Molecular Crosstalk between Chromatin Remodeling and Tumor Microenvironment in Multiple Myeloma


{kind=link}
{kind=link}
Abstract
:1. Introduction
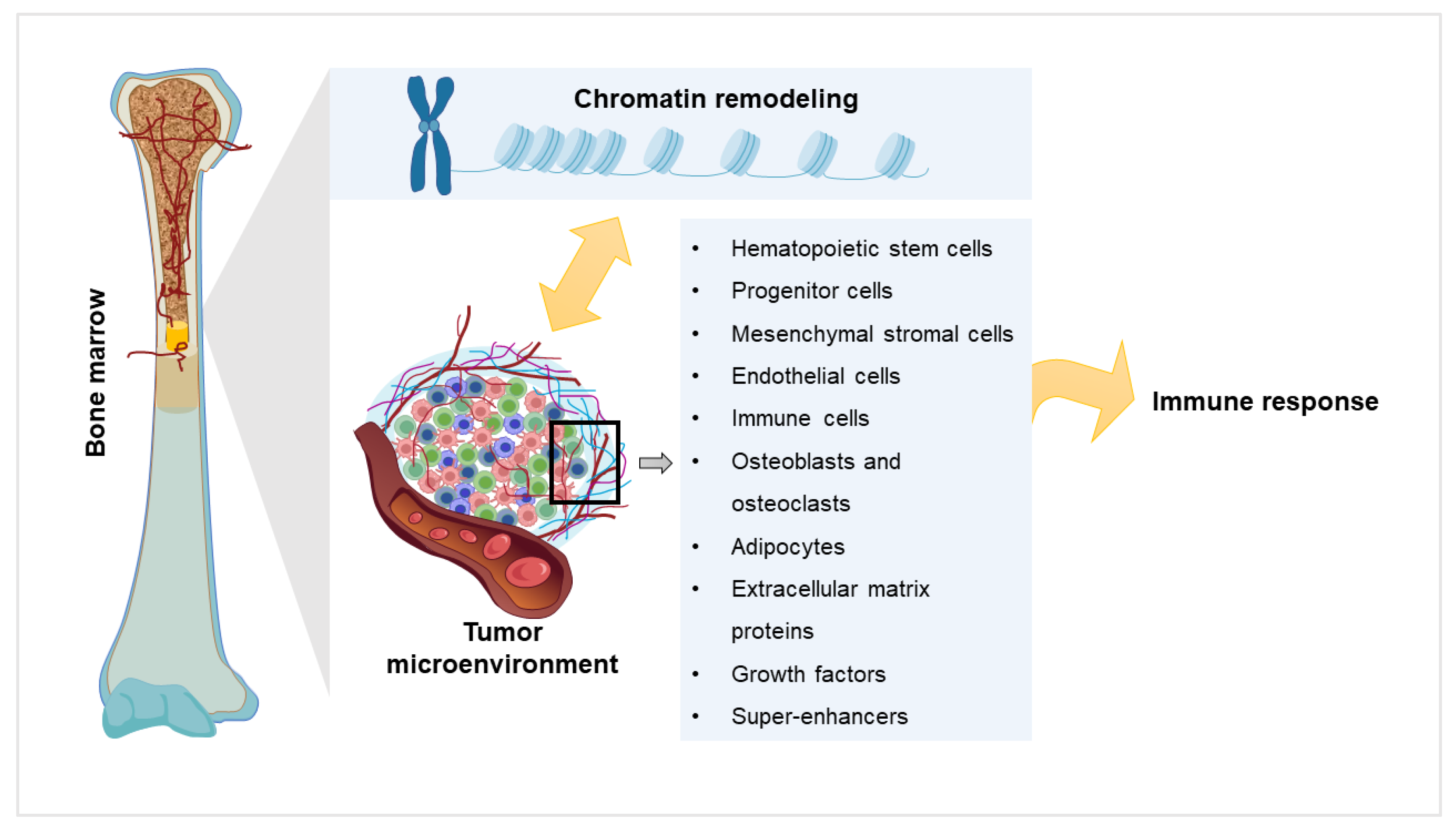
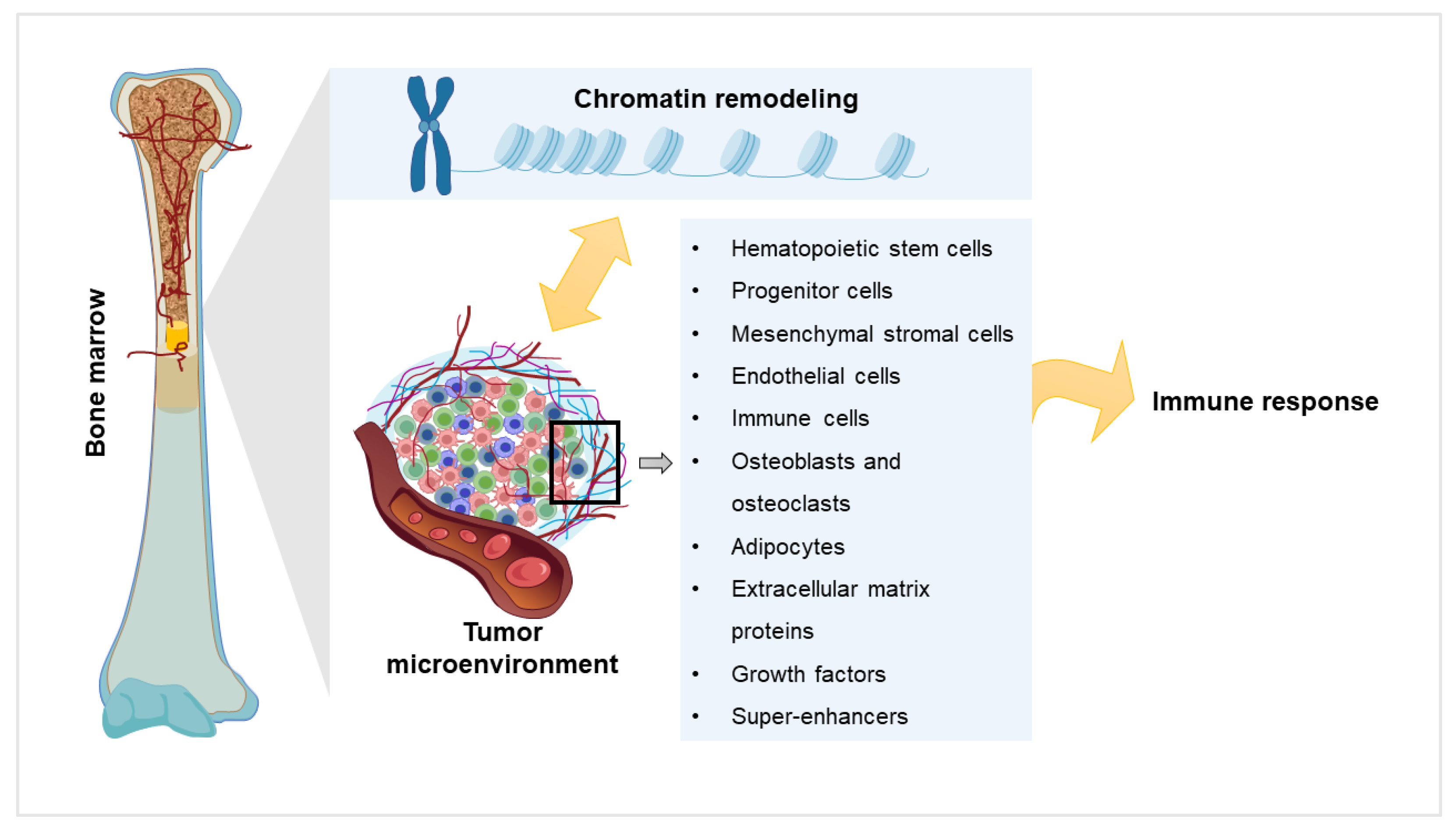
2. Chromatin Remodeling and Myeloma
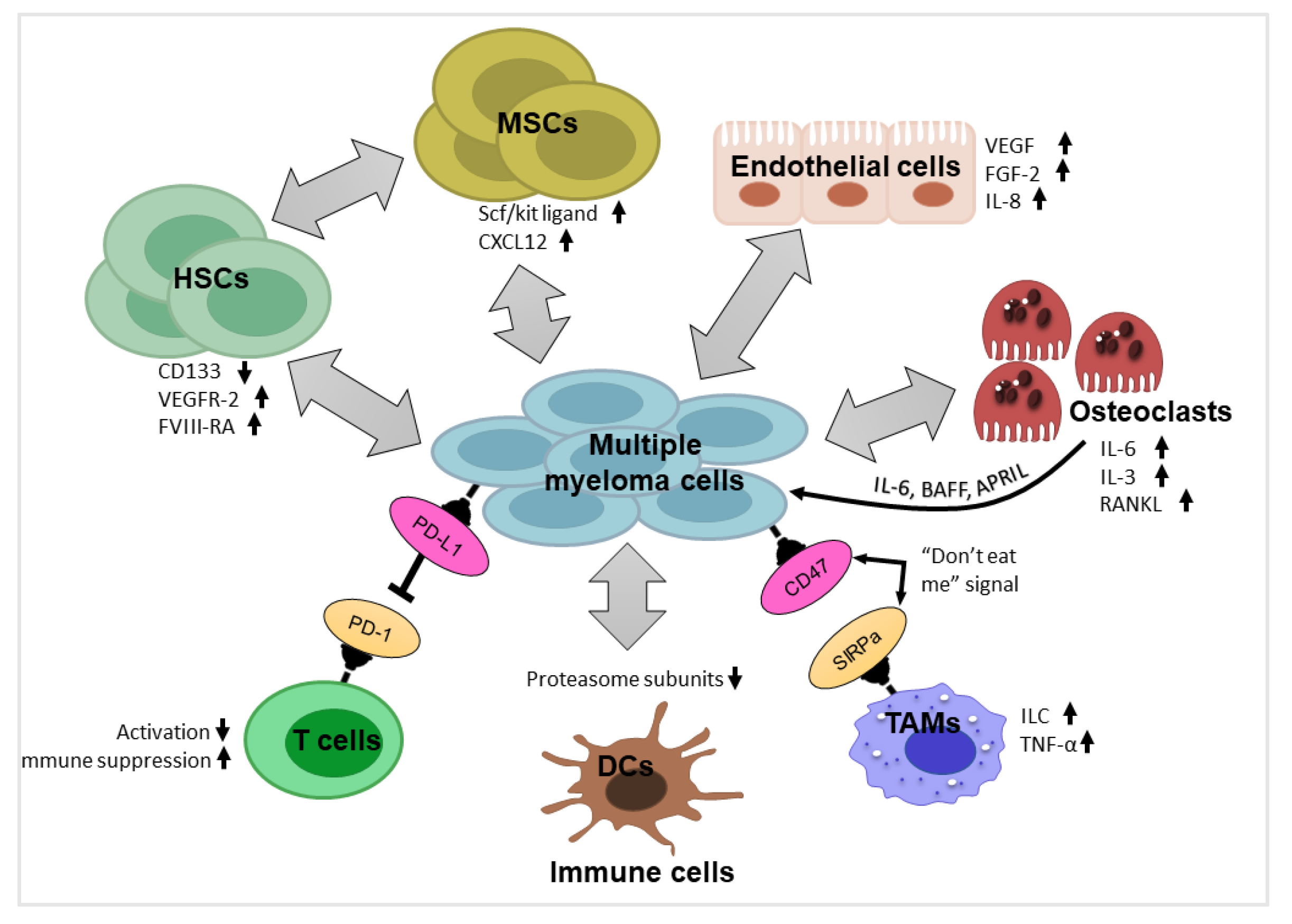
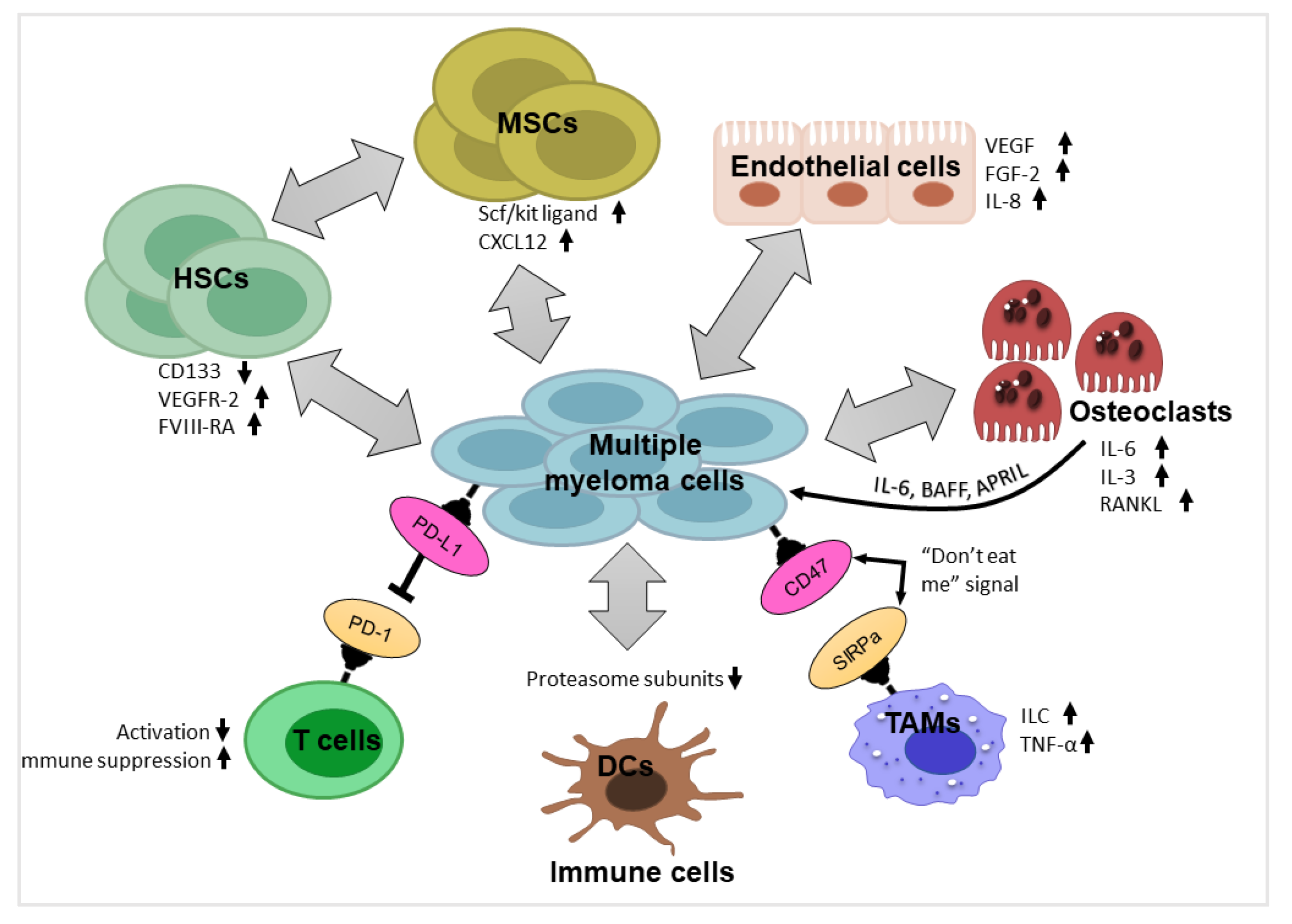
3. Components of Myeloma Tumor Microenvironment (TME) and their Impact
3.1. Hematopoietic Stem Cells and Progenitor Cells
3.2. Mesenchymal Stromal Cells
3.3. Endothelial Cells
3.4. Immune Cells
3.5. Osteoblasts and Osteoclasts
3.6. Adipocytes
3.7. Extracellular Matrix Proteins
3.8. Growth Factors
4. Components of the TME in the Light of Chromatin Remodeling
5. Super-Enhancers Affect Chromatin Remodeling and Bone Marrow Microenvironment in Myeloma
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kumar, S.K.; Rajkumar, V.; Kyle, R.A.; van Duin, M.; Sonneveld, P.; Mateos, M.V.; Gay, F.; Anderson, K.C. Multiple myeloma. Nat. Rev. Dis. Primers 2017, 3, 17046. [Google Scholar] [CrossRef] [PubMed]
- Kazandjian, D. Multiple myeloma epidemiology and survival: A unique malignancy. Semin. Oncol. 2016, 43, 676–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manojlovic, Z.; Christofferson, A.; Liang, W.S.; Aldrich, J.; Washington, M.; Wong, S.; Rohrer, D.; Jewell, S.; Kittles, R.A.; Derome, M.; et al. Comprehensive molecular profiling of 718 Multiple Myelomas reveals significant differences in mutation frequencies between African and European descent cases. PLoS Genet. 2017, 13, e1007087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, J.; Suwa, T.; Murase, Y.; Tateno, S.; Mizutome, H.; Asatsuma-Okumura, T.; Shimizu, N.; Kishi, T.; Momose, S.; Kizaki, M.; et al. ARID2 is a pomalidomide-dependent CRL4(CRBN) substrate in multiple myeloma cells. Nat. Chem. Biol. 2020, 16, 1208–1217. [Google Scholar] [CrossRef]
