
Genetic Characterization of the Belgian Nephropathogenic Infectious Bronchitis Virus (NIBV) Reference Strain B1648

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Virus Propagation
2.2. Preparation of RNA for Illumina Sequencing
2.3. Illumina Sequencing and Sequence Assembly
2.4. Genome Sequence Analysis
2.5. Recombination Analysis
2.6. GenBank Accession Number
3. Results
3.1. Genome Organization of Strain B1648

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Open Reading Frame | Frame | Nucleotide Location | Nucleotide Length (bp) | Amino Acids Size |
|---|---|---|---|---|
| 5′ UTR | - | 1–518 | 518 | - |
| 1a | +3 | 519–12368 | 11,850 | 3949 |
| 1b | +2 | 12443–20401 | 7959 | 2652 |
| Spike | +3 | 20352–23852 | 3501 | 1166 |
| 3a | +2 | 23852–24025 | 174 | 57 |
| 3b | +1 | 24025–24219 | 195 | 64 |
| Envelope | +2 | 24200–24484 | 285 | 94 |
| Membrane | +3 | 24477–25154 | 678 | 225 |
| 4b | +3 | 25155–25439 | 285 | 94 |
| 4c | +1 | 25360–25530 | 171 | 56 |
| 5a | +2 | 25514–25711 | 198 | 65 |
| 5b | +1 | 25708–25956 | 249 | 82 |
| Nucleocapsid | +3 | 25899–27128 | 1230 | 409 |
| 6b | +2 | 27137–27361 | 225 | 74 |
| 3′ UTR | - | 27362–27654 | 292 | - |
| Strain | Genome | 5′ | 1a | 1b | Spike | 3a | 3b | E (3c) | M | 4b | 4c | 5a | 5b | N | 6b | 3′ |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Beaudette | 89.7 | 96.2 | 89.7/91.7 | 91.9/96.6 | 80.7/80.7 | 91.1/94.6 | 80.1/64.6 | 88.9/81.8 | 95.1/93 | 90.2/85.8 | 87.1/84.2 | 83.4/84.8 | 95.3/90.8 | 92.4/93.6 | 93.6/93.9 | 99.1 |
| California99 | 90.4 | 98.1 | 90.1/91.9 | 93/97.1 | 79.2/84.1 | 87.8/82.8 | 96.6/96.4 | 92.3/90.7 | 93.9/91.5 | 89.6/87.5 | 89.0/89.7 | 86.5/88.4 | 93.6/86.6 | 93.4/96 | 68.9/65.2 | 98.1 |
| Cal5572003 | 90.4 | 98.4 | 90.3/91.3 | 92.9/97.2 | 79.5/83 | 93.7/94.6 | 89.8/80.7 | 93.5/91.9 | 94.4/91.5 | 83.7/76.9 | 64.4/50.5 | 87.1/86.6 | 94.4/86.6 | 93.3/94.7 | 94.3/96 | 98.1 |
| Cal56b | 90.6 | 97.8 | 90.7/92.1 | 92.9/97 | 77.8/82.8 | 89.2/85.9 | 94.8/92.7 | 91.5/88.2 | 95.1/93 | 85.5/82.4 | 66.0/50.5 | 88.3/88.4 | 94.5/90.8 | 94.1/96.2 | 93/93.9 | 98.6 |
| SAIBK | 86.8 | 95.8 | 86.4/89.4 | 89.5/95.5 | 78/81.7 | 90.9/79.6 | 76.2/67.1 | 88.5/85.7 | 92/90.4 | 83/82.4 | 64.4/46.4 | 87.2/91.9 | 93.6/89.5 | 85.7/91.8 | NA | 97.6 |
| TW2575/98 | 86.0 | 95.5 | 84.8/87.9 | 88.9/95.7 | 79.2/81.7 | 94.8/97.3 | 87.3/80.7 | 90.5/87 | 92.7/90.4 | 83.7/80.6 | 85.1/81.3 | 86.3/88.4 | 92.6/85.2 | 88.4/92 | 68.3/65.2 | 97.1 |
| ArkDPI11 | 90.8 | 98.7 | 90.6/92.2 | 93/96.9 | 80.7/83.7 | 97.4/94.6 | 96.6/94.6 | 93/91.9 | 93.6/92 | 89.6/87.5 | 89.9/92.4 | 86.5/88.4 | 94.9/89.5 | 94.2/96.5 | 94.3/96 | 98.1 |
