Mason-Pfizer Monkey Virus Envelope Glycoprotein Cycling and Its Vesicular Co-Transport with Immature Particles

 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
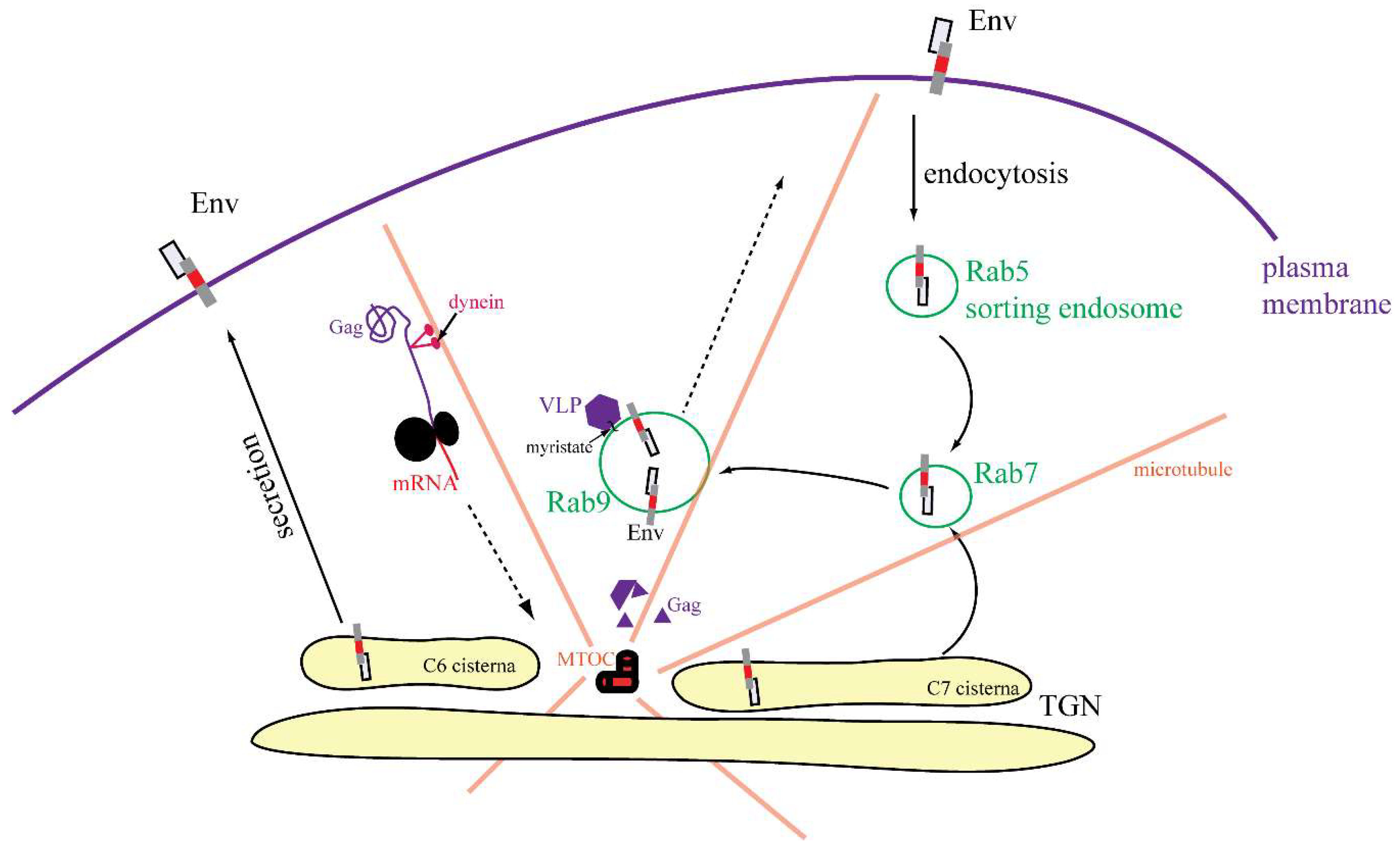
:1. Introduction
2. Materials and Methods
2.1. Plasmids
2.2. The Introduction of I18A and Y22A Mutations into pSARMXmCherryTM Construct
- 5′-AAACCTGCACAAGTCCATTATCATCGCCTTG and
- 5′-AAGAGGGCCCAATATCCGAGCAAAGACG
- 5′-GACTTGTGCAGGTTTGGCCTGGATGCTC and
- 5′-ATATTGGGCCCTCTTATCAGCAAGGCCTGG
- 5′-GTCCATGCTCATCGCCTTGAACAAGAAGACAGTGG and
- 5′-AAGAGGGCCCAATATCCGAGCAAAGACG
- 5′-GCGATGAGCATGGACTTGTATAGGTTTGGC and
- 5′-ATATTGGGCCCTCTTATCAGCAAGGC
2.3. Cultivation of Cells, Transfection of Plasmids and Imaging
2.4. Immunolabeling of Viral Proteins with Radioactive Isotopes and Immunoprecipitation (Pulse Chase Assay)
2.5. Live-Cell Imaging
2.6. Quantification of Colocalization
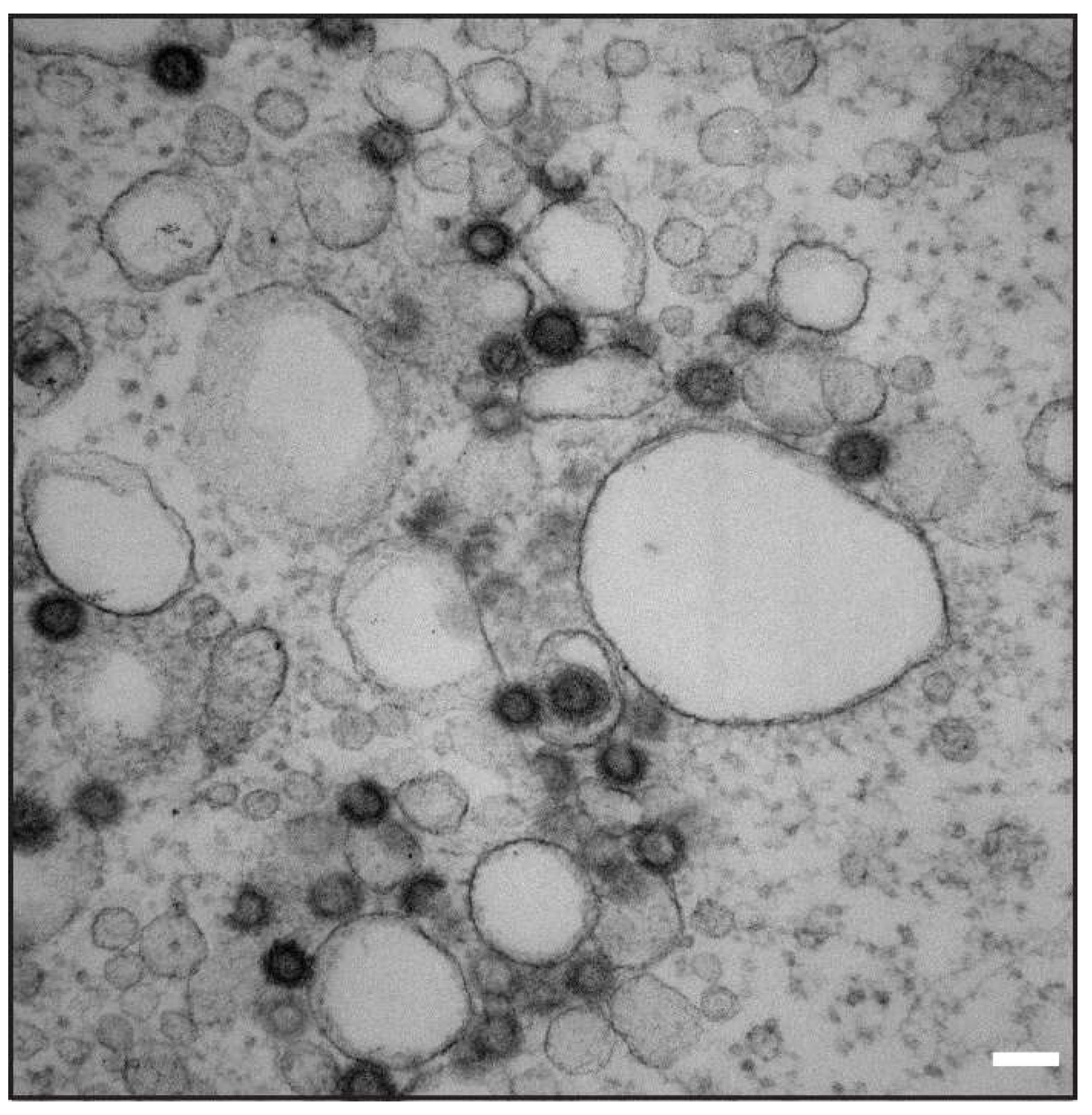
2.7. Transmission Electron Microscopy (TEM) Analysis
3. Results
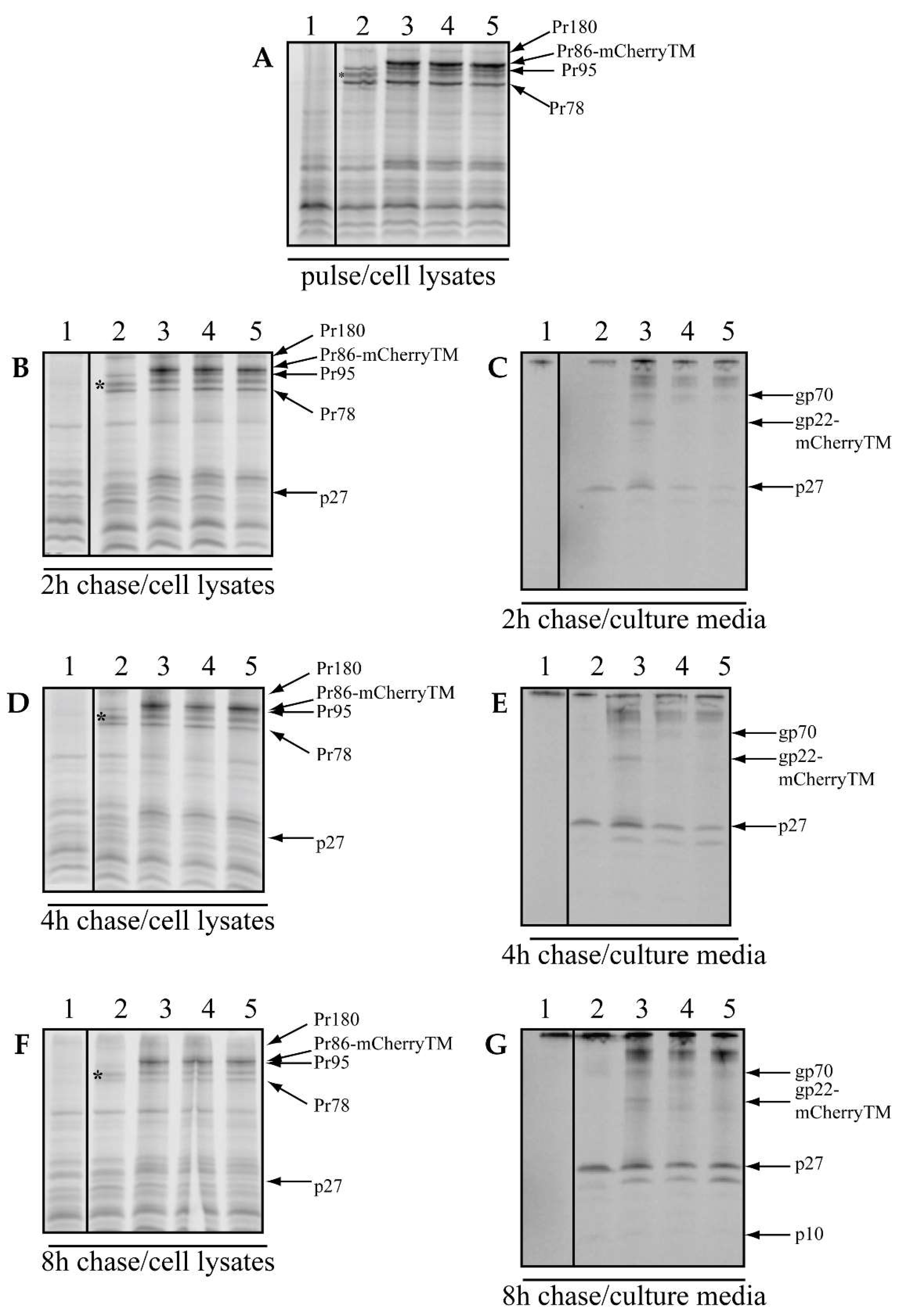
3.1. mCherryTM Protein Synthesis, Processing and Incorporation into Viral Particles
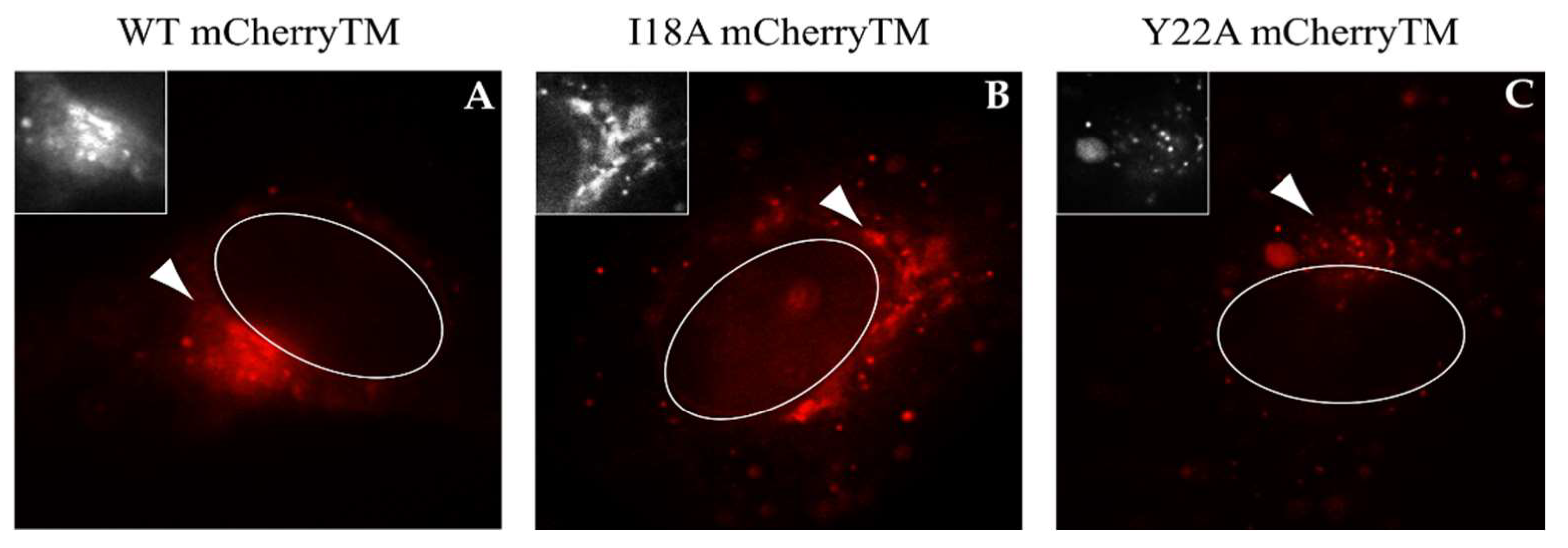
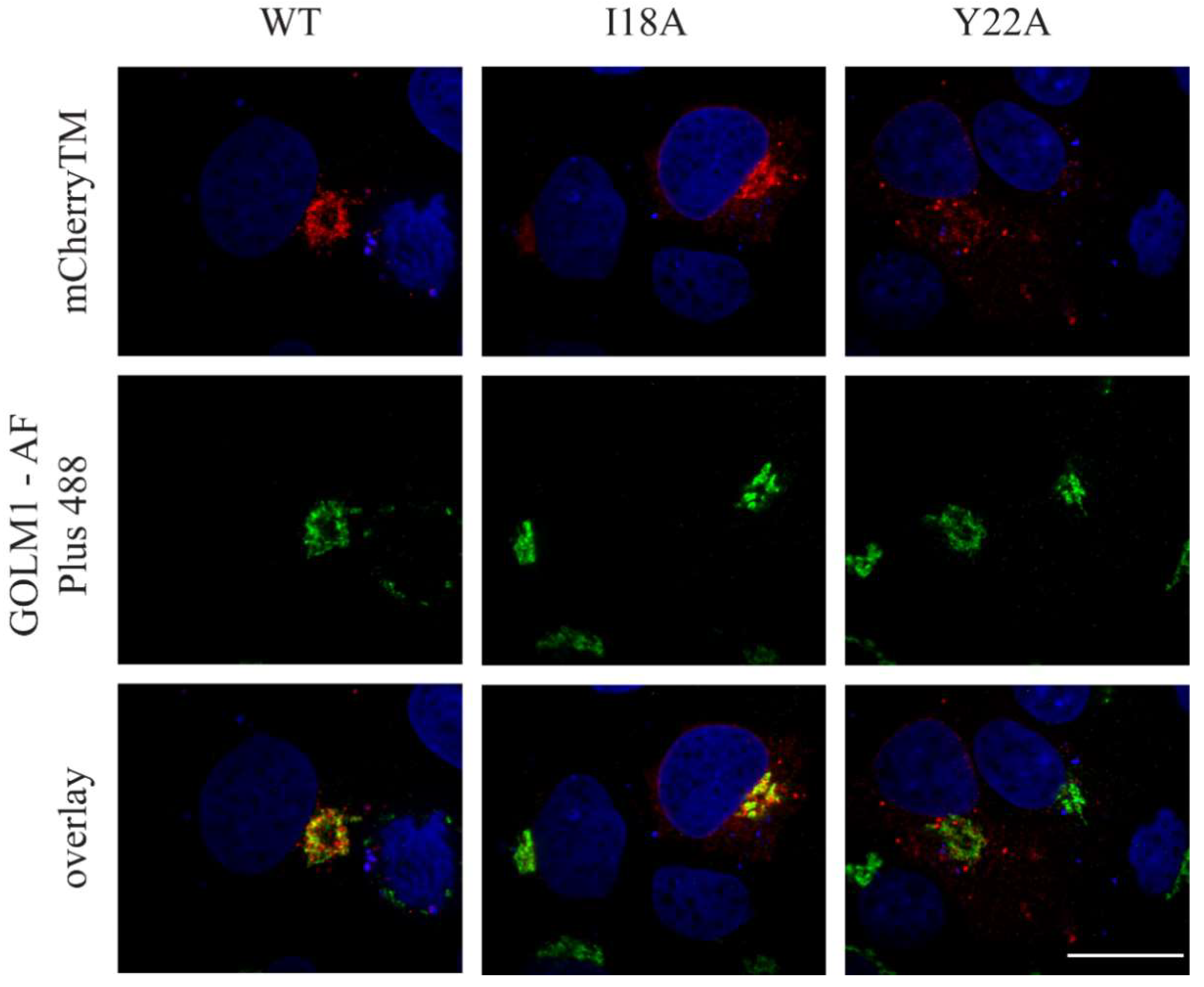
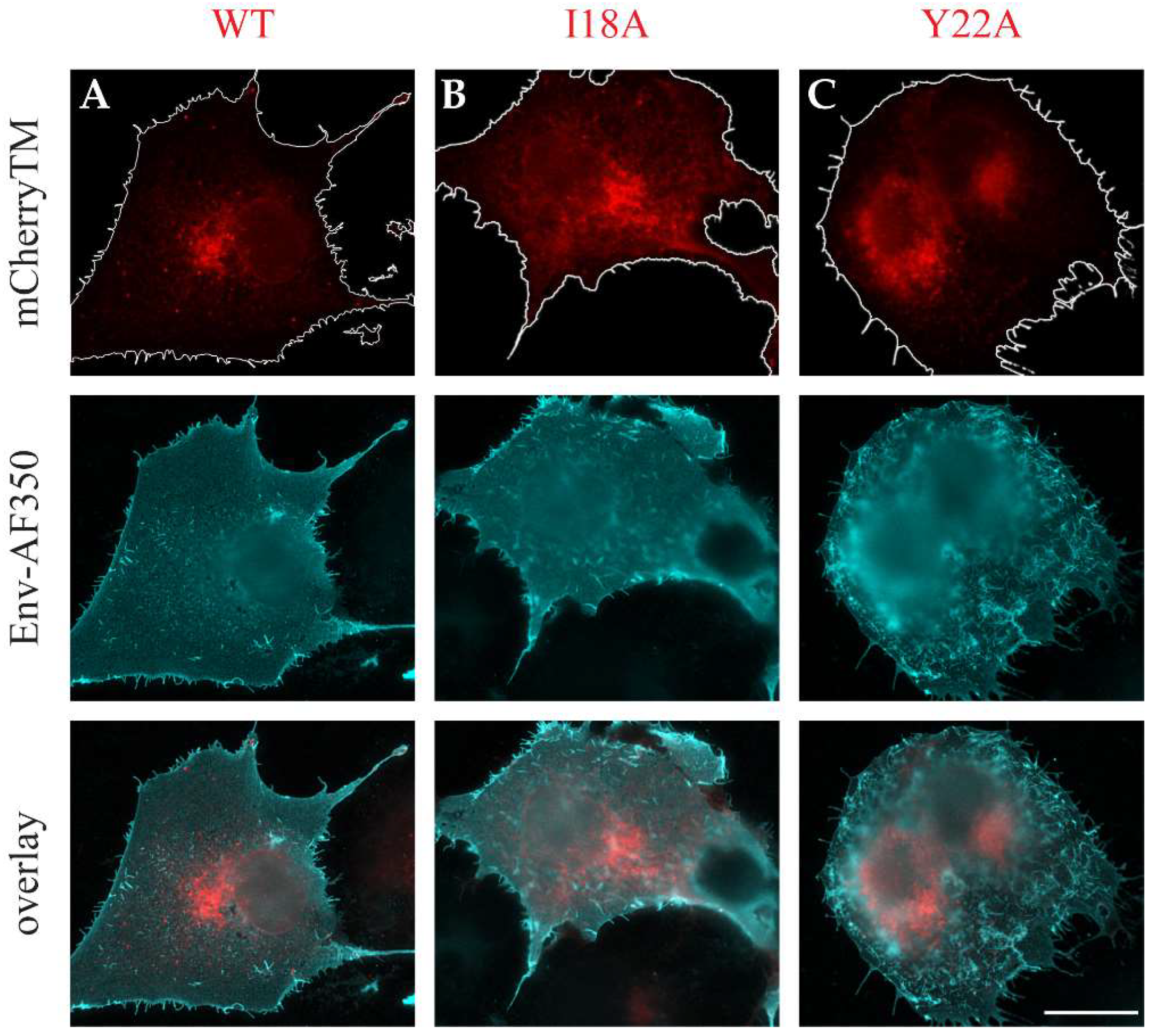
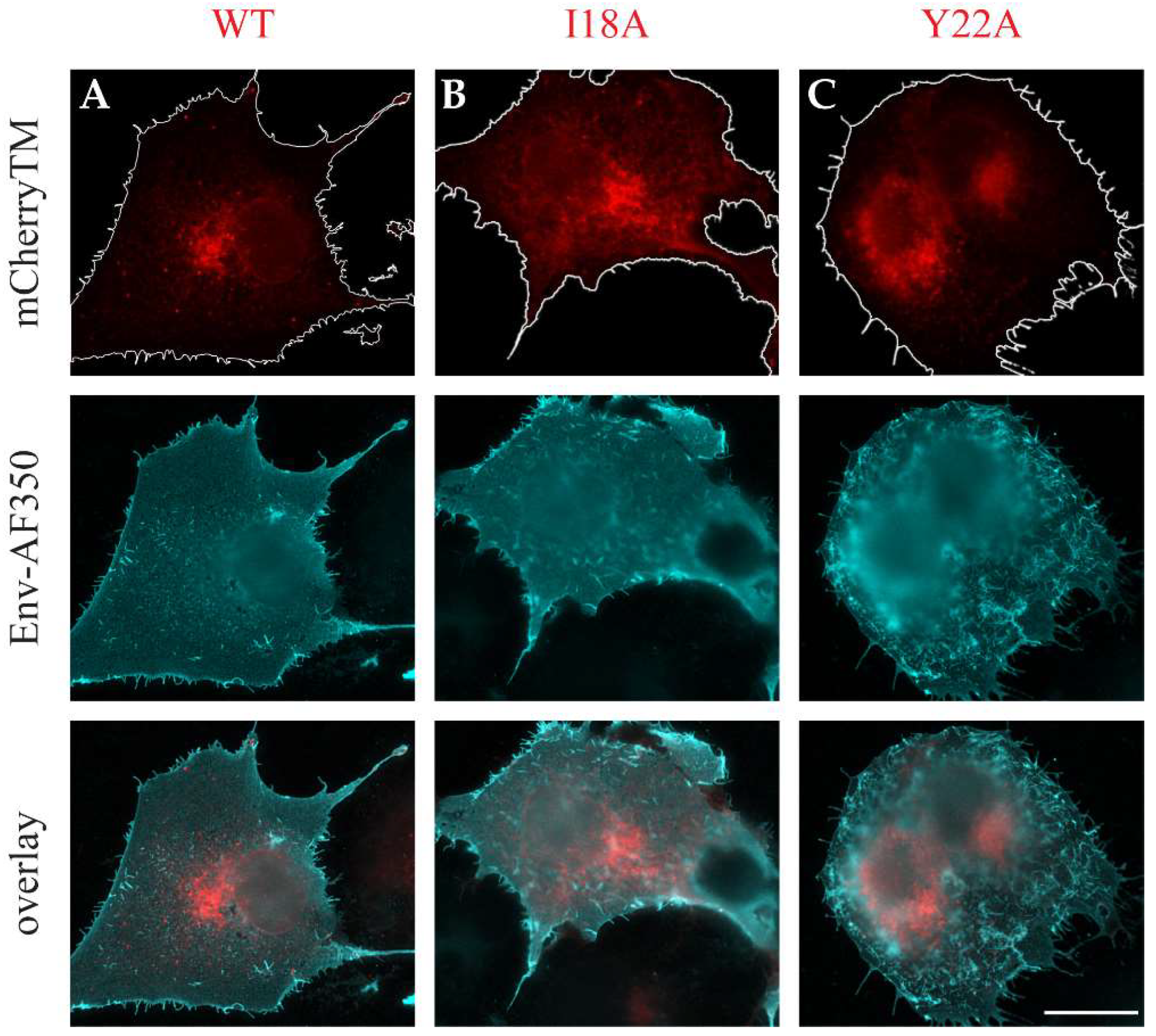
3.2. The Intracellular Localization of mCherryTM and Its I18A and Y22A Mutants
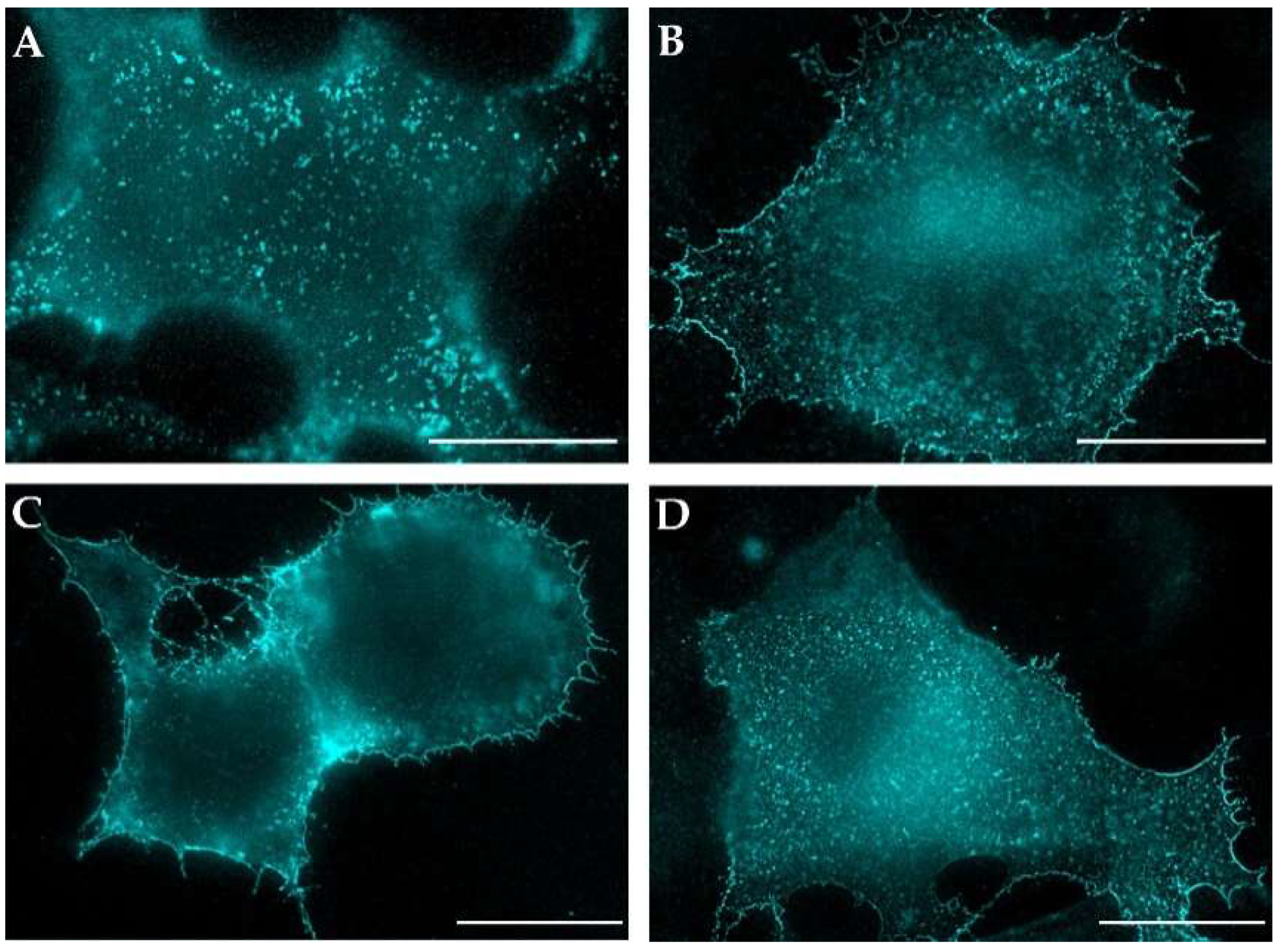
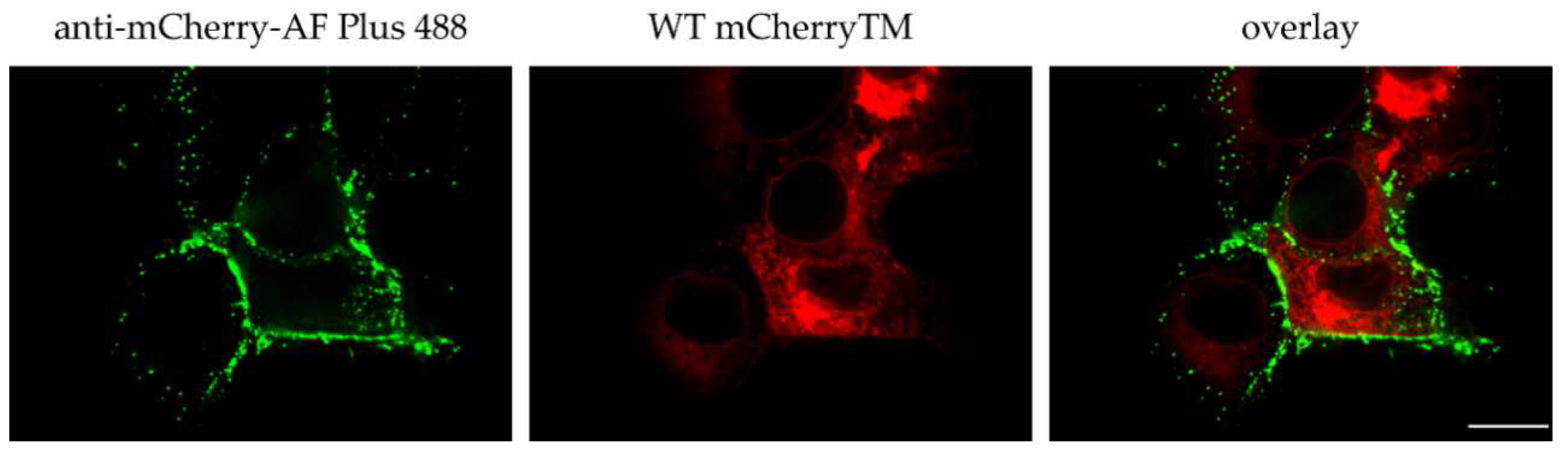
3.3. Env and mCherryTM Localization at the Plasma Membrane (PM)
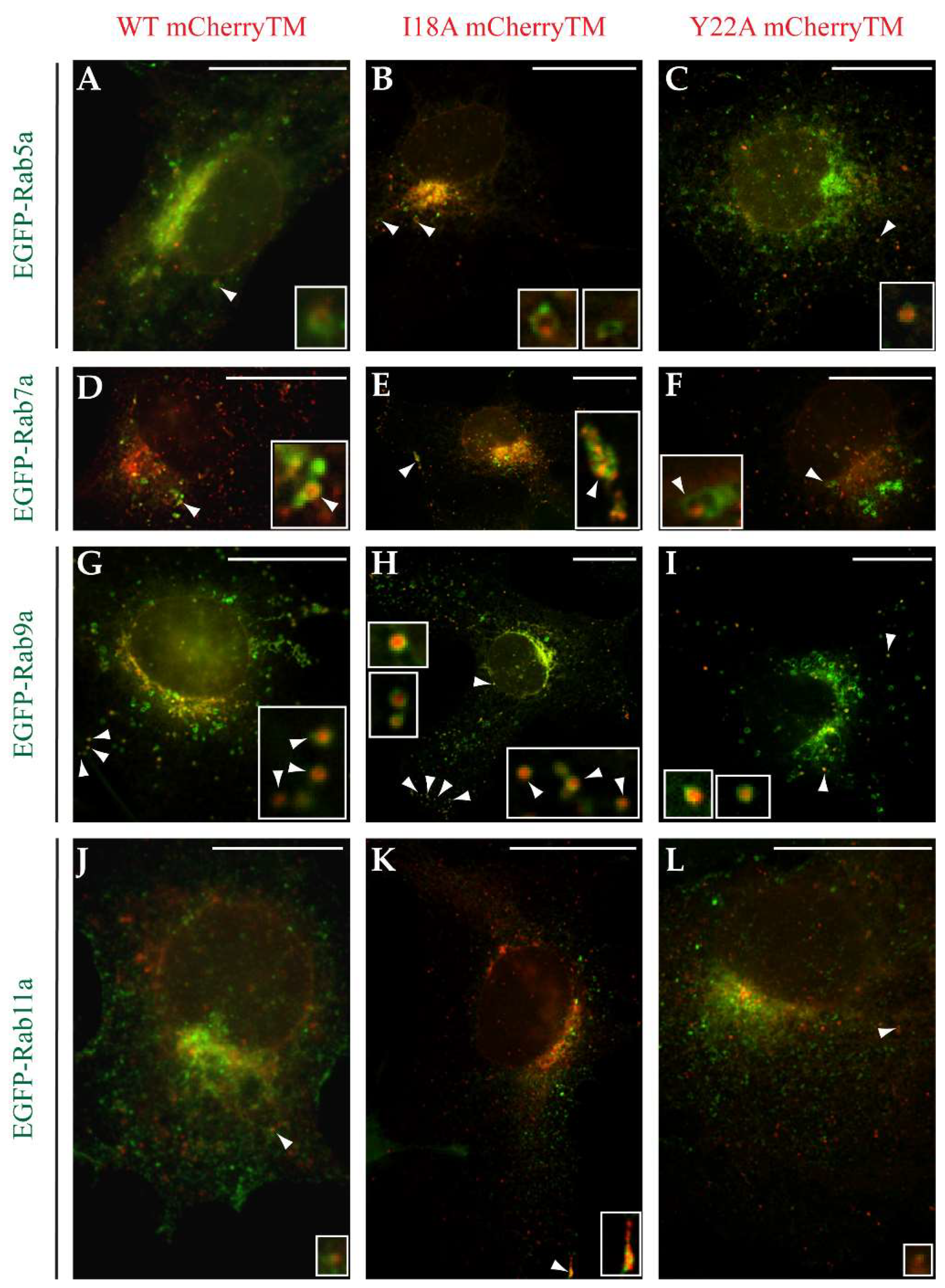
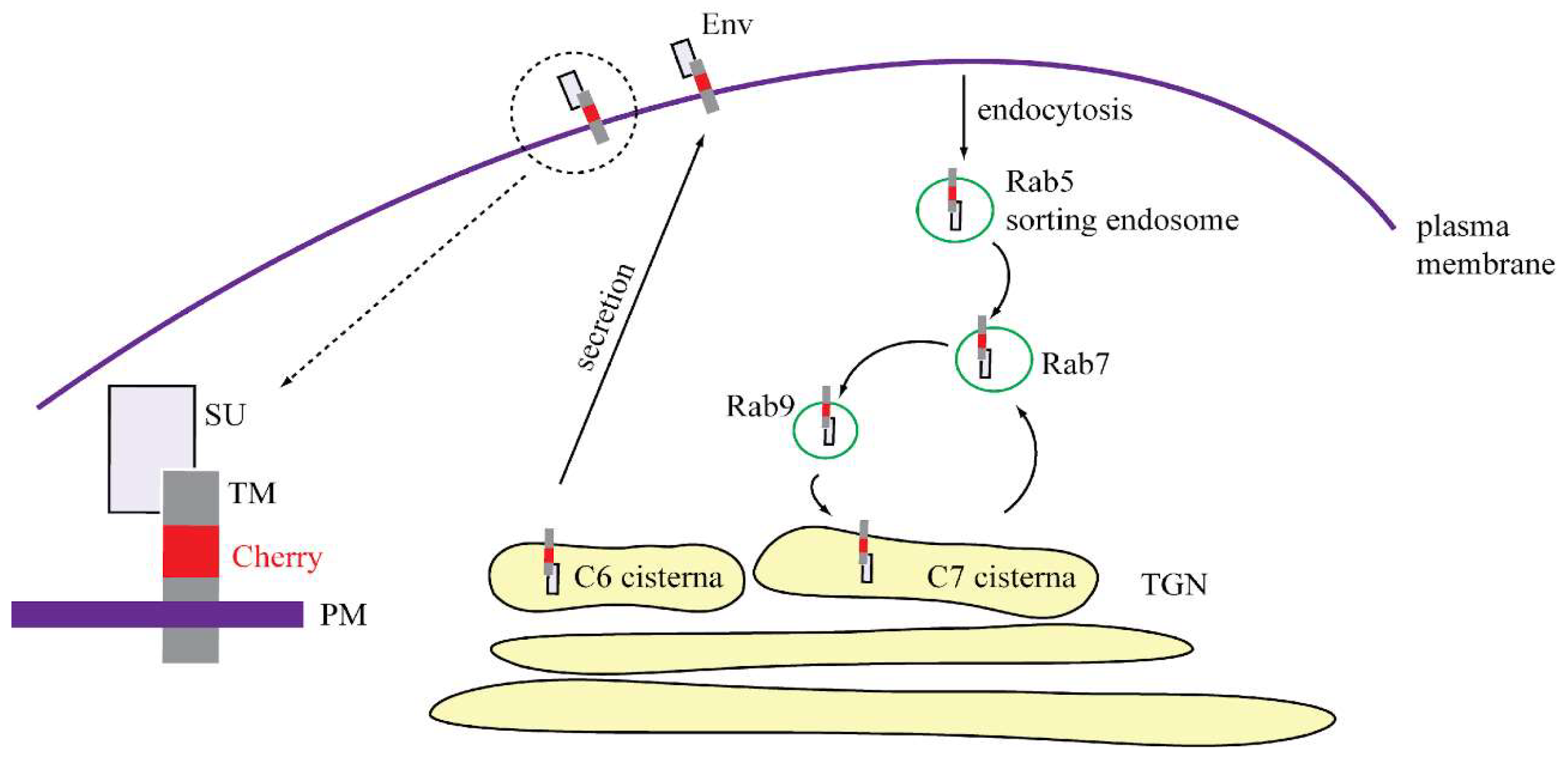
3.4. Characterization of Vesicles Carrying Mason-Pfizer Monkey Virus (M-PMV) Env
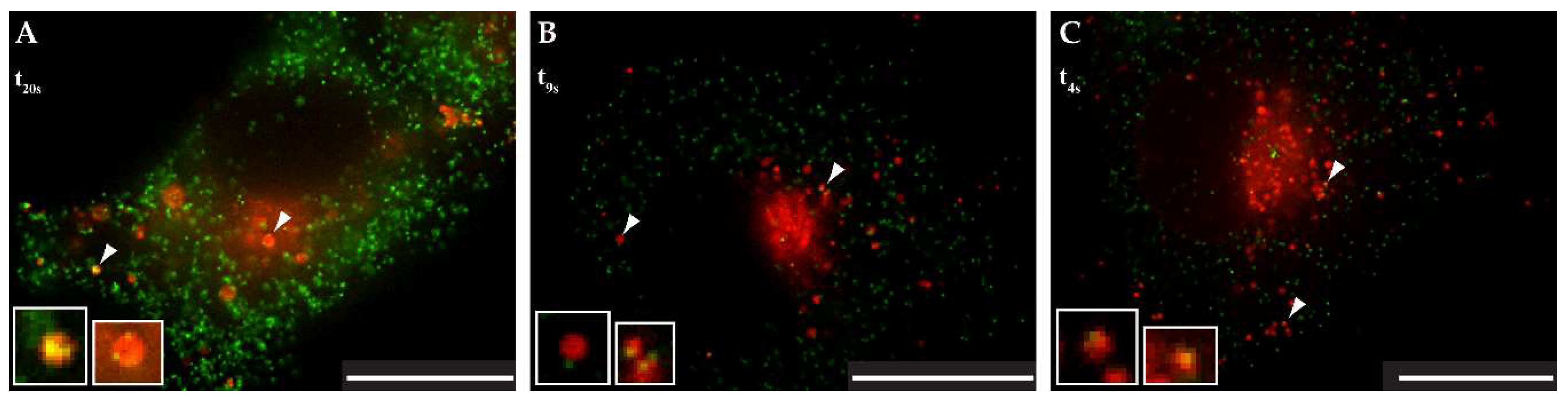
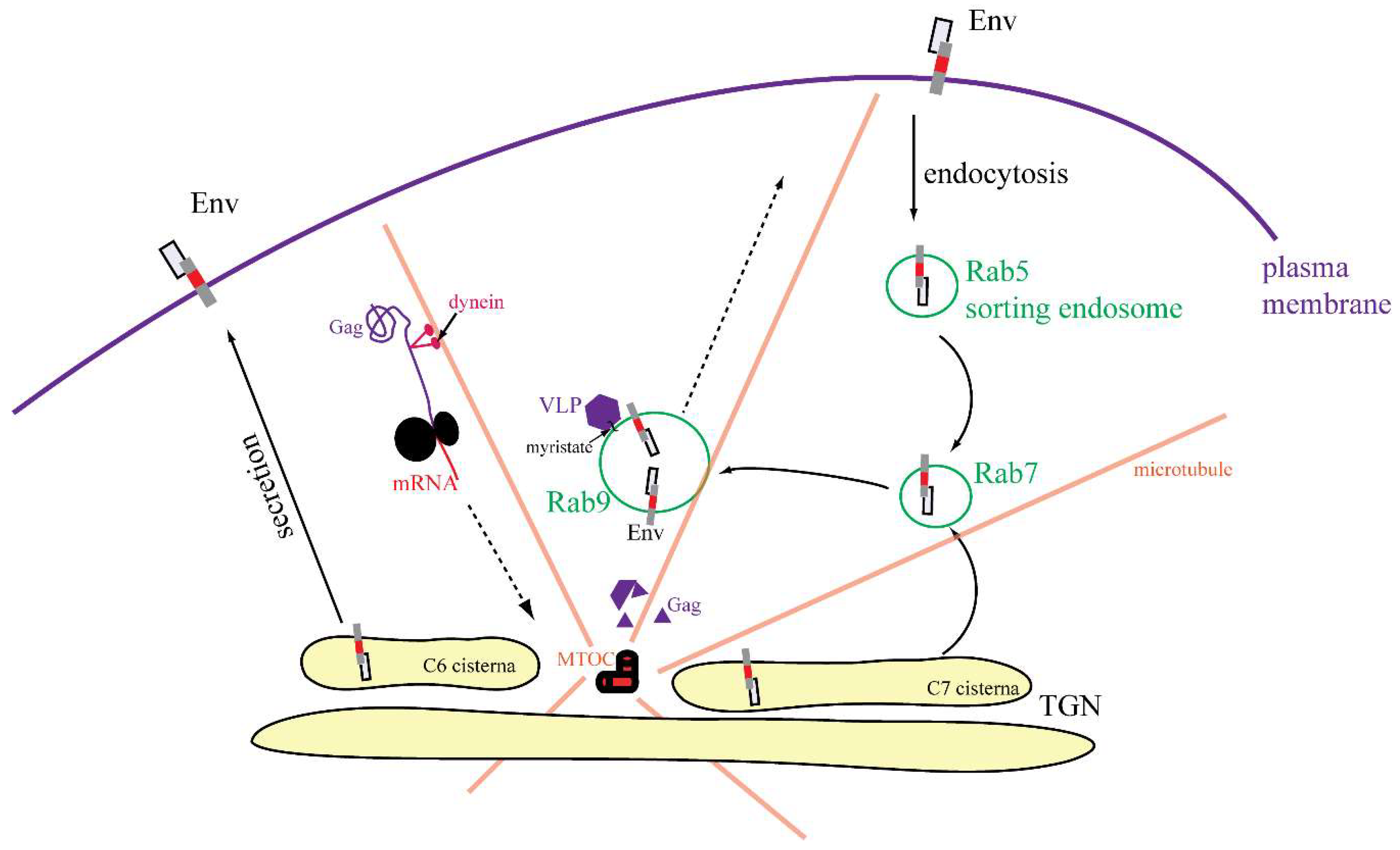
3.5. Live-Cell Imaging of mCherryTM and EGFP-Tagged M-PMV Virus Like Particle