- Ellis, L.; Atadja, P.W.; Johnstone, R.W. Epigenetics in cancer: Targeting chromatin modifications. Mol. Cancer Ther. 2009, 8, 1409–1420. [Google Scholar] [CrossRef] [Green Version]
- Lodewijk, I.; Nunes, S.P.; Henrique, R.; Jeronimo, C.; Duenas, M.; Paramio, J.M. Tackling tumor microenvironment through epigenetic tools to improve cancer immunotherapy. Clin. Epigenet. 2021, 13, 63. [Google Scholar] [CrossRef]
- Wanior, M.; Kramer, A.; Knapp, S.; Joerger, A.C. Exploiting vulnerabilities of SWI/SNF chromatin remodelling complexes for cancer therapy. Oncogene 2021, 40, 3637–3654. [Google Scholar] [CrossRef]
- Tsukiyama, T.; Wu, C. Purification and properties of an ATP-dependent nucleosome remodeling factor. Cell 1995, 83, 1011–1020. [Google Scholar] [CrossRef] [Green Version]
- Varga-Weisz, P.D.; Wilm, M.; Bonte, E.; Dumas, K.; Mann, M.; Becker, P.B. Chromatin-remodelling factor CHRAC contains the ATPases ISWI and topoisomerase II. Nature 1997, 388, 598–602. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, R.; Wang, J.; Chen, Y.; Qiao, C.; Shi, Q.; Jin, Y.; Shen, X.; Li, J.; Chen, L. Identification of clinical implications and potential prognostic models of chromatin regulator mutations in multiple myeloma. Clin Epigenet. 2022, 14, 93. [Google Scholar] [CrossRef]
