Effects of Mechanical Ball Milling Time on the Microstructure and Mechanical Properties of Mo2NiB2-Ni Cermets

Abstract
:1. Introduction
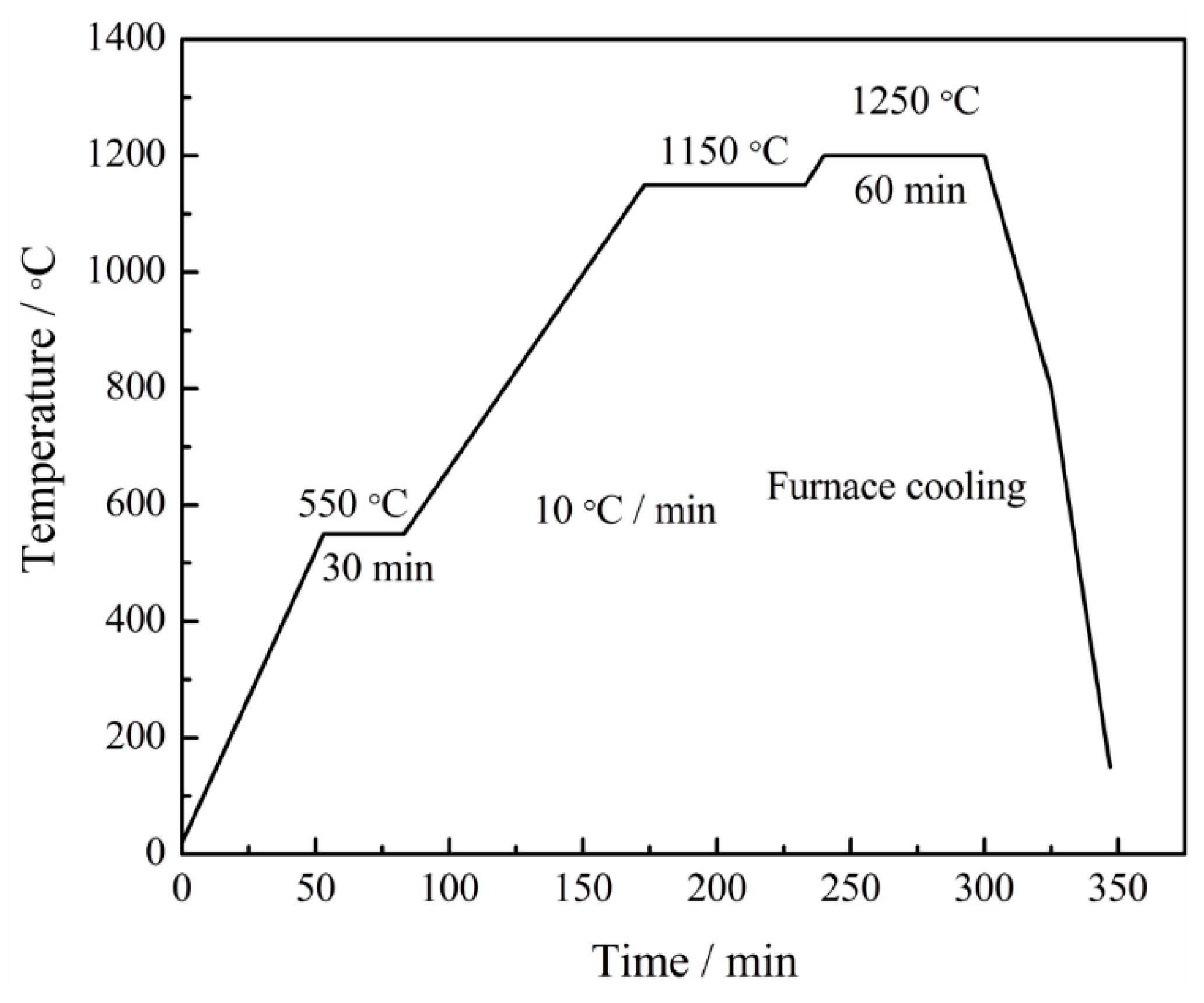
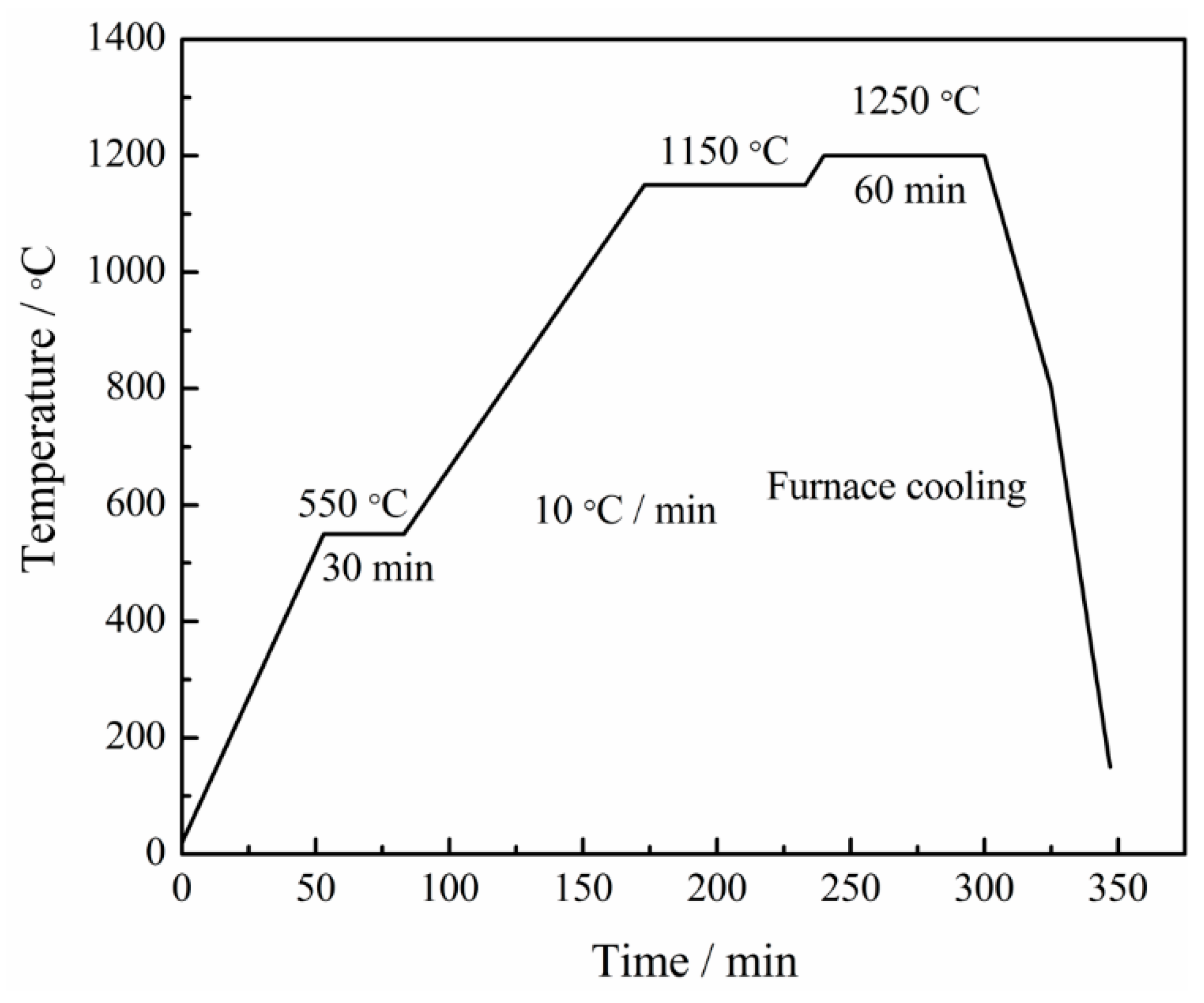
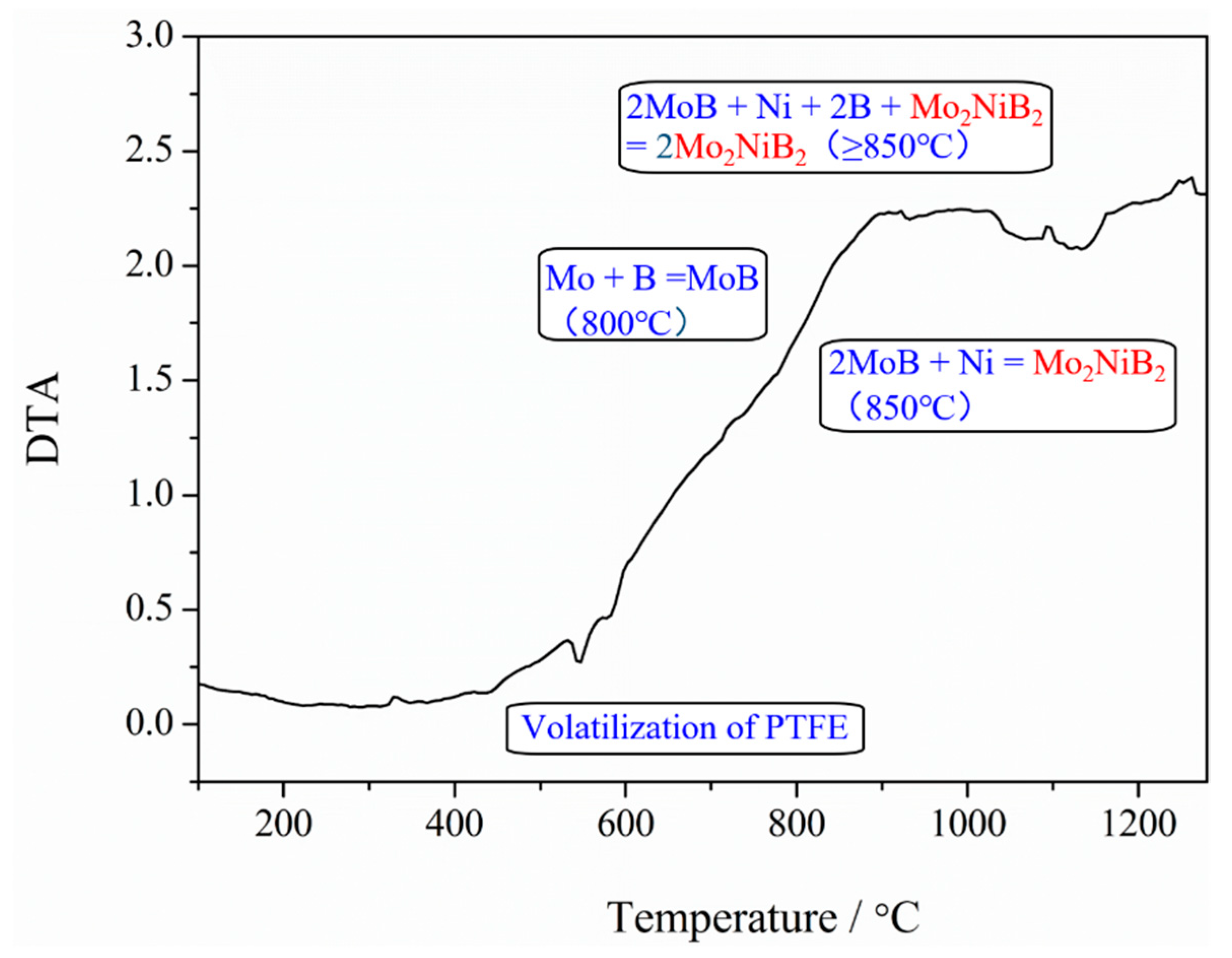
2. Materials and Methods
3. Results and Discussion
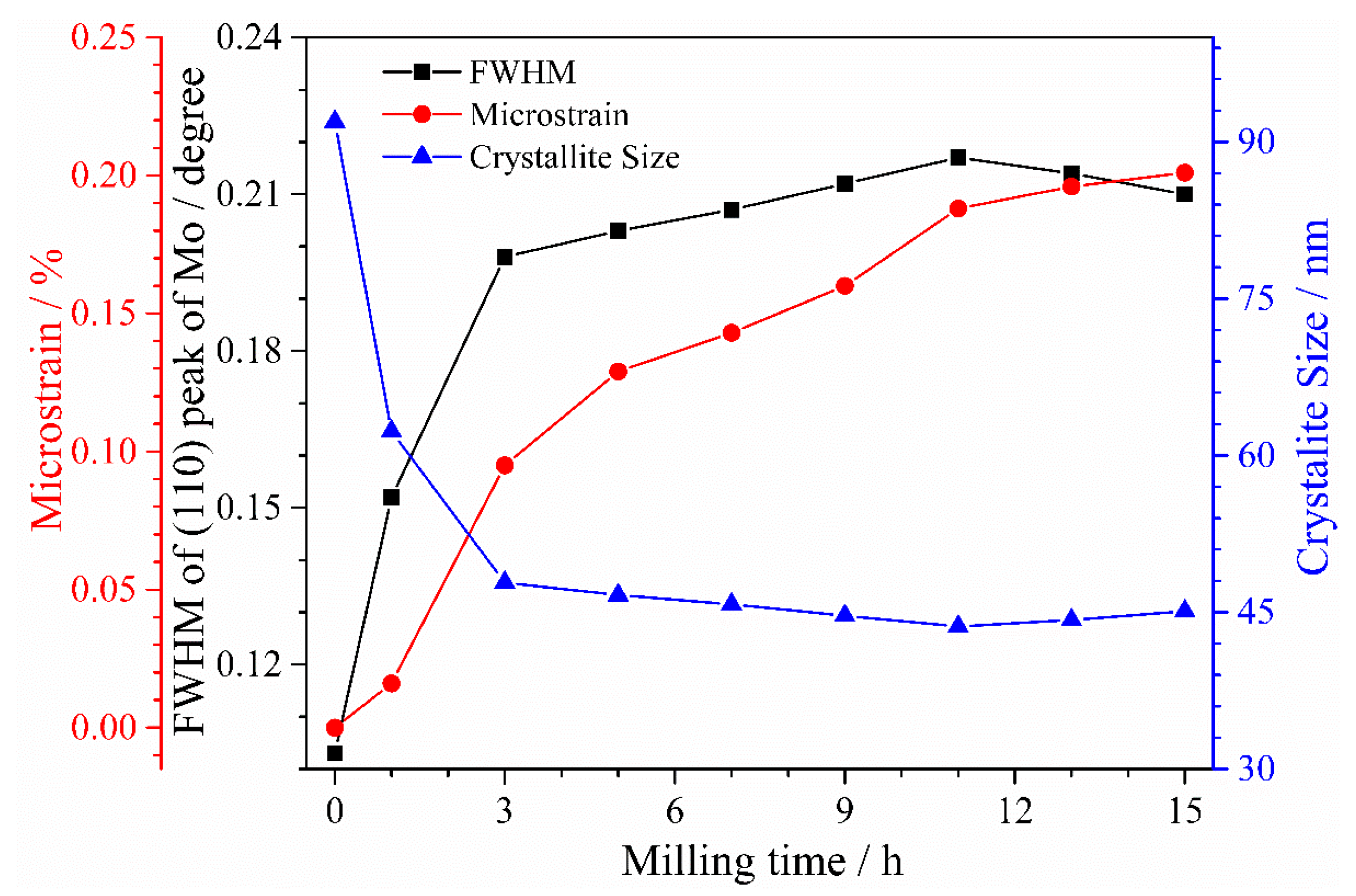
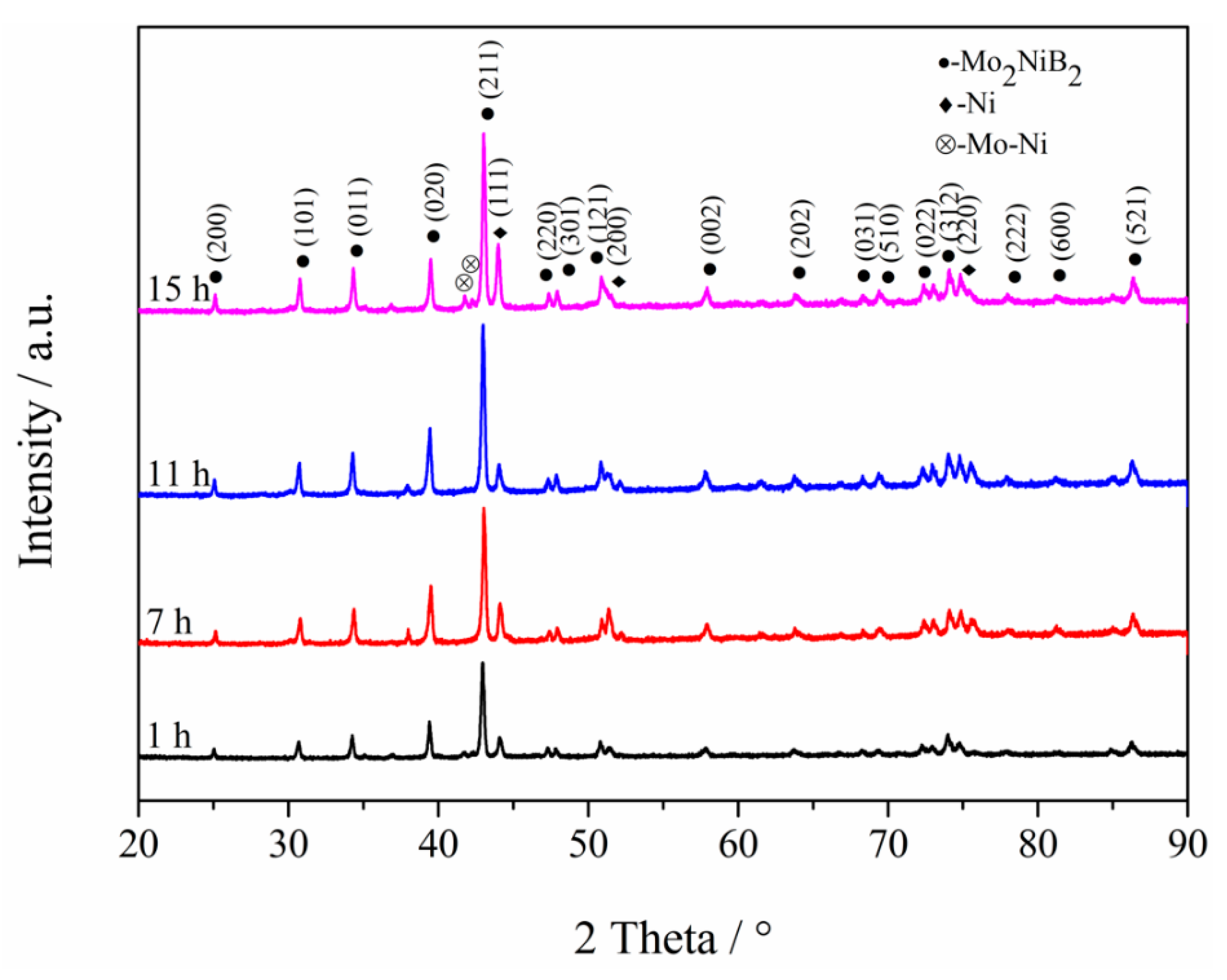
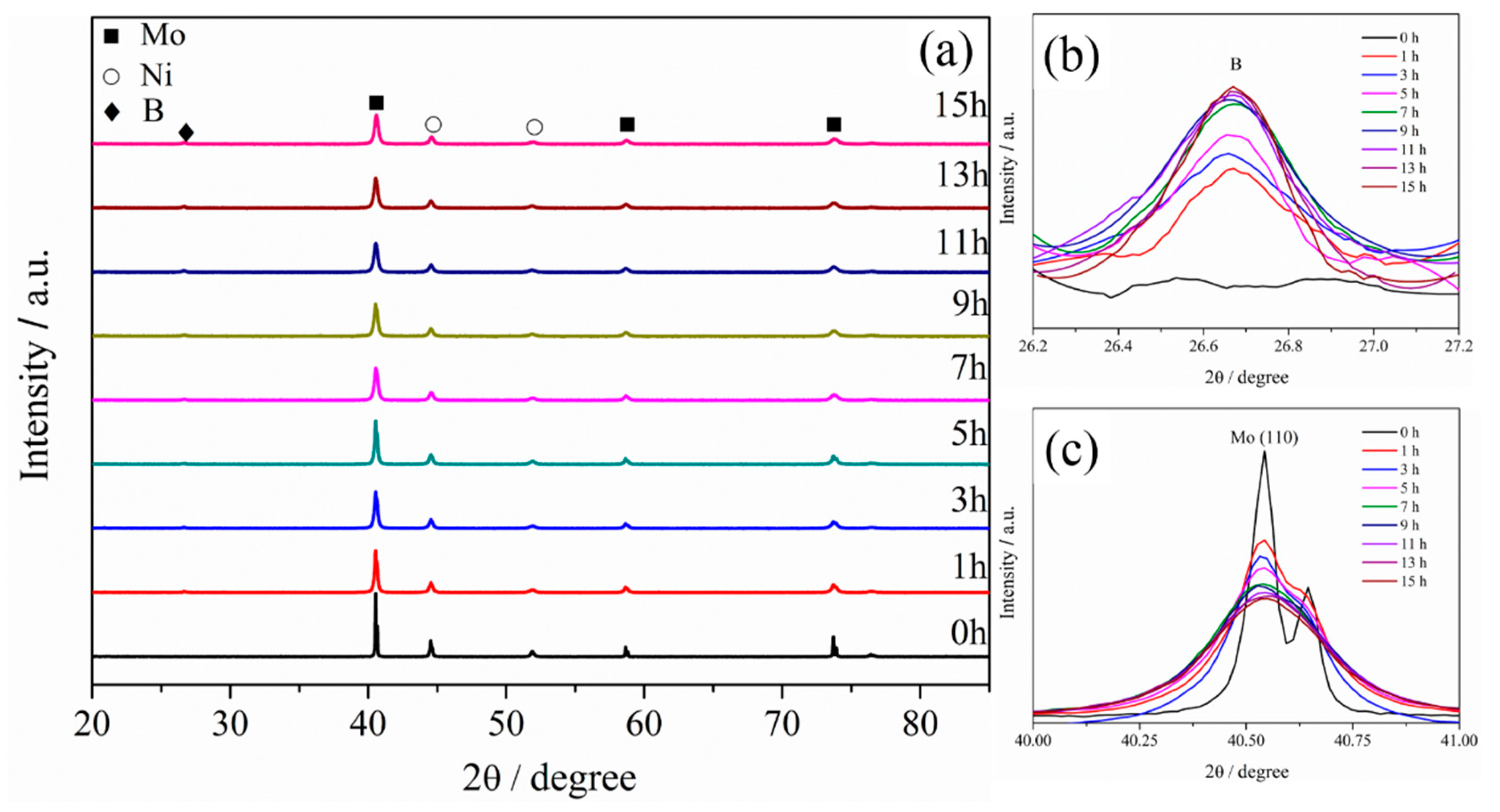
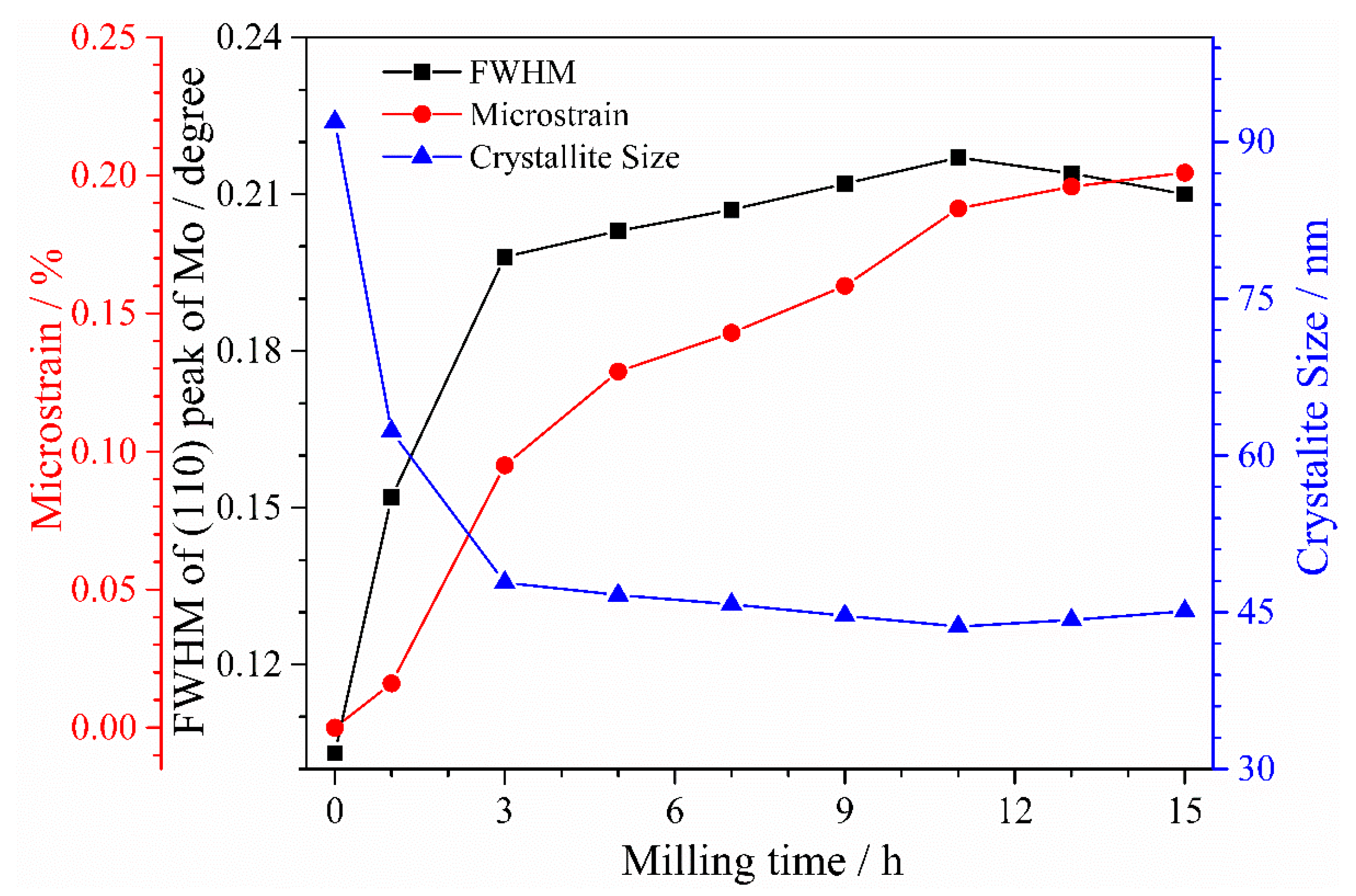
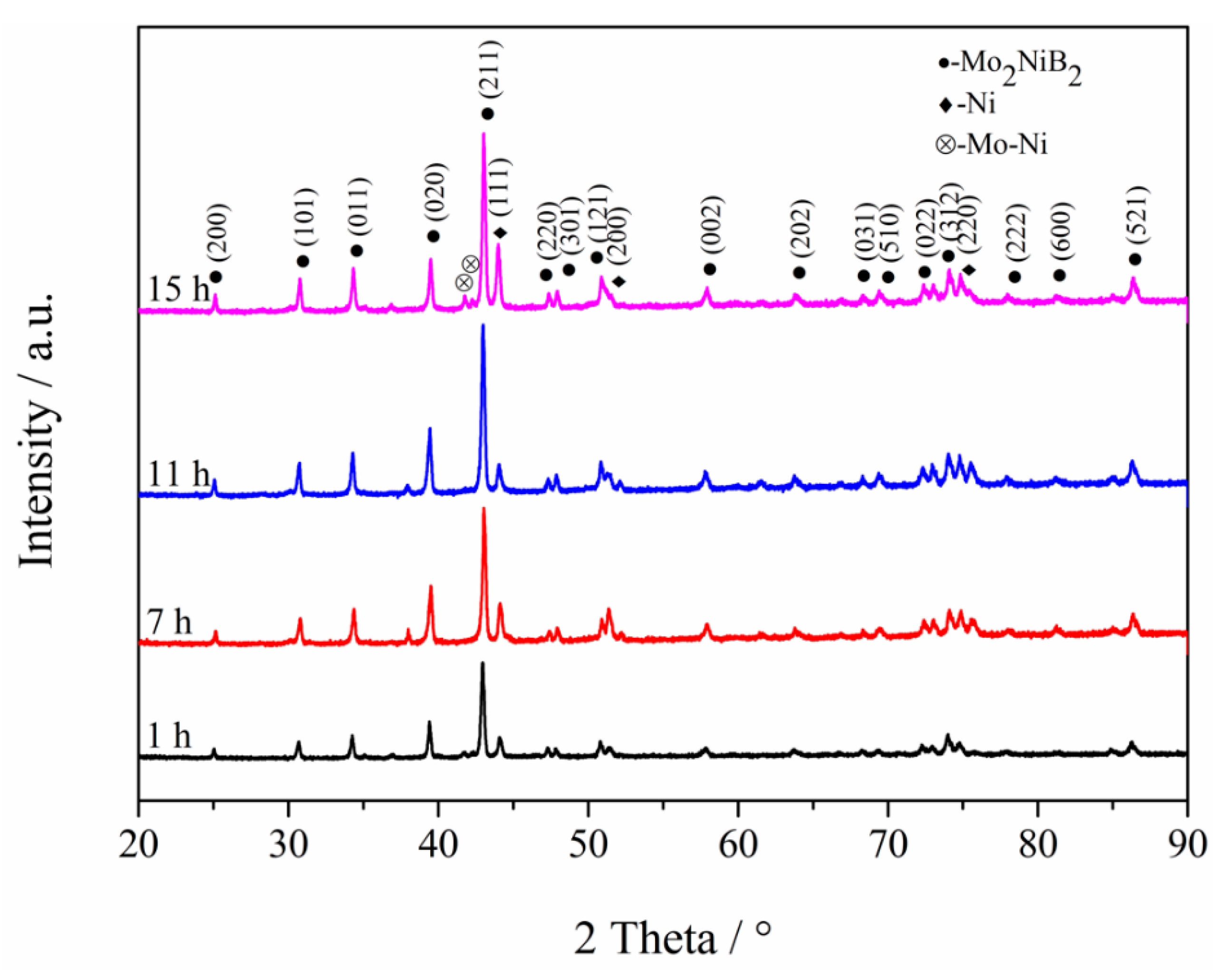
3.1. XRD Analysis of Powders at Different Milling Times
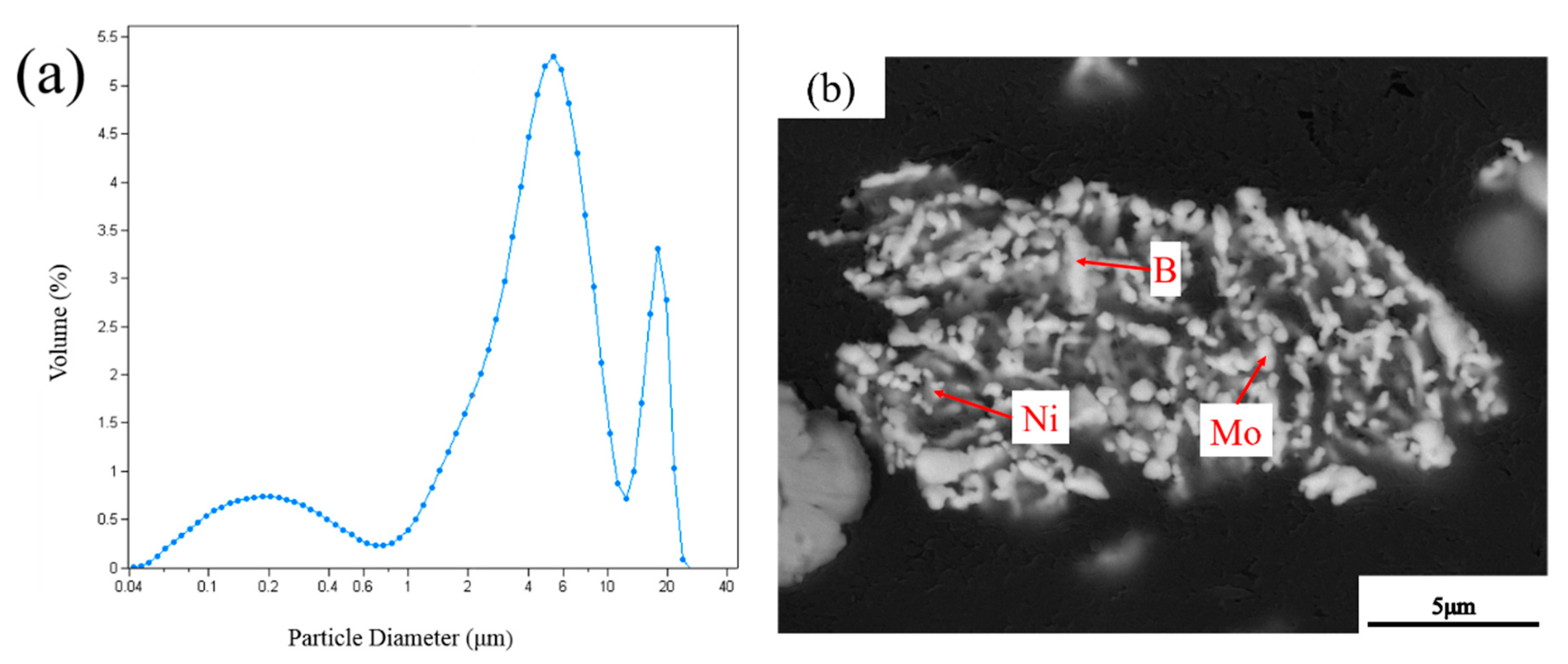
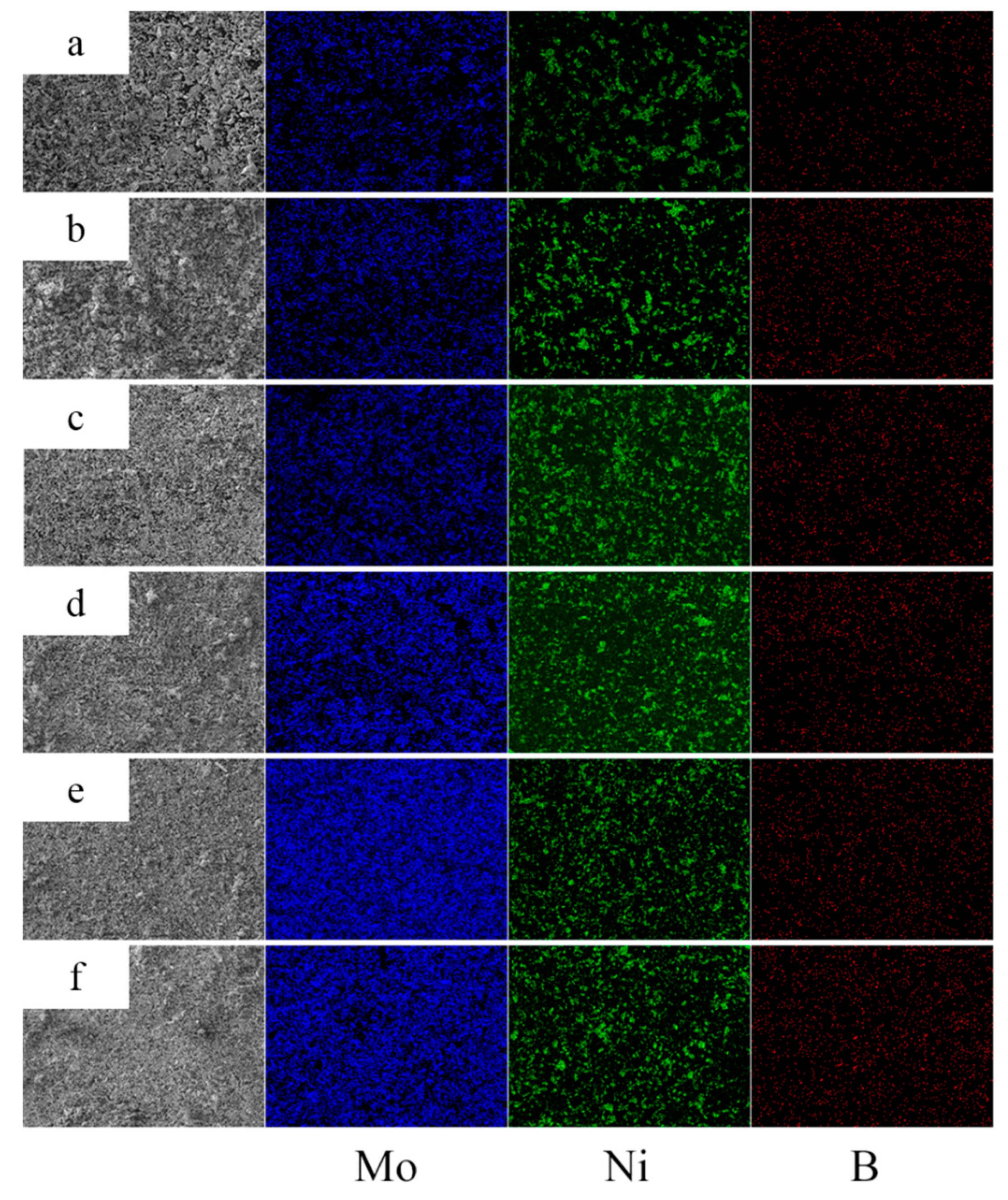
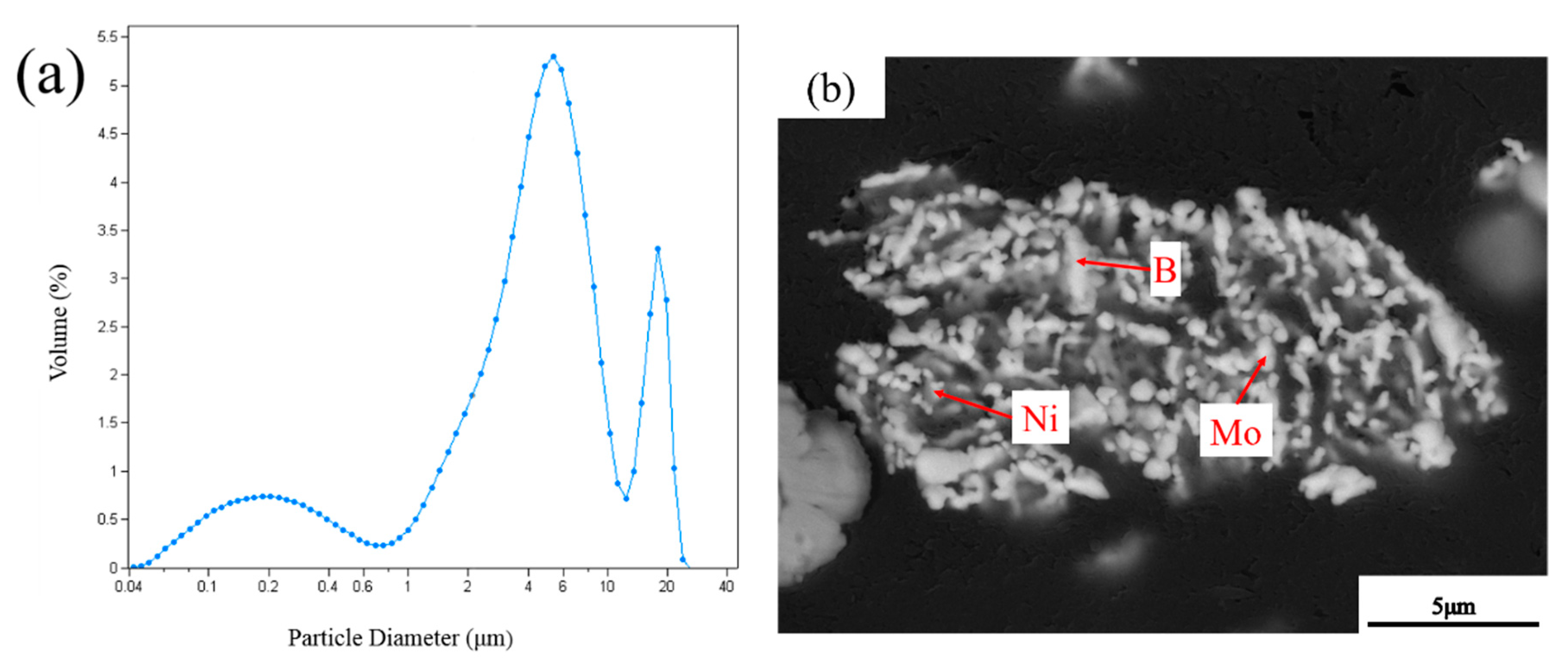
3.2. EDS Analysis of Powders at Different Milling Times
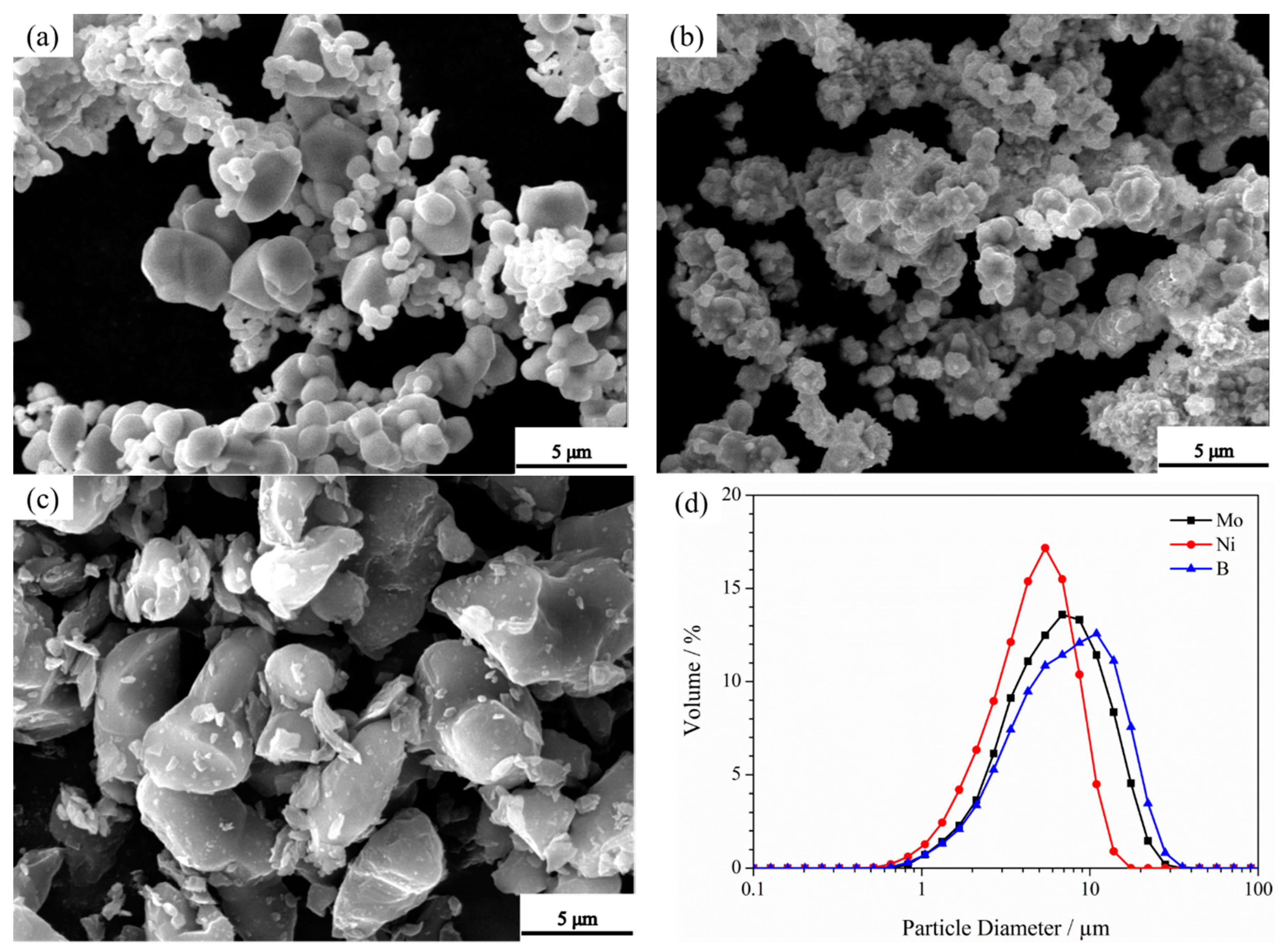
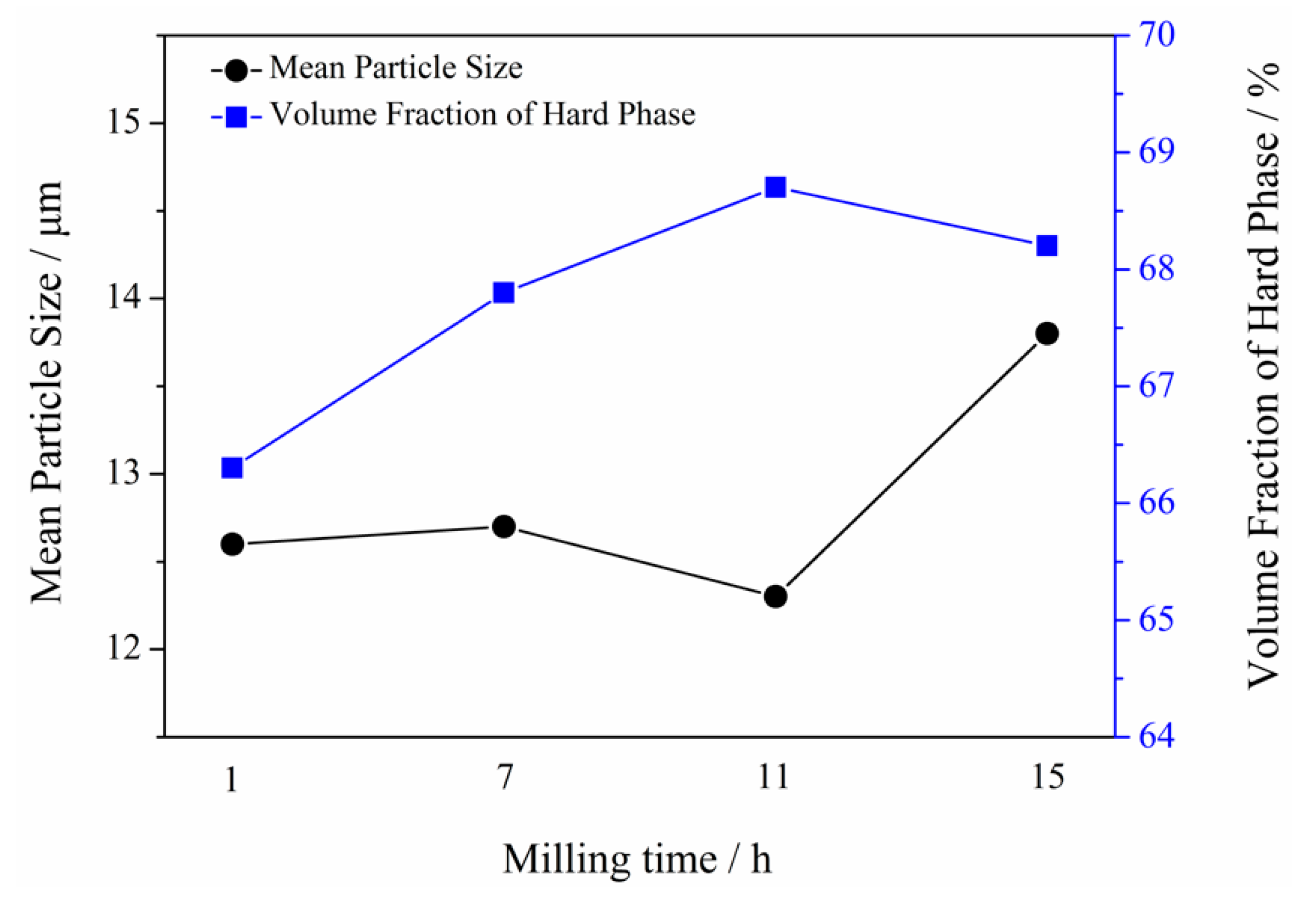
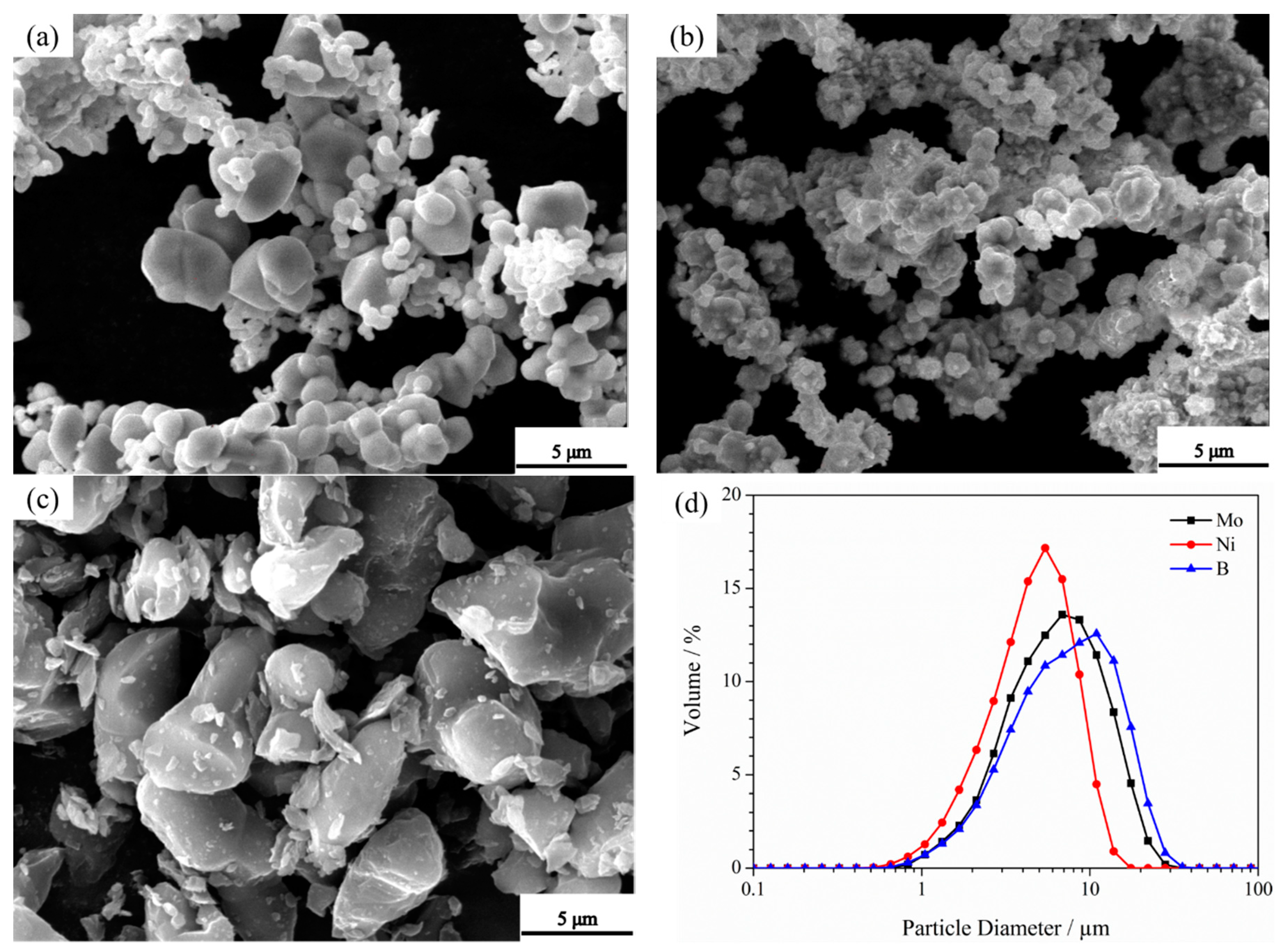