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chopra, H.C.; Mason, M.M. A new virus in a spontaneous mammary tumor of a rhesus monkey. Cancer Res. 1970, 30, 2081–2086. [Google Scholar] [PubMed]
- Kramarsky, B.; Sarkar, N.H.; Moore, D.H. Ultrastructural comparison of a virus from a Rhesus-monkey mammary carcinoma with four oncogenic RNA viruses. Proc. Natl. Acad. Sci. USA 1971, 68, 1603–1607. [Google Scholar] [CrossRef] [PubMed]
- Kohoutova, Z.; Rumlova, M.; Andreansky, M.; Sakalian, M.; Hunter, E.; Pichova, I.; Ruml, T. The impact of altered polyprotein ratios on the assembly and infectivity of Mason-Pfizer monkey virus. Virology 2009, 384, 59–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlach, J.; Lipov, J.; Rumlova, M.; Veverka, V.; Lang, J.; Srb, P.; Knejzlik, Z.; Pichova, I.; Hunter, E.; Hrabal, R.; et al. D-retrovirus morphogenetic switch driven by the targeting signal accessibility to Tctex-1 of dynein. Proc. Natl. Acad. Sci. USA 2008, 105, 10565–10570. [Google Scholar] [CrossRef] [PubMed]
- Sfakianos, J.N.; LaCasse, R.A.; Hunter, E. The M-PMV cytoplasmic targeting-retention signal directs nascent Gag polypeptides to a pericentriolar region of the cell. Traffic 2003, 4, 660–670. [Google Scholar] [CrossRef] [PubMed]
- Sakalian, M.; Hunter, E. Separate assembly and transport domains within the Gag precursor of Mason-Pfizer monkey virus. J. Virol. 1999, 73, 8073–8082. [Google Scholar] [PubMed]
- Hong, S.; Choi, G.; Park, S.; Chung, A.S.; Hunter, E.; Rhee, S.S. Type D retrovirus Gag polyprotein interacts with the cytosolic chaperonin TRiC. J. Virol. 2001, 75, 2526–2534. [Google Scholar] [CrossRef] [PubMed]
- Ulbrich, P.; Haubova, S.; Nermut, M.V.; Hunter, E.; Rumlova, M.; Ruml, T. Distinct roles for nucleic acid in in vitro assembly of purified Mason-Pfizer monkey virus CANC proteins. J. Virol. 2006, 80, 7089–7099. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Dubay, S.R.; Hunter, E. A Tyrosine Motif in the Cytoplasmic Domain of Mason-Pfizer Monkey Virus Is Essential for the Incorporation of Glycoprotein into Virions. J. Virol. 2003, 77, 5192–5200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sfakianos, J.N.; Hunter, E. M-PMV capsid transport is mediated by Env/Gag interactions at the pericentriolar recycling endosome. Traffic 2003, 4, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.E.; Clark, J.; Grznarova, P.; Wen, X.; Lacasse, R.; Ruml, T.; Spearman, P.; Hunter, E. Direct evidence for intracellular anterograde co-transport of M-PMV Gag and Env on microtubules. Virology 2014, 449, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.; Grznarova, P.; Stansell, E.; Diehl, W.; Lipov, J.; Spearman, P.; Ruml, T.; Hunter, E. A Mason-Pfizer Monkey Virus Gag-GFP Fusion Vector Allows Visualization of Capsid Transport in Live Cells and Demonstrates a Role for Microtubules. PLoS ONE 2013, 8, e83863. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, A.; Dominguez, M.; Puri, V.; Sharma, D.K.; Narita, K.; Wheatley, C.W.; Marks, D.L.; Pagano, R.E. Rab proteins mediate Golgi transport of caveola-internalized glycosphingolipids and correct lipid trafficking in Niemann-Pick C cells. J. Clin. Investig. 2002, 109, 1541–1550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, C.; Micoli, K.; Bauerova, H.; Pichova, I.; Hunter, E. Amino acid residues in the cytoplasmic domain of the Mason-Pfizer monkey virus glycoprotein critical for its incorporation into virions. J. Virol. 2005, 79, 11559–11568. [Google Scholar] [CrossRef] [PubMed]
- Rumlova, M.; Ruml, T.; Pohl, J.; Pichova, I. Specific in vitro cleavage of Mason-Pfizer monkey virus capsid protein: Evidence for a potential role of retroviral protease in early stages of infection. Virology 2003, 310, 310–318. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Rueden, C.T.; Hiner, M.C.; Eliceiri, K.W. The ImageJ ecosystem: An open platform for biomedical image analysis. Mol. Reprod. Dev. 2015, 82, 518–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costes, S.V.; Daelemans, D.; Cho, E.H.; Dobbin, Z.; Pavlakis, G.; Lockett, S. Automatic and quantitative measurement of protein-protein colocalization in live cells. Biophys. J. 2004, 86, 3993–4003. [Google Scholar] [CrossRef] [PubMed]
- Manders, E.M.M.; Verbeek, F.J.; Aten, J.A. Measurement of co-localization of objects in dual-colour confocal images. J. Microsc. 1993, 169, 375–382. [Google Scholar] [CrossRef]
- Li, Q.; Lau, A.; Morris, T.J.; Guo, L.; Fordyce, C.B.; Stanley, E.F. A syntaxin 1, Galpha(o), and N-type calcium channel complex at a presynaptic nerve terminal: Analysis by quantitative immunocolocalization. J. Neurosci. Off. J. Soc. Neurosci. 2004, 24, 4070–4081. [Google Scholar] [CrossRef] [PubMed]
- Bohmova, K.; Hadravova, R.; Stokrova, J.; Tuma, R.; Ruml, T.; Pichova, I.; Rumlova, M. Effect of dimerizing domains and basic residues on in vitro and in vivo assembly of Mason-Pfizer monkey virus and human immunodeficiency virus. J. Virol. 2010, 84, 1977–1988. [Google Scholar] [CrossRef] [PubMed]
- Muranyi, W.; Malkusch, S.; Muller, B.; Heilemann, M.; Krausslich, H.G. Super-resolution microscopy reveals specific recruitment of HIV-1 envelope proteins to viral assembly sites dependent on the envelope C-terminal tail. PLoS Pathog. 2013, 9, e1003198. [Google Scholar] [CrossRef] [PubMed]
- Blot, V.; Lopez-Verges, S.; Breton, M.; Pique, C.; Berlioz-Torrent, C.; Grange, M.P. The conserved dileucine- and tyrosine-based motifs in MLV and MPMV envelope glycoproteins are both important to regulate a common Env intracellular trafficking. Retrovirology 2006, 3, 62. [Google Scholar] [CrossRef] [PubMed]
