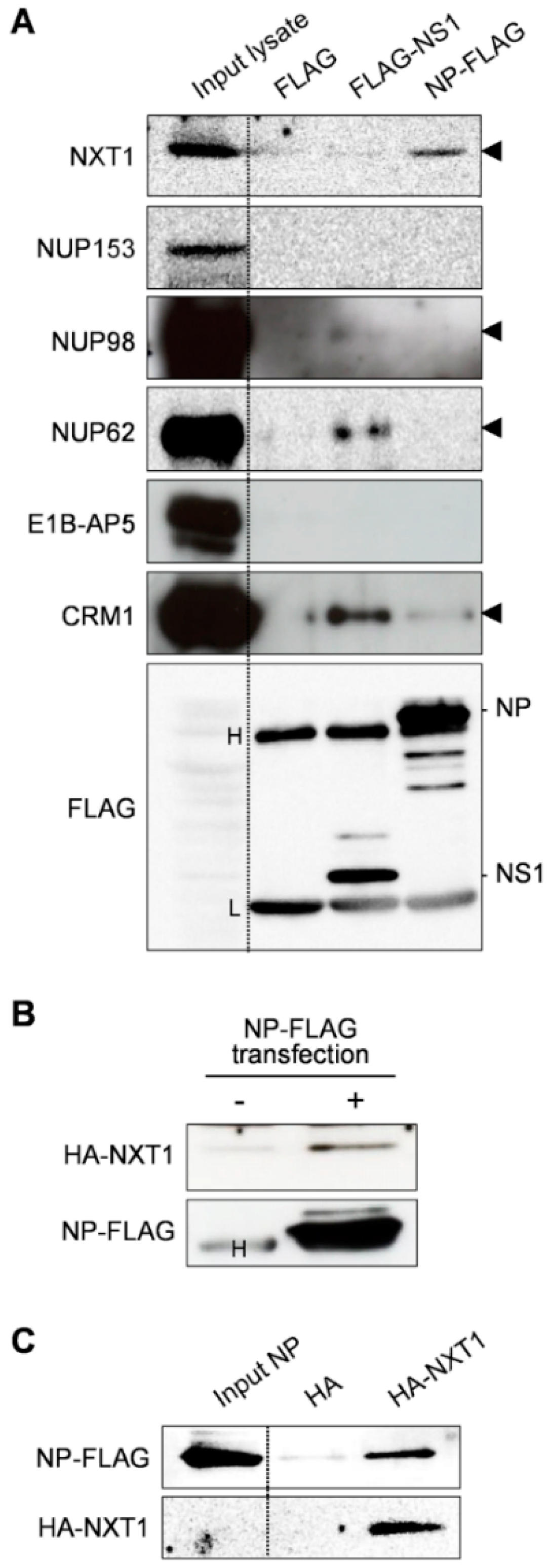
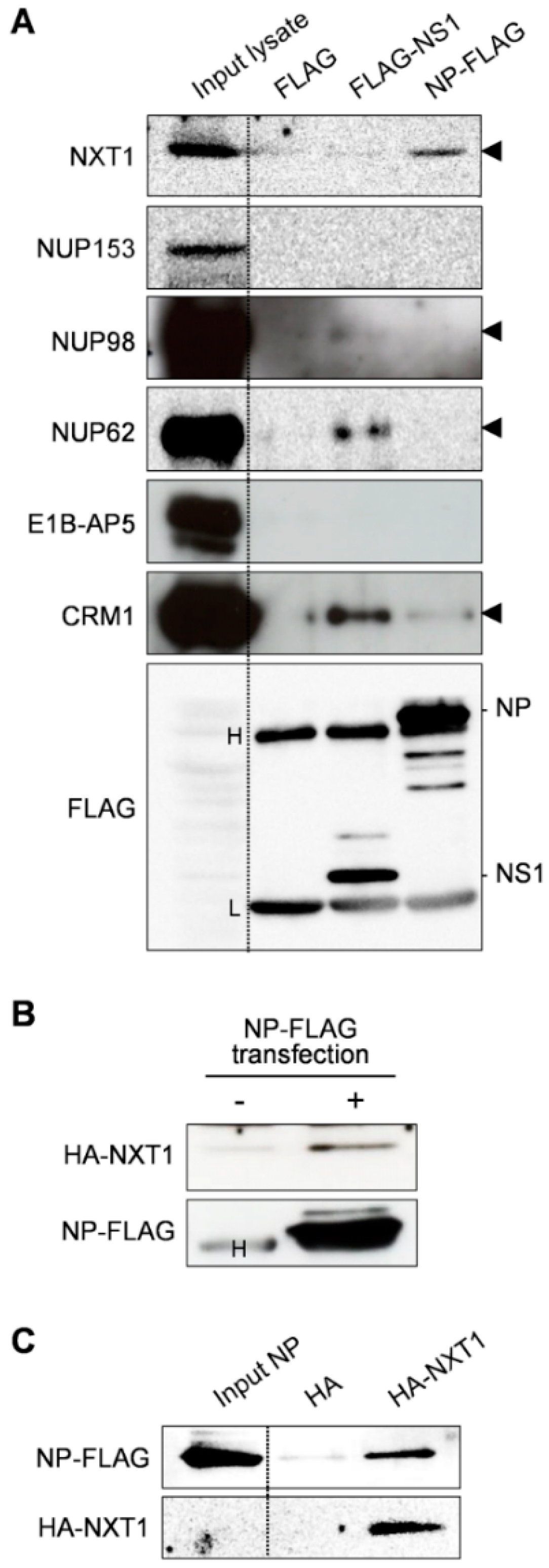
3.1. Identification of NXT1 as a Binding Partner of Influenza NP
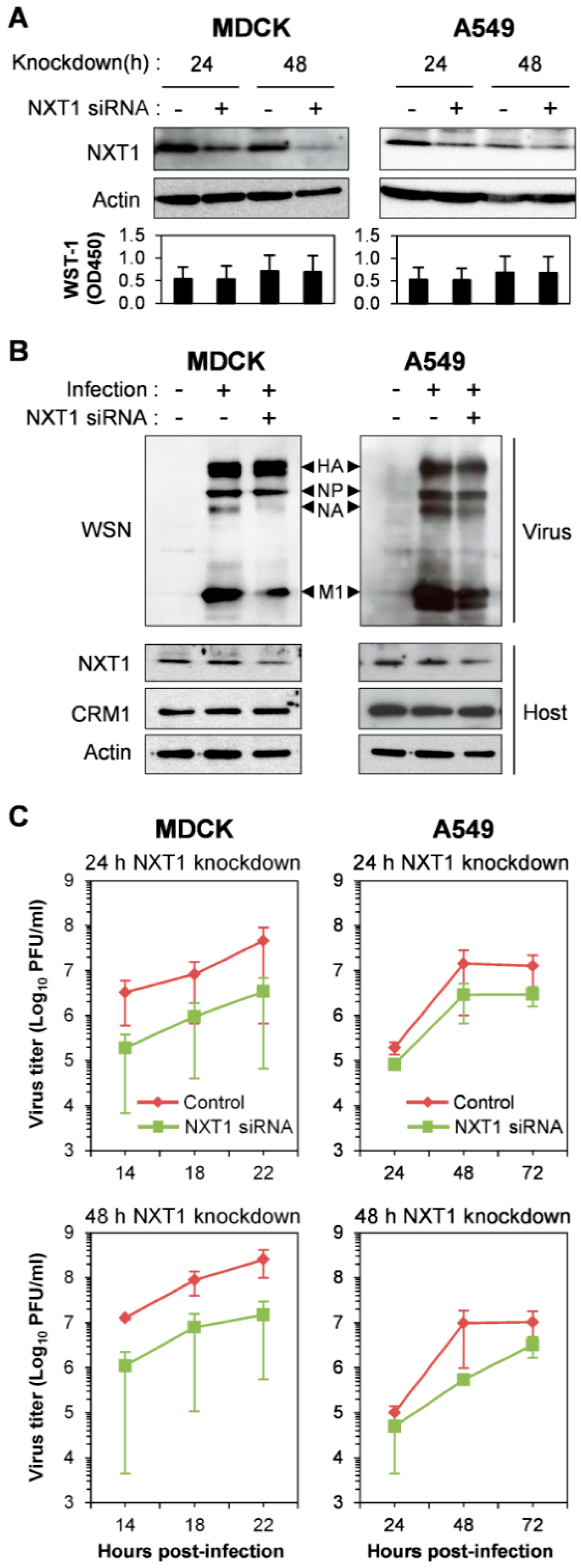
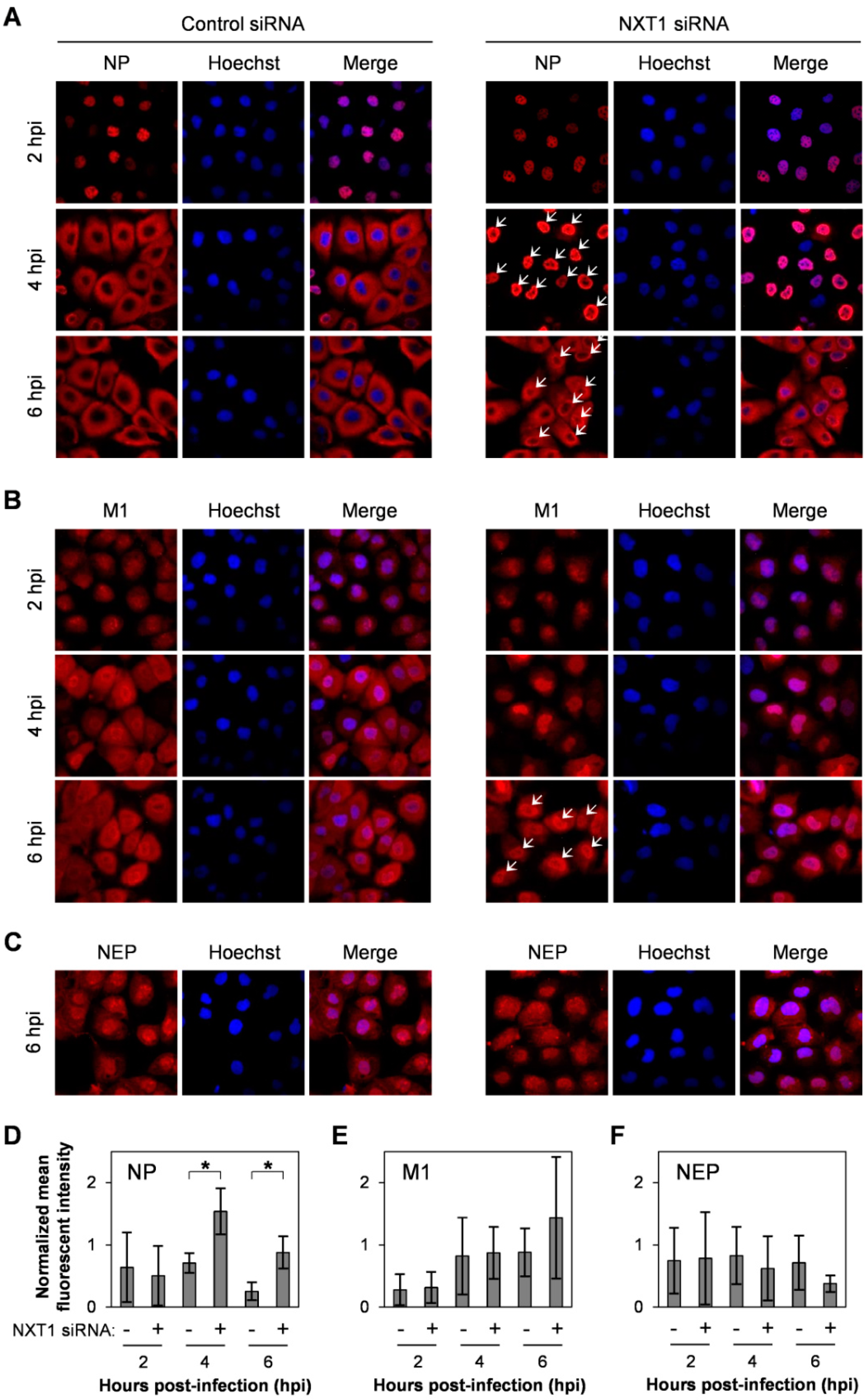
3.2. NXT1 Promotes Replication of Influenza A Virus
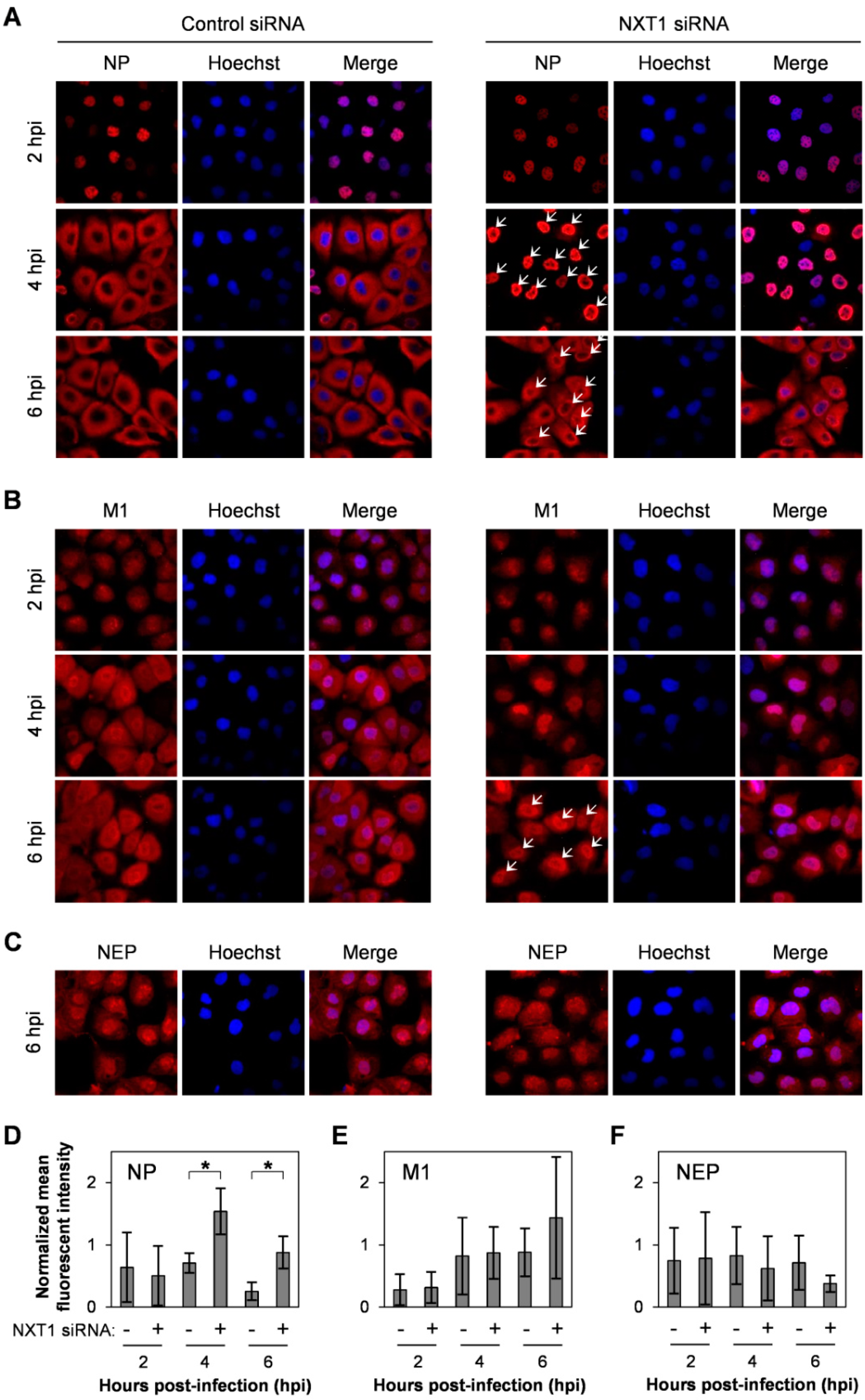
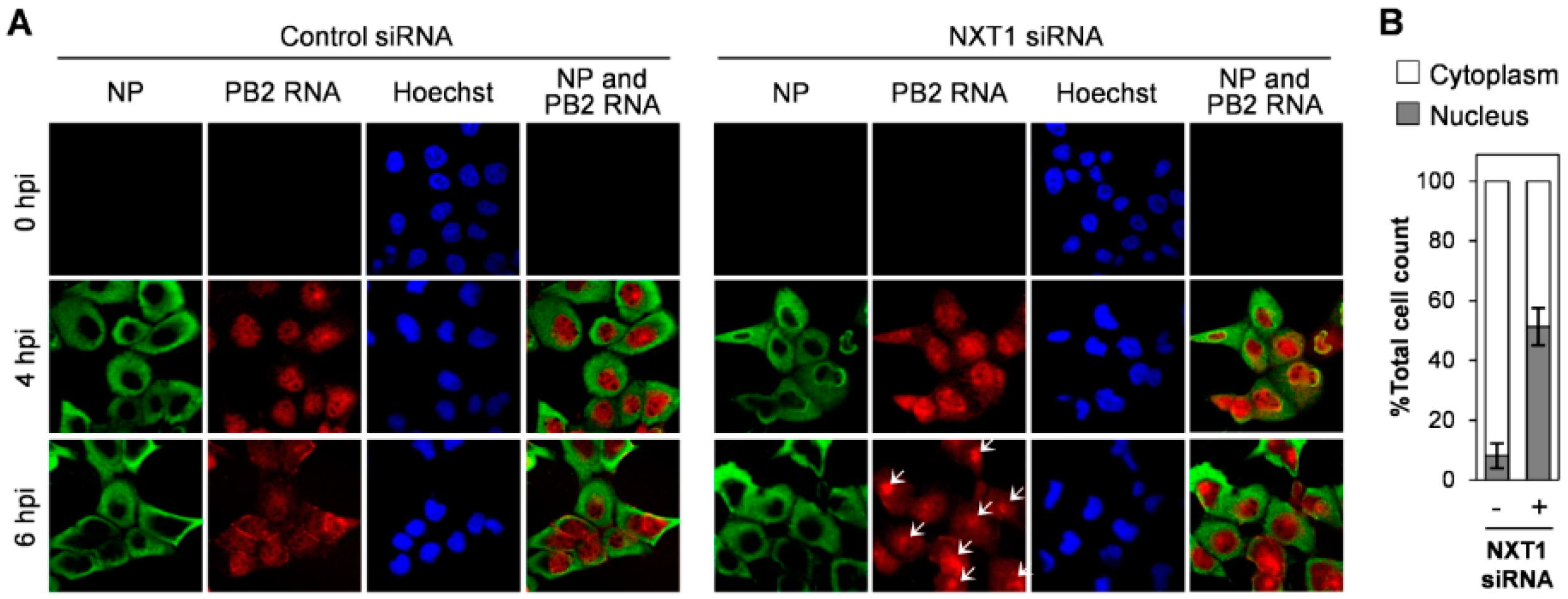
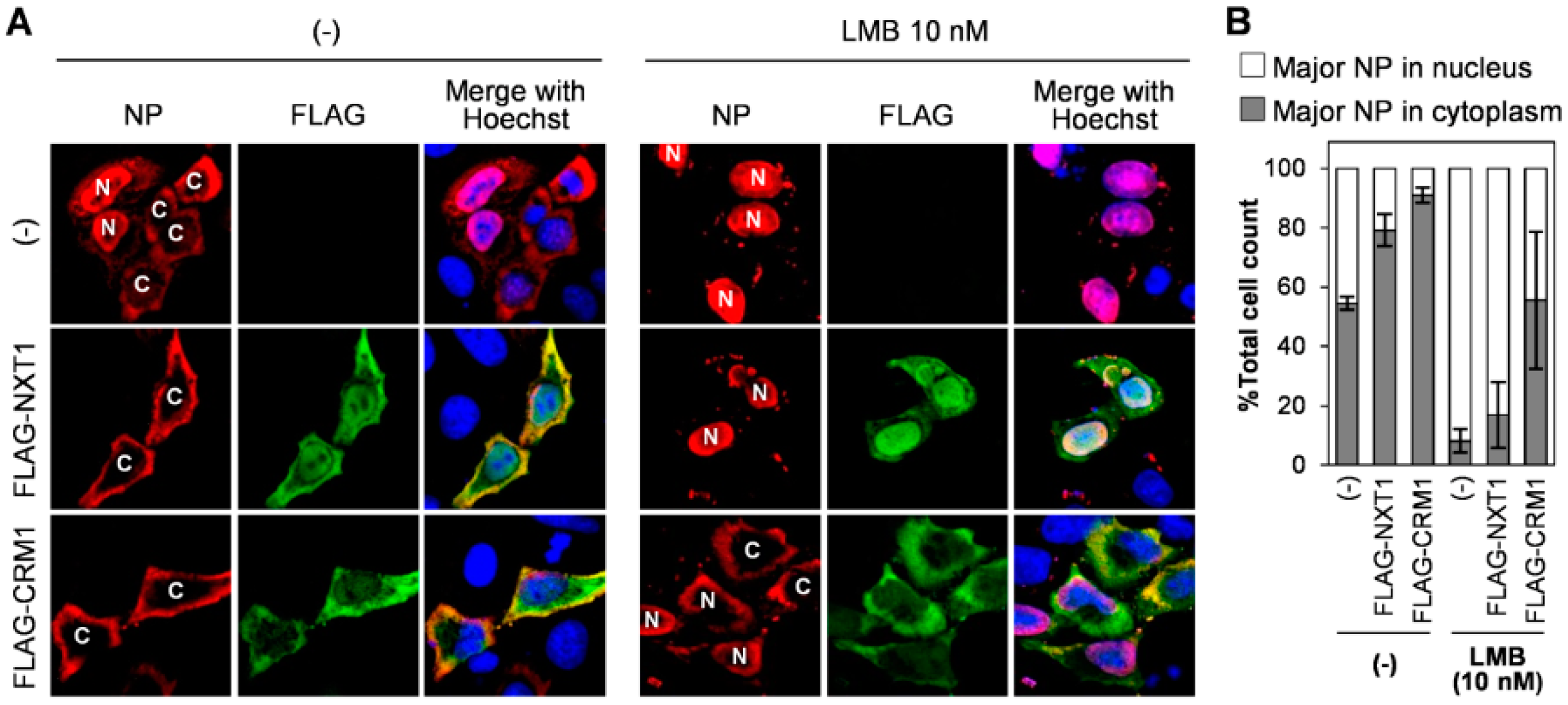
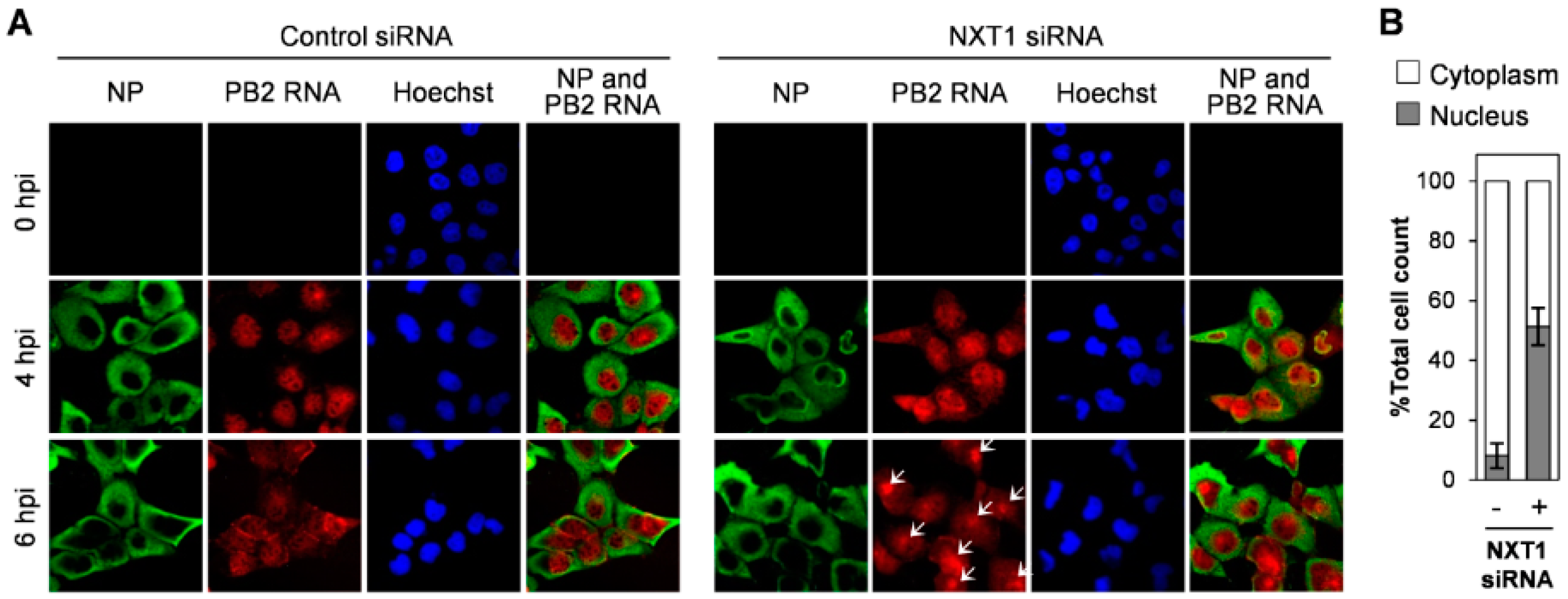
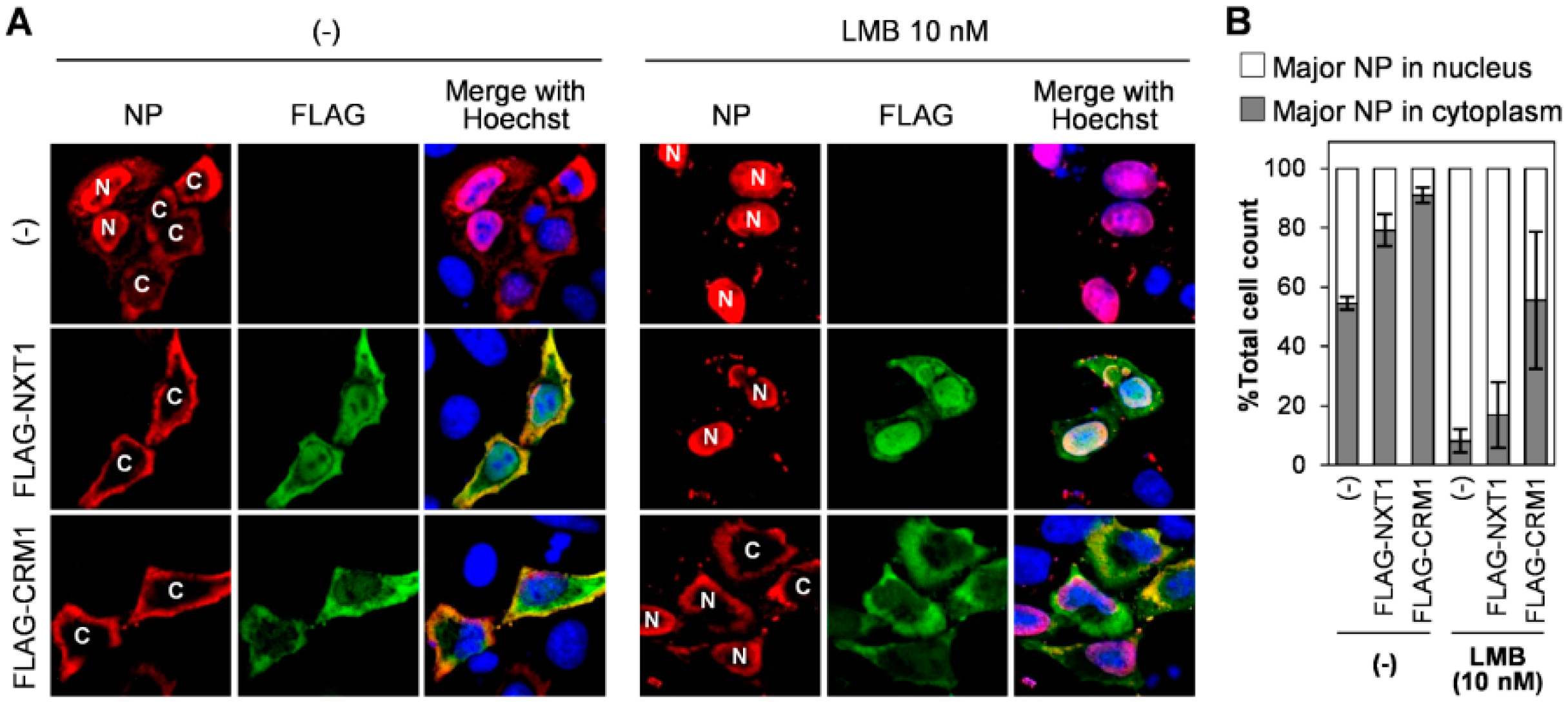
3.3. NXT1 Stimulates NP Nuclear Export Via a CRM1-Dependent Pathway
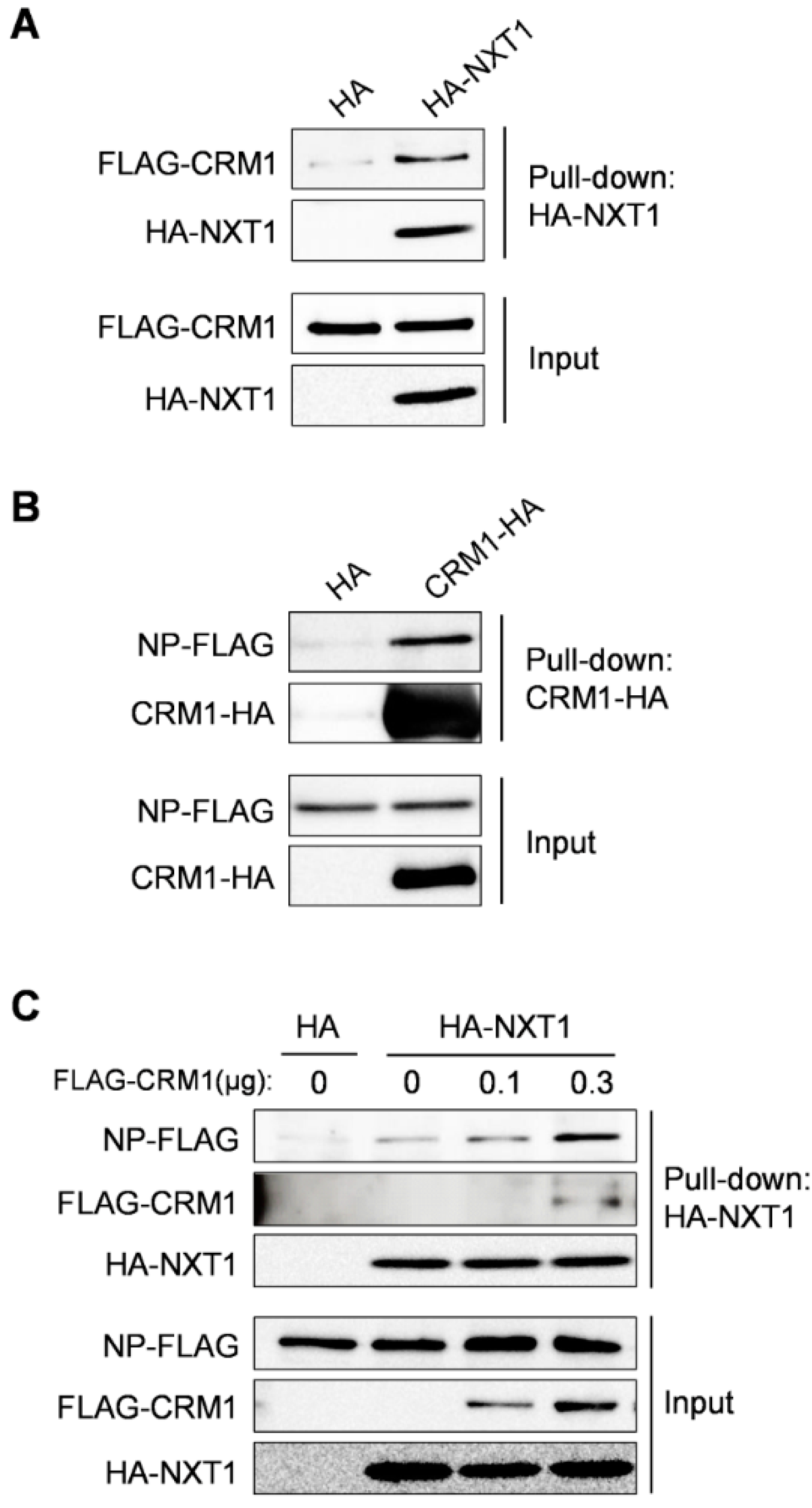
3.4. NXT1 Forms a Complex with NP and CRM1
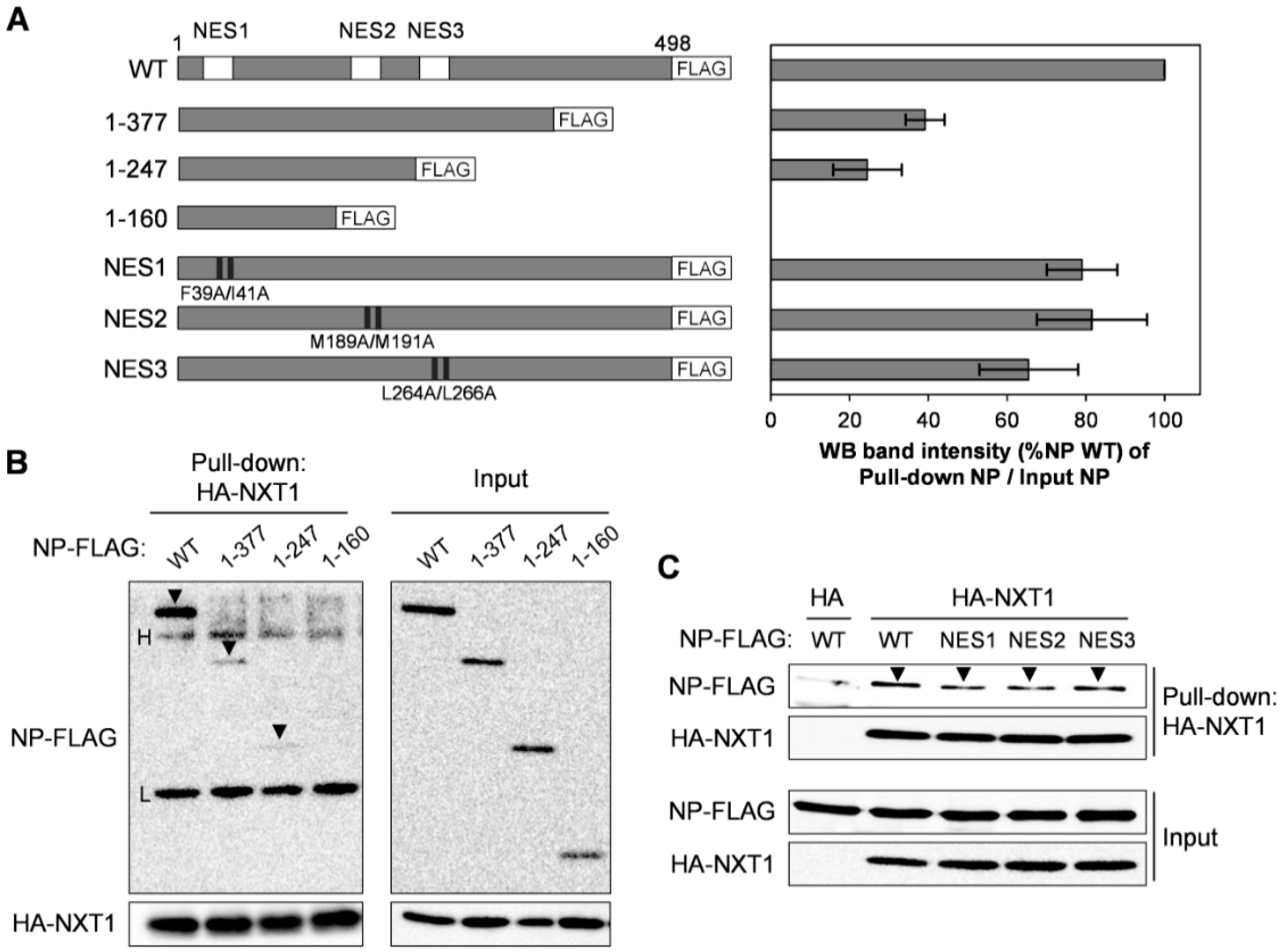
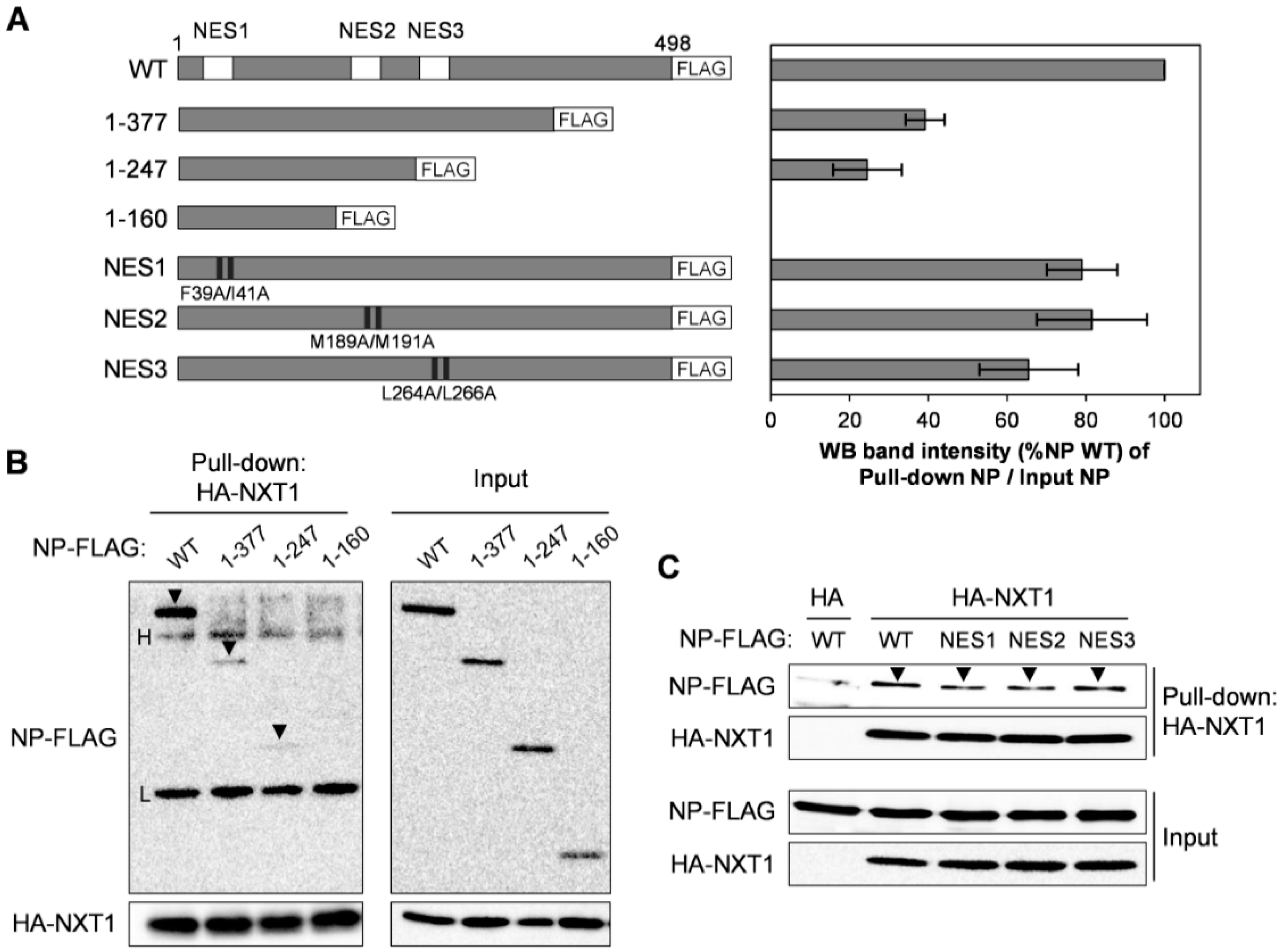
3.5. NXT1 Interacts with the C-terminal Region of NP
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Johnson, N.P.; Mueller, J. Updating the accounts: Global mortality of the 1918–1920 “Spanish” influenza pandemic. Bull. Hist. Med. 2002, 76, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Dawood, F.S.; Iuliano, A.D.; Reed, C.; Meltzer, M.I.; Shay, D.K.; Cheng, P.Y.; Bandaranayake, D.; Breiman, R.F.; Brooks, W.A.; Buchy, P.; et al. Estimated global mortality associated with the first 12 months of 2009 pandemic influenza a H1N1 virus circulation: A modelling study. Lancet Infect. Dis. 2012, 12, 687–695. [Google Scholar] [CrossRef]
- Gao, R.; Cao, B.; Hu, Y.; Feng, Z.; Wang, D.; Hu, W.; Chen, J.; Jie, Z.; Qiu, H.; Xu, K.; et al. Human infection with a novel avian-origin influenza A (H7N9) virus. N. Engl. J. Med. 2013, 368, 1888–1897. [Google Scholar] [CrossRef] [PubMed]