| H52 | 89.6 | 97.8 | 89.4/91.5 | 92.1/96.5 | 80.9/80.6 | 85.1/88.9 | 81.5/67.1 | 88.9/81.8 | 95.4/93 | 88.5/85.8 | 85.2/78.3 | 87.8/88.4 | 92.6/85.2 | 91.9/94.7 | 34.7/15.9 | 98.6 |
| H120 | 90.4 | 98.7 | 89.9/91.7 | 93.6/97.4 | 79/81.1 | 85.1/88.9 | 80.7/67.1 | 87.6/76.3 | 96.2/92.5 | 88.5/85.8 | 85.2/78.3 | 88.4/88.4 | 95.3/90.8 | 93.4/94.7 | 34.7/15.9 | 99.0 |
| Mass412006 | 90.8 | 98.7 | 91.1/92.6 | 92.5/97 | 83.1/82.3 | 87.8/82.8 | 96.6/96.4 | 93.5/93.1 | 93.7/92 | 89.6/87.5 | 89.9/92.4 | 86.5/88.4 | 94.4/88.1 | 94.4/96.7 | 93/93.9 | 98.6 |
| Mass411985 | 89.7 | 96.8 | 89.6/91.4 | 92.2/96.7 | 58.3/80.8 | 91.1/91.8 | 81.5/67.1 | 89.5/83.1 | 94.8/93 | 90.9/89.1 | 89.2/81.3 | 86.6/83 | 92.6/83.7 | 91.9/94.7 | NA | 98.1 |
| Conn461996 | 91.1 | 97.8 | 91.5/92.7 | 93.1/97 | 58.6/82.5 | 87.8/82.8 | 96.6/96.4 | 93.3/93.1 | 93.6/92 | 89.6/87.5 | 89.9/92.4 | 86.5/88.4 | 94.9/89.5 | 94.4/96.7 | 89.5/91.8 | 98.6 |
| ITA/90254/2005 | 90.2 | 97.1 | 92.2/94.2 | 90.8/96.3 | 80.5/81.8 | 85.7/79.6 | 64.1/54 | 89.2/84.4 | 95.6/94.6 | 93.4/92.4 | 92.7/89.7 | 90.1/91.9 | 92.6/86.6 | 91.3/94.9 | 97.5/96 | 99.0 |
| NGA/A116E7/2006 | 91.6 | 97.8 | 92.2/94 | 93/96.8 | 81.7/84.8 | 90/79.6 | 78.1/64.6 | 92/90.7 | 89.9/85.5 | 92.4/90.8 | 91.9/89.7 | 93.5/91.9 | 93.5/88.1 | 92.8/95.2 | 94.2/89.7 | 98.6 |
| Delaware072 | 88.1 | 98.4 | 89.1/90.5 | 91/94.2 | 81.4/57.5 | 92.8/82.8 | 96.6/96.4 | 92.9/90.7 | 94.7/93 | 88.5/85.8 | 85.1/78.3 | 85.3/88.4 | 94.9/89.5 | 92.9/94.4 | 92.9/93.9 | 98.6 |
| Gray | 91.2 | 98.7 | 91.8/93.3 | 92.6/97.2 | 78.6/82.8 | 97.4/94.6 | 96.6/94.6 | 92.6/91.9 | 88.2/81.6 | 89.6/85.8 | 89.9/92.4 | 86.5/88.4 | 93.1/86.6 | 86.1/84.5 | 94.9/96 | 96.6 |
| Holte | 90.7 | 98.7 | 91.5/92.8 | 92.2/97 | 79/80.2 | 91.9/88.9 | 94.2/88.9 | 91.4/89.5 | 94.9/92 | 87.9/82.4 | 89.1/84.2 | 83.3/86.6 | 95.8/92.2 | 91.2/93.4 | NA | NA |
| Iowa97 | 90.6 | 98.4 | 91.3/92.4 | 92.2/97 | 77.7/82.7 | 91.9/88.9 | 94.2/88.9 | 91.4/89.5 | 94.5/91.5 | 87.9/82.4 | 89.1/84.2 | 83.3/86.6 | 95.8/92.2 | 91.5/93.9 | 91.5/89.7 | 96.6 |
| JMK | 91.2 | 98.7 | 92/93.5 | 92.5/97 | 79.1/82.3 | 97.4/94.6 | 96/92.7 | 92.6/91.9 | 94.1/92 | 89.6/85.8 | 89.9/92.4 | 86.5/88.4 | 93.1/86.6 | 93.6/94.9 | NA | 98.1 |
| CK/CH/LDL/101212 | 90.4 | 98.4 | 89.8/91.6 | 93.6/97.3 | 78.8/81.2 | 85.1/88.9 | 80.7/67.1 | 87.6/76.3 | 96.2/92.5 | 88.5/85.8 | 85.2/78.3 | 87.8/86.6 | 95.3/90.8 | 93.2/94.4 | 34.7/15.9 | 98.1 |
| CK/SWE/0658946/10 | 89.5 | 96.8 | 90/91.1 | 91.3/95.8 | 80/80.8 | 83.6/76.4 | 64.9/51.2 | 93.7/88.2 | 96.1/95.6 | 91.3/92.4 | 92.0/81.3 | 94.7/98.4 | 91.2/85.2 | 89.2/90.4 | 97.5/96 | 99.0 |