- Bucci, C.; Parton, R.G.; Mather, I.H.; Stunnenberg, H.; Simons, K.; Hoflack, B.; Zerial, M. The small GTPase rab5 functions as a regulatory factor in the early endocytic pathway. Cell 1992, 70, 715–728. [Google Scholar] [CrossRef] [Green Version]
- Vanlandingham, P.A.; Ceresa, B.P. Rab7 regulates late endocytic trafficking downstream of multivesicular body biogenesis and cargo sequestration. J. Boil. Chem. 2009, 284, 12110–12124. [Google Scholar] [CrossRef] [PubMed]
- Barbero, P.; Bittova, L.; Pfeffer, S.R. Visualization of Rab9-mediated vesicle transport from endosomes to the trans-Golgi in living cells. J. Cell Boil. 2002, 156, 511–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ullrich, O.; Reinsch, S.; Urbe, S.; Zerial, M.; Parton, R.G. Rab11 regulates recycling through the pericentriolar recycling endosome. J. Cell Boil. 1996, 135, 913–924. [Google Scholar] [CrossRef] [Green Version]
- Postler, T.S.; Bixby, J.G.; Desrosiers, R.C.; Yuste, E. Systematic analysis of intracellular trafficking motifs located within the cytoplasmic domain of simian immunodeficiency virus glycoprotein gp41. PLoS ONE 2014, 9, e114753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Postler, T.S.; Desrosiers, R.C. The tale of the long tail: The cytoplasmic domain of HIV-1 gp41. J. Virol. 2013, 87, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Postler, T.S.; Desrosiers, R.C. The cytoplasmic domain of the HIV-1 glycoprotein gp41 induces NF-kappaB activation through TGF-beta-activated kinase 1. Cell Host Microbe 2012, 11, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Tedbury, P.R.; Freed, E.O. The cytoplasmic tail of retroviral envelope glycoproteins. Prog. Mol. Boil. Transl. Sci. 2015, 129, 253–284. [Google Scholar]
- Sonigo, P.; Barker, C.; Hunter, E.; Wain-Hobson, S. Nucleotide sequence of Mason-Pfizer monkey virus: An immunosuppressive D-type retrovirus. Cell 1986, 45, 375–385. [Google Scholar] [CrossRef]