- Zhang, P.; Torres, K.; Liu, X.; Liu, C.G.; Pollock, R.E. An Overview of Chromatin-Regulating Proteins in Cells. Curr. Protein Pept. Sci. 2016, 17, 401–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolli, N.; Avet-Loiseau, H.; Wedge, D.C.; Van Loo, P.; Alexandrov, L.B.; Martincorena, I.; Dawson, K.J.; Iorio, F.; Nik-Zainal, S.; Bignell, G.R.; et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat. Commun. 2014, 5, 2997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, P.S.Y.; Chooi, J.Y.; Lim, J.S.L.; Toh, S.H.M.; Tan, T.Z.; Chng, W.J. SMARCA2 Is a Novel Interactor of NSD2 and Regulates Prometastatic PTP4A3 through Chromatin Remodeling in t(4;14) Multiple Myeloma. Cancer Res 2021, 81, 2332–2344. [Google Scholar] [CrossRef] [PubMed]
- Ordonez, R.; Kulis, M.; Russinol, N.; Chapaprieta, V.; Carrasco-Leon, A.; Garcia-Torre, B.; Charalampopoulou, S.; Clot, G.; Beekman, R.; Meydan, C.; et al. Chromatin activation as a unifying principle underlying pathogenic mechanisms in multiple myeloma. Genome Res. 2020, 30, 1217–1227. [Google Scholar] [CrossRef]
- Hideshima, T.; Mitsiades, C.; Tonon, G.; Richardson, P.G.; Anderson, K.C. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat. Rev. Cancer 2007, 7, 585–598. [Google Scholar] [CrossRef]
- Bianchi, G.; Anderson, K.C. Understanding biology to tackle the disease: Multiple myeloma from bench to bedside, and back. CA Cancer J. Clin. 2014, 64, 422–444. [Google Scholar] [CrossRef]
- Krause, D.S. Regulation of hematopoietic stem cell fate. Oncogene 2002, 21, 3262–3269. [Google Scholar] [CrossRef] [Green Version]
- Lo Celso, C.; Fleming, H.E.; Wu, J.W.; Zhao, C.X.; Miake-Lye, S.; Fujisaki, J.; Cote, D.; Rowe, D.W.; Lin, C.P.; Scadden, D.T. Live-animal tracking of individual haematopoietic stem/progenitor cells in their niche. Nature 2009, 457, 92–96. [Google Scholar] [CrossRef] [Green Version]