| KM91 | 90.0 | 98.7 | 91.1/92.6 | 91.7/96.6 | 62.4/80.4 | 87.8/69.5 | 78.8/74.1 | 89.6/89.5 | 94.3/93.5 | 92.5/92.4 | 93.6/95.0 | 86/84.8 | 93.1/86.6 | 92.5/95.5 | 71.4/73.2 | 96.6 |
| CK/CH/LJL/111054 | 90.7 | 98.7 | 90.2/92.1 | 93.6/97.3 | 84.6/82.6 | 87.8/82.8 | 96.6/96.4 | 93.3/93.1 | 93.7/92 | 89.6/87.5 | 89.9/92.4 | 86.5/88.4 | 94.9/89.5 | 94.4/96.7 | 90.2/91.8 | 98.6 |
| Ukr27-11 | 91.2 | 96.5 | 89.8/91.7 | 93.6/97.2 | 80.8/86.2 | 98.3/97.3 | 93/92.7 | 92.3/91.9 | 96.9/96.6 | 97.6/97 | 96.9/100.0 | 94.7/93.5 | 94.4/89.5 | 93.2/95.2 | 97.5/96 | 99.0 |
| CK/CH/LDL/110931 | 90.6 | 98.7 | 89.9/91.7 | 93.6/97.3 | 80.7/82.4 | 87.8/82.8 | 96.6/96.4 | 93.3/93.1 | 93.7/92 | 89.6/87.5 | 89.9/92.4 | 86.5/88.4 | 94.9/89.5 | 94.4/96.7 | 90.2/91.8 | 98.6 |
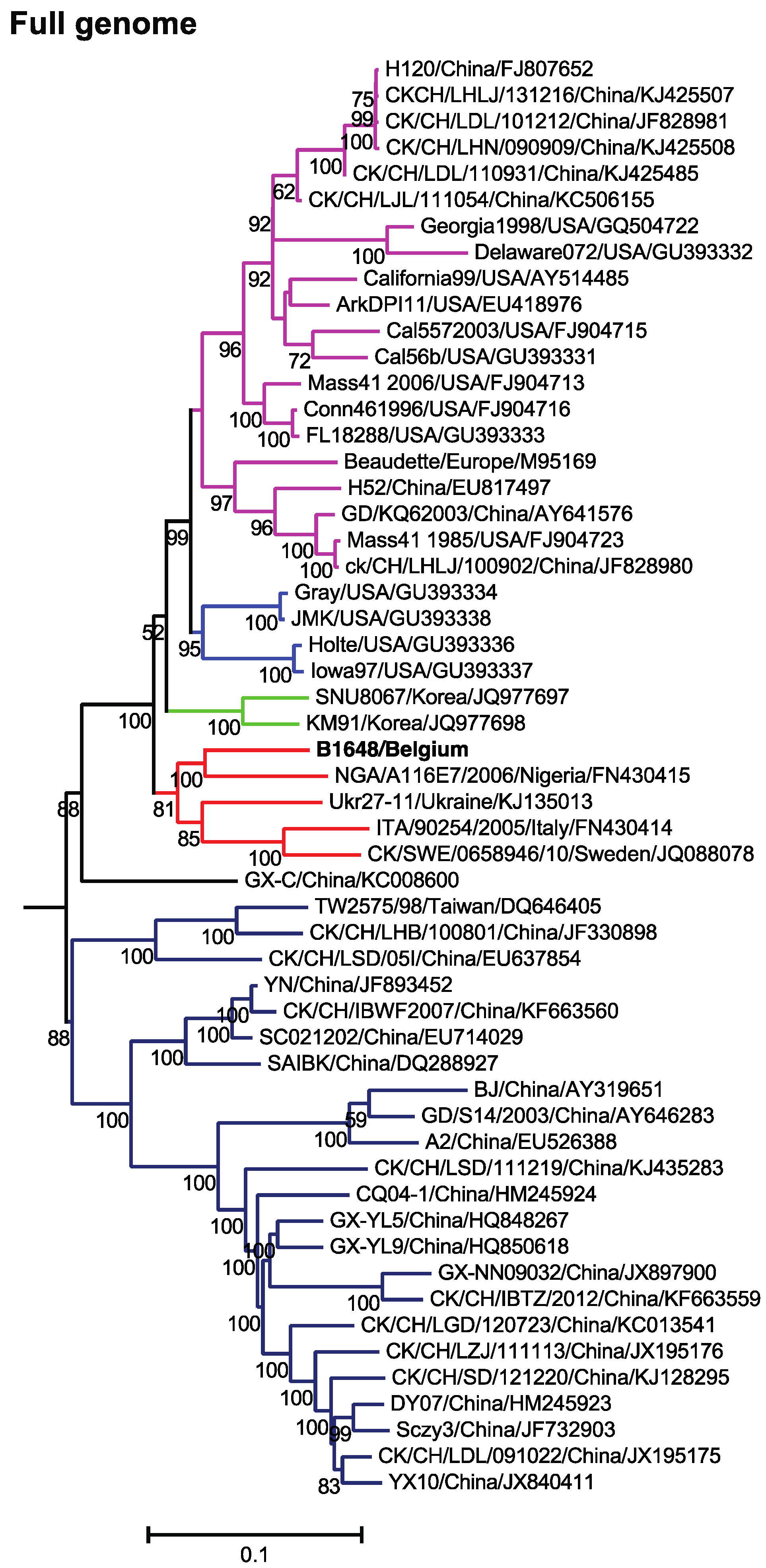
3.2. Phylogenetic Analysis and Comparison Alignments of Full Genomes of IBV Strains

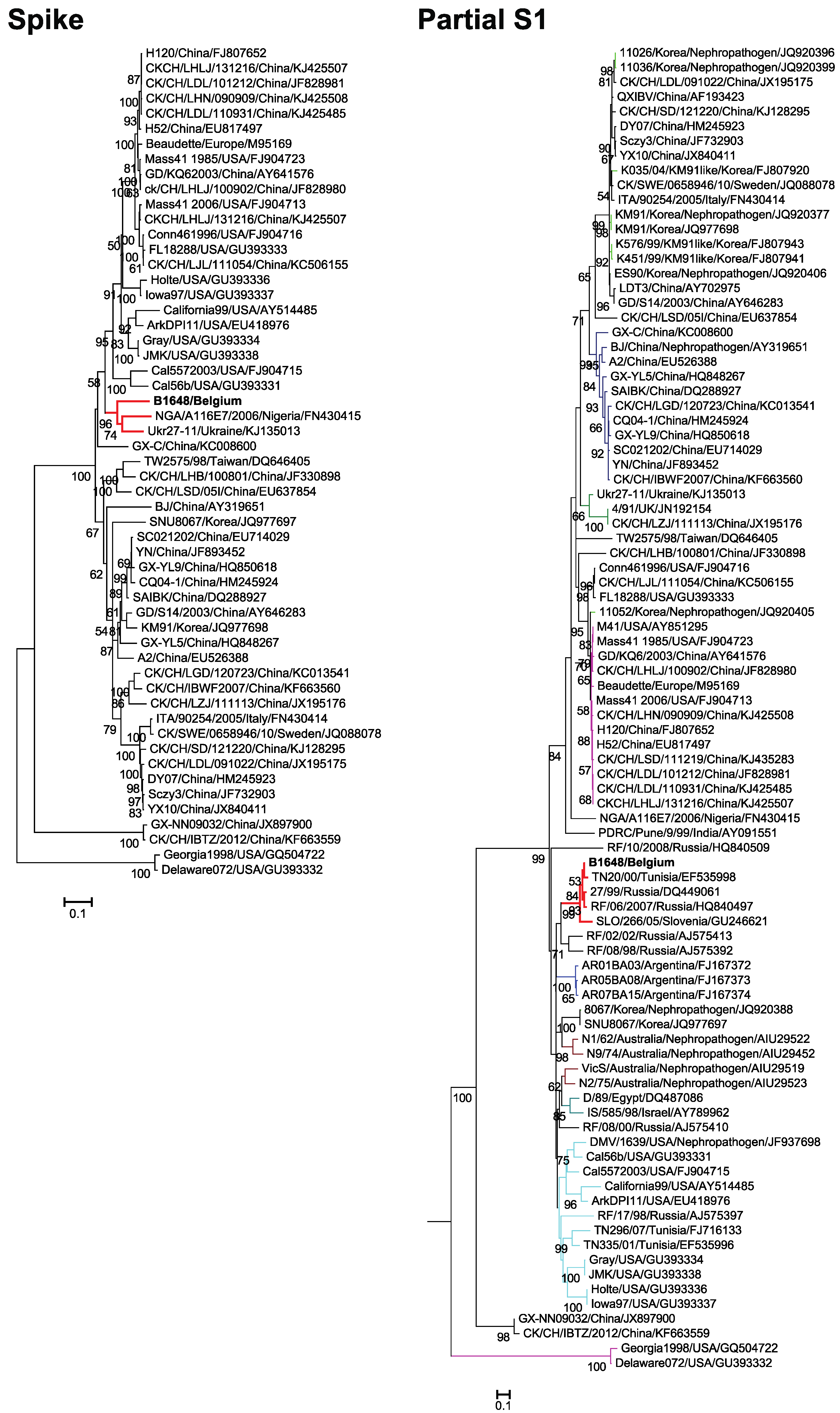
3.3. Phylogenetic Analysis and Sequence Comparison of Spike Protein (1166 aa) and Partial S1 Gene (727 nt)
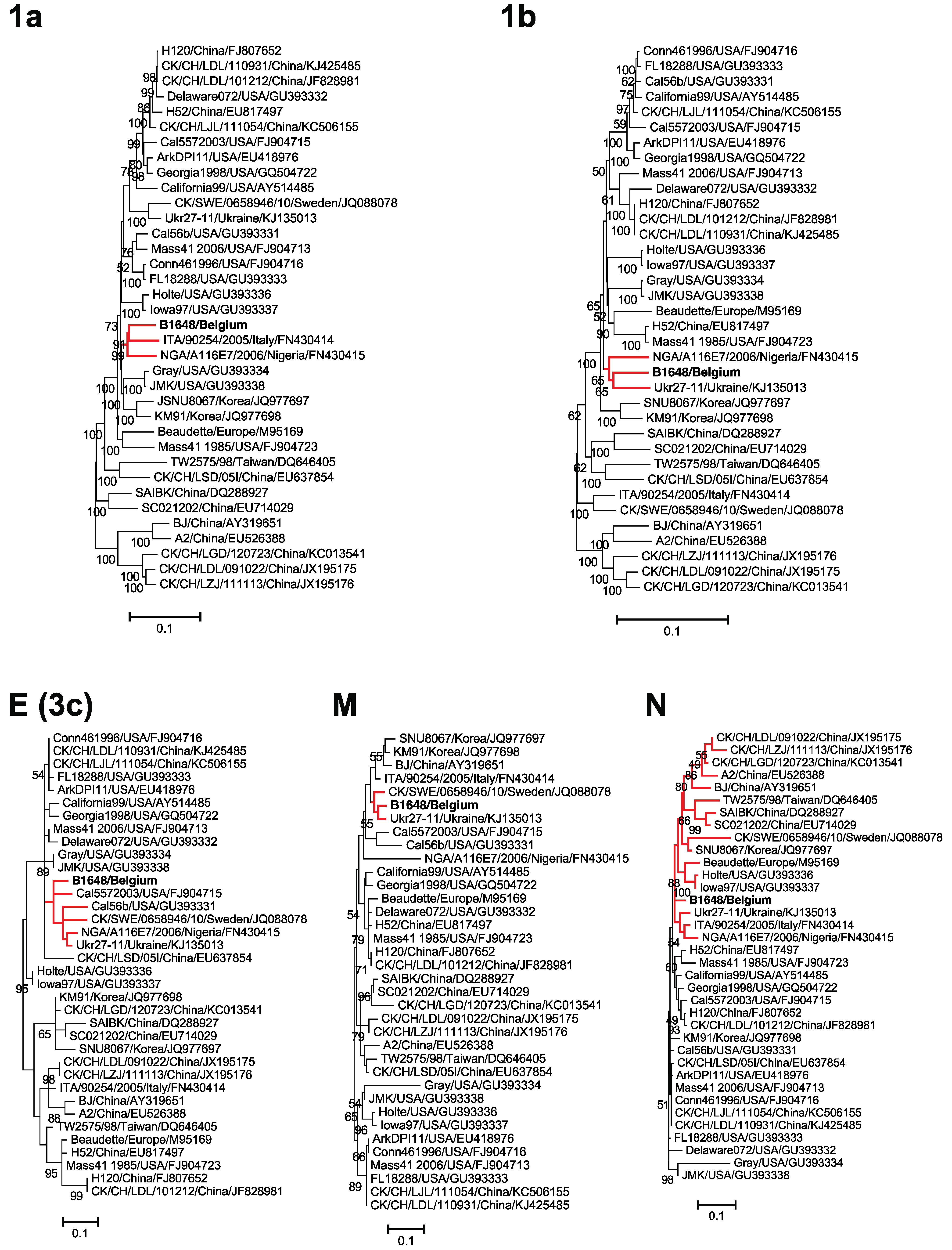
3.4. Phylogenetic Analysis and Comparison Alignments of the Replicase Transcriptase Complex (Polyprotein 1a (3949 aa) and 1b (2652 aa))
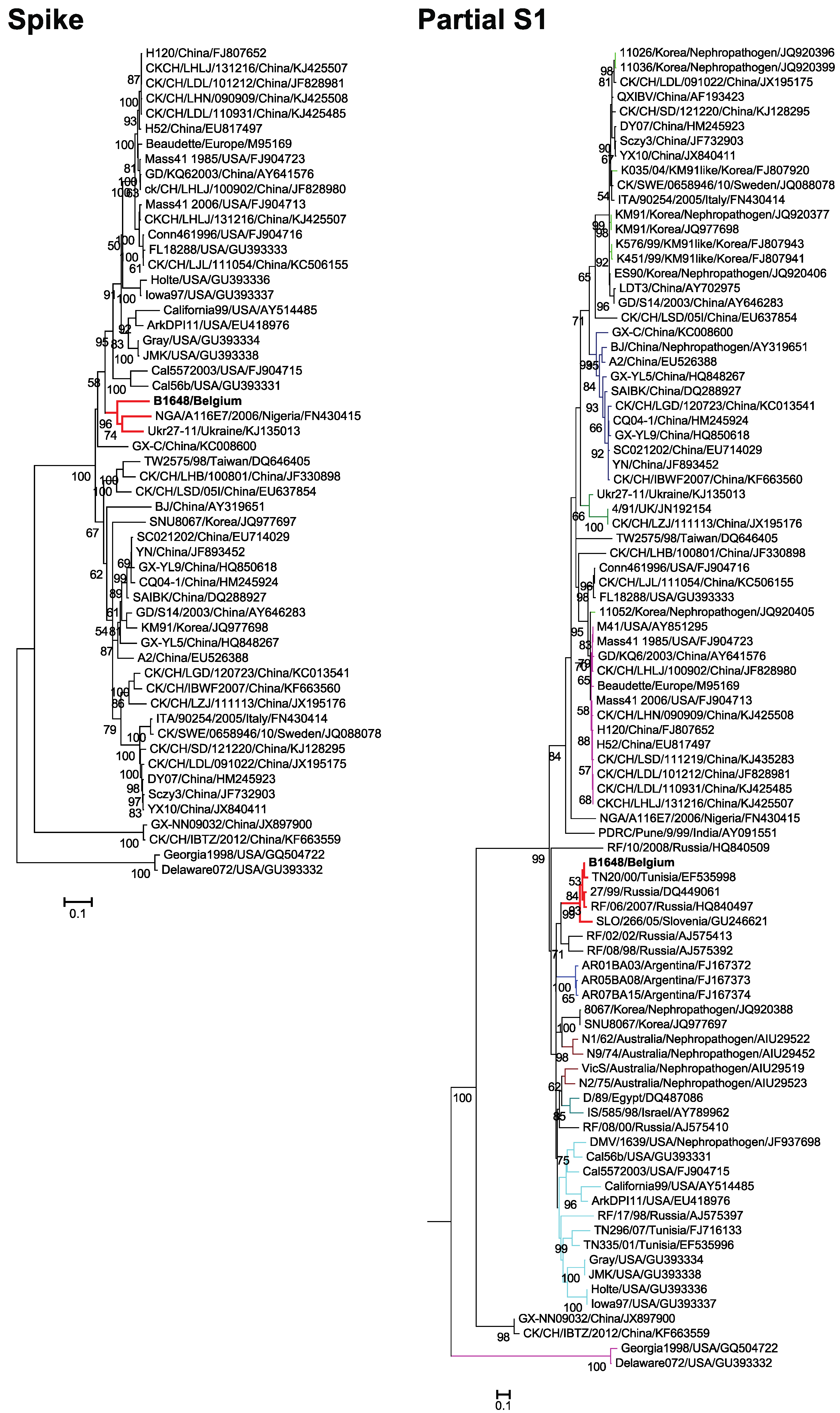

3.5. Phylogenetic Analysis and Sequence Comparison of Amino Acid Sequences of E, M and N Proteins

3.6. Accessory Proteins Alignments
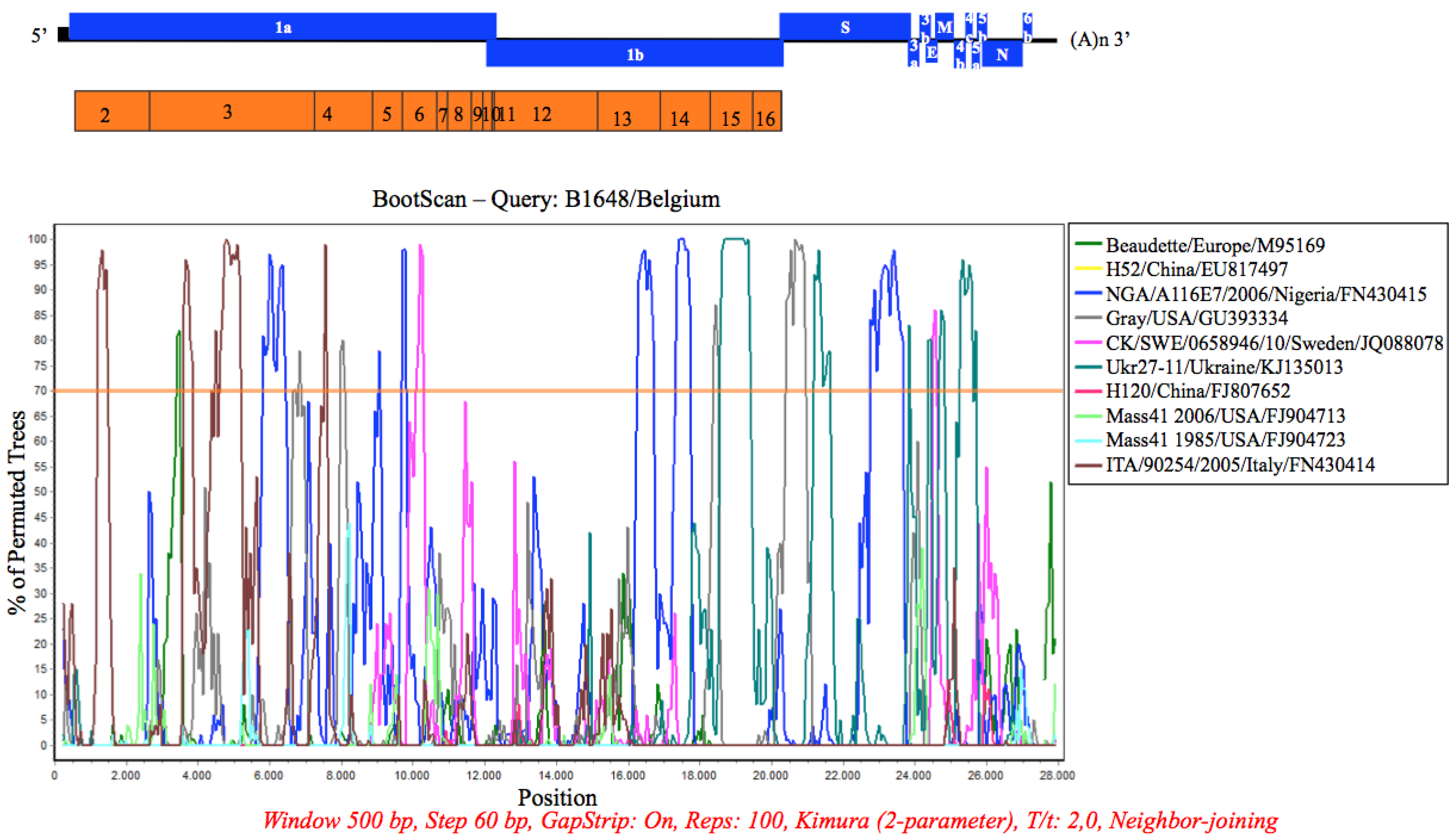
3.7. Recombination Analysis
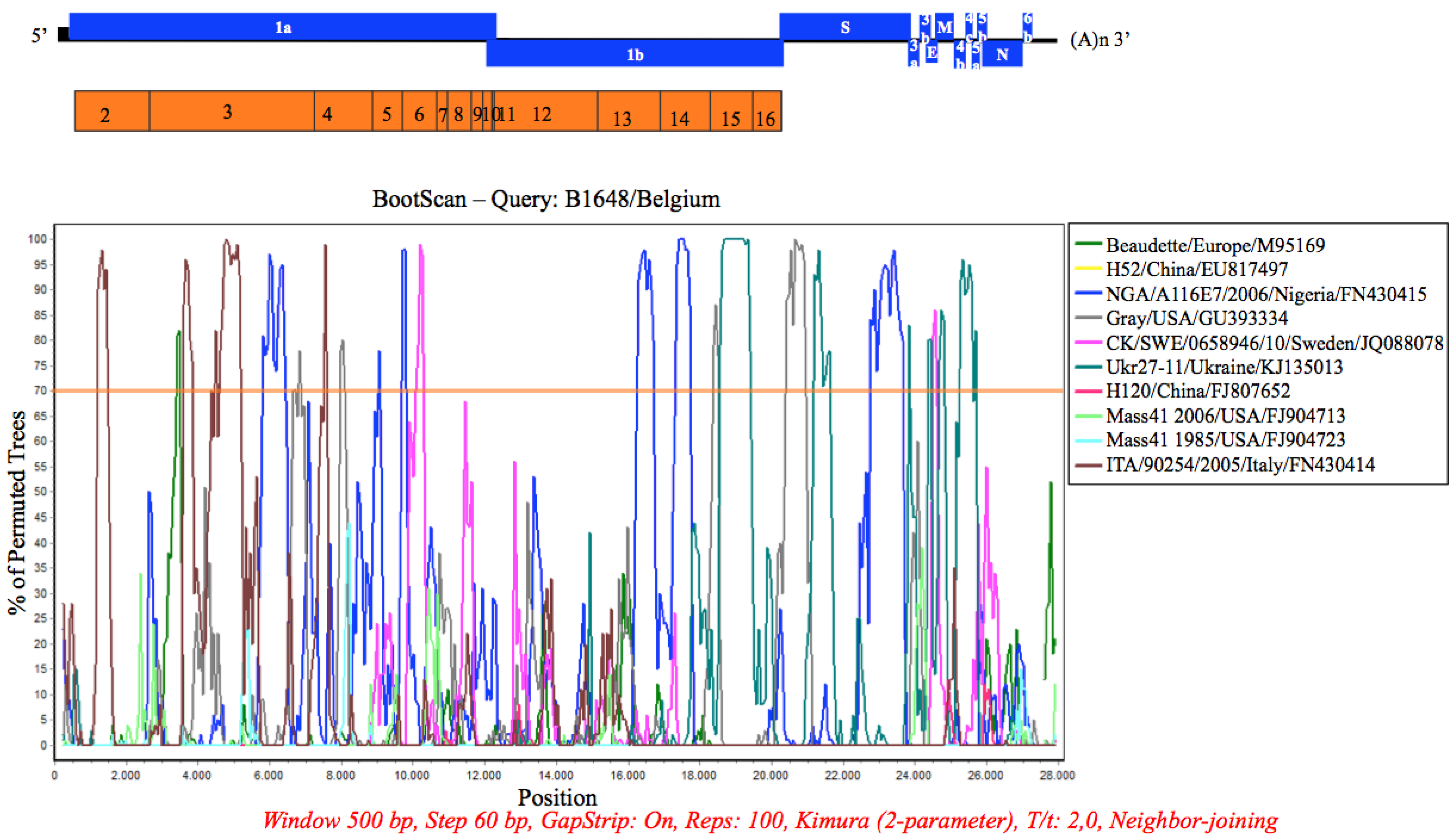
4. Discussion
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cavanagh, D. Coronaviruses in poultry and other birds. Avian Pathol. 2005, 34, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.C.; Lau, S.K.; Lam, C.S.; Tsang, A.K.; Hui, S.W.; Fan, R.Y.; Martelli, P.; Yuen, K.Y. Discovery of a novel bottlenose dolphin coronavirus reveals a distinct species of marine mammal coronavirus in Gammacoronavirus. J. Virol. 2014, 88, 1318–1331. [Google Scholar] [CrossRef] [PubMed]
- Cavanagh, D. Coronavirus avian infectious bronchitis virus. Vet. Res. 2007, 38, 281–297. [Google Scholar] [CrossRef] [PubMed]
- Kottier, S.A.; Cavanagh, D.; Britton, P. Experimental evidence of recombination in coronavirus infectious bronchitis virus. Virology 1995, 213, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Thor, S.W.; Hilt, D.A.; Kissinger, J.C.; Paterson, A.H.; Jackwood, M.W. Recombination in avian gamma-coronavirus infectious bronchitis virus. Viruses 2011, 3, 1777–1799. [Google Scholar] [CrossRef] [PubMed]