- Rhee, S.S.; Hunter, E. Amino-Acid Substitutions within the Matrix Protein of Type-D Retroviruses Affect Assembly, Transport and Membrane Association of a Capsid. EMBO J. 1991, 10, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.S.; Hunter, E. Myristylation Is Required for Intracellular-Transport but Not for Assembly of D-Type Retrovirus Capsids. J. Virol. 1987, 61, 1045–1053. [Google Scholar] [PubMed]
- Murakami, T.; Freed, E.O. Genetic evidence for an interaction between human immunodeficiency virus type 1 matrix and alpha-helix 2 of the gp41 cytoplasmic tail. J. Virol. 2000, 74, 3548–3554. [Google Scholar] [CrossRef] [PubMed]
- Freed, E.O.; Martin, M.A. Virion incorporation of envelope glycoproteins with long but not short cytoplasmic tails is blocked by specific, single amino acid substitutions in the human immunodeficiency virus type 1 matrix. J. Virol. 1995, 69, 1984–1989. [Google Scholar] [PubMed]
- Mammano, F.; Kondo, E.; Sodroski, J.; Bukovsky, A.; Gottlinger, H.G. Rescue of human immunodeficiency virus type 1 matrix protein mutants by envelope glycoproteins with short cytoplasmic domains. J. Virol. 1995, 69, 3824–3830. [Google Scholar] [PubMed]
- Ladinsky, M.S.; Kremer, J.R.; Furcinitti, P.S.; McIntosh, J.R.; Howell, K.E. HVEM tomography of the trans-Golgi network: Structural insights and identification of a lace-like vesicle coat. J. Cell Boil. 1994, 127, 29–38. [Google Scholar] [CrossRef]
- Ladinsky, M.S.; Mastronarde, D.N.; McIntosh, J.R.; Howell, K.E.; Staehelin, L.A. Golgi structure in three dimensions: Functional insights from the normal rat kidney cell. J. Cell Boil. 1999, 144, 1135–1149. [Google Scholar] [CrossRef]
- Chia, P.Z.; Gasnereau, I.; Lieu, Z.Z.; Gleeson, P.A. Rab9-dependent retrograde transport and endosomal sorting of the endopeptidase furin. J. Cell Sci. 2011, 124, 2401–2413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, J.; Keller, P.; Stelzer, E.H. Spatial partitioning of secretory cargo from Golgi resident proteins in live cells. BMC Cell Boil. 2001, 2, 19. [Google Scholar]
