Targeted Delivery of Protein Drugs by Nanocarriers

Abstract
:
| 1. Introduction................................................................................................ | 1929 |
| 2. Challenges and limitations for the delivery of protein drugs................. | 1930 |
| 3. Nanocarriers............................................................................................... | 1931 |
| 3.1. Liposomes........................................................................................... | 1933 |
| 3.2. Virosomes........................................................................................... | 1936 |
| 3.3. Solid Lipid Nanoparticles.................................................................. | 1937 |
| 3.4. Polymeric Nanoparticles................................................................... | 1940 |
| 3.5. Protein Conjugates............................................................................. | 1948 |
| 4. Targeting strategies and applications....................................................... | 1950 |
| 4.1. Topical Application............................................................................ | 1950 |
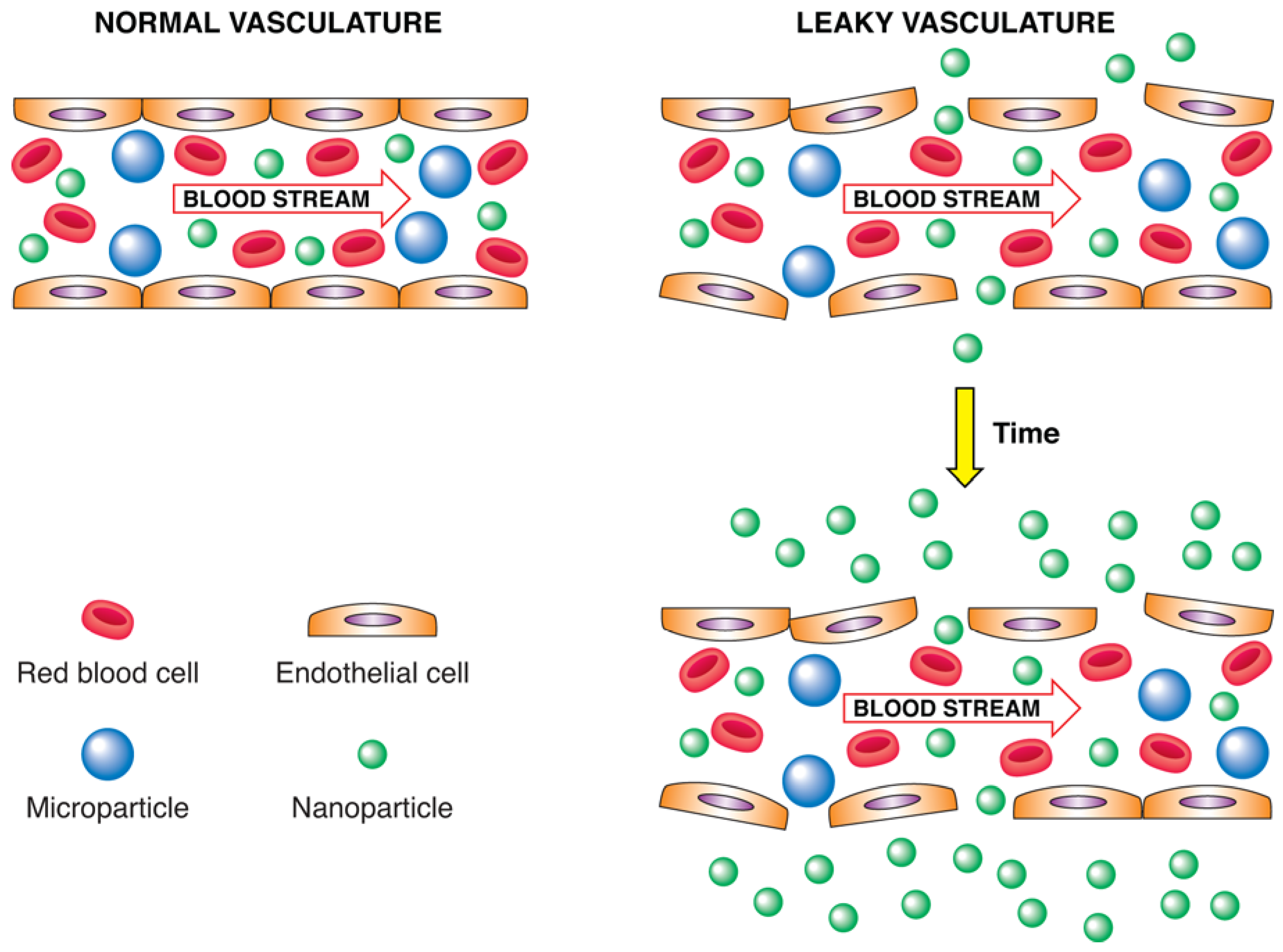
| 4.2. Enhanced Permeability and Retention Effect.................................. | 1951 |
| 4.3. Physical Targeting.............................................................................. | 1953 |
| 4.4. Molecular Targeting........................................................................... | 1956 |
| 5. Concluding remarks.................................................................................. | 1961 |
| Acknowledgements....................................................................................... | 1962 |
| References..................................................................................................... | 1962 |
1. Introduction
2. Challenges and Limitations for the Delivery of Protein Drugs
3. Nanocarriers
- ■
- The ability to encapsulate the drug without deactivating it;
- ■
- The possibility for releasing the drug under proper conditions and according to proper kinetics;
- ■
- A high stability and long circulation time after administration;
- ■
- The capability to actively or passively deliver the drug to a target area.
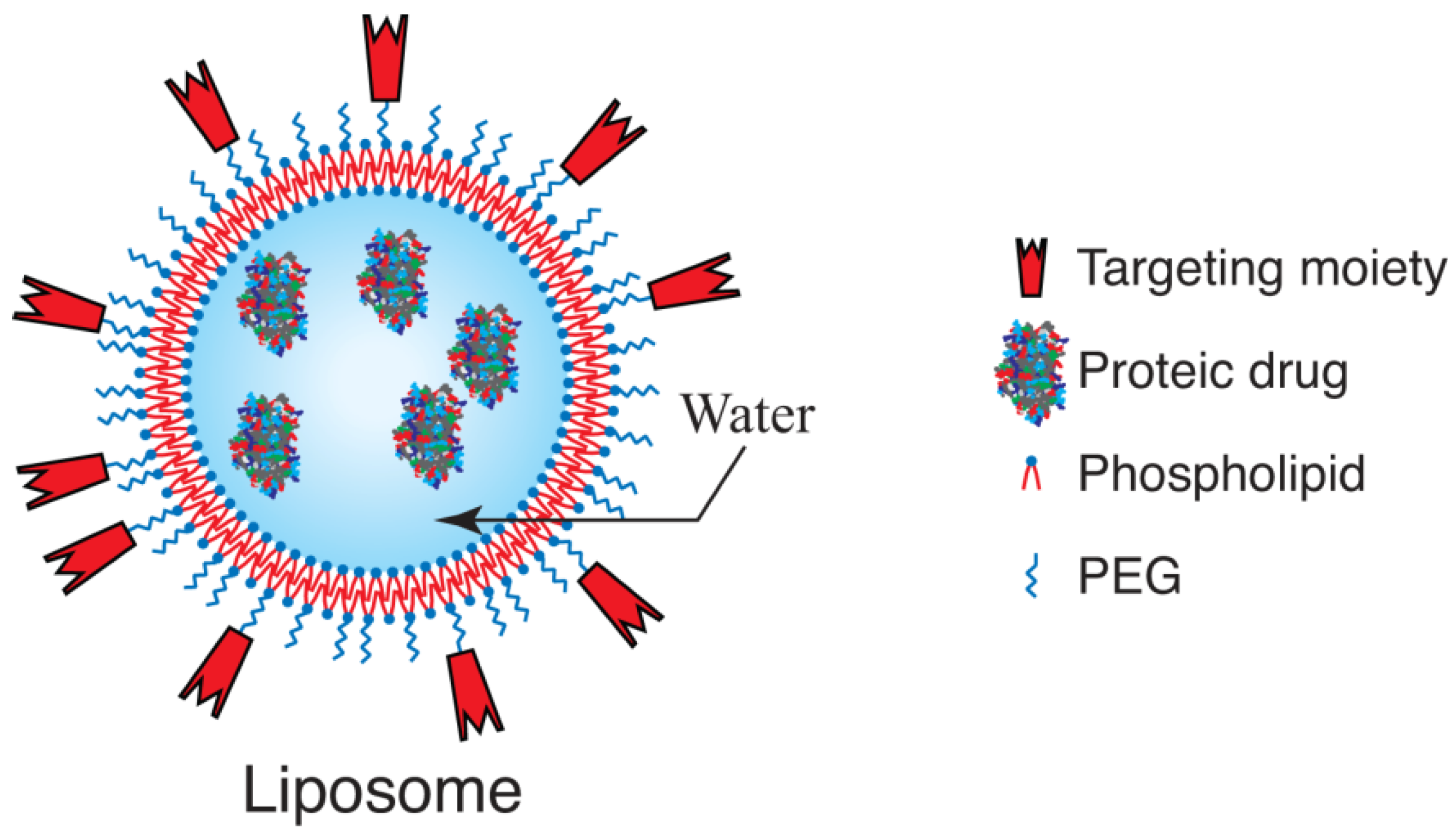
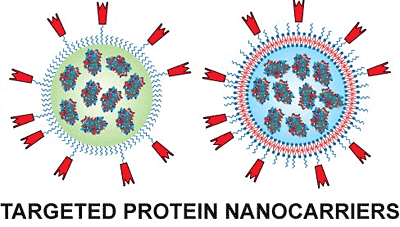
3.1. Liposomes
3.2. Virosomes
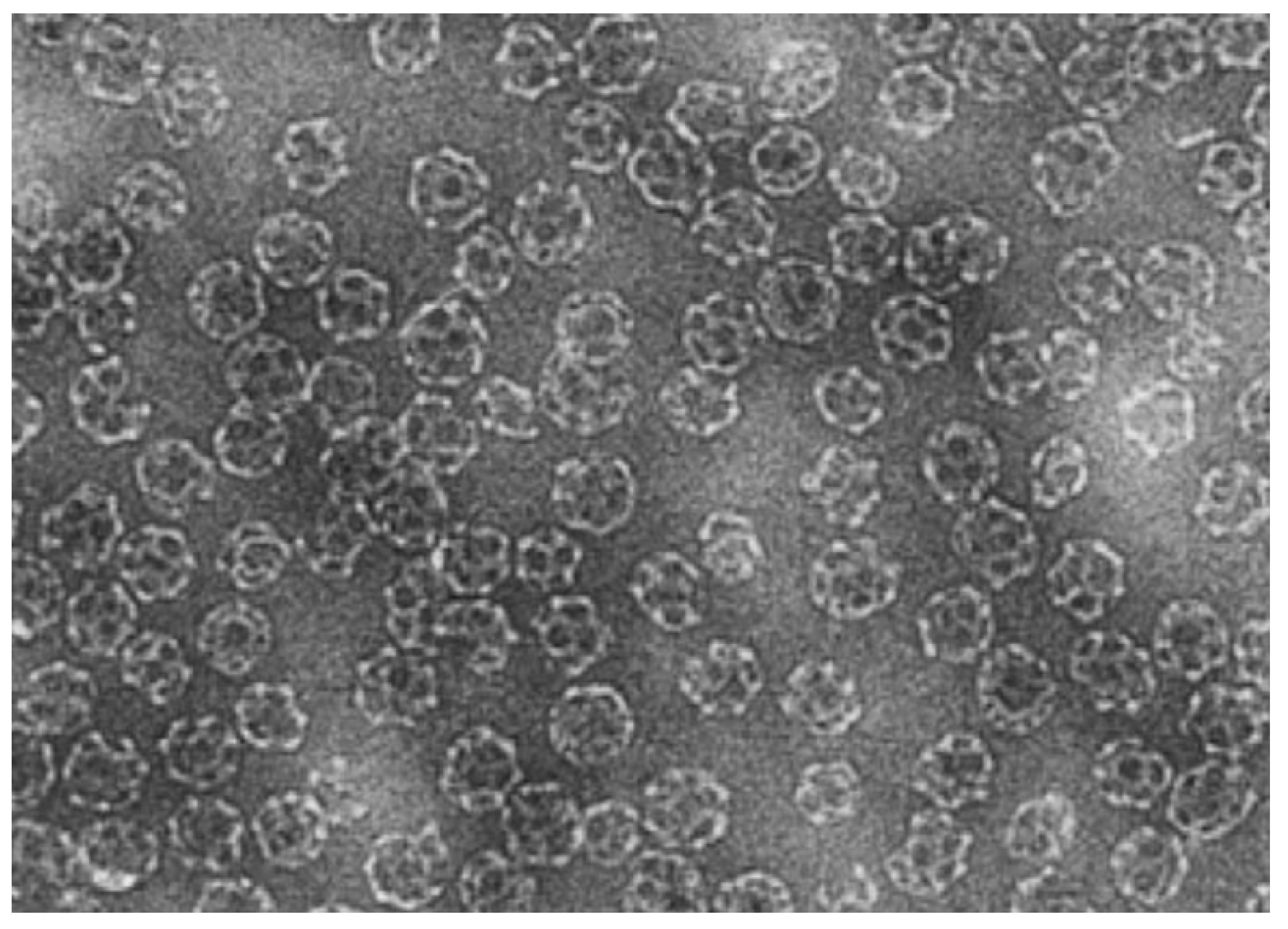
3.3. Solid Lipid Nanoparticles

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide/ Protein | Method of preparation | References |
|---|---|---|
| BSA | Adsorption onto SLN | [123] |
| Calcitonin | Solvent evaporation (w/o/w) | [124] |
| CyA | HPH hot dispersion | [96] |
| CyA | HPH cold dispersion | [125] |
| CyA | Warm microemulsion (o/w) | [126] |
| Gonadorelin | Solvent displacement | [55] |
| HSA | Adsorption onto SLN | [127] |
| Insulin | Solvent evaporation (w/o/w) | [128] |
| Insulin | Warm microemulsion | [129] |
| Insulin | Solvent displacement | [129] |
| Insulin | Supercritical CO2 (PGSS) | [130] |
| [D-Trp-6] LHRH | Warm microemulsion (w/o/w) | [131] |
| Lysozyme | HPH cold dispersion | [120] |
| Ovalbumin | Melt-dispersion (o/w) | [132] |
| Thymopentin | Warm microemulsion | [133] |
3.4. Polymeric Nanoparticles
| Material | Full name | Abbreviation or Commercial name* |
|---|---|---|
| Synthetic homopolymers | Polylactide | PLA |
| Poly(lactide-co-glycolide) | PLGA | |
| Poly(ε-caprolactone) | PCL | |
| Poly(isobutylcyanoacrylate) | PICBA | |
| Poly(isohexylcyanoacrylate) | PIHCA | |
| Poly(n-butylcyanoacrylate) | PBCA | |
| Polyacrylates and polymethacrylates | Eudragit* | |
| Natural polymers | Chitosan | |
| Alginate | ||
| Gelatin | ||
| Albumin | ||
| Copolymers | Polylactide-poly(ethylene glycol) | PLA-PEG |
| Poly(lactide-co-glycolide)-poly(ethylene glycol) | PLGA-PEG | |
| Poly(ε-caprolactone)-poly(ethylene glycol) | PCL-PEG | |
| Poly(hexadecylcyanoacrylate-co-poly(ethylene glycol) cyanoacrylate) | Poly(HDCA-PEGCA) | |
| Colloid stabilizers | Dextran | |
| Pluronic F68 | F68 | |
| Poly(vinyl alcohol) | PVA | |
| Copolymers (see above) | ||
| Tween® 20 or Tween® 80 |

| Method | Advantages | Disadvantages |
|---|---|---|
| By using a colloidal mill | Production of well characterized emulsions, uniform size. Easy to scale-up | High energy for the emulsification process |
| Emulsification–solvent evaporation | Possibility to encapsulate both hydrophilic and lipophilic drugs | Possible coalescence of the nanodroplets during the evaporation process |
| Emulsification–solvent diffusion | Control of nanoparticle size. Easy to scale-up | High volumes of water to be eliminated Leakage of water-soluble drug into the saturated-aqueous external phase |
| Emulsification–reverse salting-out | Minimal stress to fragile drugs. High loading efficiency. Easy to scale-up | Possible incompatibility between the salts and drugs. Purification needed to remove electrolytes |
| By gelation of emulsion droplets | Possibility to use natural macromolecules, hydrophilic and biocompatible | Limited to the encapsulation of hydrophilic drugs |
| Polymerization of alkyl cyanoacrylates | Easy method to obtain core-shell tuned nanoparticles. Control of nanoparticle size sby using surfactant | Possible reaction between the drug and CeVI in the case of radical emulsion polymerization. Purification needed |
| Interfacial polycondensation | Low concentrations of surfactants. Modulation of the nanocapsule thickness by varying the monomer concentration | Limited to the encapsulation of lipophilic drugs Purification needed |
| Nanoprecipitation of a polymer | Simple, fast and reproducible. Low concentrations of surfactants. Easy to scale-up | Low polymer concentration in the organic phase |
| Formation of polyelectrolyte complexes | Easy to achieve. According to the nature of the polyelectrolyte used in excess, either positively or negatively charged nanoparticles can be synthesized | Necessity to optimize the ratio between negatively and positively charged molecules |
| Formation of nanoparticles from neutral nanogels | Organic solvent free method. Controlled drug release | Not yet applicable to hydrophilic drugs |
| Methods based on ionic gelation | Organic solvent free method. Possibility to control drug release encapsulated in the nanoparticles upon the action of a pH or an ion concentration variation stimulus | Possible particle disintegration due to the weakness of the ionic interactions |


3.5. Protein Conjugates
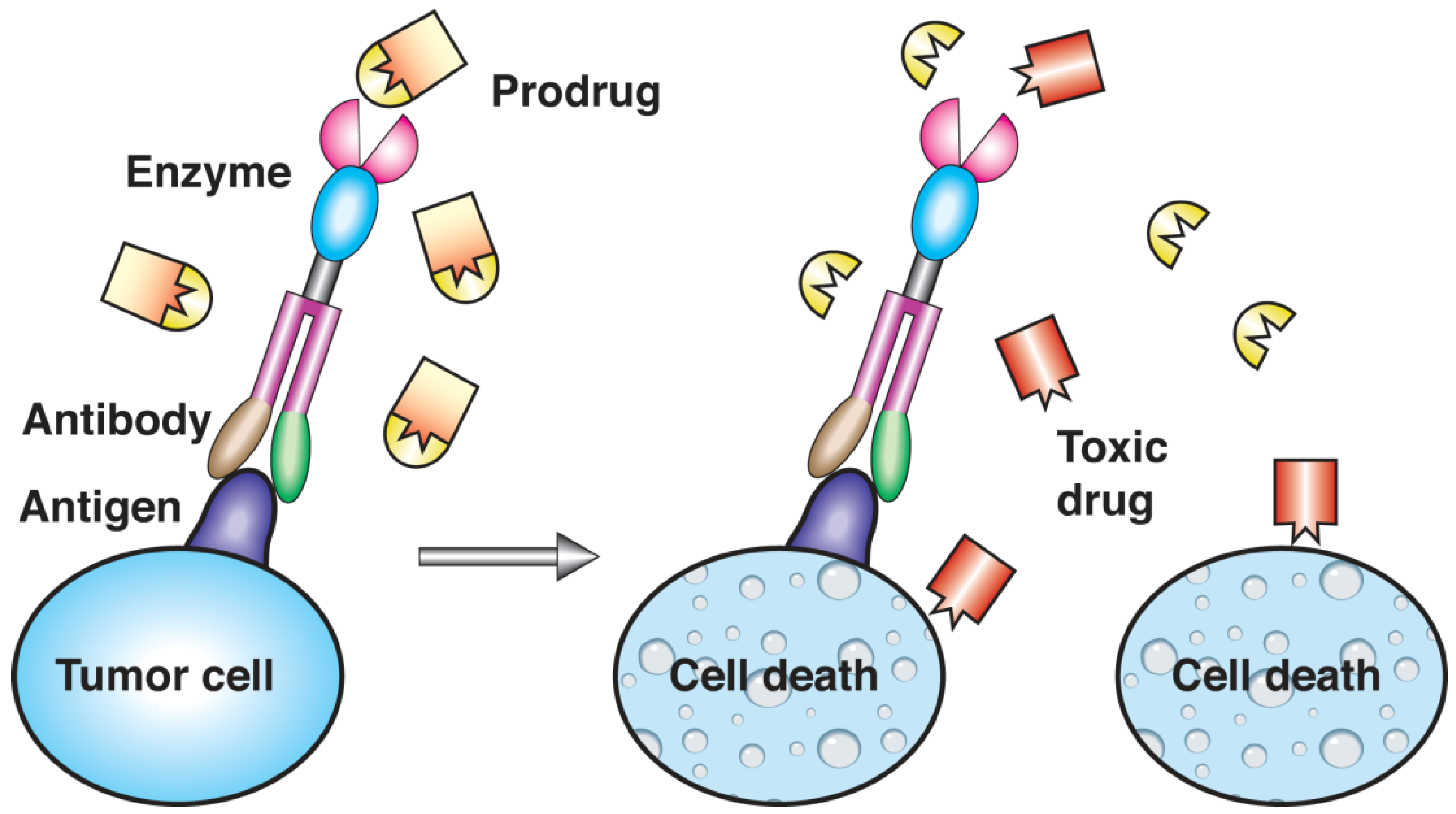
4. Targeting Strategies and Applications
4.1. Topical Application

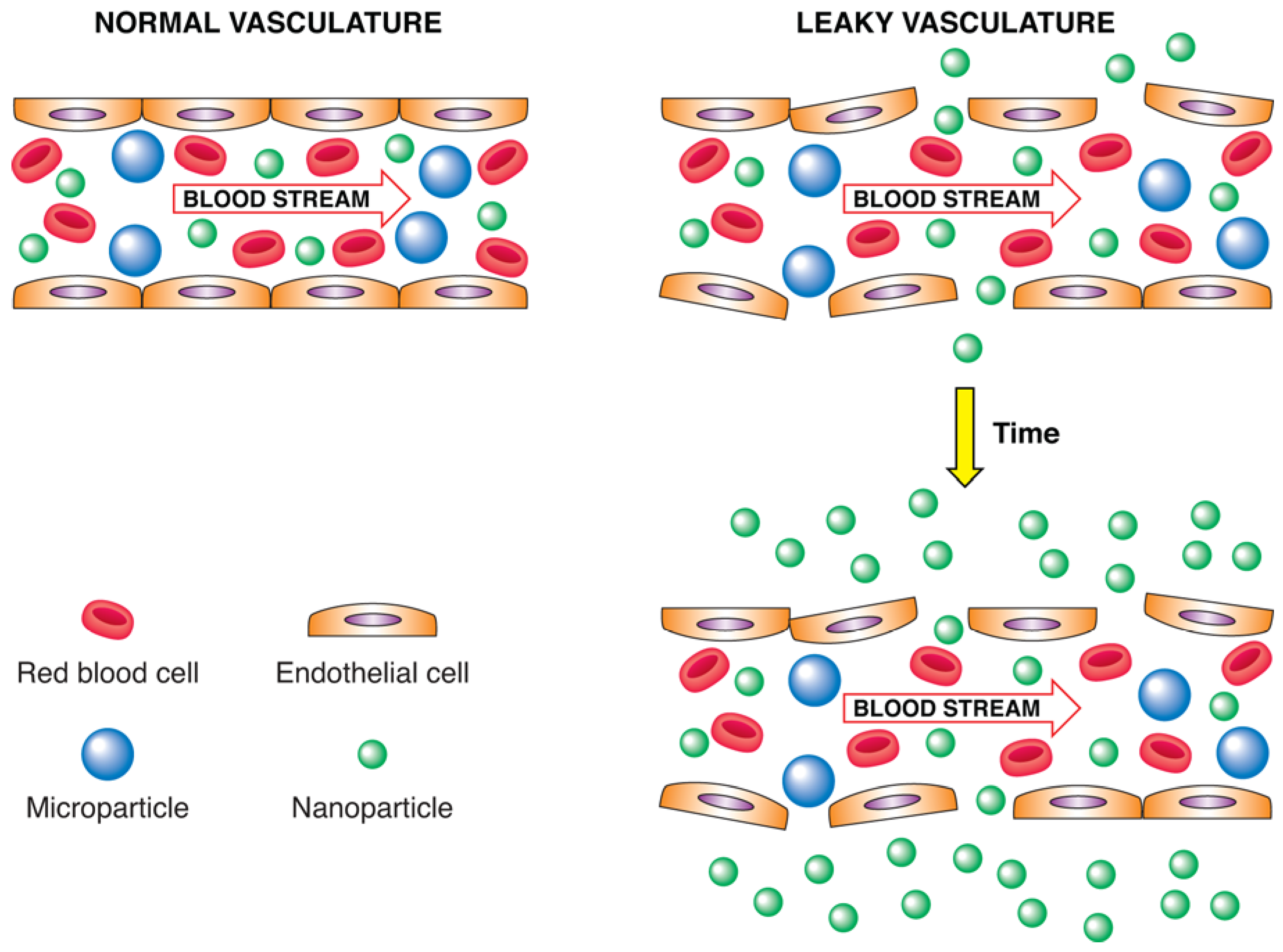
4.2. Enhanced Permeability and Retention Effect
4.3. Physical Targeting

4.4. Molecular Targeting
5. Concluding Remarks
Acknowledgements
References
- Haag, R.; Kratz, F. Polymer therapeutics: Concept and applications. Angew. Chem. Int. 2006, 45, 1198–1215. [Google Scholar] [CrossRef]
- Mizuno, N.; Niwa, T.; Yotsumoto, Y.; Sugiyama, Y. Impact of drug transporter studies on drug discovery and development. Pharm. Rev. 2003, 55, 425–461. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, P. Collected Studies in Immunity; Wiley: New York, NY, USA, 1906; p. 442. [Google Scholar]
- Crommelin, D.J. Formulation of biotech products, including biopharmaceutical considerations. In Pharmaceutical Biotechnology; Crommelin, D.J., Ed.; Taylor and Francis: London, UK, 1997; pp. 67–94. [Google Scholar]
- Salmaso, S.; Bersani, S.; Semenzato, A.; Caliceti, P.J. Nanotechnologies in protein delivery. J. Nanosci. Nanotechol. 2006, 6, 1–18. [Google Scholar] [CrossRef]
- Lu, Y.; Yang, J.; Sega, E. Issues related to targeted delivery of proteins and peptides. AAPS J. 2006, 8, E466–E478. [Google Scholar] [CrossRef] [PubMed]
- Violand, B.N.; Siegel, N.R. Peptide and Protein Drug Analysis. In Protein and Peptide Chemical and Physical Stability; Reid, R.E., Ed.; Marcel Dekker: New York, NY, USA, 2000; pp. 257–284. [Google Scholar]
- Lee, V.H.L. Peptide and Protein Drug Delivery; Marcel Dekker: New York, NY, USA, 1991. [Google Scholar]
- Rabkin, R.; Dahl, D.C. Renal Uptake and Disposal of Proteins and Peptides. In Biological Barriers in Protein Delivery; Audus, K.L., Raub, T.J., Eds.; Plenum Press: New York, NY, USA, 1993; pp. 299–338. [Google Scholar]
- Hashida, M.; Nishikawa, M.; Yamashita, F.; Takakura, Y. Cell-specific delivery of genes with glycosylated carriers. Adv. Drug Deliv. Rev. 2001, 52, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Kotto-Kome, A.C.; Fox, S.E.; Wenge, L.; Bing-Bing, Y.; Christensen, R.D.; Calhoun, D.A. Evidence that the granulocyte colony-stimulating factor (G-CSF) receptor plays a role in the pharmacokinetics of G-CSF and PegG-CSF using a G-CSF-R KO model. Pharmacol. Res. 2004, 50, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Fujimori, K.; Covell, D.G.; Fletcher, J.E.; Weinstein, J.N. Modeling analysis of the global and microscopic distribution of immunoglobulin G, F(ab’)2, and Fab in tumors. Cancer Res. 1989, 49, 5656–5663. [Google Scholar] [PubMed]
- Harris, J.M.; Chess, R.B. Effect of pegylation on pharmaceuticals. Nat. Rev. 2003, 2, 214–221. [Google Scholar]