- Cook, J.K.; Jackwood, M.; Jones, R.C. The long view: 40 years of infectious bronchitis research. Avian Pathol. 2012, 41, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Meulemans, G.; Carlier, M.C.; Gonze, M.; Petit, P.; Vandenbroeck, M. Incidence, characterisation and prophylaxis of nephropathogenic avian infectious bronchitis viruses. Vet. Rec. 1987, 120, 205–206. [Google Scholar] [CrossRef] [PubMed]
- Winterfield, R.W.; Hitchner, S.B. Etiology of an infectious nephritis-nephrosis syndrome of chickens. Am. J. Vet. Res. 1962, 23, 1273–1279. [Google Scholar] [PubMed]
- Cumming, R.B. Infectious avian nephrosis (uraemia) in Australia. Aust. Vet. J. 1963, 39, 145–147. [Google Scholar] [CrossRef]
- Wang, C.H.; Hsieh, M.C.; Chang, P.C. Isolation, pathogenicity, and H120 protection efficacy of infectious bronchitis viruses isolated in Taiwan. Avian Dis. 1996, 40, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Song, C.S.; Lee, Y.J.; Kim, J.H.; Sung, H.W.; Lee, C.W.; Izumiya, Y.; Miyazawa, T.; Jang, H.K.; Mikami, T. Epidemiological classification of infectious bronchitis virus isolated in Korea between 1986 and 1997. Avian Pathol. 1998, 27, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Lim, T.H.; Lee, H.J.; Lee, D.H.; Lee, Y.N.; Park, J.K.; Youn, H.N.; Kim, M.S.; Lee, J.B.; Park, S.Y.; Choi, I.S.; et al. An emerging recombinant cluster of nephropathogenic strains of avian infectious bronchitis virus in Korea. Infect. Genet. Evol. 2011, 11, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Meir, R.; Rosenblut, E.; Perl, S.; Kass, N.; Ayali, G.; Perk, S.; Hemsani, E. Identification of a novel nephropathogenic infectious bronchitis virus in Israel. Avian Dis. 2004, 48, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, Z.H.; Sleman, R.R.; Uthman, A.U. Isolation and molecular characterization of Sul/01/09 avian infectious bronchitis virus, indicates the emergence of a new genotype in the Middle East. Vet. Microbiol. 2011, 150, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Moneim, A.S.; El-Kady, M.F.; Ladman, B.S.; Gelb, J. S1 gene sequence analysis of a nephropathogenic strain of avian infectious bronchitis virus in Egypt. Virol. J. 2006, 3, e78. [Google Scholar] [CrossRef] [PubMed]
- Bayry, J.; Goudar, M.S.; Nighot, P.K.; Kshirsagar, S.G.; Ladman, B.S.; Gelb, J.; Ghalsasi, G.R.; Kolte, G.N. Emergence of a nephropathogenic avian infectious bronchitis virus with a novel genotype in India. J. Clin. Microbiol. 2005, 43, 916–918. [Google Scholar] [CrossRef] [PubMed]