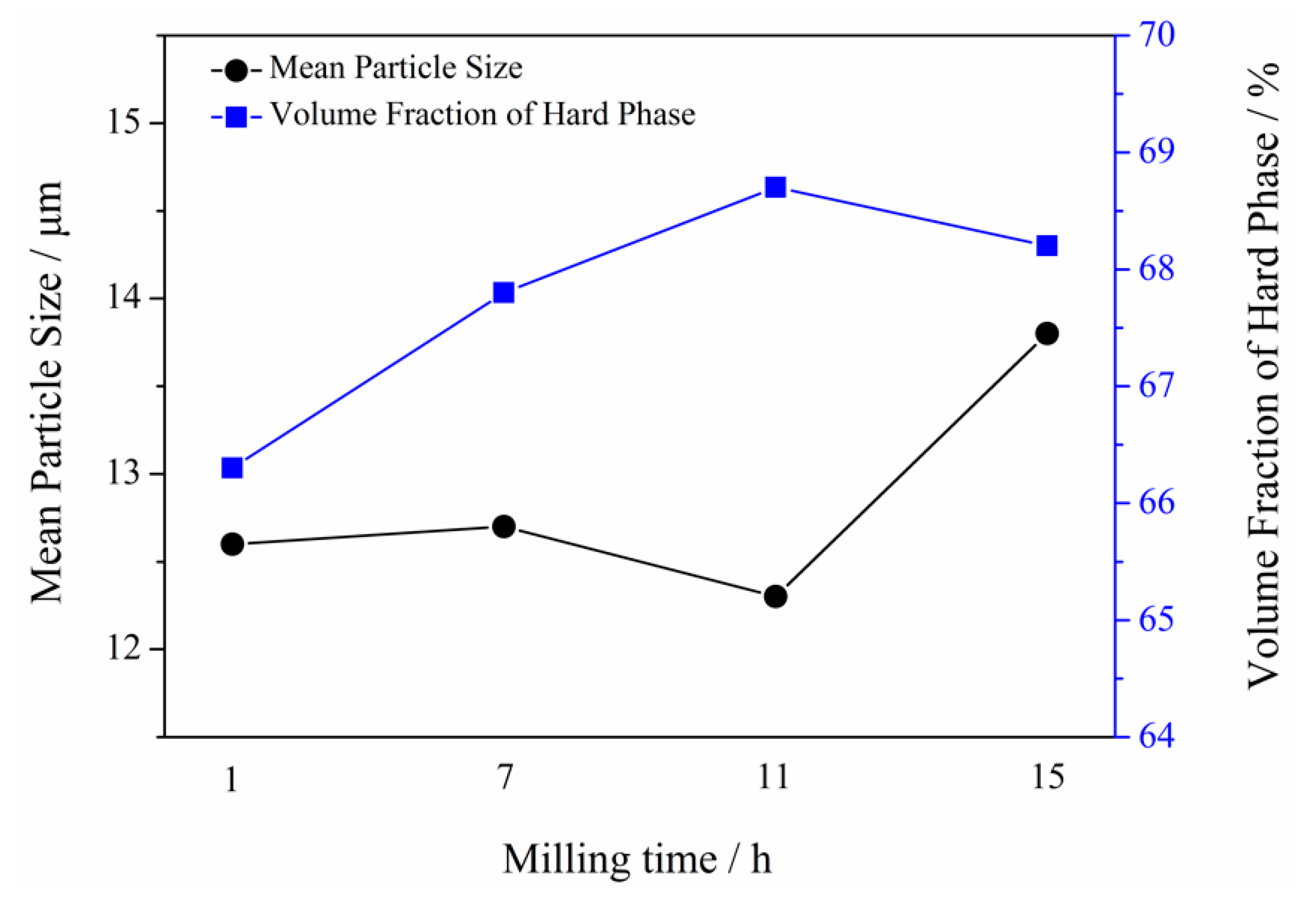
3.3. Microstructure of Powders at Different Milling Times
3.4. Phase Composition of Mo2NiB2-Ni Cermets of Powders at Different Milling Times
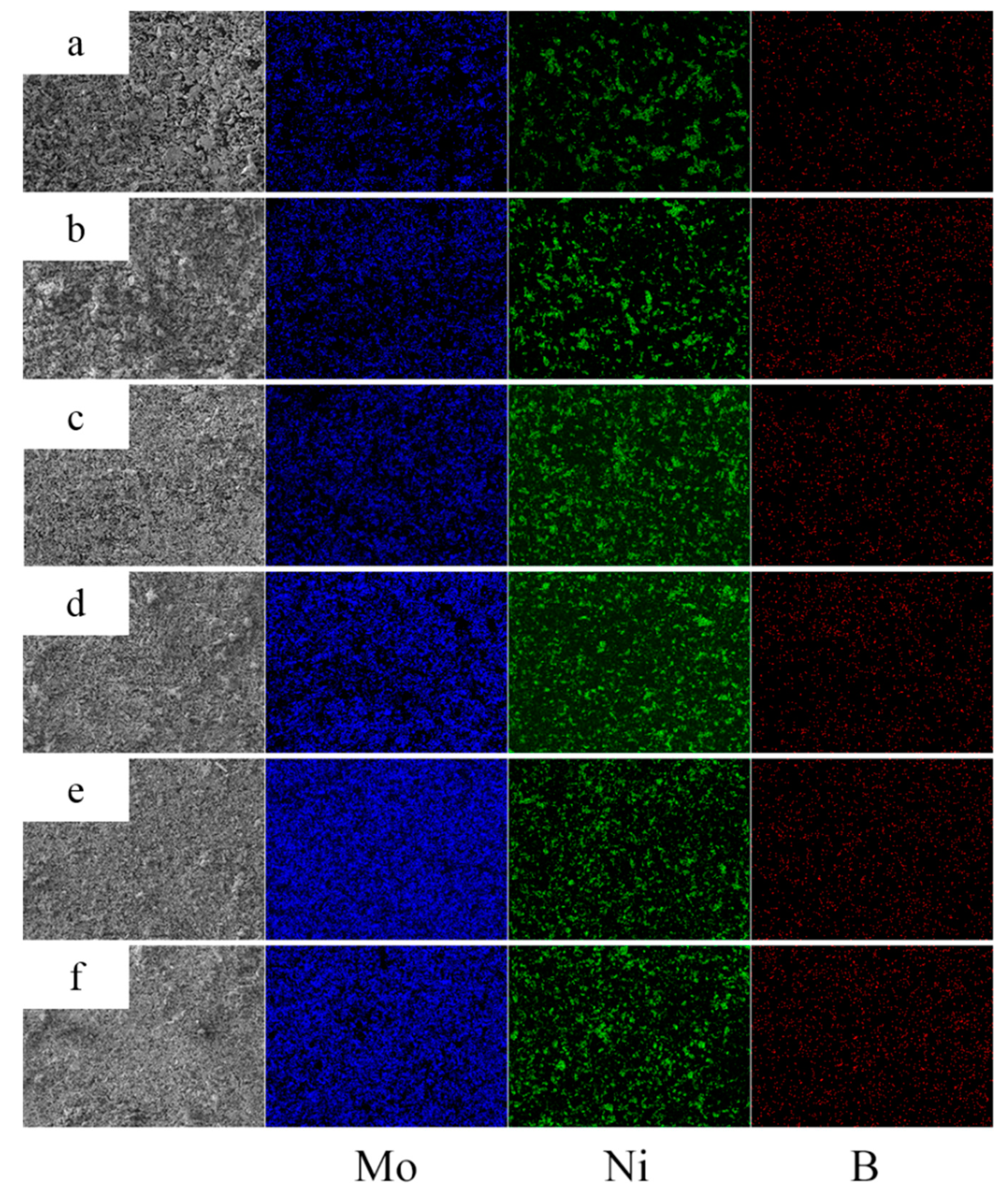
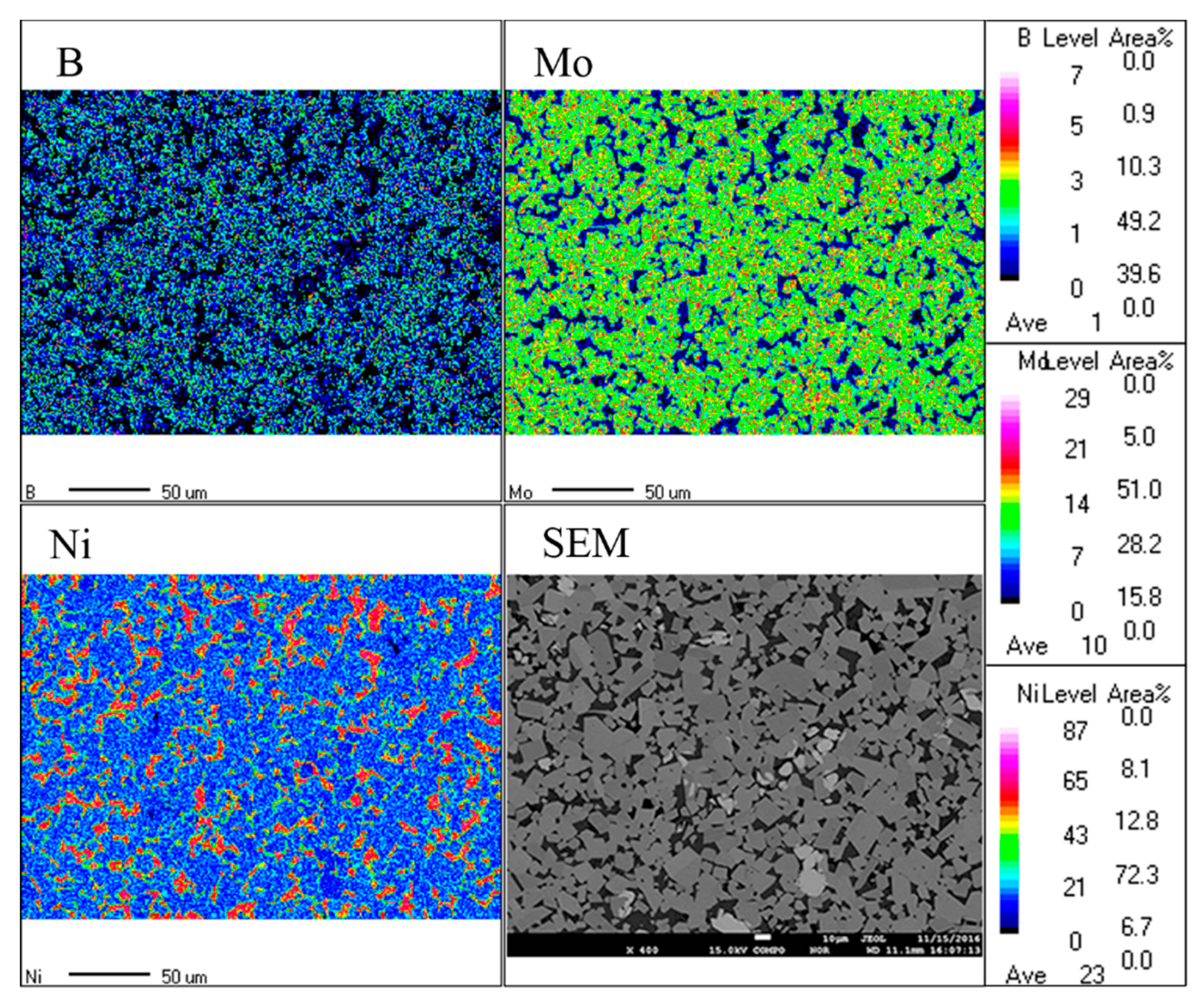
3.5. Microstructure of Mo2NiB2-Ni Cermets of Powders at Different Milling Times
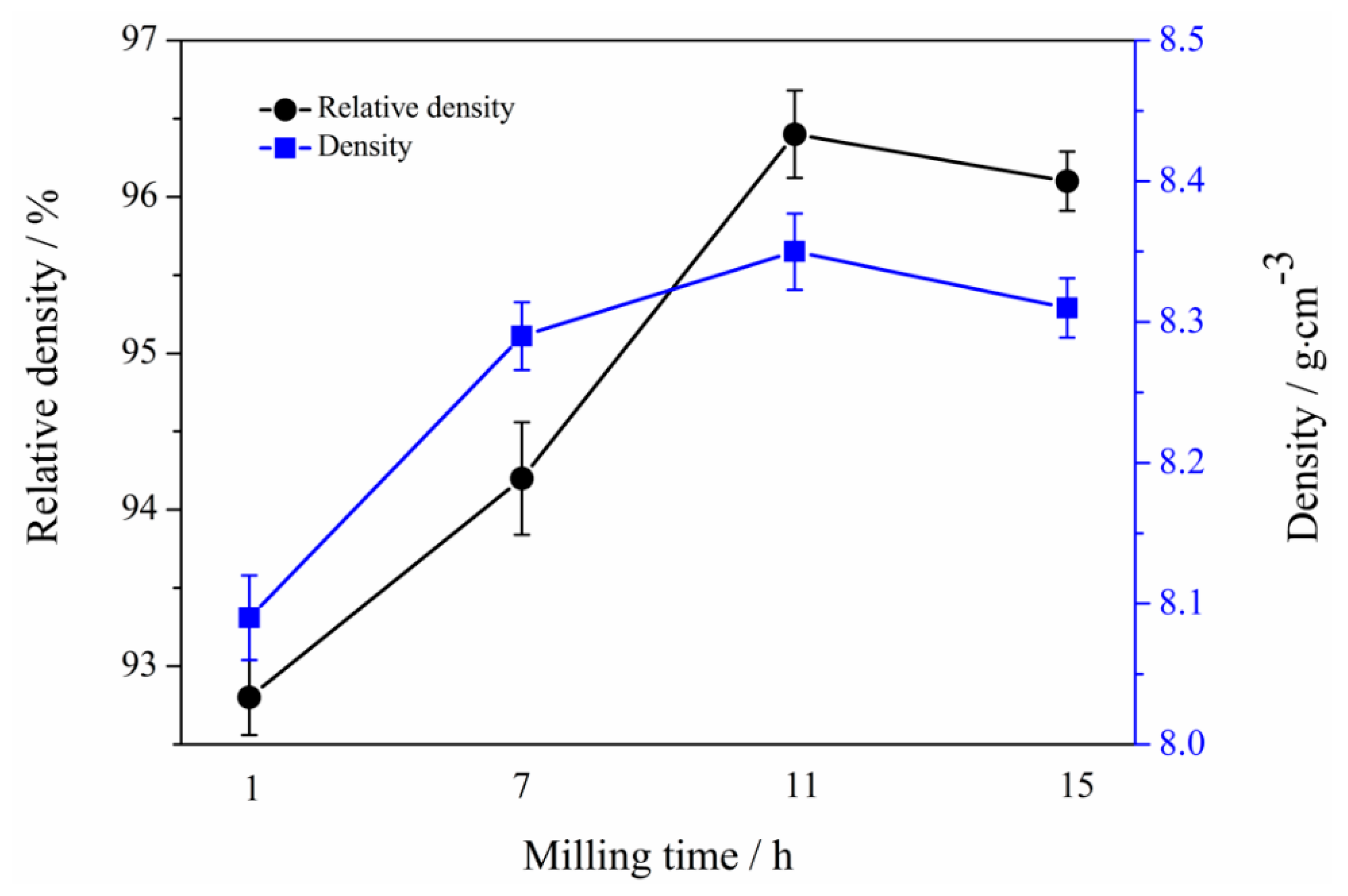
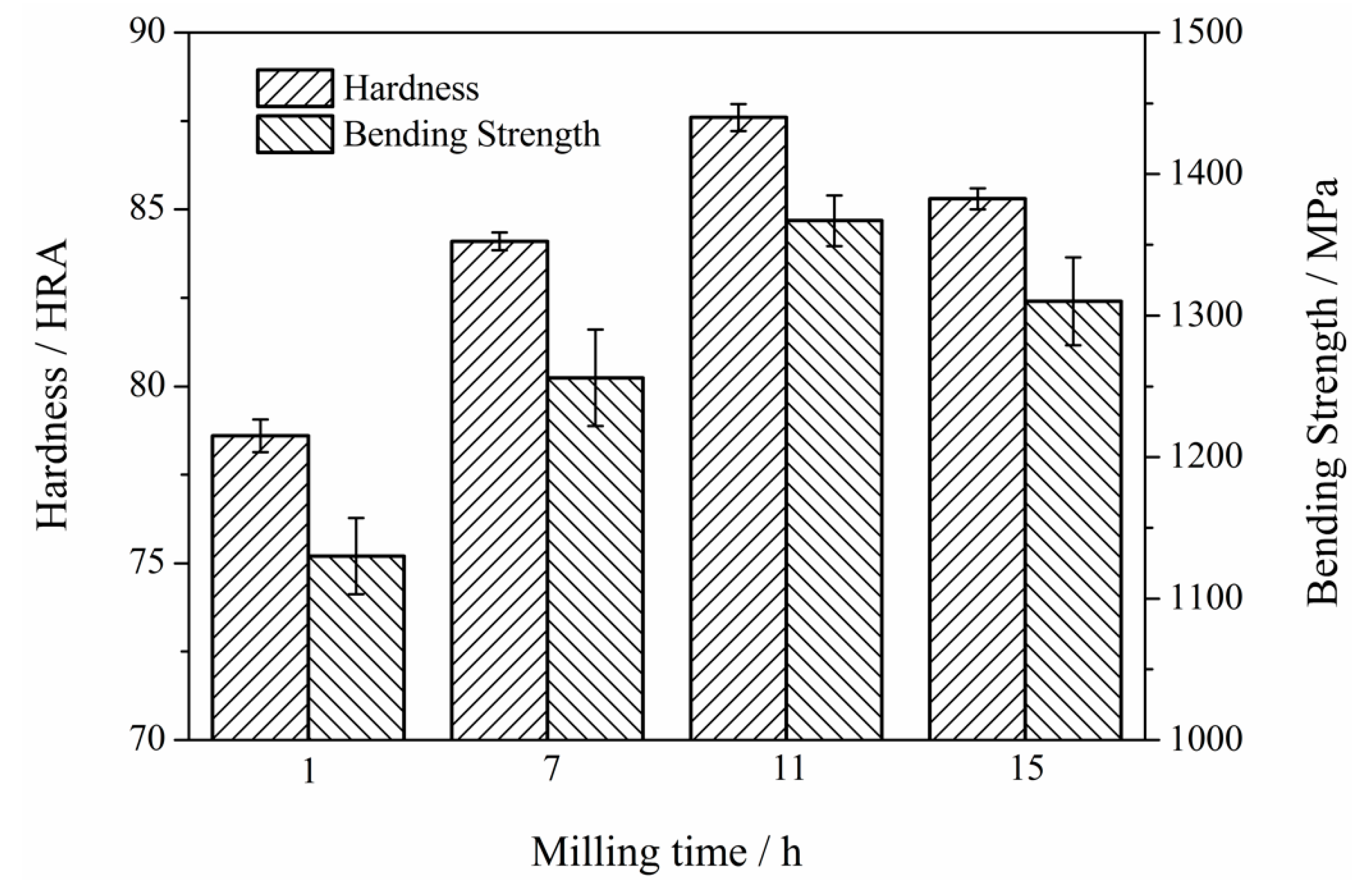
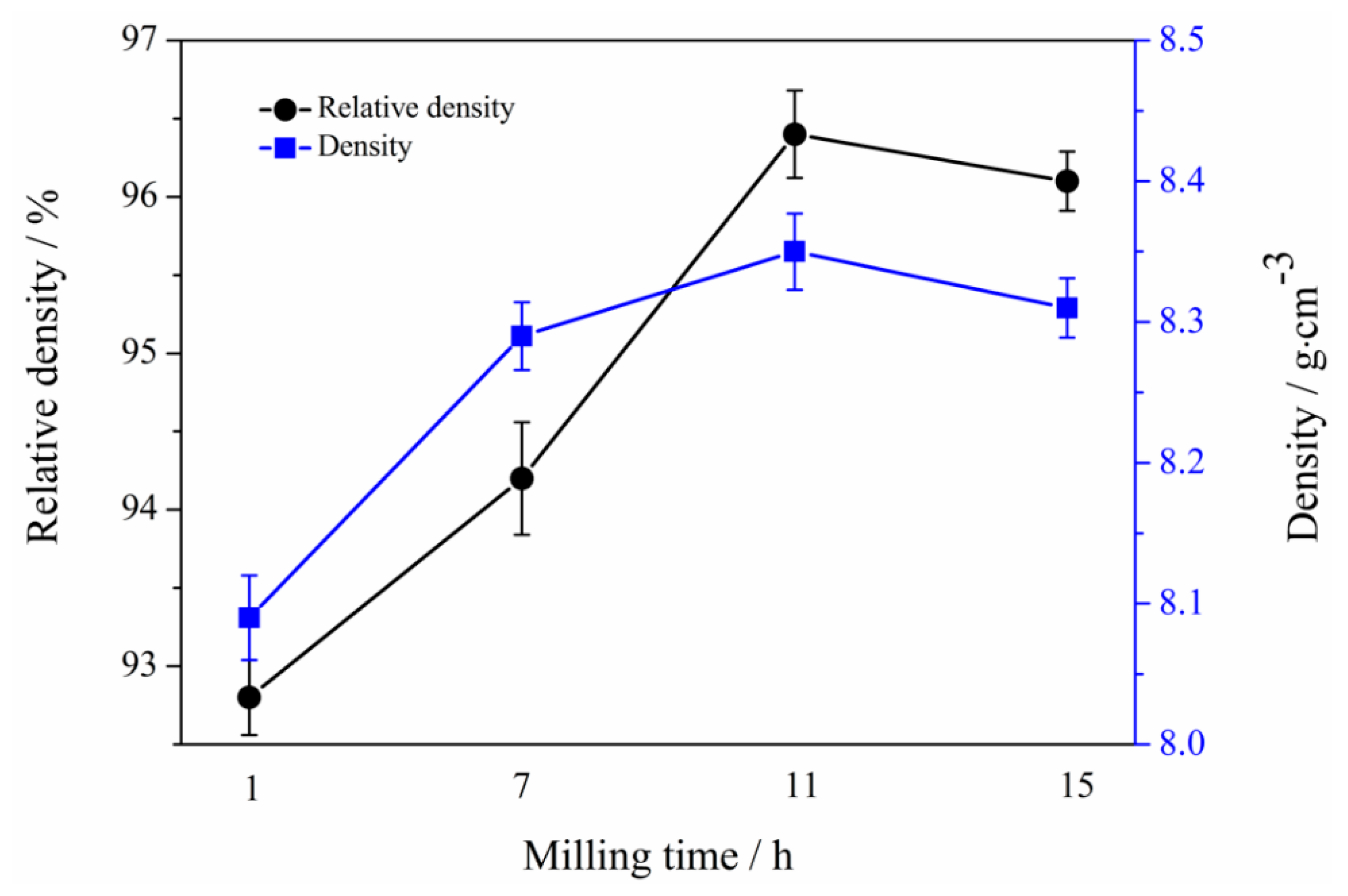
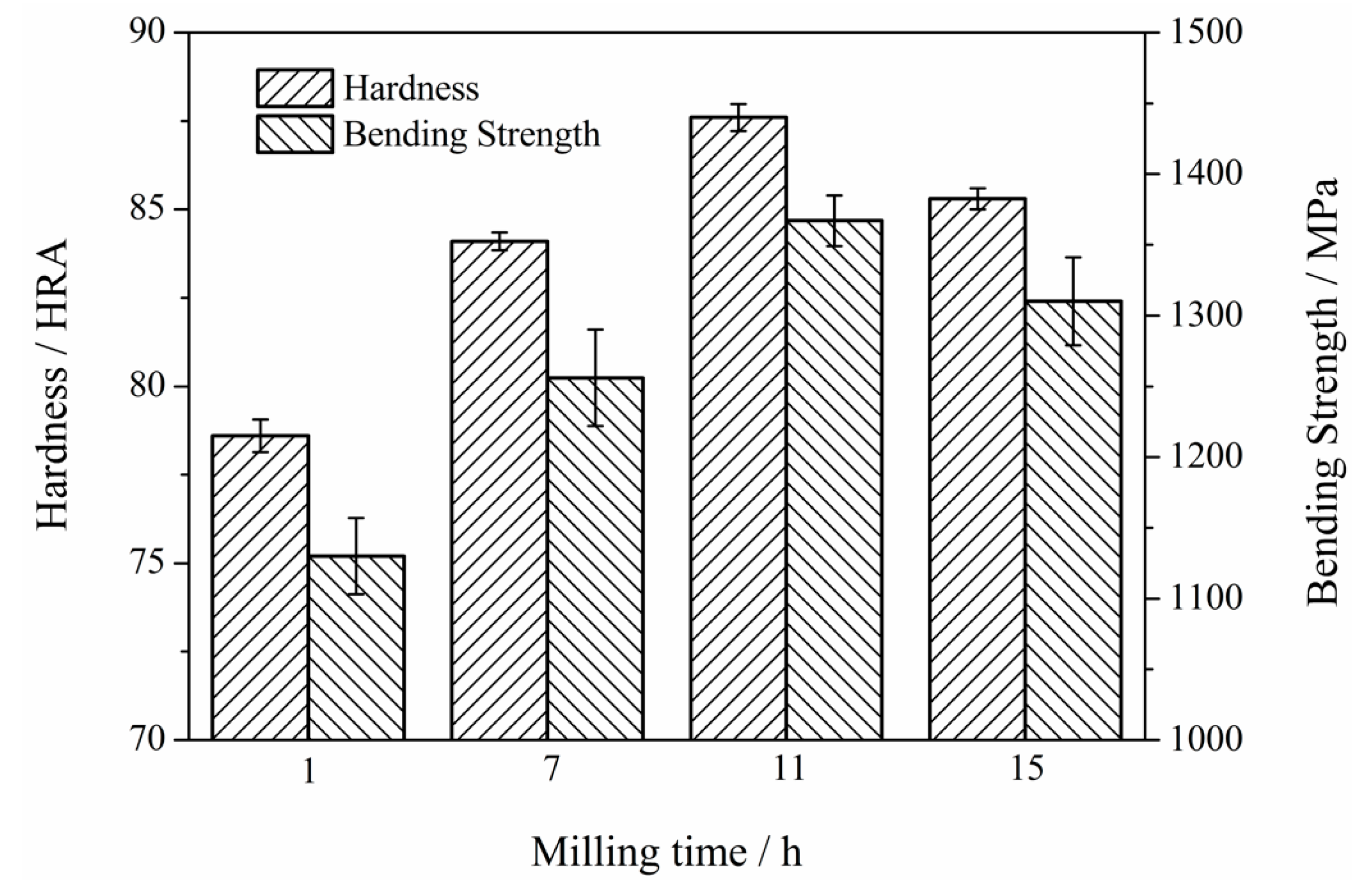
3.6. Mechanical Properties of Mo2NiB2-Ni Cermets of Powders at Different Milling Times
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Takagi, K. High tough boride base cermets produced by reaction sintering. Mater. Chem. Phys. 2001, 67, 214–219. [Google Scholar] [CrossRef]
