
NCBI’s Virus Discovery Codeathon: Building “FIVE” —The Federated Index of Viral Experiments API Index

 , , , , , , , , , , , , , , , , , , , , , add
Show full author list
, , , , , , , , , , , , , , , , , , , , , add
Show full author list
Abstract
:1. Introduction
2. Materials and Methods
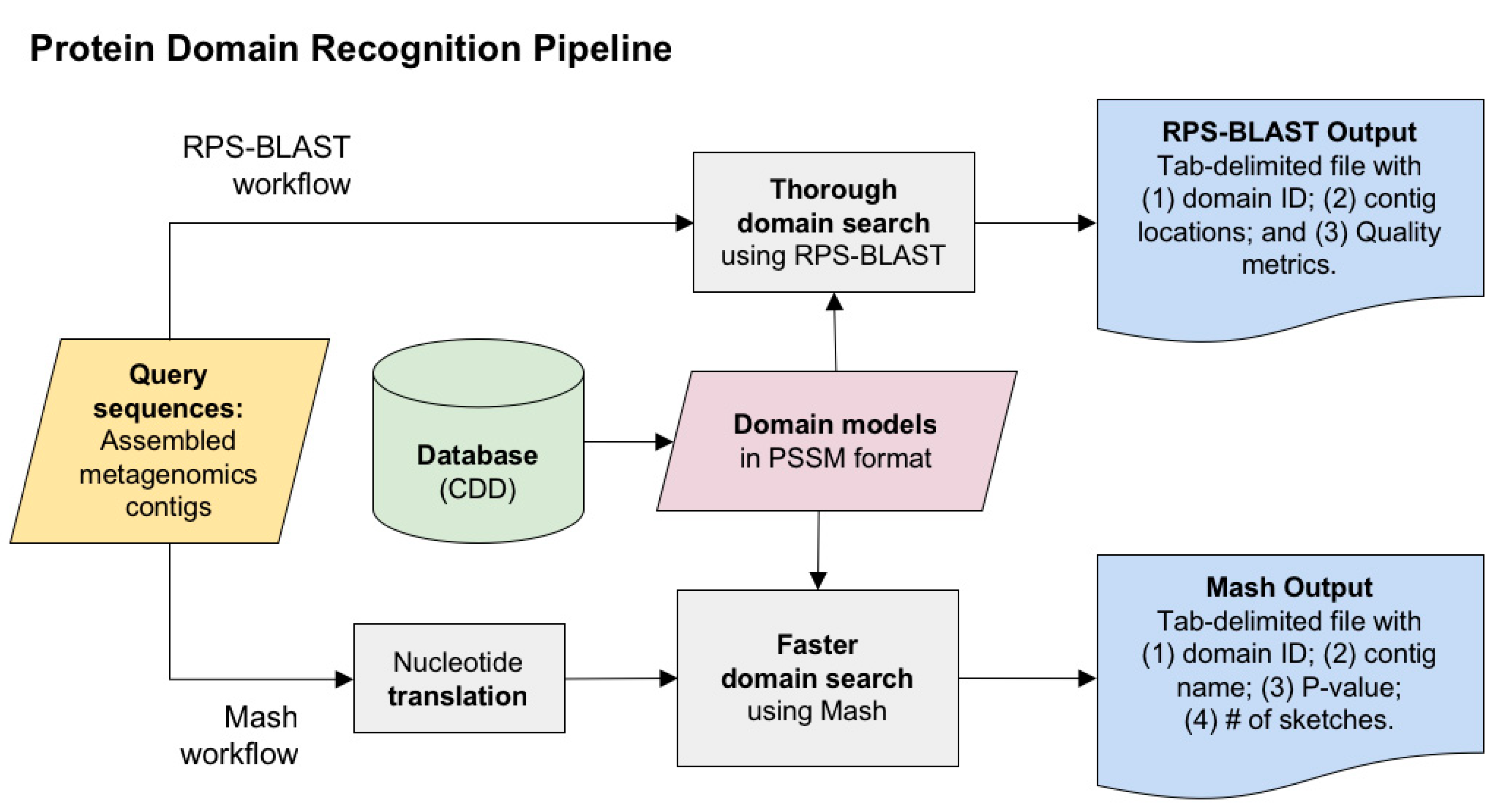
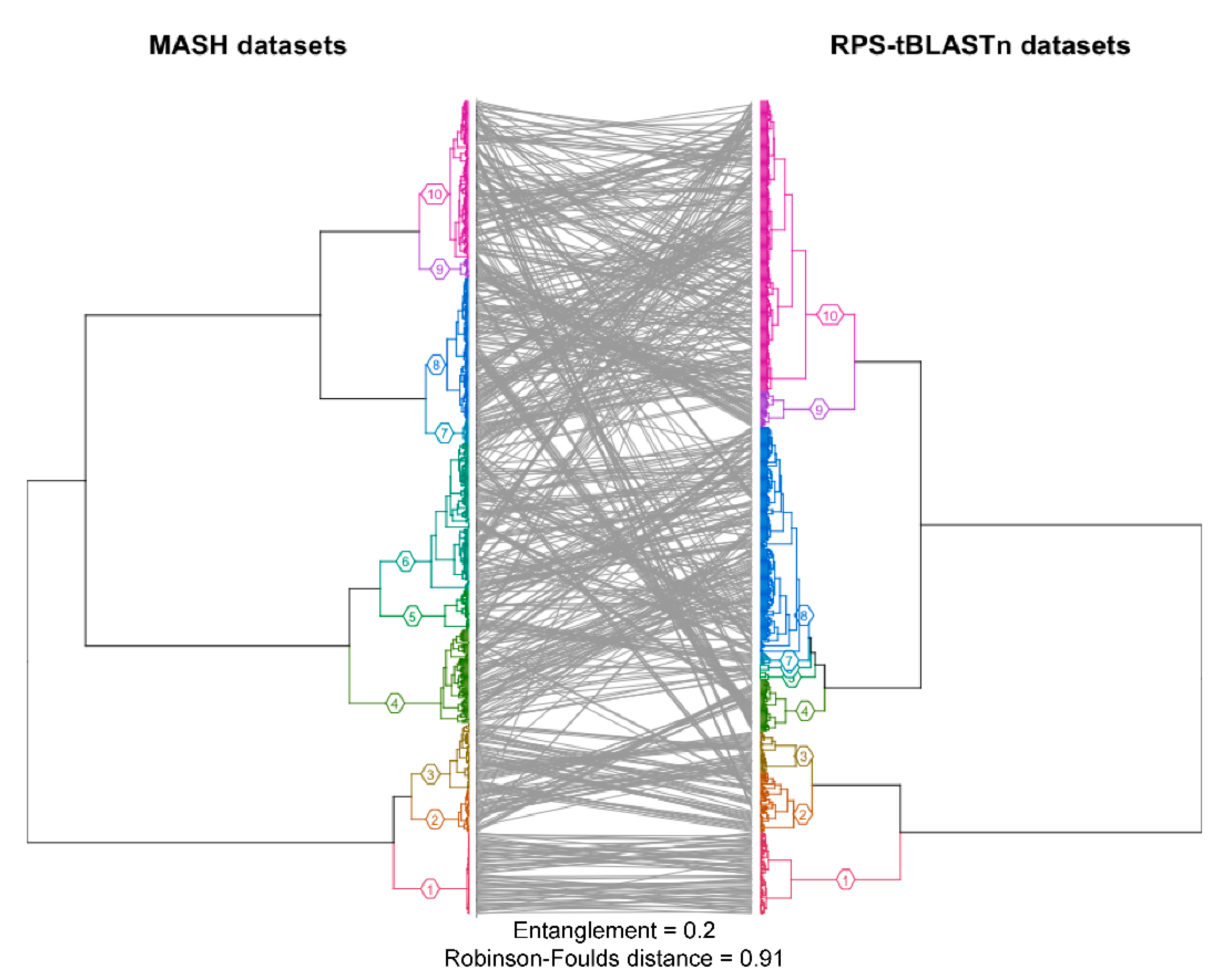
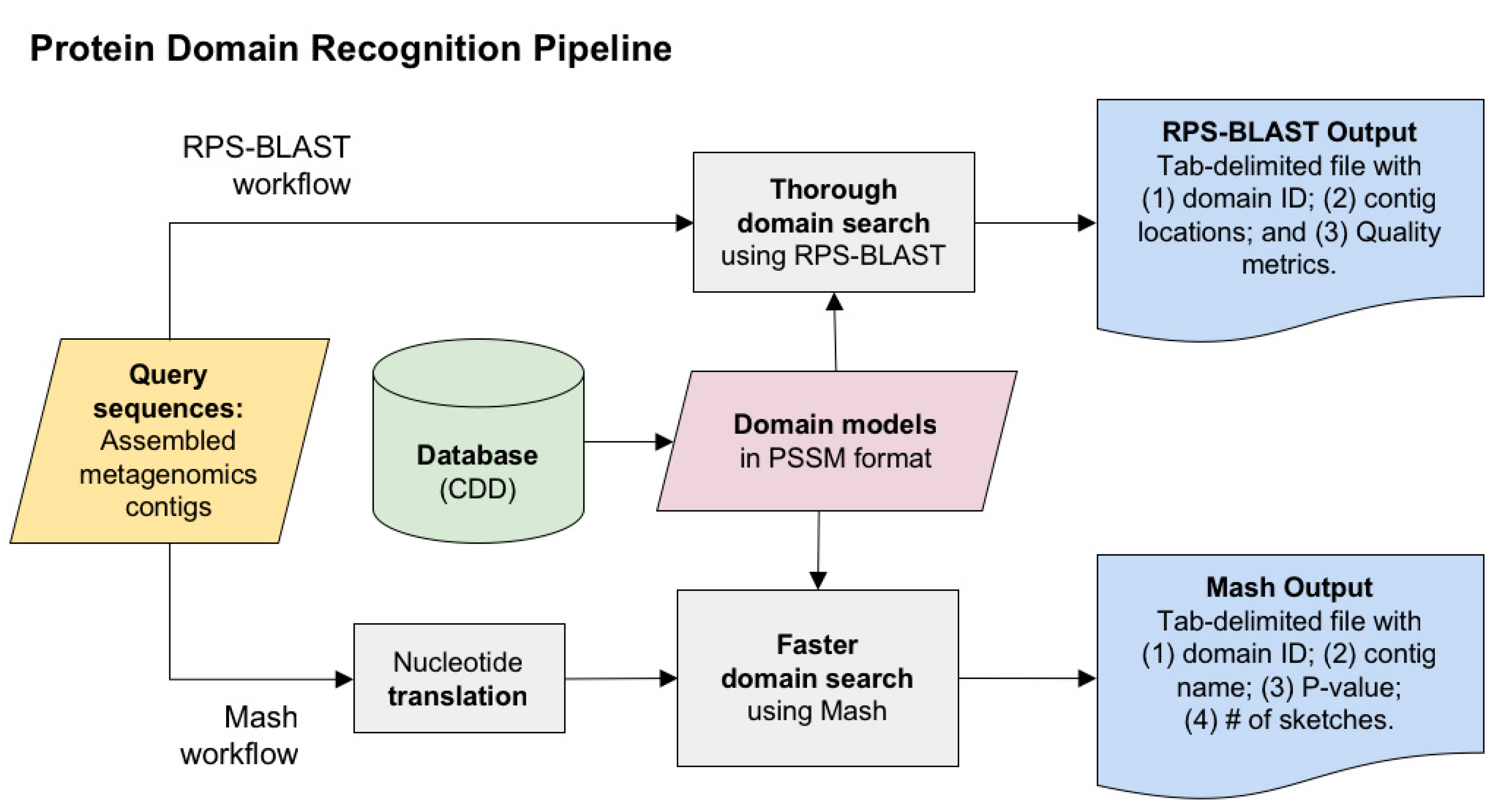
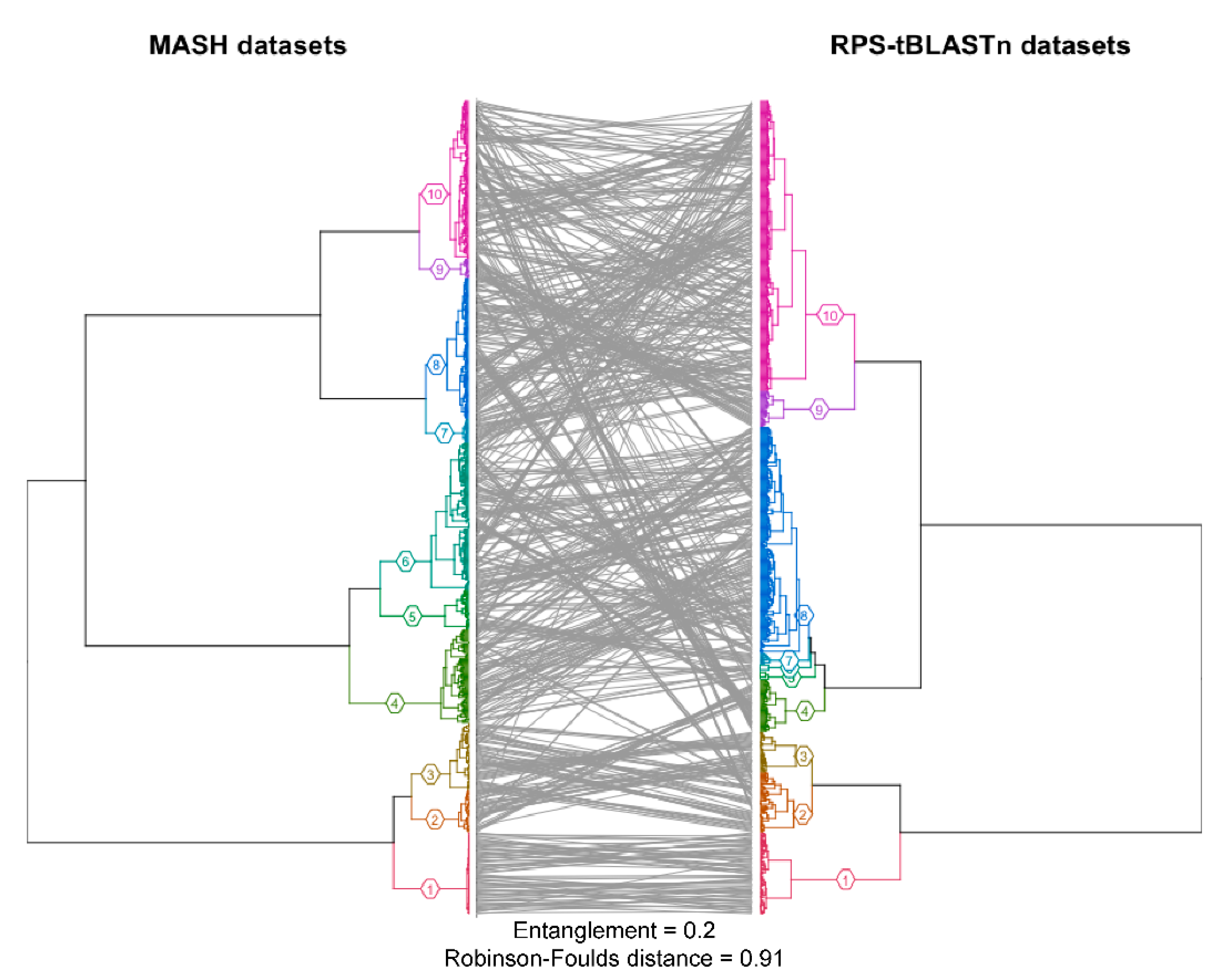
2.1. Protein Domain Recognition and Computation Scalability
2.2. Virus–Host Pairing Prediction
2.3. Viral Genome Diversity Indexing in Genome Graphs
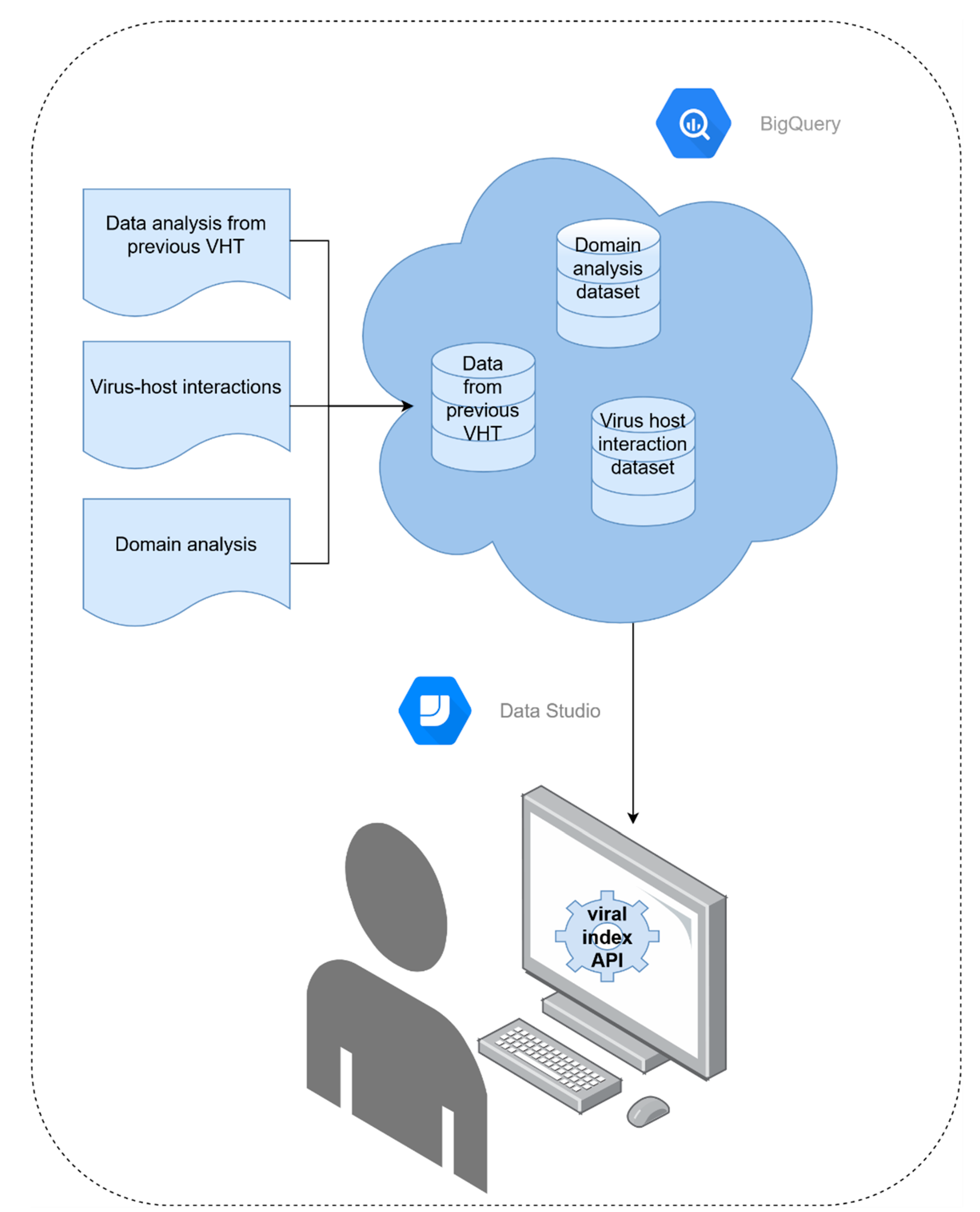
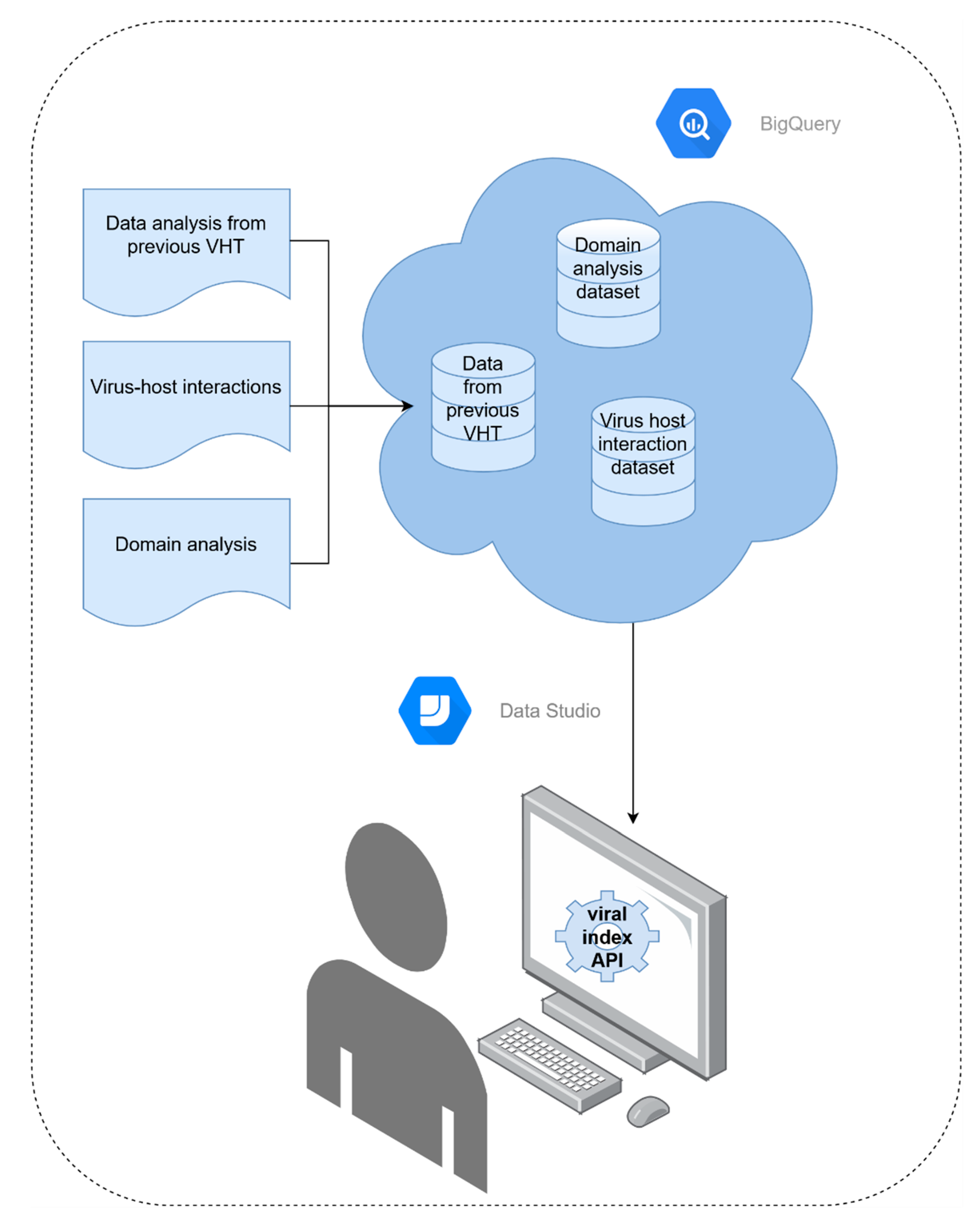
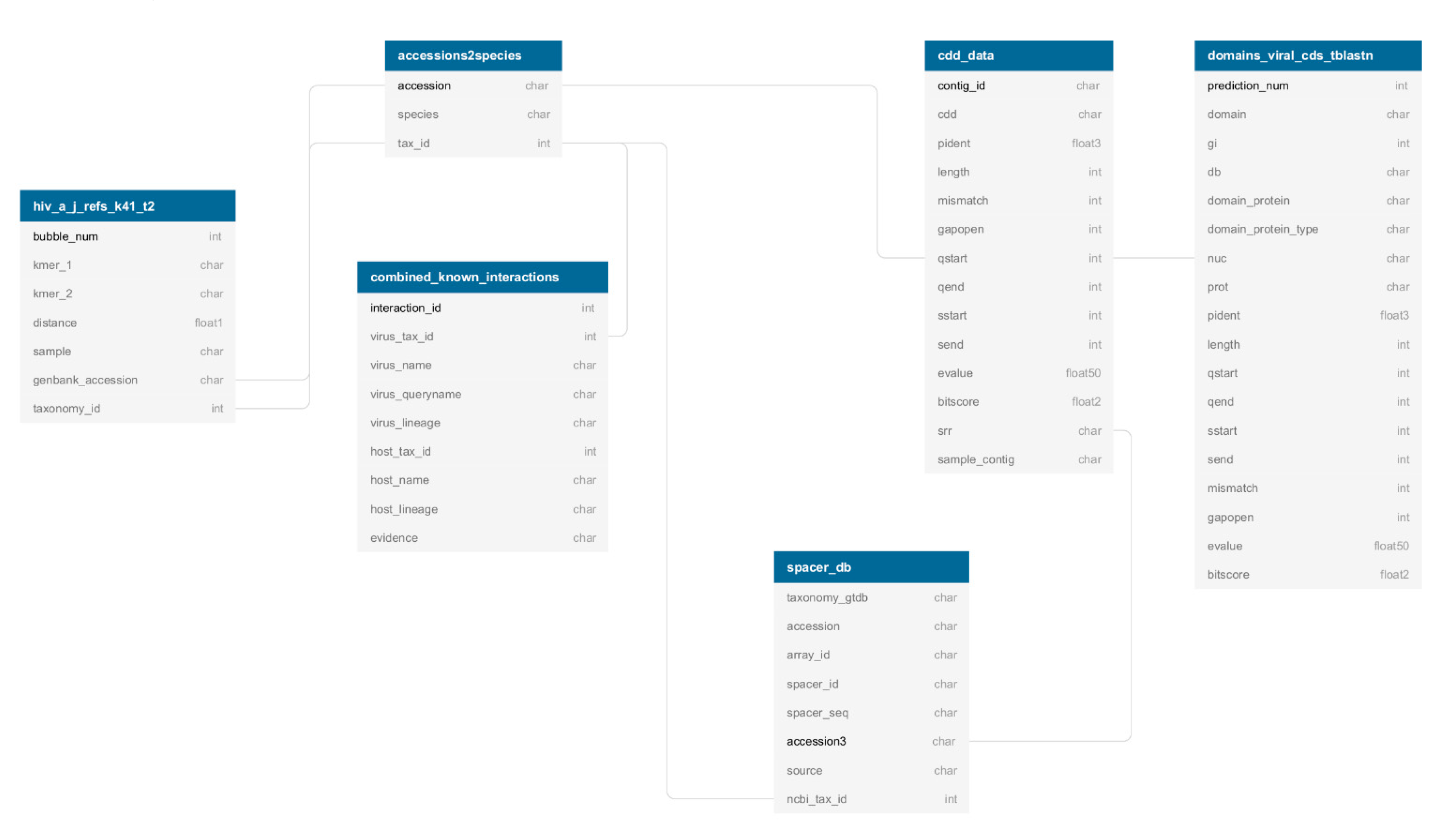