- Gubareva, L.V.; Fry, A.M. Current challenges in the risk assessment of neuraminidase inhibitor-resistant influenza viruses. J. Infect. Dis. 2010, 201, 656–658. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, S.; Sugaya, N.; Ito, M.; Yamazaki, M.; Ichikawa, M.; Kimura, K.; Kiso, M.; Shimizu, H.; Kawakami, C.; Koike, K.; et al. Emergence of influenza B viruses with reduced sensitivity to neuraminidase inhibitors. JAMA 2007, 297, 1435–1442. [Google Scholar] [CrossRef] [PubMed]
- Kiso, M.; Mitamura, K.; Sakai-Tagawa, Y.; Shiraishi, K.; Kawakami, C.; Kimura, K.; Hayden, F.G.; Sugaya, N.; Kawaoka, Y. Resistant influenza A viruses in children treated with oseltamivir: Descriptive study. Lancet 2004, 364, 759–765. [Google Scholar] [CrossRef]
- Stephenson, I.; Democratis, J.; Lackenby, A.; McNally, T.; Smith, J.; Pareek, M.; Ellis, J.; Bermingham, A.; Nicholson, K.; Zambon, M. Neuraminidase inhibitor resistance after oseltamivir treatment of acute influenza A and B in children. Clin. Infect. Dis. 2009, 48, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Aida, Y.; Sasaki, Y.; Hagiwara, K. Discovery of novel antiviral agents directed against the influenza A virus nucleoprotein. In Antiviral Drugs—Aspects of Clinical Use and Recent Advances; Arbuthnot, P., Ed.; InTech.: Rijeka, Croatia, 2012; pp. 99–120. [Google Scholar]
- Cianci, C.; Gerritz, S.W.; Deminie, C.; Krystal, M. Influenza nucleoprotein: Promising target for antiviral chemotherapy. Antivir. Chem. Chemother. 2013, 23, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Portela, A.; Digard, P. The influenza virus nucleoprotein: A multifunctional RNA-binding protein pivotal to virus replication. J. Gen. Virol. 