- Mehwar, R. Modulation of the pharmacokinetics and pharmacodynamics of proteins by polyethylene glycol conjugation. J. Pharm. Pharm. Sci. 2000, 3, 125–136. [Google Scholar] [PubMed]
- Caliceti, P.; Veronese, F.M. Pharmacokinetic and biodistribution properties of poly(ethylene glycol)-protein conjugates. Adv. Drug Delivery Rev. 2003, 55, 1261–1277. [Google Scholar] [CrossRef]
- Canelas, D.A.; Herlihy, K.P.; DeSimone, J.M. Top-down particle fabrication: Control of size and shape for diagnostic imaging and drug delivery. WIREs Nanomed. Nanobiotech. 2009, 1, 391–404. [Google Scholar] [CrossRef]
- Alexis, F.; Rhee, J.W.; Richie, J.P.; Radovic-Moreno, A.F.; Langer, R.; Farokhzad, O.C. New frontiers in nanotechnology for cancer treatment. Urol. Oncol. 2008, 26, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Koo, O.M.; Rubinstein, I.; Onyuksel, H. Role of nanotechnology in drug delivery and imaging: A concise review. Nanomedicine 2005, 1, 193–212. [Google Scholar] [CrossRef] [PubMed]
- Mundargi, R.C.; Babu, V.R.; Rangaswamy, V.; Patel, P.; Aminabhavi, T.M. Nano/microtechnologies for delivering macromolecular therapeutics using poly(d,l-lactide-co-glycolide) and its derivatives. J. Control. Release 2008, 125, 193–209. [Google Scholar] [CrossRef] [PubMed]
- Panyam, J.; Labhasetwar, V. Biodegradable nanoparticles for drug and gene delivery to cells and tissue. Adv. Drug Deliv. Rev. 2003, 55, 329–347. [Google Scholar] [CrossRef] [PubMed]
- Chiellini, F.; Piras, A.M.; Errico, C.; Chiellini, E. Micro/nanostructured polymeric systems for biomedical and pharmaceutical applications. Nanomedicine 2008, 3, 367–393. [Google Scholar] [CrossRef] [PubMed]
- Mishima, K. Biodegradable particle formation for drug and gene delivery using supercritical fluid and dense gas. Adv. Drug Deliv. Rev. 2008, 60, 411–432. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.M.; Shive, M.S. Biodegradation and biocompatibility of PLA and PLGA microspheres. Adv. Drug Deliv. Rev. 1997, 28, 5–24. [Google Scholar] [CrossRef] [PubMed]
- Harashima, H.; Kiwada, H. Liposomal targeting and drug delivery: Kinetic consideration. Adv. Drug Deliv. Rev. 1996, 19, 425–444. [Google Scholar] [CrossRef]
- Torchilin, V.P. Immobilization of specific proteins on liposome surface: Systems for drug targeting. In Liposome Technology; Gregoriadis, G., Ed.; CRC Press, Inc.: Boca Raton, FL, USA, 1984; Volume 3, pp. 75–94. [Google Scholar]
- Crommelin, D.J.A.; Daemen, T.; Scherphof, G.L.; Vingerhoeds, M.H.; Heeremans, J.L.M.; Kluft, C.; Storm, G. Liposomes: Vehicles for the targeted and controlled delivery of peptides and proteins. J. Control. Release 1997, 46, 165–175. [Google Scholar] [CrossRef]
- Gerasimov, O.V.; Boomer, J.A.; Qualls, M.M.; Thompson, D.H. Cytosolic drug delivery using pH- and light-sensitive liposomes. Adv. Drug Deliv. Rev. 1999, 38, 317–338. [Google Scholar] [CrossRef] [PubMed]
- Lian, T.; Ho, R.J.Y. Trends and developments in liposome drug delivery systems. J. Pharm. Sci. 2001, 90, 667–680. [Google Scholar] [CrossRef] [PubMed]
- Nobs, L.; Buchegger, F.; Gurny, R.; Allemann, E. Current methods for attaching targeting ligands to liposomes and nanoparticles. J. Pharm. Sci. 2004, 93, 1980–1992. [Google Scholar] [CrossRef] [PubMed]
- Vasir, J.K.; Reddy, M.K.; Labhasetwar, V.D. Nanosystems in drug targeting: Opportunities and challenges. Curr. Nanosci. 2005, 1, 47–64. [Google Scholar] [CrossRef]
- Huang, S.-L. Liposomes in ultrasonic drug and gene delivery. Adv. Drug Deliv. Rev. 2008, 60, 1167–1176. [Google Scholar] [CrossRef] [PubMed]
- Brannon-Peppas, L.; Blanchette, J.O. Nanoparticle and targeted systems for cancer therapy. Adv. Drug Deliv. Rev. 2004, 56, 1649–1659. [Google Scholar] [CrossRef] [PubMed]
- Dziubla, T.D.; Muzykantov, V.R. Synthetic carriers for vascular delivery of protein therapeutics. Biotechnol. Gen. Eng. Rev. 2006, 22, 267–298. [Google Scholar] [CrossRef]
- Roney, C.; Kulkarni, P.; Arora, V.; Antich, P.; Bonte, F.; Wu, A.; Mallikarjuana, N.N.; Manohar, S.; Liang, H.-F.; Kulkarni, A.R.; Sung, H.-W.; Sairam, M.; Aminabhavi, T.M. Targeted nanoparticles for drug delivery through the blood-brain barrier for Alzheimer's disease. J. Control. Release 2005, 108, 193–214. [Google Scholar] [CrossRef] [PubMed]
- Nahar, M.; Dutta, T.; Murgesan, S.; Asthana, A.; Mishra, D.; Rajkumar, V.; Tare, M.; Saraf, S.; Jain, N.K. Functional polymeric nanoparticles: An efficient and promising tool for active delivery of bioactives. Crit. Rev. Ther. Drug Carrier Syst. 2006, 23, 259–318. [Google Scholar] [CrossRef] [PubMed]
- Solaro, R.; Chiellini, F. Nanoparticles for the Targeted Delivery of Peptides and Proteins. In Handbook of Particulate Drug Delivery; Ravi Kumar, M.N.V., Ed.; American Scientific Publisher: New York, NY, USA, 2006; Volume 2, pp. 193–222. [Google Scholar]
- Yang, Y.Y.; Wang, Y.; Powell, R.; Chan, P. Polymeric core-shell nanoparticles for therapeutics. Clin. Exp. Pharmacol. Physiol. 2006, 33, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Husseini, G.A.; Pitt, W.G. Micelles and nanoparticles for ultrasonic drug and gene delivery. Adv. Drug Deliv. Rev. 2008, 60, 1137–1152. [Google Scholar] [CrossRef] [PubMed]
- Solaro, R. Targeted delivery of proteins by nanosized carriers. J. Polym. Sci. A Polym. Chem. 2008, 46, 1–11. [Google Scholar] [CrossRef]
- Woodle, M.C.; Lasic, D.D. Sterically stabilized liposomes. Biochim. Biophys. Acta 1992, 1113, 171–199. [Google Scholar] [CrossRef] [PubMed]
- Romberg, B.; Hennink, W.E.; Storm, G. Sheddable coatings for long-circulating nanoparticles. Pharm. Res. 2008, 25, 55–71. [Google Scholar] [CrossRef] [PubMed]
- Moghimi, S.M.; Szebeni, J. Stealth liposomes and long circulating nanoparticles: Critical issues in pharmacokinetics, opsonization and protein-binding properties. Prog. Lipid Res. 2003, 42, 463–478. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Wu, J.; Sawa, T.; Matsumura, Y.; Hori, K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J. Control. Release 2000, 65, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Hong, R.L.; Huang, C.J.; Tseng, Y.L.; Pang, V.F.; Chen, S.T.; Liu, J.J.; Chang, F.H. Direct comparison of liposomal doxorubicin with or without polyethylene glycol coating in C- 26 tumor-bearing mice: Is surface coating with polyethylene glycol beneficial? Clin. Cancer Res. 1999, 5, 3645–3652. [Google Scholar] [PubMed]
- Adiseshaiah, P.P.; Hall, J.B.; McNeil, S.E. Nanomaterial standards for efficacy and toxicity assessment. WIREs Nanobiomed. Nanotechnol. 2009, 2, 99–112. [Google Scholar] [CrossRef]
- Kong, G.; Braun, R.D.; Dewhirst, M.W. Hyperthermia enables tumor-specific nanoparticle delivery: Effect of particle size. Cancer Res. 2000, 60, 4440–4445. [Google Scholar] [PubMed]
- Maeda, H.; Matsumura, Y. Tumoritropic and lymphotropic principles of macromolecular drugs. Crit. Rev. Ther. Drug Carrier Syst. 1989, 6, 193–210. [Google Scholar] [PubMed]
- Misselwitz, B.; Platzek, J.; Raduchel, B.; Oellinger, J.J.; Weinmann, H.J. Gadofluorine 8: Initial experience with a new contrast medium for interstitial MR lymphography. Magma 1999, 8, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Harivardhan Reddy, L.; Sharma, R.K.; Chuttani, K.; Mishra, A.K.; Murthy, R.S. Influence of administration route on tumor uptake and biodistribution of etoposide loaded solid lipid nanoparticles in Dalton’s lymphoma tumor bearing mice. J. Control. Release 2005, 105, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Visser, C.C.; Stevanović, S.; Voorwinden, L.H.; van Bloois, L.; Gaillard, P.J.; Danhof, M.; Crommelin, D.J.A.; de Boer, A.G. Targeting liposomes with protein drugs to the blood–brain barrier in vitro. Eur. J. Pharm. Sci. 2005, 25, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Rezler, E.M.; Khan, D.R.; Lauer-Fields, J.; Cudic, M.; Baronas-Lowell, D.; Fields, G.B. Targeted drug delivery utilizing protein-like molecular architecture. J. Am. Chem. Soc. 2007, 129, 4961–4972. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.E.; Stevenson, B.R.; Rogers, J.A. Folic acid-PEO-labeled liposomes to improve gastrointestinal absorption of encapsulated agents. J. Control. Release 1999, 60, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.K.; Liu, D.; Maruyama, K.; Takizawa, T. Antibody mediated lung targeting of long-circulating emulsions. J. Pharm. Sci. Technol. 1996, 50, 372–377. [Google Scholar]
- Storm, G.; Oussoren, C.; Peeters, P.A.M.; Barenholz, Y.B. Tolerability of liposomes in vivo. In Liposome Technology, 2nd ed.; Gregoriadis, G., Ed.; CRC Press, Inc.: Boca Raton, FL, USA, 1993; pp. 345–383. [Google Scholar]
- Bajoria, R.; Sooranna, S.R. Liposome as a drug carrier system: Prospects for safer prescribing during pregnancy: A review. Placenta 1998, 19, 265–287. [Google Scholar]
- Bi, R.; Shao, W.; Wang, Q.; Zhang, N. Spray-freeze-dried dry powder inhalation of insulin-loaded liposomes for enhanced pulmonary delivery. J. Drug Target. 2008, 16, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Liguori, L.; Marques, B.; Villegas-Mendez, A.; Rothe, R.; Lenormand, J.-L. Liposomes-mediated delivery of pro-apoptotic therapeutic membrane proteins. J. Control. Release 2008, 126, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Harashima, H; Kiwada, H. Liposome clearance. Biosci. Rep. 2002, 22, 197–224. [Google Scholar] [CrossRef] [PubMed]