2.4. Index Structure and Accessibility
2.5. Data and Software Availability
3. Results and Discussion
3.1. Protein Domain Recognition and Computation Scalability
3.2. Virus–Host Pairing Prediction
3.3. Viral Genome Diversity Indexing in Genome Graphs
3.4. Index Structure and Accessibility
4. Conclusions and Future Directions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mardis, E.R. A decade’s perspective on DNA sequencing technology. Nature 2011, 470, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Kodama, Y.; Shumway, M.; Leinonen, R. The sequence read archive: Explosive growth of sequencing data. Nucleic Acids Res. 2012, 40, D54–D56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- SRA Database Growth. Available online: https://www.ncbi.nlm.nih.gov/sra/docs/sragrowth/ (accessed on 3 December 2020).
- Connor, R.; Brister, R.; Buchmann, J.; Deboutte, W.; Edwards, R.; Martí-Carreras, J.; Tisza, M.; Zalunin, V.; Andrade-Martínez, J.; Cantu, A.; et al. NCBI’s Virus Discovery Hackathon: Engaging Research Communities to Identify Cloud Infrastructure Requirements. Genes (Basel). 2019, 10, 714. [Google Scholar] [CrossRef] [Green Version]
- STRIDES Initiative. Available online: https://datascience.nih.gov/strides (accessed on 3 December 2020).
- NIH Strategic Plan for Data Science. Available online: https://datascience.nih.gov/strategicplan (accessed on 3 December 2020).