- Hunter, E.; Swanstrom, R. Retrovirus envelope glycoproteins. Curr. Top. Microbiol. Immunol. 1990, 157, 187–253. [Google Scholar] [PubMed]
- Prchal, J.; Srb, P.; Hunter, E.; Ruml, T.; Hrabal, R. The structure of myristoylated Mason-Pfizer monkey virus matrix protein and the role of phosphatidylinositol-(4,5)-bisphosphate in its membrane binding. J. Mol. Boil. 2012, 423, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, D.; Soldati, T.; Riederer, M.A.; Goda, Y.; Zerial, M.; Pfeffer, S.R. Rab9 functions in transport between late endosomes and the trans Golgi network. EMBO J. 1993, 12, 677–682. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.K.; Choudhury, A.; Singh, R.D.; Wheatley, C.L.; Marks, D.L.; Pagano, R.E. Glycosphingolipids internalized via caveolar-related endocytosis rapidly merge with the clathrin pathway in early endosomes and form microdomains for recycling. J. Boil. Chem. 2003, 278, 7564–7572. [Google Scholar] [CrossRef] [PubMed]
- Cheryl Chia, P.Z.; Gleeson, P.A. The Regulation of Endosome-to-Golgi Retrograde Transport by Tethers and Scaffolds. Traffic 2011, 12, 939–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patterson, G.H.; Hirschberg, K.; Polishchuk, R.S.; Gerlich, D.; Phair, R.D.; Lippincott-Schwartz, J. Transport through the Golgi apparatus by rapid partitioning within a two-phase membrane system. Cell 2008, 133, 1055–1067. [Google Scholar] [CrossRef] [PubMed]
- Bonifacino, J.S.; Glick, B.S. The mechanisms of vesicle budding and fusion. Cell 2004, 116, 153–166. [Google Scholar] [CrossRef]
- Bonifacino, J.S.; Traub, L.M. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu. Rev. Biochem. 2003, 72, 395–447. [Google Scholar] [CrossRef] [PubMed]