- Senior, J.H. Fate and behavior of liposomes in vivo: A review of controlling factors. Crit. Rev. Ther. Drug Carrier Syst. 1987, 3, 123–193. [Google Scholar] [PubMed]
- Fretz, M.M.; Høgset, A.; Koning, G.A.; Jiskoot, W.; Storm, G. Cytosolic delivery of liposomally targeted proteins induced by photochemical internalization. Pharm. Res. 2007, 24, 2040–2047. [Google Scholar] [CrossRef] [PubMed]
- Scherphof, G.; Roerdink, F.; Waite, M.; Parks, J. Disintegration of phosphatidylcholine liposomes in plasma as a result of interaction with high-density lipoproteins. BBA–Gen. Subjects 1978, 542, 296–307. [Google Scholar] [CrossRef]
- Dhoot, N.O.; Wheatley, M.A. Microencapsulated liposomes in controlled drug delivery: Strategies to modulate drug release and eliminate the burst effect. J. Pharm. Sci. 2003, 92, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Veronese, F.M.; Pasut, G. PEGylation, successful approach to drug delivery. Drug Discov. Today 2005, 10, 1451–1458. [Google Scholar] [CrossRef] [PubMed]
- Owens, D.E., III; Peppas, N.A. Opsonization, biodistribution and pharmacokinetics of polymeric nanoparticles. Int. J. Pharm. 2006, 307, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Van Vlerken, L.E.; Vyas, T.K.; Amiji, M.M. Poly(ethylene glycol)-modified nanocarriers for tumor-targeted and intracellular delivery. Pharm. Res. 2007, 24, 1405–1414. [Google Scholar] [CrossRef] [PubMed]
- Käsbauer, M.; Lasic, D.D.; Winterhalter, M. Polymer induced fusion and leakage of small unilamellar phospholipids vescicles: Effect of surface grafted polyethylene-glycol in the presence of free PEG. Chem. Phys. Lipids 1997, 86, 153–159. [Google Scholar] [CrossRef]
- Bedu-Addo, F.K.; Huang, L. Interaction of PEG-phospholipid conjugates with phospholipid implications in liposomal drug delivery. Adv. Drug Deliv. Rev. 1995, 16, 235–247. [Google Scholar] [CrossRef]
- Lasic, D.D.; Needham, D. The ‘stealth’ liposome: A prototypical biomaterial. Chem. Rev. 1995, 95, 2601–2628. [Google Scholar] [CrossRef]
- Mayhew, E.G.; Lasic, D.; Babbar, S.; Martin, F.J. Pharmacokinetics and antitumor activity of epirubicin encapsulated in long-circulating liposomes incorporating a polyethylene glycol-derivatized phospholipid. Int. J. Cancer 1992, 51, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, T.; Chen, F.-A.; Kida, H.; Kunieda, K.; Cuenca, R.E.; Martin, F.J.; Bankert, R.B. Doxorubicin encapsulated in sterically stabilized liposomes is superior to free drug or drug-containing conventional liposomes at suppressing growth and metastases of human lung tumor xenografts. Cancer Res. 1996, 56, 3743–3746. [Google Scholar] [PubMed]
- Judge, A.; McClintock, K.; Phelps, J.R.; MacLachlan, I. Hypersensitivity and loss of disease site targeting caused by antibody responses to PEGylated liposomes. Mol. Ther. 2006, 13, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Woodle, M.C.; Storm, G.; Newman, M.S.; Jekot, J.J.; Collins, L.R.; Martin, F.J.; Szoka, F.C., Jr. Prolonged systemic delivery of peptide drugs by long-circulating liposomes: Illustration with vasopressin in the brattleboro rat. Pharm. Res. 1992, 9, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Kedar, E.; Braun, E.; Rutkowski, Y.; Emanuel, N.; Barenholz, Y. Delivery of cytokines by liposomes. II. Interleukin-2 encapsulated in long-circulating sterically stabilized liposomes: Immunomodulatory and anti-tumor activity in mice. J. Immunother. Emphasis Tumor. Immunol. 1994, 16, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Boerman, O.; Storm, G.; Van Oyen, W.J.G.; Van Bloois, L.; Van Der Meer, J.W.M.; Claessens, R.A.M.J.; Crommelin, D.J.A.; Corstens, F.H.M. Sterically stabilized liposomes labeled with Indium-111 to image focal infection. J. Nucl. Med. 1995, 36, 1639–1644. [Google Scholar] [PubMed]
- Yagi, N.; Yano, Y.; Hatanaka, K.; Yokoyama, Y.; Okuno, H. Synthesis and evaluation of a novel lipid–peptide conjugate for functionalized liposome. Bioorg. Med. Chem. Lett. 2007, 17, 2590–2593. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Akita, H.; Kamiya, H.; Kogure, K.; Yamamoto, T.; Shinohara, Y.; Yamashita, K.; Kobayashi, H.; Kikuchi, H.; Harashima, H. MITO-Porter: A liposome-based carrier system for delivery of macromolecules into mitochondria via membrane fusion. Biochim. Biophys. Acta 2008, 1778, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Kepczyński, M.; Nawalany, K.; Kumorek, M.; Kobierska, A.; Jachimska, B.; Nowakowska, M. Which physical and structural factors of liposome carriers control their drug-loading efficiency? Chem. Phys. Lipids 2008, 155, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.H.; Schaffer, D.V. Advanced targeting strategies for murine retroviral and adeno-associated viral vectors. Adv. Biochem. Eng. Biotech. 2005, 99, 147–167. [Google Scholar]
- Kost, T.A.; Condreay, J.P.; Ames, R.S.; Rees, S.; Romanos, M.A. Implementation of BacMam virus gene delivery technology in a drug discovery setting. Drug Discov. Today 2007, 12, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Eisele, F.; Kuhlmann, J.; Waldmann, H. Synthesis and membrane-binding properties of a characteristic lipopeptide from the membrane-anchoring domain of influenza virus A hemagglutinin. Angew. Chem. Int. Ed. Engl. 2001, 40, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Daemen, T.; De Mare, A.; Bungener, L.; De Jonge, J.; Huckriede, A.; Wilschut, J. Virosomes for antigen and DNA delivery. Adv. Drug Deliv. Rev. 2005, 57, 451–463. [Google Scholar] [CrossRef] [PubMed]
- De Jonge, J.; Holtrop, M.; Wilschut, J.; Huckriede, A. Reconstituted influenza virus envelopes as an efficient carrier system for cellular delivery of small-interfering RNAs. Gene Ther. 2006, 13, 400–411. [Google Scholar] [CrossRef] [PubMed]
- Angel, J.; Chaperot, L.; Molens, J.-P.; Mezin, P.; Amacker, M.; Zurbriggen, R.; Grichine, A.; Plumas, J. Virosome-mediated delivery of tumor antigen to plasmacytoid dendritic cells. Vaccine 2007, 25, 3913–3921. [Google Scholar] [CrossRef] [PubMed]
- Thompson, F.M.; Porter, D.W.; Okitsu, S.L.; Westerfeld, N.; Vogel, D.; Todryk, S.; Poulton, I.; Correa, S.; Hutchings, C.; Berthoud, T.; Dunachie, S.; Andrews, L.; Williams, J.L.; Sinden, R.; Gilbert, S.C.; Pluschke, G.; Zurbriggen, R.; Hill, A.V.S. Evidence of blood stage efficacy with a virosomal malaria vaccine in a phase IIa clinical trial. PLoS ONE 2008, 3, e1493. [Google Scholar] [CrossRef] [PubMed]
- Morein, B.; Hu, K.-F.; Abusugra, I. Current status and potential application of ISCOMs in veterinary medicine. Adv. Drug Deliv. Rev. 2004, 56, 1367–1382. [Google Scholar] [CrossRef] [PubMed]
- Bron, R.; Ortiz, A.; Dijkstra, J.; Stegmann, T.; Wilschut, J. Preparation, properties, and applications of reconstituted influenza virus envelopes (virosomes). Methods Enzymol. 1993, 220, 313–31. [Google Scholar] [PubMed]
- Schoen, P.; Chonn, A.; Cullis, P.R.; Wilschut, J.; Scherrer, P. Gene transfer mediated by fusion protein hemagglutinin reconstituted in cationic lipid vesicles. Gene Ther. 1999, 6, 823–832. [Google Scholar] [CrossRef] [PubMed]
- Fujii, G. To fuse or not to fuse: The effects of electrostatic interactions, hydrophobic forces, and structural amphiphilicity on protein-mediated membrane destabilization. Adv. Drug Deliv. Rev. 1999, 38, 257–277. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E. Application of membrane-active peptides for nonviral gene delivery. Adv. Drug Deliv. Rev. 1999, 38, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Baru, M.; Nahum, O.; Jaaro, H.; Sha’anani, J.; Nur, I. Lysosome-disrupting peptide increases the efficiency of in-vivo gene transfer by liposome-encapsulated DNA. J. Drug Target. 1998, 6, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Kaneda, Y. Virosomes: Evolution of the liposome as a targeted drug delivery system. Adv. Drug Deliv. Rev. 2000, 43, 197–205. [Google Scholar] [CrossRef] [PubMed]
- De Jonge, J.; Leenhouts, J.M.; Holtrop, M.; Schoen, P.; Scherrer, P.; Cullis, P.R.; Wilschut, J.; Huckriede, A. Cellular gene transfer mediated by influenza virosomes with encapsulated plasmid DNA. Biochem. J. 2007, 405, 41–49. [Google Scholar] [PubMed]
- Müller, R.H.; Mehnert, W.; Lucks, J.-S.; Schwarz, C.; Zur Mühlen, A.; Meyhers, H.; Freitas, C.; Rühl, D. Solid lipid nanoparticles (SLN): An alternative colloidal carrier system for controlled drug delivery. Eur. J. Pharm. Biopharm. 1995, 41, 62–69. [Google Scholar]