- Takagi, K.; Yamasaki, Y.; Komai, M. High-strength boride base hard materials. J. Solid State Chem. 1997, 133, 243–248. [Google Scholar] [CrossRef]
- Takagi, K. Development and application of high strength ternary boride base cermets. J. Solid State Chem. 2006, 179, 2809–2818. [Google Scholar] [CrossRef]
- Takagi, K.; Yamasaki, Y.; Hirata, K. Development of high strength complex boride base hard materials produced by reaction boronizing sintering. J. Jpn. Soc. Powder Powder Met. 1998, 45, 507–514. [Google Scholar] [CrossRef]
- Takagi, K.; Ohira, S.; Ide, T. New multiple-boride base hard alloy. Met. Powder Rep. 1987, 42, 483–490. [Google Scholar]
- Takagi, K.; Komai, M.; Kondo, Y. Microstructure and properties of sintered Mo2FeB2 base hard alloys. Sci. Technol. Sinter. Tokyo 1987, 11, 1296–1301. [Google Scholar]
- Yamasaki, Y.; Nishi, M.; Takagi, K. Development of very high strength Mo2NiB2 complex boride base hard alloy. J. Solid State Chem. 2004, 177, 551–555. [Google Scholar] [CrossRef]
- Komai, M.; Isobe, Y.; Ozaki, S. Effects of Cr on the microstructure and mechanical properties of WCoB base hard alloys. J. Jpn. Soc. Powder Powder Met. 1993, 40, 38–43. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Z. Development of ternary-boride-based hard cladding material. Mater. Res. Bull. 2002, 37, 417–423. [Google Scholar]
- Koyama, K.; Morishita, M.; Zhang, G. Calcualated Phase Diagrams of the Ni-Mo-B and Ni-W-B Ternary Systems on the Basis of the Data Obtained by Thermodynamic Measurement of the Related Materials. J. Jpn. Soc. Powder Powder Met. 2006, 53, 419–429. [Google Scholar] [CrossRef]
- Hirata, K.; Iwanaga, K.; Yamasaki, Y. Development of Mo2NiB2 base cermets with high strength and excellent corrosion resistance for plastic injection molding machine parts. J. Jpn. Soc. Powder Powder Met. 2006, 53, 447–451. [Google Scholar] [CrossRef]