- Sayers, E.W.; Agarwala, R.; Bolton, E.E.; Brister, J.R.; Canese, K.; Clark, K.; Connor, R.; Fiorini, N.; Funk, K.; Hefferon, T.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2019, 47, D23–D28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- NCBI Codeathons. Available online: https://ncbi-codeathons.github.io/ (accessed on 3 December 2020).
- Busby, B.; Saha, S.; Martí-Carreras, J. Virus Discovery Project 2019. Available online: https://osf.io/g9w8r/ (accessed on 4 December 2019).
- NCBI-Codeathons/Virus_Graphs. Available online: https://github.com/NCBI-Codeathons/Virus_Graphs/tree/master/data (accessed on 3 December 2020).
- Paten, B.; Novak, A.M.; Eizenga, J.M.; Garrison, E. Genome graphs and the evolution of genome inference. Genome Res. 2017, 27, 665–676. [Google Scholar] [CrossRef] [Green Version]
- Pickett, B.E.; Sadat, E.L.; Zhang, Y.; Noronha, J.M.; Squires, R.B.; Hunt, V.; Liu, M.; Kumar, S.; Zaremba, S.; Gu, Z.; et al. ViPR: An Open Bioinformatics Database and Analysis Resource for Virology Research. Nucleic Acids Res. 2012, 40, D593–D598. [Google Scholar] [CrossRef]
- NCBI-Hackathons/VirusDiscoveryProject. Available online: https://github.com/NCBI-Hackathons/VirusDiscoveryProject (accessed on 3 December 2020).
- NCBI-Codeathons/Domain_HMM_Boundaries. Available online: https://github.com/NCBI-Codeathons/Domain_HMM_Boundaries/tree/master/viral-cdd-models (accessed on 3 December 2020).
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST plus: Architecture and applications. BMC Bioinformatics 2009, 10, 1. [Google Scholar] [CrossRef] [Green Version]
- Ondov, B.D.; Treangen, T.J.; Melsted, P.; Mallonee, A.B.; Bergman, N.H.; Koren, S.; Phillippy, A.M. Mash: Fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016, 17, 132. [Google Scholar] [CrossRef] [Green Version]
- NCBI-Codeathons/Domain_HMM_Boundaries. Available online: https://github.com/NCBI-Codeathons/Domain_HMM_Boundaries/blob/master/dataset_accessions/Mash_accessions.txt (accessed on 3 December 2020).
- R Core Team R: A Language and Environment for Statistical Computing 2019. Available online: https://www.r-project.org/ (accessed on 10 December 2020).