2002, 83, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, K.; Kondoh, Y.; Ueda, A.; Yamada, K.; Goto, H.; Watanabe, T.; Nakata, T.; Osada, H.; Aida, Y. Discovery of novel antiviral agents directed against the influenza A virus nucleoprotein using photo-cross-linked chemical arrays. Biochem. Biophys. Res. Commun. 2010, 394, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Gerritz, S.W.; Cianci, C.; Kim, S.; Pearce, B.C.; Deminie, C.; Discotto, L.; McAuliffe, B.; Minassian, B.F.; Shi, S.; Zhu, S.; et al. Inhibition of influenza virus replication via small molecules that induce the formation of higher-order nucleoprotein oligomers. Proc. Natl. Acad. Sci. USA 2011, 108, 15366–15371. [Google Scholar] [CrossRef] [PubMed]
- Kao, R.Y.; Yang, D.; Lau, L.S.; Tsui, W.H.; Hu, L.; Dai, J.; Chan, M.P.; Chan, C.M.; Wang, P.; Zheng, B.J.; et al. Identification of influenza A nucleoprotein as an antiviral target. Nat. Biotechnol. 2010, 28, 600–605. [Google Scholar] [CrossRef] [PubMed]
- Chutiwitoonchai, N.; Kakisaka, M.; Yamada, K.; Aida, Y. Comparative analysis of seven viral nuclear export signals (NESs) reveals the crucial role of nuclear export mediated by the third NES consensus sequence of nucleoprotein (NP) in influenza A virus replication. PLoS ONE 2014, 9, e105081. [Google Scholar] [CrossRef] [PubMed]
- Kakisaka, M.; Mano, T.; Aida, Y. A high-throughput screening system targeting the nuclear export pathway via the third nuclear export signal of influenza A virus nucleoprotein. Virus Res. 2016, 217, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Kakisaka, M.; Sasaki, Y.; Yamada, K.; Kondoh, Y.; Hikono, H.; Osada, H.; Tomii, K.; Saito, T.; Aida, Y. A novel antiviral target structure involved in the RNA binding, dimerization, and nuclear export functions of the influenza A virus nucleoprotein. PLoS Pathog. 2015, 11, e1005062. [Google Scholar] [CrossRef] [PubMed]