- Hirata, K.; Iwanaga, K.; Yamasaki, Y. Development of High Corrosion Resistant Mo2NiB2 Boride Base Cermets for Plastic Injection Molding Machine Parts. In Proceedings of the Materials Science Forum, Herceg-Novi, Montenegro, 10–14 September 2007. [Google Scholar]
- Huang, P.Y. Principle of Powder Metallurgy, 2nd ed.; Machine Press: Beijing, China, 2012. [Google Scholar]
- Yuan, B.; Zhang, G.; Kan, Y. Reactive synthesis and mechanical properties of Mo2NiB2 based hard alloy. Int. J. Refract. Met. Hard Mater. 2010, 28, 291–296. [Google Scholar] [CrossRef]
- Shen, Y.; Huang, Z.F.; Zhang, L. Effect of milling time on the microstructure and mechanical properties of Mo2FeB2 based cermets. Mater. Res. Express 2017, 4, 106518. [Google Scholar] [CrossRef]
- Zhang, L.; Huang, Z.F.; Shen, Y. Bulk Mo2FeB2 based cermets fabricated by mechanical ball milling and reaction boronizing sintering. Powder Metall. Met. Ceram. 2017, 55, 665–675. [Google Scholar] [CrossRef]
- Dorset, D.L. X-ray Diffraction: A Practical Approach. Microsc. Microanal. 1998, 4, 513–515. [Google Scholar] [CrossRef]
- Khorsand, Z.A. X-ray analysis of ZnO nanoparticles by Williamson–Hall and size–strain plot methods. Solid State Sci. 2011, 13, 251–256. [Google Scholar] [CrossRef]
- Ozkaya, S.; Canakci, A. Effect of the B4C content and the milling time on the synthesis, consolidation and mechanical properties of AlCuMg-B4C nanocomposites synthesized by mechanical milling. Powder Technol. 2016, 297, 8–16. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Powder | Chemical Composition (wt%) | Particle Size (μm) | Manufacturer |
|---|---|---|---|