- Mukherjee, S.; Ray, S.; Thakur, R.S. Solid lipid nanoparticles: A modern formulation approach in drug delivery system. Indian J. Pharm. Sci. 2009, 71, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Hu, W.; Chen, H.; Ni, Q.; Xu, H.; Yang, X. Isotretinoin-loaded solid lipid nanoparticles with skin targeting for topical delivery. Int. J. Pharm. 2007, 328, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Müller, R.H.; Runge, S.A.; Ravelli, V.; Thünemann, A.F.; Mehnert, W.; Souto, E.B. Cyclosporine-loaded solid lipid nanoparticles (SLN®): Drug-lipid physicochemical interactions and characterization of drug incorporation. Eur. J. Pharm. Biopharm. 2008, 68, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chang, X.; Du, D.; Liu, W.; Liu, J.; Weng, T.; Yang, Y.; Xu, H.; Yang, H. Podophyllotoxin-loaded solid lipid nanoparticles for epidermal targeting. J. Control. Release 2006, 110, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Müller, R.H.; Mäder, K.; Gohla, S. Solid lipid nanoparticles (SLN) for controlled drug delivery–A review of the state of the art. Eur. J. Pharm. Biopharm. 2000, 50, 161–177. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Gong, T.; Wang, C.; Zhong, Z.; Zhang, Z. Solid lipid nanoparticles loaded with insulin by sodium cholate-phosphatidylcholine-based mixed micelles: Preparation and characterization. Int. J. Pharm. 2007, 340, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Mehnert, W.; Mäder, K. Solid lipid nanoparticles–Production, characterization and applications. Adv. Drug Deliv. Rev. 2001, 47, 165–197. [Google Scholar] [CrossRef] [PubMed]
- Sjöström, B.; Bergenstahl, B. Preparation of submicron drug particles in lecithin-stabilized o/w emulsions. I. Model studies of the precipitation of cholesteryl acetate. Int. J. Pharm. 1992, 88, 53–62. [Google Scholar] [CrossRef]
- Schubert, M.A.; Müller-Goymann, C.C. Solvent injection as a new approach for manufacturing lipid nanoparticles–Evaluation of the method and process parameters. Eur. J. Pharm. Biopharm. 2003, 55, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.Q.; Yuan, H.; Zhang, H.H.; Fang, M. Preparation of solid lipid nanoparticles with clobetasol propionate by a novel solvent diffusion method in aqueous system and physicochemical characterization. Int. J. Pharm. 2002, 239, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Trotta, M.; Debernardi, F.; Caputo, O. Preparation of solid lipid nanoparticles by a solvent emulsification-diffusion technique. Int. J. Pharm. 2003, 257, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Puglia, C.; Blasi, P.; Rizza, L.; Schoubben, A.; Bonina, F.; Rossi, C.; Ricci, M. Lipid nanoparticles for prolonged topical delivery: An in vitro and in vivo investigation. Int. J. Pharm. 2008, 357, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Joshi, M.D.; Muller, R.H. Lipid nanoparticles for parenteral delivery of actives. Eur. J. Pharm. Biopharm. 2009, 71, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Salmaso, S.; Bersani, S.; Elvassore, N.; Bertucco, A.; Caliceti, P. Biopharmaceutical characterisation of insulin and recombinant human growth hormone loaded lipid submicron particles produced by supercritical gas. Int. J. Pharm. 2009, 379, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Caliceti, P.; Salmaso, S.; Elvassore, N.; Bertucco, A. Effective protein release from PEG/PLA nano-particles produced by compressed gas anti-solvent precipitation techniques. J. Control. Release 2004, 94, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Davies, O.R.; Lewis, A.L.; Whitaker, M.J.; Tai, H.; Shakesheff, K.M.; Howdle, S.M. Applications of supercritical CO2 in the fabrication of polymer systems for drug delivery and tissue engineering. Adv. Drug Deliv. Rev. 2008, 60, 373–387. [Google Scholar] [CrossRef] [PubMed]
- Reverchon, E.; Adami, R.; Cardea, S.; Della Porta, G. Supercritical fluids processing of polymers for pharmaceutical and medical applications. J. Supercrit. Fluids 2009, 47, 484–492. [Google Scholar] [CrossRef]
- Salmaso, S.; Elvassore, N.; Bertucco, A.; Caliceti, P. Production of solid lipid submicron particles for protein delivery using a novel supercritical gas-assisted melting atomization process. J. Pharm. Sci. 2009, 98, 640–650. [Google Scholar] [CrossRef] [PubMed]
- Lippacher, A.; Müller, R.H.; Mäder, K. Liquid and semisolid SLN™ dispersions for topical application: Rheological characterization. Eur. J. Pharm. Biopharm. 2004, 58, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Siekmann, B.; Bunjes, H.; Koch, M.H.J.; Westesen, K. Preparation and structural investigations of colloidal dispersions prepared from cubic monoglyceride-water phases. Int. J. Parm. 2002, 244, 33–43. [Google Scholar] [CrossRef]
- Liedtke, S.; Wissing, S.; Müller, R.H.; Mäder, K. Influence of high pressure homogenisation equipment on nanodispersions characteristics. Int. J. Pharm. 2000, 196, 183–185. [Google Scholar] [CrossRef] [PubMed]
- Gasco, M.R. Method for producing solid lipid microspheres having a narrow size distribution. US Pat. No. 5,250,236, 1993. [Google Scholar]
- Zur Mühlen, A.; Schwarz, C.; Mehnert, W. Solid lipid nanoparticles (SLN) for controlled drug delivery–Drug release and release mechanism. Eur. J. Pharm. Biopharm. 1998, 45, 149–155. [Google Scholar] [CrossRef] [PubMed]
- AL-Haj, N.A.; Abdullah, R.; Ibrahim, S.; Bustamam, A. Tamoxifen drug loading solid lipid nanoparticles prepared by hot high pressure homogenization techniques. Am. J. Pharmacol. Toxicol. 2008, 3, 219–224. [Google Scholar] [CrossRef]
- Harivardhan Reddy, L.; Vivek, K.; Bakshi, N.; Murthy, R.S.R. Tamoxifen citrate loaded solid lipid nanoparticles (SLN™): Preparation, characterization, in vitro drug release, and pharmacokinetic evaluation. Pharm. Dev. Technol. 2006, 11, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Almeida, A.J.; Souto, E. Solid lipid nanoparticles as a drug delivery system for peptides and proteins. Adv. Drug Deliv. Rev. 2007, 59, 478–490. [Google Scholar] [CrossRef] [PubMed]
- Almeida, A.J.; Runge, S.; Müller, R.H. Peptide-loaded solid lipid nanoparticles (SLN): Influence of production parameters. Int. J. Pharm. 1997, 149, 255–265. [Google Scholar] [CrossRef]
- Hu, F.Q.; Hong, Y.; Yuan, H. Preparation and characterization of solid lipid nanoparticles containing peptide. Int. J. Pharm. 2004, 273, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhao, B.; Wang, F.; Wang, M.; Xie, S.; Wang, S.; Han, C.; Zhu, L.; Zhou, W. Yak interferon-alpha loaded solid lipid nanoparticles for controlled release. Res. Vet. Sci. 2010, 88, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Gualbert, J.; Shahgaldian, P.; Coleman, A.W. Interactions of amphiphilic calyx[4]arene-based solid lipid nanoparticles with bovine serum albumin. Int. J. Pharm. 2003, 257, 69–73. [Google Scholar] [CrossRef]
- Olbrich, C.; Geßner, A.; Kayser, O.; Müller, R.H. Lipid-drug conjugate (LDC) nanoparticles as novel carrier system for the hydrophilic antitrypanosomal drug diminazenediaceturate. J. Drug Target. 2002, 10, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Penkler, L.; Müller, R.H.; Runge, S.; Ravelli, V. Pharmaceutical cyclosporin formulation with improved biopharmaceutical properties, improved physical quality and greater stability, and method for producing said formulation. US Pat. No. 6,551,619, 2003. [Google Scholar]
- Ugazio, E.; Cavalli, R.; Gasco, M.R. Incorporation of cyclosporine A in solid lipid nanoparticles (SLN). Int. J. Pharm. 2002, 24, 341–344. [Google Scholar] [CrossRef]
- Cavalli, R.; Bocca, C.; Miglietta, A.; Caputo, O.; Gasco, M.R. Albumin adsorption on stealth and non-stealth solid lipid nanoparticles. STP Pharma. Sci. 1999, 9, 183–189. [Google Scholar]
- García-Fuentes, M.; Torres, D.; Alonso, M.J. Design of lipid nanoparticles for the oral delivery of hydrophilic macromolecules. Colloids Surf. B Biointerf. 2002, 27, 159–168. [Google Scholar] [CrossRef]
- Zhang, N.; Ping, Q.; Huang, G.; Xu, W.; Cheng, Y.; Han, X. Lectin-modified solid lipid nanoparticles as carriers for oral administration of insulin. Int. J. Pharm. 2006, 327, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Caliceti, P.; Brossa, A.; Salmaso, S.; Bersani, S.; Elvassore, N.; Bertucco, A. Preparation of protein loaded solid lipid nano-particles by compressed fluid process. Proc. Int. Symp. Control. Rel. Bioact. Mater. 2006, 23, 383. [Google Scholar]
- Morel, S.; Gasco, M.R.; Cavalli, R. Incorporation in lipospheres of [D-Trp-6] LHRH. Int. J. Pharm. 1994, 105, R1–R3. [Google Scholar] [CrossRef]
- Videira, M.; Azevedo, A.F.; Almeida, A.J. Entrapment of a high molecular weight protein into solid lipid nanoparticles. In Proc. 2nd World Meeting APV/APGI, Paris, May 1998; pp. 629–630.
- Morel, S.; Ugazio, E.; Cavalli, R.; Gasco, M.R. Thymopentin in solid lipid nanoparticles. Int. J. Pharm. 1996, 132, 259–261. [Google Scholar] [CrossRef]
- Schöler, N.; Hahn, H.; Müller, R.H.; Liesenfeld, O. Effect of lipid matrix and size of solid lipid nanoparticles (SLN) on the viability and cytokine production of macrophages. Int. J. Pharm. 2002, 231, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wu, W. In situ evading of phagocytic uptake of stealth solid lipid nanoparticles by mouse peritoneal macrophages. Drug Deliv. 2006, 13, 189–192. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Fuentes, M.; Torres, D.; Alonso, M.J. New surface-modified lipid nanoparticles as delivery vehicles for salmon calcitonin. Int. J. Pharm. 2005, 296, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Constantinides, P.P.; Scalart, J.P.; Lancastar, S.; Marcello, J.; Marks, G.; Ellens, H.; Smith, P.L. Formulation and intestinal absorption enhancement evaluation of water-in-oil microemulsions incorporating medium-chain glycerides. Pharm. Res. 1994, 11, 1385–1390. [Google Scholar] [CrossRef] [PubMed]