- Stansell, E.; Apkarian, R.; Haubova, S.; Diehl, W.E.; Tytler, E.M.; Hunter, E. Basic residues in the Mason-Pfizer monkey virus gag matrix domain regulate intracellular trafficking and capsid-membrane interactions. J. Virol. 2007, 81, 8977–8988. [Google Scholar] [CrossRef] [PubMed]
- Dostalkova, A.; Kaufman, F.; Krizova, I.; Kultova, A.; Strohalmova, K.; Hadravova, R.; Ruml, T.; Rumlova, M. Mutations in the Basic Region of the Mason-Pfizer Monkey Virus Nucleocapsid Protein Affect Reverse Transcription, Genomic RNA Packaging, and the Virus Assembly Site. J. Virol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Sbalzarini, I.F.; Koumoutsakos, P. Feature point tracking and trajectory analysis for video imaging in cell biology. J. Struct. Boil. 2005, 151, 182–195. [Google Scholar] [CrossRef] [PubMed]
- Chenouard, N.; Smal, I.; de Chaumont, F.; Maska, M.; Sbalzarini, I.F.; Gong, Y.; Cardinale, J.; Carthel, C.; Coraluppi, S.; Winter, M.; et al. Objective comparison of particle tracking methods. Nat. Methods 2014, 11, 281–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]









© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prokšová, P.G.; Lipov, J.; Zelenka, J.; Hunter, E.; Langerová, H.; Rumlová, M.; Ruml, T. Mason-Pfizer Monkey Virus Envelope Glycoprotein Cycling and Its Vesicular Co-Transport with Immature Particles. Viruses 2018, 10, 575. https://doi.org/10.3390/v10100575
Prokšová PG, Lipov J, Zelenka J, Hunter E, Langerová H, Rumlová M, Ruml T. Mason-Pfizer Monkey Virus Envelope Glycoprotein Cycling and Its Vesicular Co-Transport with Immature Particles. Viruses. 2018; 10(10):575. https://doi.org/10.3390/v10100575
Chicago/Turabian StyleProkšová, Petra Grznárová, Jan Lipov, Jaroslav Zelenka, Eric Hunter, Hana Langerová, Michaela Rumlová, and Tomáš Ruml. 2018. "Mason-Pfizer Monkey Virus Envelope Glycoprotein Cycling and Its Vesicular Co-Transport with Immature Particles" Viruses 10, no. 10: 575. https://doi.org/10.3390/v10100575