- Pensaert, M.; Lambrechts, C. Vaccination of chickens against a Belgian nephropathogenic strain of infectious bronchitis virus B1648 using attenuated homologous and heterologous strains. Avian Pathol. 1994, 23, 631–641. [Google Scholar] [CrossRef] [PubMed]
- Bochkov, Y.A.; Batchenko, G.V.; Shcherbakova, L.O.; Borisov, A.V.; Drygin, V.V. Molecular epizootiology of avian infectious bronchitis in Russia. Avian Pathol. 2006, 35, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Ducatez, M.F.; Martin, A.M.; Owoade, A.A.; Olatoye, I.O.; Alkali, B.R.; Maikano, I.; Snoeck, C.J.; Sausy, A.; Cordioli, P.; Muller, C.P. Characterization of a new genotype and serotype of infectious bronchitis virus in Western Africa. J. Gen. Virol. 2009, 90, 2679–2685. [Google Scholar] [CrossRef] [PubMed]
- Krapež, U.; Slavec, B.; Barlič-Maganja, D.; Rojs, O.Z. Molecular analysis of infectious bronchitis viruses isolated in Slovenia between 1990 and 2005: A retrospective study. Virus Genes 2010, 41, 414–416. [Google Scholar] [CrossRef] [PubMed]
- Tosi, G.; Taddei, R.; Barbieri, I.; Fiorentini, L.; Massi, P. Caratterizzazione molecolare dei ceppi di virus della bronchite infettiva aviare isolati in Italia nel periodo 2007–2009 e nel primo bimestre del 2010. In Proceedings of the 49th Annual Conference of Acts Italian society of Avian Pathology (SIPA), Forli, Italy, 29–30 April 2010; pp. 217–224.
- Bourogaa, H.; Hellal, I.; Hassen, J.; Fathallah, I.; Ghram, A. S1 gene sequence analysis of new variant isolates of avian infectious bronchitis virus in Tunisia. Vet. Med. 2012, 3, 41–48. [Google Scholar] [CrossRef]
- Toffan, A.; Bonci, M.; Bano, L.; Valastro, V.; Vascellari, M.; Capua, I.; Terregino, C. Diagnostic and clinical observation on the infectious bronchitis virus strain Q1 in Italy. Vet. Ital. 2013, 49, 347–355. [Google Scholar] [PubMed]
- Shaw, K.; Britton, P.; Cavanagh, D. Sequence of the spike protein of the Belgian B164S isolate of nephropathogenic infectious bronchitis virus. Avian Pathol. 1996, 25, 607–611. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Huang, Y.; Lau, S.K.; Woo, P.C.; Yuen, K.Y. CoVDB: A comprehensive database for comparative analysis of coronavirus genes and genomes. Nucleic Acids Res. 2008, 36, D504–D511. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Sundaram, J.P.; Spiro, D. VIGOR, an annotation program for small viral genomes. BMC Bioinform. 2010, 11, e451. [Google Scholar] [CrossRef] [PubMed]
- Lambrechts, C.; Pensaert, M.; Ducatelle, R. Challenge experiments to evaluate cross-protection induced at the trachea and kidney level by vaccine strains and Belgian nephropathogenic isolates of avian infectious bronchitis virus. Avian Pathol. 1993, 22, 577–590. [Google Scholar] [CrossRef] [PubMed]