- Vauthier, C.; Bouchemal, K. Methods for the preparation and manufacture of polymeric nanoparticles. Pharm. Res. 2009, 26, 1025–1058. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.Y.; Bae, Y.H. Polymer architecture and drug delivery. Pharm. Res. 2006, 23, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Vauthier, C.; Labarre, D. Modular biomimetic drug delivery systems. J. Drug Deliv. Sci. Technol. 2008, 18, 59–68. [Google Scholar] [CrossRef]
- van de Weert, M.; Hennink, W.E.; Jiskoot, W. Protein instability in poly(lactic-co-glycolic acid) microparticles. Pharm. Res. 2000, 17, 1159–1167. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Teply, B.A.; Sherifi, I.; Sung, J.; Luther, G.; Gu, F.X.; Levy-Nissenbaum, E.; Radovic-Moreno, A.F.; Langer, R.; Farokhzad, O.C. Formulation of functionalized PLGA–PEG nanoparticles for in vivo targeted drug delivery. Biomaterials 2007, 28, 869–876. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Zhang, Y.; Chen, W.; Shen, C.; Liao, M.; Pan, Y.; Wang, J.; Deng, X.; Zhao, J. Cationic polybutyl cyanoacrylate nanoparticles for DNA delivery. J. Biomed. Biotechnol. 2009, 149254. [Google Scholar]
- Soppimath, K.S.; Aminabhavi, T.M.; Kulkarni, A.R.; Rudzinski, W.E. Biodegradable polymeric nanoparticles as drug delivery devices. J. Control. Release 2001, 70, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Rytting, E.; Nguyen, J.; Wang, X.; Kissel, T. Biodegradable polymeric nanocarriers for pulmonary drug delivery. Expert Opin. Drug Deliv. 2008, 5, 629–639. [Google Scholar] [CrossRef] [PubMed]
- Akin, D.; Sturgis, J.; Ragheb, K.; Sherman, D.; Burkholder, K.; Robinson, J.P.; Bhunia, A.K.; Mohammed, S.; Bashir, R. Bacteria-mediated delivery of nanoparticles and cargo into cells. Nat. Nanotech. 2007, 2, 441–449. [Google Scholar] [CrossRef]
- Minigo, G.; Scholzen, A.; Tang, C.K.; Hanley, J.C.; Kalkanidis, M.; Pietersz, G.A.; Apostolopoulos, V.; Plebanski, M. Poly-l-lysine-coated nanoparticles: A potent delivery system to enhance DNA vaccine efficacy. Vaccine 2007, 25, 1316–1327. [Google Scholar] [CrossRef] [PubMed]
- Hasadsri, L.; Kreuter, J.; Hattori, H.; Iwasaki, T.; George, J.M. Functional protein delivery into neurons using polymeric nanoparticles. J. Biol. Chem. 2009, 284, 6972–6981. [Google Scholar] [CrossRef] [PubMed]
- Weiss, C.K.; Kohnle, M.-V.; Landfester, K.; Hauk, T.; Fischer, D.; Schmitz-Wienke, J.; Mailänder, V. The first step into the brain: Uptake of NIO-PBCA nanoparticles by endothelial cells in vitro and in vivo, and direct evidence for their blood-brain barrier permeation. Chem. Med. Chem. 2008, 3, 1395–1403. [Google Scholar] [CrossRef] [PubMed]
- Vauthier, C.; Dubernet, C.; Chauvierre, C.; Brigger, I.; Couvreur, P. Drug delivery to resistant tumors: The potential of poly(alkyl cyanoacrylate) nanoparticles. J. Control. Release 2003, 93, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Stella, B.; Arpicco, S.; Peracchia, M.T.; Desmaele, D.; Hoebeke, J.; Renoir, M.; D’Angelo, J.; Cattel, L.; Couvreur, P. Design of folic acid-conjugated nanoparticles for drug targeting. J. Pharm. Sci. 2000, 89, 1452–1464. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.R.; Andrieux, K.; Gil, S.; Taverna, M.; Chacun, H.; Desmaële, D.; Taran, F.; Georgin, D.; Couvreur, P. Translocation of poly(ethylene glycol-co-hexadecyl) cyanoacrylate nanoparticles into rat brain endothelial cells: Role of apolipoproteins in receptor-mediated endocytosis. Biomacromolecules 2007, 8, 793–799. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Shi, B.; Pei, Y.-Y.; Hong, M.-H.; Wu, J.; Chen, H.-Z. In vivo tumor targeting of tumor necrosis factor-α-loaded stealth nanoparticles: Effect of MePEG molecular weight and particle size. Eur. J. Pharm. Sci. 2006, 27, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-P.; Pei, Y.-Y.; Zhou, Z.-H.; Zhang, X.-Y.; Gu, Z.-H.; Ding, J.; Zhou, J.-J.; Gao, X.-J.; Zhu, J.-H. Stealth polycyanoacrylate nanoparticles as tumor necrosis Factor-α carriers: Pharmacokinetics and anti-tumor effects. Biol. Pharm. Bull. 2001, 24, 662–665. [Google Scholar] [CrossRef] [PubMed]
- Calvo, P.; Gouritin, B.; Brigger, I.; Lasmezas, C.; Deslys, J.-P.; Williams, A.; Andreux, J.P.; Dormont, D.; Couvreur, P. PEGylated polycyanoacrylate nanoparticles as vector for drug delivery in prion diseases. J. Neurosci. Methods 2001, 111, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Lukyanov, A.N.; Torchilin, V. Micelles from lipid derivatives of water-soluble polymers as delivery systems for poorly soluble drugs. Adv. Drug Deliv. Rev. 2004, 56, 1273–1289. [Google Scholar] [CrossRef] [PubMed]
- Bisht, S.; Feldmann, G.; Soni, S.; Ravi, R.; Karikar, C.; Maitra, A.; Maitra, A. Polymeric nanoparticle-encapsulated curcumin (“nanocurcumin”): A novel strategy for human cancer therapy. J. Nanobiotechnol. 2007, 5. [Google Scholar] [CrossRef]
- Kim, J.H.; Park, K.; Nam, H.Y.; Lee, S.; Kim, K.; Kwon, I.C. Polymers for bioimaging. Prog. Polym. Sci. 2007, 32, 1031–1053. [Google Scholar] [CrossRef]
- Kim, K.; Lee, M.; Park, H.; Kim, J.-H.; Kim, S.; Chung, H.; Choi, K.; Kim, I.-S.; Seong, B.L.; Kwon, I.C. Cell-permeable and biocompatible polymeric nanoparticles for apoptosis imaging. J. Am. Chem. Soc. 2006, 128, 3490–3491. [Google Scholar] [CrossRef] [PubMed]
- Bala, I.; Hariharan, S.; Ravi Kumar, M.N.V. PLGA nanoparticles in drug delivery: The state of the art. Crit. Rev. Ther. Drug Carrier Syst. 2004, 21, 387–422. [Google Scholar] [CrossRef] [PubMed]
- Vasir, J.K.; Labhasetwar, V. Biodegradable nanoparticles for cytosolic delivery of therapeutics. Adv. Drug Deliv. Rev. 2007, 59, 718–728. [Google Scholar] [CrossRef] [PubMed]
- Freitas, S.; Hielscher, G.; Merkle, H.P.; Gander, B. A fast and simple method for producing biodegradable nanospheres. Eur. Cell. Mater. 2004, 7, 28. [Google Scholar]
- Damgé, C.; Maincent, P.; Ubrich, N. Oral delivery of insulin associated to polymeric nanoparticles in diabetic rats. J. Control. Release 2007, 117, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Cui, F.; Shi, K.; Zhang, L.; Tao, A.; Kawashima, Y. Biodegradable nanoparticles loaded with insulin–phospholipid complex for oral delivery: Preparation, in vitro characterization and in vivo evaluation. J. Control. Release 2006, 114, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Vila, A.; Sànchez, A.; Tobìo, M.; Calvo, P.; Alonso, M.J. Design of biodegradable particles for protein delivery. J. Control. Release 2002, 78, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Dziubla, T.D.; Karim, A.; Muzykantov, V.R. Polymer nanocarriers protecting active enzyme cargo against proteolysis. J. Control. Release 2005, 102, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Tobio, M.; Sanchez, A.; Vila, A.; Soriano, I.; Evora, C.; Vila-Jato, J.L.; Alonso, M.J. The role of PEG on the stability in digestive fluids and in vivo fate of PEG-PLA nanoparticles following oral administration. Colloid. Surfaces B Biointerf. 2000, 18, 315–323. [Google Scholar] [CrossRef]
- Vila, A.; Sanchez, A.; Evora, C.; Soriano, I.; McCallion, O.; Alonso, M.J. PLA-PEG particles as nasal protein carriers: The influence of the particle size. Int. J. Pharm. 2005, 292, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Simone, E.A.; Dziubla, T.D.; Colon-Gonzalez, F.; Discher, D.E.; Muzykantov, V.R. Effect of polymer amphiphilicity on loading of a therapeutic enzyme into protective filamentous and spherical polymer nanocarriers. Biomacromolecules 2007, 8, 3914–3921. [Google Scholar] [CrossRef] [PubMed]
- Simone, E.A.; Dziubla, T.D.; Discher, D.E.; Muzykantov, V.R. Filamentous polymer nanocarriers of tunable stiffness that encapsulate the therapeutic enzyme catalase. Biomacromolecules 2009, 10, 1324–1330. [Google Scholar] [CrossRef] [PubMed]
- Chiellini, F.; Bartoli, C.; Dinucci, D.; Piras, A.M.; Anderson, R.; Croucher, T. Bioeliminable polymeric nanoparticles for proteic drug delivery. Int. J. Pharm. 2007, 343, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Coué, G.; Feijen, J.; Engbersen, J.F.J. Development of biodegradable poly(amidoamine)s for protein delivery. J. Control. Release 2008, 132, e2–e3. [Google Scholar] [CrossRef]
- Akagi, T.; Wang, X.; Uto, T.; Baba, M.; Akashi, M. Protein direct delivery to dendritic cells using nanoparticles based on amphiphilic poly(amino acid) derivatives. Biomaterials 2007, 28, 3427–3436. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Zhang, Z.-R.; Yuan, F.; Qin, X.; Wang, M.; Huang, Y. In vitro and in vivo study of N-trimethyl chitosan nanoparticles for oral protein delivery. Int. J. Pharm. 2008, 349, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Sarmento, B.; Ribeiro, A.; Veiga, F.; Sampaio, P.; Neufeld, R.; Ferreira, D. Alginate/chitosan nanoparticles are effective for oral insulin delivery. Pharm. Res. 2007, 24, 2198–2206. [Google Scholar] [CrossRef] [PubMed]
- Thakkar, H.; Sharma, R.K.; Mishra, A.K.; Chuttani, K.; Murthy, R.R. Albumin microspheres as carriers for the antiarthritic drug celecoxib. AAPS Pharm. Sci. Tech. 2005, 6, E65–E73. [Google Scholar] [CrossRef]
- Chiellini, E.; Chiellini, E.E.; Chiellini, F.; Solaro, R. Targeted administration of proteic drugs. I. preparation of polymeric nanoparticles. J. Bioact. Compat. Polym. 2001, 16, 441–465. [Google Scholar] [CrossRef]
- Chiellini, E.E.; Chiellini, F.; Solaro, R. Bioerodible polymeric nanoparticles for targeted administration of proteic drugs. J. Nanosci. Nanotechnol. 2006, 6, 3040–3047. [Google Scholar] [CrossRef] [PubMed]