- Akarsu, H.; Burmeister, W.P.; Petosa, C.; Petit, I.; Muller, C.W.; Ruigrok, R.W.; Baudin, F. Crystal structure of the M1 protein-binding domain of the influenza A virus nuclear export protein (NEP/NS2). EMBO J. 2003, 22, 4646–4655. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, R.E.; Talon, J.; Palese, P. The influenza virus NEP (NS2 protein) mediates the nuclear export of viral ribonucleoproteins. EMBO J. 1998, 17, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Brunotte, L.; Flies, J.; Bolte, H.; Reuther, P.; Vreede, F.; Schwemmle, M. The nuclear export protein of H5N1 influenza a viruses recruits matrix 1 (M1) protein to the viral ribonucleoprotein to mediate nuclear export. J. Biol. Chem. 2014, 289, 20067–20077. [Google Scholar] [CrossRef] [PubMed]
- Kudo, N.; Matsumori, N.; Taoka, H.; Fujiwara, D.; Schreiner, E.P.; Wolff, B.; Yoshida, M.; Horinouchi, S. Leptomycin B inactivates CRM1/exportin 1 by covalent modification at a cysteine residue in the central conserved region. Proc. Natl. Acad. Sci. USA 1999, 96, 9112–9117. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Takizawa, N.; Katoh, M.; Hoshida, K.; Kobayashi, N.; Nagata, K. Inhibition of nuclear export of ribonucleoprotein complexes of influenza virus by leptomycin B. Virus Res. 2001, 77, 31–42. [Google Scholar] [CrossRef]
- Perwitasari, O.; Johnson, S.; Yan, X.; Howerth, E.; Shacham, S.; Landesman, Y.; Baloglu, E.; McCauley, D.; Tamir, S.; Tompkins, S.M.; et al. Verdinexor, a novel selective inhibitor of nuclear export, reduces influenza A virus replication in vitro and in vivo. J. Virol. 2014, 88, 10228–10243. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Fuse, T.; Asano, I.; Tsukahara, F.; Maru, Y.; Nagata, K.; Kitazato, K.; Kobayashi, N. Identification of Hsc70 as an influenza virus matrix protein (M1) binding factor involved in the virus life cycle. FEBS Lett. 2006, 580, 5785–5790. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Shimizu, T.; Noda, S.; Tsukahara, F.; Maru, Y.; Kobayashi, N. Nuclear export of the influenza virus ribonucleoprotein complex: Interaction of Hsc70 with viral proteins M1 and NS2. FEBS Open Bio 2014, 4, 683–688. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Liu, X.; Zhang, A.; Zhou, H.; Liu, Z.; Chen, H.; Jin, M. CHD3 facilitates vRNP nuclear export by interacting with NES1 of influenza A virus NS2. Cell. Mol. Life Sci. 2015, 72, 971–982. [Google Scholar] [CrossRef] [PubMed]
- Morita, M.; Kuba, K.; Ichikawa, A.; Nakayama, M.; Katahira, J.; Iwamoto, R.; Watanebe, T.; Sakabe, S.; Daidoji, T.; Nakamura, S.; et al. The lipid mediator protectin D1 inhibits influenza virus replication and improves severe influenza. Cell 2013, 153, 112–125. [Google Scholar] [CrossRef] [PubMed]
- Munier, S.; Rolland, T.; Diot, C.; Jacob, Y.; Naffakh, N. Exploration of binary virus-host interactions using an infectious protein complementation assay. Mol. Cell. Proteom. 2013, 12, 2845–2855. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Huang, S.; Chen, Z. Human cellular protein nucleoporin hNup98 interacts with influenza A virus NS2/nuclear export protein and overexpression of its GLFG repeat domain can inhibit virus propagation. J. Gen. Virol. 2010, 91, 2474–2484. [Google Scholar] [CrossRef] [PubMed]
- Satterly, N.; Tsai, P.L.; van Deursen, J.; Nussenzveig, D.R.; Wang, Y.; Faria, P.A.; Levay, A.; Levy, D.E.; Fontoura, B.M. Influenza virus targets the mRNA export machinery and the nuclear pore complex. Proc. Natl. Acad. Sci. USA 2007, 104, 1853–1858. [Google Scholar] [CrossRef] [PubMed]