- Cook, J.K.; Chesher, J.; Baxendale, W.; Greenwood, N.; Huggins, M.B.; Orbell, S.J. Protection of chickens against renal damage caused by a nephropathogenic infectious bronchitis virus. Avian Pathol. 2001, 30, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Hewson, K.A.; Ignjatovic, J.; Browning, G.F.; Devlin, J.M.; Noormohammadi, A.H. Infectious bronchitis viruses with naturally occurring genomic rearrangement and gene deletion. Arch. Virol. 2011, 156, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Xie, Q.; Yan, Z.; Ji, J.; Chen, F.; Qin, J.; Sun, B.; Ma, J.; Bi, Y. Complete genome sequence of a recombinant nephropathogenic infectious bronchitis virus strain in China. J. Virol. 2012, 86, 13812–13813. [Google Scholar] [CrossRef] [PubMed]
- Abolnik, C. Genomic and single nucleotide polymorphism analysis of infectious bronchitis coronavirus. Infect. Genet. Evol. 2015, 32, 416–424. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Wu, C.C.; Lin, T.L. Complete nucleotide sequence of polyprotein gene 1 and genome organization of turkey coronavirus. Virus Res. 2008, 136, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Bentley, K.; Keep, S.M.; Armesto, M.; Britton, P. Identification of a noncanonically transcribed subgenomic mRNA of infectious bronchitis virus and other gammacoronaviruses. J. Virol. 2013, 87, 2128–2136. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhang, L.; Geng, H.; Deng, Y.; Huang, B.; Guo, Y.; Zhao, Z.; Tan, W. The structural and accessory proteins M, ORF 4a, ORF 4b, and ORF 5 of Middle East respiratory syndrome coronavirus (MERS-CoV) are potent interferon antagonists. Protein Cell 2013, 4, 951–961. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Wong, C.K.; Li, P.; Xie, Y. A SARS-CoV protein, ORF-6, induces caspase-3 mediated, ER stress and JNK-dependent apoptosis. Biochim. Biophys. Acta 2008, 1780, 1383–1387. [Google Scholar] [CrossRef] [PubMed]
- Gough, R.E.; Cox, W.J.; de B Welchman, D.; Worthington, K.J.; Jones, R.C. Chinese QX strain of infectious bronchitis virus isolated in the UK. Vet. Rec. 2008, 162, 99–100. [Google Scholar] [CrossRef] [PubMed]