- Solaro, R.; Chiellini, F.; Signori, F.; Fiumi, C.; Bizzarri, R.; Chiellini, E. Nanoparticle systems for the targeted release of active principles of proteic nature. J. Mat. Sci. Mater. Med. 2003, 14, 705–711. [Google Scholar] [CrossRef]
- Merkel, T.J.; Herlihy, K.P.; Nunes, J.; Orgel, R.M.; Rolland, J.P.; DeSimone, J.M. Scalable, shape-specific, top-down fabrication methods for the synthesis of engineered colloidal particles. Langmuir 2010. [Google Scholar] [CrossRef]
- Kelly, J.Y.; Desimone, J.M. Shape-specific, monodisperse nano-molding of protein particles. J. Am. Chem. Soc. 2008, 130, 5438–5439. [Google Scholar] [CrossRef] [PubMed]
- Yu-Su, S.Y.; Thomas, D.R.; Alford, J.E.; LaRue, I.; Pitsikalis, M.; Hadjichristidis, N.; DeSimone, J.M.; Dobrynin, A.V.; Sheiko, S.S. Molding block copolymer micelles: A framework for molding of discrete objects on surfaces. Langmuir 2008, 24, 12671–12679. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Nunes, J.K.; Gratton, S.E.A.; Herlihy, K.P.; Pohlhaus, P.D.; DeSimone, J.M. Fabrication of multiphasic and regio-specifically functionalized PRINT® particles of controlled size and shape. New J. Phys. 2009, 11, 075018. [Google Scholar] [CrossRef]
- Anton, N.; Saulnier, P.; Gaillard, C.; Porcher, E.; Vrignaud, S.; Benoit, J.-P. Aqueous-core lipid nanocapsules for encapsulating fragile hydrophilic and/or lipophilic molecules. Langmuir 2009, 25, 11413–11419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daoud-Mahammed, S.; Ringard-Lefebvre, C.; Razzouq, N.; Rosilio, V.; Gillet, B.; Couvreur, P.; Amiel, C.; Gref, R. Spontaneous association of hydrophobized dextran and poly-β-cyclodextrin into nanoassemblies. Formation and interaction with a hydrophobic drug. J. Colloid Interf. Sci. 2007, 307, 83–93. [Google Scholar] [CrossRef]
- Nishikawa, T.; Akiyoshi, K.; Sunamoto, J. Supramolecular assembly between nanoparticles of hydrophobized polysaccharide and soluble protein complexation between the self-aggregate of cholesterol-bearing pullulan and α-chymotrypsin. Macromolecules 1994, 27, 7654–7659. [Google Scholar] [CrossRef]
- Akiyoshi, K.; Nishikawa, T.; Mitsui, Y.; Miyata, T.; Kodama, M.; Sunamoto, J. Self-assembly of polymer amphiphiles: Thermodynamics of complexation between bovine serum albumin and self-aggregate of cholesterol-bearing pullulan. Colloids Surf. A Physicochem. Eng. Asp. 1996, 112, 91–95. [Google Scholar] [CrossRef]
- Chattopadhyay, P.; Gupta, R.B. Protein nanoparticles formation by supercritical antisolvent with enhanced mass transfer. AIChE J. 2002, 48, 235–244. [Google Scholar] [CrossRef]
- Tozuka, Y.; Miyazaki, Y.; Takeuchi, H. A combinational supercritical CO2 system for nanoparticle preparation of indomethacin. Int. J. Pharm. 2010, 386, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.I. Polymer synthesis and processing using supercritical carbon dioxide. J. Mater. Chem. 2000, 10, 207–234. [Google Scholar] [CrossRef]
- Schrama, D.; Reisfeld, R.A.; Becker, J.C. Antibody targeted drugs as cancer therapeutics. Nat. Rev. Drug Discov. 2006, 5, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Akita, H.; Kogure, K.; Kamiya, H.; Harashima, H. Mitochondrial drug delivery and mitochondrial disease therapy—An approach to liposome-based delivery targeted to mitochondria. Mitochondrion 2007, 7, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Borgeld, H.M.; Zhang, J.; Muramatsu, S.; Gong, J.S.; Yoneda, M.; Maruyama, W.; Naoi, M.; Ibi, T.; Sahashi, K.; Shamoto, M.; Fuku, N.; Kurata, M.; Yamada, Y.; Nishizawa, K.; Akao, Y.; Ohishi, N.; Miyabayashi, S.; Umemoto, H.; Muramatsu, T.; Furukawa, K.; Kikuchi, A.; Nakano, I.; Ozawa, K.; Yagi, K. Gene therapy for mitochondrial disease by delivering restriction endonuclease SmaI into mitochondria. J. Biomed. Sci. 2002, 9, 534–541. [Google Scholar] [PubMed]
- Futaki, S. Arginine-rich peptides: Potential for intracellular delivery of macromolecules and the mystery of the translocation mechanisms. Int. J. Pharm. 2002, 245, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, M.; Lorberboum-Galski, H. TAT-based drug delivery system—New directions in protein delivery for new hopes? Expert Opin. Drug Deliv. 2009, 6, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Schwarze, S.R.; Hruska, K.A.; Dowdy, S.F. Protein transduction: Unrestricted delivery into all cells? Trends Cell Biol. 2002, 10, 290–295. [Google Scholar] [CrossRef]
- Shokolenko, I.N.; Alexeyev, M.F.; LeDoux, S.P.; Wilson, G.L. TAT-mediated protein transduction and targeted delivery of fusion proteins into mitochondria of breast cancer cells. DNA Repair (Amst) 2005, 4, 511–518. [Google Scholar] [CrossRef]
- Middlebrook, J.L.; Dorland, R.B. Bacterial toxins: Cellular mechanisms of action. Microbiol. Rev. 1984, 48, 199–221. [Google Scholar] [PubMed]
- Bade, S.; Rummel, A.; Reisinger, C.; Karnath, T.; Ahnert-Hilger, G.; Bigalke, H.; Binz, T. Botulinum neurotoxin type D enables cytosolic delivery of enzymatically active cargo proteins to neurones via unfolded translocation intermediates. J. Neurochem. 2004, 91, 1461–1472. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Adams, R.; Sun, W.; Lee, I.; Debromilskaya, I.; Drachman, D.B. Targeting of viral vectors to motor neurons by botulinum toxin binding domain to treat ALS. Mol. Ther. 2006, 13, S347. [Google Scholar] [CrossRef]
- Chen, S.; Barbieri, J.T. Engineering botulinum neurotoxin to extend therapeutic intervention. Proc. Natl. Acad. Sci. USA 2009, 106, 9180–9184. [Google Scholar] [CrossRef] [PubMed]
- Foster, K.A. Engeneered toxins: New therapeutics. Toxicon 2009, 54, 587–592. [Google Scholar] [CrossRef] [PubMed]
- Sokoloff, A.V.; Wong, S.C.; Ludtke, J.J.; Sebestyen, M.G.; Subbotin, V.M.; Zhang, G.; Budker, T.; Bachhuber, M.; Sumita, Y.; Wolff, J.A. A new peptide ligand that targets particles and heterologous proteins to hepatocytes in vivo. Mol. Ther. 2003, 8, 867–872. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.C.; Wakefield, D.; Klein, J.; Monahan, S.D.; Rozema, D.B.; Lewis, D.L.; Higgs, L.; Ludtke, J.J.; Sokoloff, A.V.; Wolff, J.A. Hepatocyte targeting of nucleic acid complexes and liposomes by a T7 phage p17 peptide. Mol. Pharm. 2006, 3, 386–397. [Google Scholar] [CrossRef] [PubMed]
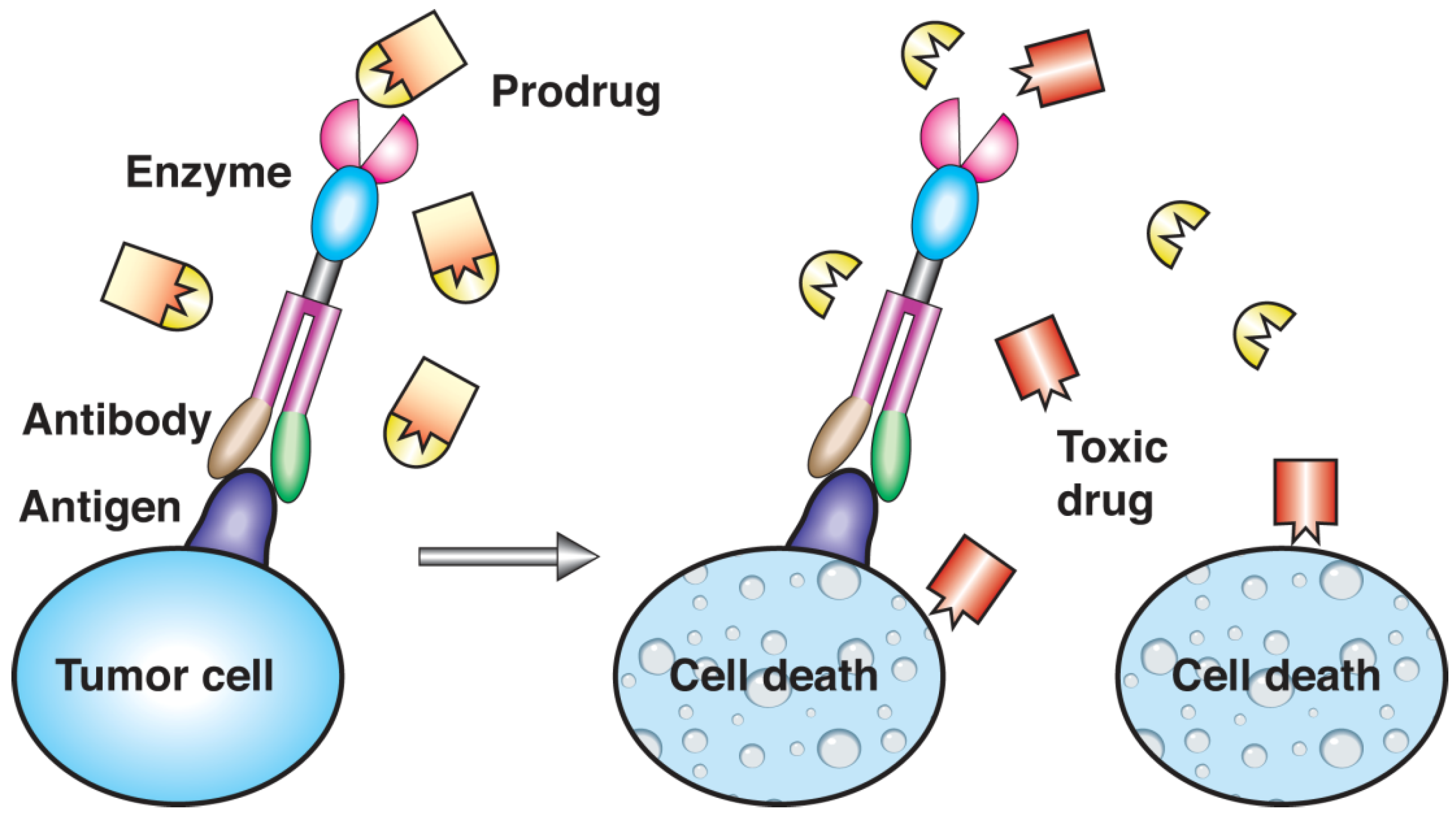
- Sharma, S.K.; Pedley, R.B.; Bhatia, J.; Boxer, G.M.; El-Emir, E.; Qureshi, U.; Tolner, B.; Lowe, H.; Michael, N.P.; Minton, N.; Begent, R.H.J.; Chester, K.A. Sustained tumor regression of human colorectal cancer xenografts using a multifunctional mannosylated fusion protein in antibody-directed enzyme prodrug therapy. Clin. Cancer Res. 2005, 11, 814–825. [Google Scholar] [PubMed]
- Williams, A.S.; Camilleri, J.P.; Goodfellow, R.M.; Williams, B.D. A single intra-articular injection of liposomally conjugated methotrexate suppresses joint inflammation in rat antigen-induced arthritis. Br. J. Rheumatol. 1996, 35, 719–724. [Google Scholar]
- Chazov, E.I.; Alexeev, A.V.; Antonov, A.S.; Koteliansky, V.E.; Leytin, V.L.; Ljubimova, A.V.; Repin, V.S.; Sviridov, D.D.; Torchilin, V.P.; Smirnov, V.N. Endothelial cell culture on fibrillar collagen: Model to study plateled adhesion and liposome targeting to intercellular collagen matrix. Procl. Natl. Acad. Sci. USA 1981, 78, 5603–5607. [Google Scholar] [CrossRef]
- Himber, J.; Sallee, V.L.; Andermann, G.; Bouzoubaa, M.; Leclerc, G.; De Santis, L. Effects of topically applied falintolol: A new beta-adrenergic antagonist for treatment of glaucoma. J. Ocul. Pharmacol. Ther. 1987, 3, 111–120. [Google Scholar] [CrossRef]
- Ma, L.; Wu, X.; Ling-Ling, E.; Wang, D.-S.; Liu, H.-C. The transmembrane transport of metformin by osteoblasts from rat mandible. Arch. Oral Biol. 2009, 54, 951–962. [Google Scholar] [CrossRef] [PubMed]
- Jenning, V.; Gysler, A.; Schäfer-Korting, M.; Gohla, S.H. Vitamin A loaded solid lipid nanoparticles for topical use: Occlusive properties and drug targeting to the upper skin. Eur. J. Pharm. Biopharm. 2000, 49, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Santos Maia, C.; Mehnert, W; Schäfer-Korting, M. Solid lipid nanoparticles as drug carriers for topical glucocorticoids. Int. J. Pharm. 2000, 196, 165–167. [Google Scholar] [CrossRef] [PubMed]
- Bhaskar, K.; Anbu, J.; Ravichandiran, V.; Venkateswarlu, V.; Madhusudan Rao, Y. Lipid nanoparticles for transdermal delivery of flurbiprofen: Formulation, in vitro, ex vivo and in vivo studies. Lipids Health Dis. 2009, 8. [Google Scholar] [CrossRef] [PubMed]
- Seymour, L.W. Passive tumor targeting of soluble macromolecules and drug conjugates. Crit. Rev. Ther. Drug Carrier Syst. 1992, 9, 135–187. [Google Scholar] [PubMed]
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent SMANCS. Cancer Res. 1986, 46, 6387–6392. [Google Scholar] [PubMed]
- Maeda, H.; Matsumura, Y. Tumoritropic and lymphotropic principles of macromolecular drugs. Crit. Rev. Ther. Drug Carrier Syst. 1989, 6, 193–210. [Google Scholar] [PubMed]
- Maeda, H.; Wu, J.; Sawa, T.; Matsumura, Y.; Hori, K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J. Control. Release 2000, 65, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Sawa, T.; Konno, T. Mechanism of tumor-targeted delivery of macromolecular drugs including the EPR effect in solid tumor and clinical overview of the prototype polymeric drug SMANCS. J. Control. Release 2001, 74, 47–61. [Google Scholar] [CrossRef] [PubMed]