| Mo | Fe < 0.005, Si < 0.002, Mg < 0.002 | 3.4–8.6 | Changsha Tianjiu Metal Material Corp., Ltd., Changsha, China |
| Ni | Fe < 0.006, Mg < 0.002 | 2.8–6.8 | Changsha Tianjiu Metal Material Corp., Ltd., Changsha, China |
| B | Fe < 0.005, Mn < 0.003 | 5.4–10.1 | Baoding Zhongpuruituo Technology Corp., Ltd., Baoding, China |
| Mo | Ni | B |
|---|---|---|
| 60.5 | 33.5 | 6.0 |
| Milled Powders | Compositions (wt%) | |||||
|---|---|---|---|---|---|---|
| Mo | Ni | B | C | F | Si | |
| 0 h | 61.26 | 33.02 | 5.72 | - | - | - |
| 1 h | 60.50 | 32.66 | 5.48 | 1.36 | - | - |
| 3 h | 57.43 | 30.24 | 5.20 | 4.28 | 1.17 | 1.68 |
| 7 h | 56.75 | 28.64 | 5.15 | 6.07 | 1.52 | 1.87 |
| 11 h | 55.86 | 27.21 | 5.07 | 7.62 | 2.18 | 2.06 |
| 15 h | 53.97 | 25.83 | 4.69 | 10.04 | 3.16 | 2.31 |
| Point | Composition (at%) | ||
|---|---|---|---|
| Mo | Ni | B | |
| 1 | 40.31 | 21.36 | 38.33 |
| 2 | 3.98 | 96.02 | - |
| 3 | 64.19 | 35.81 | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Huang, Z.; Liu, Y.; Shen, Y.; Li, K.; Cao, Z.; Ren, Z.; Jian, Y. Effects of Mechanical Ball Milling Time on the Microstructure and Mechanical Properties of Mo2NiB2-Ni Cermets. Materials 2019, 12, 1926. https://doi.org/10.3390/ma12121926
Zhang L, Huang Z, Liu Y, Shen Y, Li K, Cao Z, Ren Z, Jian Y. Effects of Mechanical Ball Milling Time on the Microstructure and Mechanical Properties of Mo2NiB2-Ni Cermets. Materials. 2019; 12(12):1926. https://doi.org/10.3390/ma12121926
Chicago/Turabian StyleZhang, Lei, Zhifu Huang, Yangzhen Liu, Yupeng Shen, Kemin Li, Zhen Cao, Zijun Ren, and Yongxin Jian. 2019. "Effects of Mechanical Ball Milling Time on the Microstructure and Mechanical Properties of Mo2NiB2-Ni Cermets" Materials 12, no. 12: 1926. https://doi.org/10.3390/ma12121926