- Bougeard, S.; Dray, S. Supervised Multiblock Analysis in R with the ade4 Package. J. Stat. Softw. 2018, 86. [Google Scholar] [CrossRef] [Green Version]
- Schliep, K.P. phangorn: Phylogenetic analysis in R. Bioinformatics 2011, 27, 592–593. [Google Scholar] [CrossRef] [Green Version]
- Galili, T. dendextend: An R package for visualizing, adjusting and comparing trees of hierarchical clustering. Bioinformatics 2015, 31, 3718–3720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eddy, S.R. Accelerated profile HMM searches. PLoS Comput. Biol. 2011, 7, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domain_HMM_Boundaries/viral-cdd-models/virus_models.txt. Available online: https://github.com/NCBI-Codeathons/Domain_HMM_Boundaries/blob/master/viral-cdd-models/virus_models.txt (accessed on 3 December 2020).
- Conserved Domains and Protein Classification Help. Available online: https://www.ncbi.nlm.nih.gov/Structure/cdd/cdd_help.shtml (accessed on 3 December 2020).
- Domain_HMM_Boundaries/scripts/tReads.py. Available online: https://github.com/NCBI-Codeathons/Domain_HMM_Boundaries/tree/master/scripts/tReads.py (accessed on 3 December 2020).
- Hatcher, E.L.; Zhdanov, S.A.; Bao, Y.; Blinkova, O.; Nawrocki, E.P.; Ostapchuck, Y.; Schäffer, A.A.; Brister, J.R. Virus Variation Resource—Improved response to emergent viral outbreaks. Nucleic Acids Res. 2017, 45, D482–D490. [Google Scholar] [CrossRef]
- Russell, D.A.; Hatfull, G.F. PhagesDB: The actinobacteriophage database. Bioinformatics 2017, 33, 784–786. [Google Scholar] [CrossRef] [Green Version]
- Sayers, E.W.; Barrett, T.; Benson, D.A.; Bryant, S.H.; Canese, K.; Chetvernin, V.; Church, D.M.; DiCuccio, M.; Edgar, R.; Federhen, S.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2009, 37, D5–D15. [Google Scholar] [CrossRef] [Green Version]
- NCBI-Codeathons/Host_Phage_Interactions. Available online: https://github.com/NCBI-Codeathons/Host_Phage_Interactions/tree/development/src (accessed on 3 December 2020).
- CRISPR-Cas++. Available online: https://crisprcas.i2bc.paris-saclay.fr/Home/Download (accessed on 3 December 2020).
- Couvin, D.; Bernheim, A.; Toffano-Nioche, C.; Touchon, M.; Michalik, J.; Néron, B.; Rocha, E.P.C.; Vergnaud, G.; Gautheret, D.; Pourcel, C. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 2018, 46, W246–W251. [Google Scholar] [CrossRef] [Green Version]
- Weissman, J.L.; Fagan, W.F.; Johnson, P.L.F. Selective Maintenance of Multiple CRISPR Arrays Across Prokaryotes. Cris. J. 2018, 1, 405–413. [Google Scholar] [CrossRef]
- Biswas, A.; Staals, R.H.J.; Morales, S.E.; Fineran, P.C.; Brown, C.M. CRISPRDetect: A flexible algorithm to define CRISPR arrays. BMC Genom. 2016, 17, 356. [Google Scholar] [CrossRef] [Green Version]
- Nayfach, S.; Shi, Z.J.; Seshadri, R.; Pollard, K.S.; Kyrpides, N.C. New insights from uncultivated genomes of the global human gut microbiome. Nature 2019, 568, 505–510. [Google Scholar] [CrossRef] [Green Version]
- ctSkennerton/minced. Available online: https://github.com/ctSkennerton/minced (accessed on 3 December 2020).
- Bland, C.; Ramsey, T.L.; Sabree, F.; Lowe, M.; Brown, K.; Kyrpides, N.C.; Hugenholtz, P. CRISPR Recognition Tool (CRT): A tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinform. 2007, 8, 209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parks, D.H.; Chuvochina, M.; Chaumeil, P.A.; Rinke, C.; Mussig, A.J.; Hugenholtz, P. Selection of representative genomes for 24,706 bacterial and archaeal species clusters provide a complete genome-based taxonomy. bioRxiv 2019, 771964. [Google Scholar]
- NCBI-Codeathons/Virus_Graphs. Available online: https://github.com/NCBI-Codeathons/Virus_Graphs (accessed on 3 December 2020).
- Virus_Graphs/Reference_Seq.fasta. Available online: https://github.com/NCBI-Codeathons/Virus_Graphs/blob/master/Reference_Seq.fasta (accessed on 3 December 2020).
- HIV Databases. Available online: http://www.hiv.lanl.gov/ (accessed on 3 December 2020).
- HIV Sequence Database. Available online: https://www.hiv.lanl.gov/content/sequence/HIV/mainpage.html (accessed on 3 December 2020).
- Hagberg, A.A.; Schult, D.A.; Swart, P.J. Exploring Network Structure, Dynamics, and Function using NetworkX. In Proceedings of the 7th Python in Science Conference, Pasadena, CA, USA, 19–24 August 2008; Varoquaux, G., Vaught, T., Millman, J., Eds.; pp. 11–15. [Google Scholar]
- NCBI-Codeathons/The_Virus_Index. Available online: https://github.com/NCBI-Codeathons/The_Virus_Index/tree/master/python (accessed on 3 December 2020).