- Greish, K.; Iyer, A.K.; Fan, J.; Maeda, H. Enhanced permeability and retention (EPR) effect and tumor selective delivery of anticancer drugs. In Delivery of Protein and Peptide Drugs to Cancer; Torchilin, V.P., Ed.; Imperial College Press: London, UK, 2006; pp. 37–52. [Google Scholar]
- Maeda, H.; Greish, K.; Fang, J. The EPR effect and polymeric drugs: A paradigm shift for cancer chemotherapy in the 21st century. Adv. Polym. Sci. 2006, 193, 103–121. [Google Scholar]
- Greish, K. Enhanced permeability and retention of macromolecular drugs in solid tumors: A royal gate for targeted anticancer nanomedicines. J. Drug Target. 2007, 15, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Gabizon, A.A. Selective tumor localization and improved therapeutic index of anthracyclines encapsulated in long-circulating liposomes. Cancer Res. 1992, 52, 891–896. [Google Scholar] [PubMed]
- Jun, Y.J.; Kim, J.I.; Jun, M.J.; Sohn, Y.S. Selective tumor targeting by enhanced permeability and retention effect. Synthesis and antitumor activity of polyphosphazene–platinum (II) conjugates. J. Inorg. Biochem. 2005, 99, 1593–1601. [Google Scholar] [CrossRef] [PubMed]
- Palframan, R.; Vugle, A.; Moore, A.; Baker, M.; Lightwood, D.; Nesbitt, A.; Foulkes, R.; Gozzard, N. Certolizumab pegol and adalimumab accumulation in the inflammed paws of mice with collagen-induced arthritis compared to noninflammed tissue in vivo biofluorescence imaging of Alexa 680-labeled antibodies. Clin. Immunol. 2007, 123, S170. [Google Scholar] [CrossRef]
- Seow, W.Y.; Xue, J.M.; Yang, Y.-Y. Targeted and intracellular delivery of paclitaxel using multi-functional polymeric micelles. Biomaterials 2007, 28, 1730–1740. [Google Scholar] [CrossRef] [PubMed]
- Soppimath, K.S.; Liu, L.-H.; Seow, W.Y.; Liu, S.-Q.; Powell, R.; Chan, P.; Yang, Y.-Y. Multifunctional core/shell nanoparticles self-assembled from pH-induced thermosensitive polymers for targeted intracellular anticancer drug delivery. Adv. Funct. Mater. 2007, 17, 355–362. [Google Scholar] [CrossRef]
- Dai, J.; Nagai, T.; Wang, X.; Zhang, T.; Meng, M.; Zhang, Q. pH-sensitive nanoparticles for improving the oral bioavailability of cyclosporine A. Int. J. Pharm. 2004, 280, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, Y.; Niwa, T.; Handa, T.; Takeuchi, H.; lwamoto, T.; Itoh, K. Preparation of controlled-release microspheres of ibuprofen with acrylic polymers by a novel quasi-emulsion solvent diffusion method. J. Pharm. Sci. 1989, 78, 68–72. [Google Scholar] [CrossRef] [PubMed]
- El-Sayed, M.E.H.; Hoffman, A.S.; Stayton, P.S. Smart polymeric carriers for enhanced intracellular delivery of therapeutic macromolecules. Expert Opin. Biol. Ther. 2005, 5, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Freeman, R.W.; Arrott, A.; Watson, J.H.L. Magnetism in medicine. J. Appl. Phys. 1960, 31, S404. [Google Scholar] [CrossRef]
- Portet, D.; Denizot, B.; Rump, E.; Lejeune, J.-J.; Jallet, P. Nonpolymeric coatings of iron oxide colloids for biological use as magnetic resonance imaging contrast agents. J. Colloid Interface Sci. 2001, 238, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.K.; Wells, S. Surface-modified superparamagnetic nanoparticles for drug delivery: Preparation, characterization, and cytotoxicity studies. IEEE Trans. Nanobiosci. 2004, 3, 66–73. [Google Scholar] [CrossRef]
- Ström, V.; Hultenby, K.; Grüttner, C.; Teller, J.; Xu, B.; Holgersson, J. A novel and rapid method for quantification of magnetic nanoparticle-cell interactions using a desktop susceptometer. Nanotechnology 2004, 15, 457–466. [Google Scholar] [CrossRef]
- Neuberger, T.; Schöpf, B.; Hofmann, H.; Hofmann, M.; von Rechenberg, B. Superparamagnetic nanoparticles for biomedical applications: Possibilities and limitations of a new drug delivery system. J. Magn. Magn. Mater. 2005, 293, 483–496. [Google Scholar] [CrossRef]
- Dobson, J. Magnetic nanoparticles for drug delivery. Drug Dev. Res. 2006, 67, 55–60. [Google Scholar] [CrossRef]
- Alexiou, C.; Schmid, R.J.; Jurgons, R.; Kremer, M.; Wanner, G.; Bergemann, G.; Huenges, E.; Nawroth, T.; Arnold, W.; Parak, F.G. Targeting cancer cells: Magnetic nanoparticles as drug carriers. Eur. Biophys. J. 2006, 35, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Alexiou, C.; Jurgons, R.; Schmid, R.J.; Bergemann, G.; Henke, J.; Erhardt, W.; Huenges, E.; Parak, F.G. Magnetic drug targeting-biodistribution of the magnetic carrier and the chemotherapeutic agent Mitoxantrone after locoregional cancer treatment. J. Drug Target. 2003, 11, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Jurgons, R.; Seliger, C.; Hilpert, A.; Trahms, L.; Odenbach, S.; Alexiou, C. Drug loaded magnetic nanoparticles for cancer therapy. J. Phys.: Condens. Matter 2006, 18, S2893–S2902. [Google Scholar] [CrossRef]
- Torchilin, V.P.; Paptov, M.I.; Orekova, N.M.; Belyaev, A.A.; Petrov, A.D.; Ragimov, S.E. Magnetically driven thrombolytic preparation containing immobilized streptokinase-targeted transport and action. Haemostasis 1988, 18, 113–116. [Google Scholar] [PubMed]
- Mikhaylova, M.; Kim, D.K.; Berry, C.C.; Zagorodni, A.; Toprak, M.; Curtis, A.S.G.; Muhammed, M. BSA immobilization on amine-functionalized superparamagnetic iron oxide nanoparticles. Chem. Mater. 2004, 16, 2344–2354. [Google Scholar] [CrossRef]
- Xu, L.; Kim, M.-J.; Kim, K.-D.; Choa, Y.-H.; Kim, H.-T. Surface modified Fe3O4 nanoparticles as a protein delivery vehicle. Colloid Surf. A-Physicochem. Eng. Asp. 2009, 350, 8–12. [Google Scholar] [CrossRef]
- Kohler, N.; Fryxell, G.E.; Zhang, M.Q. A bifunctional poly(ethylene glycol) silane immobilized on metallic oxide-based nanoparticles for conjugation with cell targeting agents. J. Am. Chem. Soc. 2004, 126, 7206–7211. [Google Scholar] [CrossRef] [PubMed]
- Lecommandoux, S.; Sandre, O.; Chécot, F.; Perzynski, R. Smart hybrid magnetic self-assembled micelles and hollow capsules. Prog. Solid State Chem. 2006, 34, 171–179. [Google Scholar] [CrossRef]
- Mastrobattista, E.; Koning, G.A.; Storm, G. Immunoliposomes for the targeted delivery of antitumor drugs. Adv. Drug Deliv. Rev. 1999, 40, 103–127. [Google Scholar] [CrossRef] [PubMed]
- Farokhzad, O.C.; Jon, S.; Khademhosseini, A.; Tran, T.-N.T.; LaVan, D.A.; Langer, R. Nanoparticle-aptamer bioconjugates: A new approach for targeting prostate cancer cells. Cancer Res. 2004, 74, 7668–7672. [Google Scholar] [CrossRef]
- Farokhzad, O.C.; Khademhosseini, A.; Jon, S.; Hermmann, A.; Cheng, J.; Chin, C.; Kiselyuk, A.; Teply, B.; Eng, G.; Langer, R. Microfluidic system for studying the interaction of nanoparticles and microparticles with cells. Anal. Chem. 2005, 77, 5453–5459. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-F.; Kim, Y.; Meng, L.; Tan, W. Assembly of aptamer conjugates as molecular tools in therapeutics. Chem. Today 2009, 27, 52–54. [Google Scholar]
- Vivès, E. Present and future of cell-penetrating peptide mediated delivery systems: “Is the Trojan horse too wild to go only to Troy?”. J. Control. Release 2005, 109, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Vivès, E.; Schmidt, J.; Pèlegrin, A. Cell-penetrating and cell-targeting peptides in drug delivery. BBA–Rev. Cancer 2008, 1786, 126–138. [Google Scholar]
- Shin, S.U.; Friden, P.; Moran, M.; Olson, T.; Kang, Y.S.; Pardridge, W.M.; Morrison, S.L. Transferrin-antibody fusion proteins are effective in brain targeting. Proc. Natl. Acad. Sci. USA 1995, 92, 2820–2824. [Google Scholar] [CrossRef] [PubMed]
- Quan, C.-Y.; Chang, C.; Wei, H.; Chen, C.-S.; Xu, X.-D.; Cheng, S.-X.; Zhang, X.Z.; Zhuo, R.X. Dual targeting of a thermosensitive nanogel conjugated with transferrin and RGD-containing peptide for effective cell uptake and drug release. Nanotechnology 2009, 20, 335101. [Google Scholar] [CrossRef]
- Minko, T. Drug targeting to the colon with lectins and neoglycoconjugates. Adv. Drug Deliv. Rev. 2004, 56, 491–509. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, N.; Kojima, S.; Bovin, N.V.; André, S.; Gabius, S.; Gabius, H.-J. Endogenous lectins as targets for drug delivery. Adv. Drug Deliv. Rev. 2000, 43, 225–244. [Google Scholar] [CrossRef] [PubMed]
- Tulsani, N.B.; Kumar, A.; Pasha, Q.; Kumar, H.; Sarma, U.P. Immobilization of hormones for drug targeting. Artif. Cells Blood Substit. Immobil. Biotechnol. 2000, 28, 503–519. [Google Scholar] [CrossRef] [PubMed]
- Daniels, T.R.; Delgado, T.; Helguera, G.; Penichet, M.L. The transferrin receptor part II: Targeted delivery of therapeutic agents into cancer cells. Clin. Immunol. 2006, 121, 159–176. [Google Scholar] [CrossRef] [PubMed]
- Sudimak, J.; Lee, R.J. Targeted drug delivery via the folate receptor. Adv. Drug Deliv. Rev. 2000, 41, 147–162. [Google Scholar] [CrossRef] [PubMed]
- Andresen, T.L.; Jensen, S.S.; Jorgensen, K. Advanced strategies in liposomal cancer therapy: Problems and prospects of active and tumor specific drug release. Prog. Lipid Res. 2004, 44, 68–97. [Google Scholar] [CrossRef]
- Barenholz, Y. Liposome application: Problems and prospects. Curr. Opin. Colloid Interface Sci. 2000, 6, 66–77. [Google Scholar] [CrossRef]