- Morrison, S.J.; Scadden, D.T. The bone marrow niche for haematopoietic stem cells. Nature 2014, 505, 327–334. [Google Scholar] [CrossRef] [Green Version]
- Kiel, M.J.; Yilmaz, O.H.; Iwashita, T.; Yilmaz, O.H.; Terhorst, C.; Morrison, S.J. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell 2005, 121, 1109–1121. [Google Scholar] [CrossRef]
- Koechlein, C.S.; Harris, J.R.; Lee, T.K.; Weeks, J.; Fox, R.G.; Zimdahl, B.; Ito, T.; Blevins, A.; Jung, S.H.; Chute, J.P.; et al. High-resolution imaging and computational analysis of haematopoietic cell dynamics in vivo. Nat. Commun. 2016, 7, 12169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunisaki, Y.; Bruns, I.; Scheiermann, C.; Ahmed, J.; Pinho, S.; Zhang, D.; Mizoguchi, T.; Wei, Q.; Lucas, D.; Ito, K.; et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature 2013, 502, 637–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ria, R.; Piccoli, C.; Cirulli, T.; Falzetti, F.; Mangialardi, G.; Guidolin, D.; Tabilio, A.; Di Renzo, N.; Guarini, A.; Ribatti, D.; et al. Endothelial differentiation of hematopoietic stem and progenitor cells from patients with multiple myeloma. Clin. Cancer Res. 2008, 14, 1678–1685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruns, I.; Cadeddu, R.P.; Brueckmann, I.; Frobel, J.; Geyh, S.; Bust, S.; Fischer, J.C.; Roels, F.; Wilk, C.M.; Schildberg, F.A.; et al. Multiple myeloma-related deregulation of bone marrow-derived CD34(+) hematopoietic stem and progenitor cells. Blood 2012, 120, 2620–2630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendez-Ferrer, S.; Michurina, T.V.; Ferraro, F.; Mazloom, A.R.; Macarthur, B.D.; Lira, S.A.; Scadden, D.T.; Ma’ayan, A.; Enikolopov, G.N.; Frenette, P.S. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 2010, 466, 829–834. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Saunders, T.L.; Enikolopov, G.; Morrison, S.J. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 2012, 481, 457–462. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Morrison, S.J. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature 2013, 495, 231–235. [Google Scholar] [CrossRef] [Green Version]
- Kfoury, Y.; Scadden, D.T. Mesenchymal cell contributions to the stem cell niche. Cell Stem Cell 2015, 16, 239–253. [Google Scholar] [CrossRef] [Green Version]
- Asada, N.; Kunisaki, Y.; Pierce, H.; Wang, Z.; Fernandez, N.F.; Birbrair, A.; Ma’ayan, A.; Frenette, P.S. Differential cytokine contributions of perivascular haematopoietic stem cell niches. Nat. Cell Biol. 2017, 19, 214–223. [Google Scholar] [CrossRef] [Green Version]
- Ribatti, D.; Nico, B.; Crivellato, E.; Roccaro, A.M.; Vacca, A. The history of the angiogenic switch concept. Leukemia 2007, 21, 44–52. [Google Scholar] [CrossRef]
- Azab, A.K.; Hu, J.; Quang, P.; Azab, F.; Pitsillides, C.; Awwad, R.; Thompson, B.; Maiso, P.; Sun, J.D.; Hart, C.P.; et al. Hypoxia promotes dissemination of multiple myeloma through acquisition of epithelial to mesenchymal transition-like features. Blood 2012, 119, 5782–5794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coluccia, A.M.; Cirulli, T.; Neri, P.; Mangieri, D.; Colanardi, M.C.; Gnoni, A.; Di Renzo, N.; Dammacco, F.; Tassone, P.; Ribatti, D.; et al. Validation of PDGFRbeta and c-Src tyrosine kinases as tumor/vessel targets in patients with multiple myeloma: Preclinical efficacy of the novel, orally available inhibitor dasatinib. Blood 2008, 112, 1346–1356. [Google Scholar] [CrossRef] [PubMed]