- Neumann, G.; Watanabe, T.; Ito, H.; Watanabe, S.; Goto, H.; Gao, P.; Hughes, M.; Perez, D.R.; Donis, R.; Hoffmann, E.; et al. Generation of influenza A viruses entirely from cloned cDNAs. Proc. Natl. Acad. Sci. USA 1999, 96, 9345–9350. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, K.; Murakami, T.; Xue, G.; Shimizu, Y.; Takeda, E.; Hashimoto, Y.; Honda, K.; Kondoh, Y.; Osada, H.; Tsunetsugu-Yokota, Y.; et al. Identification of a novel Vpr-binding compound that inhibits HIV-1 multiplication in macrophages by chemical array. Biochem. Biophys. Res. Commun. 2010, 403, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Chou, Y.Y.; Heaton, N.S.; Gao, Q.; Palese, P.; Singer, R.H.; Lionnet, T. Colocalization of different influenza viral RNA segments in the cytoplasm before viral budding as shown by single-molecule sensitivity FISH analysis. PLoS Pathog. 2013, 9, e1003358. [Google Scholar] [CrossRef]
- Sasaki, Y.; Kakisaka, M.; Chutiwitoonchai, N.; Tajima, S.; Hikono, H.; Saito, T.; Aida, Y. Identification of a novel multiple kinase inhibitor with potent antiviral activity against influenza virus by reducing viral polymerase activity. Biochem. Biophys. Res. Commun. 2014, 450, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Hao, L.; Sakurai, A.; Watanabe, T.; Sorensen, E.; Nidom, C.A.; Newton, M.A.; Ahlquist, P.; Kawaoka, Y. Drosophila RNAi screen identifies host genes important for influenza virus replication. Nature 2008, 454, 890–893. [Google Scholar] [CrossRef] [PubMed]