- Abro, S.H.; Renström, L.H.; Ullman, K.; Isaksson, M.; Zohari, S.; Jansson, D.S.; Belák, S.; Baule, C. Emergence of novel strains of avian infectious bronchitis virus in Sweden. Vet. Microbiol. 2012, 155, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Benyeda, Z.; Mató, T.; Süveges, T.; Szabó, E.; Kardi, V.; Abonyi-Tóth, Z.; Rusvai, M.; Palya, V. Comparison of the pathogenicity of QX-like, M41 and 793/B infectious bronchitis strains from different pathological conditions. Avian Pathol. 2009, 38, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Domanska-Blicharz, K.; Minta, Z.; Smietanka, K.; Porwan, T. New variant of IBV in Poland. Vet. Rec. 2006, 158, e808. [Google Scholar] [CrossRef]
- Krapez, U.; Slavec, B.; Rojs, O.Z. Circulation of infectious bronchitis virus strains from Italy 02 and QX genotypes in Slovenia between 2007 and 2009. Avian Dis. 2011, 55, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Valastro, V.; Monne, I.; Fasolato, M.; Cecchettin, K.; Parker, D.; Terregino, C.; Cattoli, G. QX-type infectious bronchitis virus in commercial flocks in the UK. Vet. Rec. 2010, 167, 865–866. [Google Scholar] [CrossRef] [PubMed]
- Worthington, K.J.; Currie, R.J.; Jones, R.C. A reverse transcriptase-polymerase chain reaction survey of infectious bronchitis virus genotypes in Western Europe from 2002 to 2006. Avian Pathol. 2008, 37, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Jiang, Y.; Low, S.; Wang, Z.; Nam, S.J.; Liu, W.; Kwangac, J. Characterization of three infectious bronchitis virus isolates from China associated with proventriculus in vaccinated chickens. Avian Dis. 2001, 45, 416–424. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.L. Recombinational histories of avian infectious bronchitis virus and turkey coronavirus. Arch. Virol. 2011, 156, 1823–1829. [Google Scholar] [CrossRef] [PubMed]
- Ammayappan, A.; Upadhyay, C.; Gelb, J.; Vakharia, V.N. Identification of sequence changes responsible for the attenuation of avian infectious bronchitis virus strain Arkansas DPI. Arch. Virol. 2009, 154, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Armesto, M.; Cavanagh, D.; Britton, P. The replicase gene of avian coronavirus infectious bronchitis virus is a determinant of pathogenicity. PLoS ONE 2009, 4, e7384. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.E.; Jackwood, M.W.; McKinley, E.T.; Thor, S.W.; Hilt, D.A.; Acevedol, N.D.; Williams, S.M.; Kissinger, J.C.; Paterson, A.H.; Robertson, J.S.; et al. Changes in nonstructural protein 3 are associated with attenuation in avian coronavirus infectious bronchitis virus. Virus Genes 2012, 44, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Züst, R.; Cervantes-Barragán, L.; Kuri, T.; Blakqori, G.; Weber, F.; Ludewig, B.; Thiel, V. Coronavirus non-structural protein 1 is a major pathogenicity factor: Implications for the rational design of coronavirus vaccines. PLoS Pathog. 2007, 3, e109. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, K.K.; Cervantes-Barragán, L.; Ludewig, B.; Thiel, V. Mouse hepatitis virus liver pathology is dependent on ADP-ribose-1′′-phosphatase, a viral function conserved in the alpha-like supergroup. J. Virol. 2008, 82, 12325–12334. [Google Scholar] [CrossRef] [PubMed]