- The_Virus_Index/schema. Available online: https://github.com/NCBI-Codeathons/The_Virus_Index/tree/master/schema (accessed on 3 December 2020).
- viral-index 0.0.3. Available online: https://test.pypi.org/project/viral-index/ (accessed on 3 December 2020).
- NCBI-Codeathons/The_Virus_Index. Available online: https://github.com/NCBI-Codeathons/The_Virus_Index (accessed on 3 December 2020).
- Viral-index API. Available online: https://github.com/NCBI-Codeathons/The_Virus_Index#api (accessed on 3 December 2020).
- Broder, A.Z. On the Resemblance and Containment of Documents. In Proceedings of the Compression and Complexity of SEQUENCES 1997 (Cat. No.97TB100171), Washington, DC, USA, 13 June 1997; pp. 21–29. [Google Scholar]
- Domain_HMM_Boundaries/dataset_accessions/RPStbln_accessions.txt. Available online: https://github.com/NCBI-Codeathons/Domain_HMM_Boundaries/blob/master/dataset_accessions/RPStbln_accessions.txt (accessed on 3 December 2020).
- The Open Graph Viz Platform. Available online: https://gephi.org (accessed on 3 December 2020).
- Cytoscape. Available online: https://cytoscape.org/ (accessed on 3 December 2020).
- Shannon, P. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Google Cloud Platform. Available online: https://console.cloud.google.com/bigquery?p=virus-hunting-2-codeathon&d=viasq&page=dataset (accessed on 3 December 2020).
- Sample Code. Available online: https://github.com/NCBI-Codeathons/The_Virus_Index#sample-code (accessed on 3 December 2020).
- Guirimand, T.; Delmotte, S.; Navratil, V. VirHostNet 2.0: Surfing on the web of virus/host molecular interactions data. Nucleic Acids Res. 2015, 43, D583–D587. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, M.D.; Dumontier, M.; Aalbersberg, I.J.; Appleton, G.; Axton, M.; Baak, A.; Blomberg, N.; Boiten, J.-W.; da Silva Santos, L.B.; Bourne, P.E.; et al. The FAIR Guiding Principles for scientific data management and stewardship. Sci. Data 2016, 3, 160018. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Relevant Repositories | GitHub Project | Dataset Citation |
|---|---|---|
| Connor et al., 2019 VHT [4] | https://github.com/NCBI-Hackathons/VirusDiscoveryProject | 10.3390/genes10090714 |
| VHT contig list | https://github.com/NCBI-Hackathons/VirusDiscoveryProject/blob/master/contigs_readme.md | 10.17605/osf.io/g9w8r |
| VHT contig repository | https://storage.googleapis.com/experimental-sra-metagenome-contigs | 10.17605/osf.io/g9w8r |
| Protein domain recognition and computation scalability | https://github.com/NCBI-Codeathons/Domain_HMM_Boundaries | 10.5281/zenodo.4027168 |
| Virus–Host pairing prediction | https://github.com/NCBI-Codeathons/Host_Phage_Interactions | 10.5281/zenodo.4027172 |
| Viral genome diversity indexing in genome graphs | https://github.com/NCBI-Codeathons/Virus_Graphs | 10.5281/zenodo.4027629 |
| Index structure and accessibility | https://github.com/NCBI-Codeathons/The_Virus_Index | 10.5281/zenodo.4027617 |
| FIVE | https://console.cloud.google.com/bigquery?p=virus-hunting-2-codeathon&d=viasq&page=dataset | - |
| Function | Description |
|---|---|
| get_viruses_for_host_taxonomy | Retrieve host(s) for a given virus taxonomy ID |
| get_host_from_virus_taxonomy | Retrieve virus(es) that can infect a given host |
| get_potential_hosts_for_virus_domain | Get all potential host(s) given a domain that is found in viruses |
| get_virus_host_interactions_from_confidence_level | Get all virus–host interactions for specified confidence level |
| get_SRAs_where_CDD_is_found | Get Sequence Read Archive (SRA) accessions of studies wherein a viral protein domain is found |
| get_domains | Find all domains present in a virus |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martí-Carreras, J.; Gener, A.R.; Miller, S.D.; Brito, A.F.; Camacho, C.E.; Connor, R.; Deboutte, W.; Glickman, C.; Kristensen, D.M.; Meyer, W.K.; et al. NCBI’s Virus Discovery Codeathon: Building “FIVE” —The Federated Index of Viral Experiments API Index. Viruses 2020, 12, 1424. https://doi.org/10.3390/v12121424
Martí-Carreras J, Gener AR, Miller SD, Brito AF, Camacho CE, Connor R, Deboutte W, Glickman C, Kristensen DM, Meyer WK, et al. NCBI’s Virus Discovery Codeathon: Building “FIVE” —The Federated Index of Viral Experiments API Index. Viruses. 2020; 12(12):1424. https://doi.org/10.3390/v12121424
Chicago/Turabian StyleMartí-Carreras, Joan, Alejandro Rafael Gener, Sierra D. Miller, Anderson F. Brito, Christiam E. Camacho, Ryan Connor, Ward Deboutte, Cody Glickman, David M. Kristensen, Wynn K. Meyer, and et al. 2020. "NCBI’s Virus Discovery Codeathon: Building “FIVE” —The Federated Index of Viral Experiments API Index" Viruses 12, no. 12: 1424. https://doi.org/10.3390/v12121424