- Braun, I.C.; Herold, A.; Rode, M.; Conti, E.; Izaurralde, E. Overexpression of TAP/p15 heterodimers bypasses nuclear retention and stimulates nuclear mRNA export. J. Biol. Chem. 2001, 276, 20536–20543. [Google Scholar] [CrossRef] [PubMed]
- Levesque, L.; Guzik, B.; Guan, T.; Coyle, J.; Black, B.E.; Rekosh, D.; Hammarskjold, M.L.; Paschal, B.M. RNA export mediated by TAP involves NXT1-dependent interactions with the nuclear pore complex. J. Biol. Chem. 2001, 276, 44953–44962. [Google Scholar] [CrossRef] [PubMed]
- Wiegand, H.L.; Coburn, G.A.; Zeng, Y.; Kang, Y.; Bogerd, H.P.; Cullen, B.R. Formation of TAP/NXT1 heterodimers activates TAP-dependent nuclear mRNA export by enhancing recruitment to nuclear pore complexes. Mol. Cell. Biol. 2002, 22, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Ossareh-Nazari, B.; Maison, C.; Black, B.E.; Levesque, L.; Paschal, B.M.; Dargemont, C. RanGTP-binding protein NXT1 facilitates nuclear export of different classes of RNA in vitro. Mol. Cell. Biol. 2000, 20, 4562–4571. [Google Scholar] [CrossRef] [PubMed]
- Black, B.E.; Holaska, J.M.; Levesque, L.; Ossareh-Nazari, B.; Gwizdek, C.; Dargemont, C.; Paschal, B.M. NXT1 is necessary for the terminal step of CRM1-mediated nuclear export. J. Cell Biol. 2001, 152, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Liu, X.; Cao, S.; Zhao, Z.; Zhang, K.; Xie, Q.; Chen, C.; Gao, S.; Bi, Y.; Sun, L.; et al. Identification and characterization of three novel nuclear export signals in the influenza A virus nucleoprotein. J. Virol. 2012, 86, 4970–4980. [Google Scholar] [CrossRef] [PubMed]
- Black, B.E.; Levesque, L.; Holaska, J.M.; Wood, T.C.; Paschal, B.M. Identification of an NTF2-related factor that binds Ran-GTP and regulates nuclear protein export. Mol. Cell. Biol. 1999, 19, 8616–8624. [Google Scholar] [CrossRef] [PubMed]