- Crane, G.M.; Jeffery, E.; Morrison, S.J. Adult haematopoietic stem cell niches. Nat. Rev. Immunol. 2017, 17, 573–590. [Google Scholar] [CrossRef] [PubMed]
- Winkler, I.G.; Barbier, V.; Nowlan, B.; Jacobsen, R.N.; Forristal, C.E.; Patton, J.T.; Magnani, J.L.; Levesque, J.P. Vascular niche E-selectin regulates hematopoietic stem cell dormancy, self renewal and chemoresistance. Nat. Med. 2012, 18, 1651–1657. [Google Scholar] [CrossRef] [PubMed]
- Golstein, P.; Griffiths, G.M. An early history of T cell-mediated cytotoxicity. Nat. Rev. Immunol. 2018, 18, 527–535. [Google Scholar] [CrossRef]
- Bardhan, K.; Anagnostou, T.; Boussiotis, V.A. The PD1:PD-L1/2 Pathway from Discovery to Clinical Implementation. Front. Immunol. 2016, 7, 550. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.H.; Park, Y.; Kim, J.H.; Kang, K.W.; Lee, S.J.; Kim, S.J.; Kim, B.S. PD-L1 expression in bone marrow plasma cells as a biomarker to predict multiple myeloma prognosis: Developing a nomogram-based prognostic model. Sci. Rep. 2020, 10, 12641. [Google Scholar] [CrossRef]
- Radpour, R.; Stucki, M.; Riether, C.; Ochsenbein, A.F. Epigenetic Silencing of Immune-Checkpoint Receptors in Bone Marrow- Infiltrating T Cells in Acute Myeloid Leukemia. Front. Oncol. 2021, 11, 663406. [Google Scholar] [CrossRef]
- Tan, J.; Chen, S.; Huang, J.; Chen, Y.; Yang, L.; Wang, C.; Zhong, J.; Lu, Y.; Wang, L.; Zhu, K.; et al. Increased exhausted CD8(+) T cells with programmed death-1, T-cell immunoglobulin and mucin-domain-containing-3 phenotype in patients with multiple myeloma. Asia Pac. J. Clin. Oncol. 2018, 14, e266–e274. [Google Scholar] [CrossRef]