- Sperry, S.M.; Kazi, L.; Graham, R.L.; Baric, R.S.; Weiss, S.R.; Denison, M.R. Single-amino-acid substitutions in open reading frame (ORF) 1b-nsp14 and ORF 2a proteins of the coronavirus mouse hepatitis virus are attenuating in mice. J. Virol. 2005, 79, 3391–3400. [Google Scholar] [CrossRef] [PubMed]
- Rimondi, A.; Craig, M.I.; Vagnozzi, A.; König, G.; Delamer, M.; Pereda, A. Molecular characterization of avian infectious bronchitis virus strains from outbreaks in Argentina (2001–2008). Avian Pathol. 2009, 38, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Moneim, A.S. Middle East respiratory syndrome coronavirus (MERS-CoV): Evidence and speculations. Arch. Virol. 2014, 159, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Chastel, C. Middle East respiratory syndrome (MERS): Bats or dromedary, which of them is responsible? Bull. Soc. Pathol. Exot. 2014, 107, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.R.; Oem, J.K. Surveillance of avian coronaviruses in wild bird populations of Korea. J. Wildl. Dis. 2014, 50, 964–968. [Google Scholar] [CrossRef] [PubMed]
- Muradrasoli, S.; Bálint, A.; Wahlgren, J.; Waldenström, J.; Belák, S.; Blomberg, J.; Olsen, B. Prevalence and phylogeny of coronaviruses in wild birds from the Bering Strait area (Beringia). PLoS ONE 2010, 5, e13640. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reddy, V.R.A.P.; Theuns, S.; Roukaerts, I.D.M.; Zeller, M.; Matthijnssens, J.; Nauwynck, H.J. Genetic Characterization of the Belgian Nephropathogenic Infectious Bronchitis Virus (NIBV) Reference Strain B1648. Viruses 2015, 7, 4488-4506. https://doi.org/10.3390/v7082827
Reddy VRAP, Theuns S, Roukaerts IDM, Zeller M, Matthijnssens J, Nauwynck HJ. Genetic Characterization of the Belgian Nephropathogenic Infectious Bronchitis Virus (NIBV) Reference Strain B1648. Viruses. 2015; 7(8):4488-4506. https://doi.org/10.3390/v7082827
Chicago/Turabian StyleReddy, Vishwanatha R.A.P., Sebastiaan Theuns, Inge D.M. Roukaerts, Mark Zeller, Jelle Matthijnssens, and Hans J. Nauwynck. 2015. "Genetic Characterization of the Belgian Nephropathogenic Infectious Bronchitis Virus (NIBV) Reference Strain B1648" Viruses 7, no. 8: 4488-4506. https://doi.org/10.3390/v7082827