- Chowdhury, P.S.; Wu, R. Tailor-made antibody therapeutics. Methods 2005, 36, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Van Osdol, W.; Fujimori, K.; Weinstein, J.N. An analysis of monoclonal antibody distribution in microscopic tumor nodules: Consequences of a “binding site barrier”. Cancer Res. 1991, 51, 4776–4784. [Google Scholar] [PubMed]
- Fischman, A.J.; Babich, J.W.; Strauss, H.W. A ticket to ride: Peptide radiopharmaceuticals. J. Nucl. Med. 1993, 34, 2253–2263. [Google Scholar] [PubMed]
- Wu, D.; Yang, J.; Pardridge, W.M. Drug targeting of a peptide radiopharmaceutical through the primate blood-brain barrier in vivo with a monoclonal antibody to the human insulin receptor. J. Clin. Invest. 1997, 100, 1804–1812. [Google Scholar] [CrossRef] [PubMed]
- Cegnar, M.; Kocbek, P.; Obermajer, N.; Kos, J.; Kristl, J. Immunonanoparticles for targeting and delivery of protein drugs into tumour cells. J. Control. Release 2008, 132, e60–e61. [Google Scholar] [CrossRef]
- Messerschmidt, S.K.E.; Musyanovych, A.; Altvater, M.; Scheurich, P.; Pfizenmaier, K.; Landfester, K.; Kontermann, R.E. Targeted lipid-coated nanoparticles: Delivery of tumor necrosis factor-functionalized particles to tumor cells. J. Control. Release 2009, 137, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Kontermann, R.E. Immunoliposomes for cancer therapy. Curr. Opin. Mol. Ther. 2006, 8, 39–45. [Google Scholar] [PubMed]
- Yamada, T.; Iwasaki, Y.; Tada, H.; Iwabuki., H; Chuah, M.K.; VandenDriessche, T.; Fukuda, H.; Kondo, A.; Ueda, M.; Seno, M.; Tanizawa, K.; Kuroda, S. Nanoparticles for the delivery of genes and drugs to human hepatocytes. Nat. Biotechnol. 2003, 21, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Kurata, N.; Shishido, T.; Muraoka, M.; Tanaka, T.; Ogino, C.; Fukuda, H.; Kondo, A. Specific protein delivery to target cells by antibody-displaying bionanocapsules. J. Biochem. 2008, 144, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Desnick, R.J.; Schuchman, E.H. Enzyme replacement and enhancementtherapies: Lessons from lysosomal disorders. Nat. Rev. Genet. 2002, 3, 954–966. [Google Scholar] [CrossRef] [PubMed]
- Garnacho, C.; Dhami, R.; Simone, E.; Dziubla, T.; Leferovich, J.; Schuchman, E.H.; Muzykantov, V.; Muro, S. Delivery of acid sphingomyelinase in normal and Niemann-Pick disease mice using intercellular adhesion molecule-1-targeted polymer nanocarriers. J. Pharm. Exp. Ther. 2008, 325, 400–408. [Google Scholar] [CrossRef]
- Muro, S.; Cui, X.; Gajewski, C.; Murciano, J.C.; Muzykantov, V.R.; Koval, M. Slow intracellular trafficking of catalase nanoparticles targeted to ICAM-1 protects endothelial cells from oxidative stress. Am. J. Physiol. Cell Physiol. 2003, 285, C1339–C1347. [Google Scholar] [CrossRef] [PubMed]
- Muro, S.; Gajewski, C.; Koval, M.; Muzykantov, V.R. ICAM-1 recycling in endothelial cells: A novel pathway for sustained intracellular delivery and prolonged effects of drugs. Blood 2005, 105, 650–658. [Google Scholar] [CrossRef] [PubMed]
- Muro, S.; Dziubla, T.; Qiu, W.; Leferovich, J.; Cui, X.; Berk, E.; Muzykantov, V.R. Endothelial targeting of high-affinity multivalent polymer nanocarriers directed to intercellular adhesion molecule 1. J. Pharmacol. Exp. Ther. 2006, 317, 1161–1169. [Google Scholar] [CrossRef] [PubMed]
- Muro, S.; Mateescu, M.; Gajewski, C.; Robinson, M.; Muzykantov, V.R.; Koval, M. Control of intracellular trafficking of ICAM-1–targeted nanocarriers by endothelial Na+/H+ exchanger proteins. Am. J. Physiol. Lung Cell Mol. Physiol. 2006, 290, L809–L817. [Google Scholar] [CrossRef] [PubMed]
- Garnacho, C.; Albelda, S.M.; Muzykantov, V.R.; Muro, S. Differential intra-endothelial delivery of polymer nanocarriers targeted to distinct PECAM-1 epitopes. J. Control. Release 2008, 130, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Dhar, S.; Gu, F.X.; Langer, R.; Farokhzad, O.C.; Lippard, S.J. Targeted delivery of cisplatin to prostate cancer cells by aptamer functionalized Pt(IV) prodrug-PLGA-PEG nanoparticles. Proc. Natl. Acad. Sci. USA 2008, 105, 17356–17361. [Google Scholar] [CrossRef] [PubMed]
- Terada, T.; Mizobata, M.; Kawakami, S.; Yamashita, F.; Hashida, M. Optimization of tumor-selective targeting by basic fibroblast growth factor-binding peptide grafted PEGylated liposomes. J. Control. Release 2007, 119, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Kogure, K.; Moriguchi, R.; Sasaki, K.; Ueno, M.; Futaki, S.; Harashima, H. Development of a non-viral multifunctional envelope-type nano device by a novel lipid film hydration method. J. Control. Release 2004, 98, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Khalil, I.A.; Kogure, K.; Futaki, S.; Harashima, H. High density of octaarginine stimulates macropinocytosis leading to efficient intracellular trafficking for gene expression. J. Biol. Chem. 2006, 281, 3544–3551. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, R.; Yamada, Y.; Harashima, H. Efficient cytoplasmic protein delivery by means of a multifunctional envelope-type nano device. Biol. Pharm. Bull. 2007, 30, 758–762. [Google Scholar] [CrossRef] [PubMed]
- Demirgöz, D.; Pangburn, T.O.; Davis, K.P.; Lee, S.; Bates, F.S.; Kokkoli, E. PR_b-Targeted delivery of tumor necrosis factor-α by polymersomes for the treatment of prostate cancer. Soft Matter 2009, 5, 2011–2019. [Google Scholar] [CrossRef]
- Schwarze, S.R.; Dowdy, S.F. In vivo protein transduction; intracellular delivery of biologically active proteins, compounds and DNA. Trend Pharmacol. Sci. 2000, 21, 45–48. [Google Scholar] [CrossRef]
- Nam, Y.S.; Park, J.Y.; Han, S.-H.; Chang, I.-S. Intracellular drug delivery using poly(D,L-lactide-co-glycolide) nanoparticles derivatized with a peptide from a transcriptional activator protein of HIV-1. Biotechnol. Lett. 2002, 24, 2093–2098. [Google Scholar] [CrossRef]
- Huwyler, J.; Wu, D.; Pardridge, W.M. Brain drug delivery of small molecules using immunoliposomes. Proc. Natl. Acad. Sci. USA 1996, 93, 14164–14169. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Qian, Z.M. Transferrin/transferrin receptor-mediated drug delivery. Med. Res. Rev. 2002, 22, 225–250. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Gong, T.; Wang, C.; Zhong, Z.; Zhang, Z. Solid lipid nanoparticles loaded with insulin by sodium cholate-phosphatidylcholine-based mixed micelles: Preparation and characterization. Int. J. Pharm. 2007, 340, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Issa, M.M.; Köping-Höggård, M.; Tømmeraas, K.; Vårum, K.M.; Christensen, B.E.; Strand, S.P.; Artursson, P. Targeted gene delivery with trisaccharide-substituted chitosan oligomers in vitro and after lung administration in vivo. J. Control. Release 2006, 115, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Ehrhardt, C.; Kneuer, C.; Bakowsky, U. Selectins-an emerging target for drug delivery. Adv. Drug Deliv. Rev. 2004, 56, 527–549. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Kováč, P.; Sinaÿ, P.; Glaudemans, C.P.J. Synthetic C-oligosaccharides mimic their natural, analogous immunodeterminants in binding to three monoclonal immunoglobulins. Carbohydr. Res. 1998, 308, 191–193. [Google Scholar] [CrossRef] [PubMed]
- Machut-Binkowski, C.; Hapiot, F.; Cecchelli, R.; Martin, P.; Monflier, E. A versatile liposome/cyclodextrin supramolecular carrier for drug delivery through the blood-brain barrier. J. Incl. Phenom. Macrocycl. Chem. 2007, 57, 567–572. [Google Scholar] [CrossRef]
- Saad, M.; Garbuzenko, O.B.; Ber, E.; Chandna, P.; Khandare, J.J.; Pozharov, V.P.; Minko, T. Receptor targeted polymers, dendrimers, liposomes: Which nanocarrier is the most efficient for tumor-specific treatment and imaging? J. Control. Release 2008, 130, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Dziubla, T.D.; Shuvaev, V.V.; Hong, N.K.; Hawkins, B.J.; Madesh, M.; Takano, H.; Simone, E.; Nakada, M.T.; Fisher, A.; Albelda, S.M.; Muzykantov, V.R. Endothelial targeting of semi-permeable polymer nanocarriers for enzyme therapies. Biomaterials 2008, 29, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Leamon, C.P.; Reddy, J.A.; Vetzel, M.; Dorton, R.; Westrick, E.; Parker, N.; Wang, Y.; Vlahov, I. Folate targeting enables durable and specific antitumor responses from a therapeutically null tubulysin B analogue. Cancer Res. 2008, 68, 9839–9844. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Cai, Z.; Song, X.; Chen, Q.; Bi, Y.; Li, Y.; Hou, S. Preparation and characterization of folate conjugated N-trimethyl chitosan nanoparticles as protein carrier targeting folate receptor: In vitro studies. J. Drug Target. 2009, 17, 294–303. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors. Licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Solaro, R.; Chiellini, F.; Battisti, A. Targeted Delivery of Protein Drugs by Nanocarriers. Materials 2010, 3, 1928-1980. https://doi.org/10.3390/ma3031928
Solaro R, Chiellini F, Battisti A. Targeted Delivery of Protein Drugs by Nanocarriers. Materials. 2010; 3(3):1928-1980. https://doi.org/10.3390/ma3031928
Chicago/Turabian StyleSolaro, Roberto, Federica Chiellini, and Antonella Battisti. 2010. "Targeted Delivery of Protein Drugs by Nanocarriers" Materials 3, no. 3: 1928-1980. https://doi.org/10.3390/ma3031928