- Katahira, J.; Strasser, K.; Podtelejnikov, A.; Mann, M.; Jung, J.U.; Hurt, E. The Mex67p-mediated nuclear mRNA export pathway is conserved from yeast to human. EMBO J. 1999, 18, 2593–2609. [Google Scholar] [CrossRef] [PubMed]
- Guzik, B.W.; Levesque, L.; Prasad, S.; Bor, Y.C.; Black, B.E.; Paschal, B.M.; Rekosh, D.; Hammarskjold, M.L. NXT1 (p15) is a crucial cellular cofactor in TAP-dependent export of intron-containing RNA in mammalian cells. Mol. Cell. Biol. 2001, 21, 2545–2554. [Google Scholar] [CrossRef] [PubMed]
- Elton, D.; Simpson-Holley, M.; Archer, K.; Medcalf, L.; Hallam, R.; McCauley, J.; Digard, P. Interaction of the influenza virus nucleoprotein with the cellular CRM1-mediated nuclear export pathway. J. Virol. 2001, 75, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Read, E.K.; Digard, P. Individual influenza A virus mRNAs show differential dependence on cellular NXF1/TAP for their nuclear export. J. Gen. Virol. 2010, 91, 1290–1301. [Google Scholar] [CrossRef] [PubMed]


© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chutiwitoonchai, N.; Aida, Y. NXT1, a Novel Influenza A NP Binding Protein, Promotes the Nuclear Export of NP via a CRM1-Dependent Pathway. Viruses 2016, 8, 209. https://doi.org/10.3390/v8080209
Chutiwitoonchai N, Aida Y. NXT1, a Novel Influenza A NP Binding Protein, Promotes the Nuclear Export of NP via a CRM1-Dependent Pathway. Viruses. 2016; 8(8):209. https://doi.org/10.3390/v8080209
Chicago/Turabian StyleChutiwitoonchai, Nopporn, and Yoko Aida. 2016. "NXT1, a Novel Influenza A NP Binding Protein, Promotes the Nuclear Export of NP via a CRM1-Dependent Pathway" Viruses 8, no. 8: 209. https://doi.org/10.3390/v8080209