- Ribrag, V.; Avigan, D.E.; Green, D.J.; Wise-Draper, T.; Posada, J.G.; Vij, R.; Zhu, Y.; Farooqui, M.Z.H.; Marinello, P.; Siegel, D.S. Phase 1b trial of pembrolizumab monotherapy for relapsed/refractory multiple myeloma: KEYNOTE-013. Br. J. Haematol. 2019, 186, e41–e44. [Google Scholar] [CrossRef]
- Ma, X.; Zhang, Y.; Wang, S.; Wei, H.; Yu, J. Immune checkpoint inhibitor (ICI) combination therapy compared to monotherapy in advanced solid cancer: A systematic review. J. Cancer 2021, 12, 1318–1333. [Google Scholar] [CrossRef] [PubMed]
- Leone, P.; Berardi, S.; Frassanito, M.A.; Ria, R.; De Re, V.; Cicco, S.; Battaglia, S.; Ditonno, P.; Dammacco, F.; Vacca, A.; et al. Dendritic cells accumulate in the bone marrow of myeloma patients where they protect tumor plasma cells from CD8+ T-cell killing. Blood 2015, 126, 1443–1451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, C.; Sheng, L.; Pan, D.; Jiang, S.; Ding, L.; Ma, X.; Liu, Y.; Jia, D. Single-Cell Transcriptomic Analysis Revealed a Critical Role of SPP1/CD44-Mediated Crosstalk Between Macrophages and Cancer Cells in Glioma. Front. Cell Dev. Biol. 2021, 9, 779319. [Google Scholar] [CrossRef] [PubMed]
- Valencia, J.C.; Erwin-Cohen, R.A.; Clavijo, P.E.; Allen, C.; Sanford, M.E.; Day, C.P.; Hess, M.M.; Johnson, M.; Yin, J.; Fenimore, J.M.; et al. Myeloid-Derived Suppressive Cell Expansion Promotes Melanoma Growth and Autoimmunity by Inhibiting CD40/IL27 Regulation in Macrophages. Cancer Res. 2021, 81, 5977–5990. [Google Scholar] [CrossRef]
- Panneerselvam, J.; Madka, V.; Rai, R.; Morris, K.T.; Houchen, C.W.; Chandrakesan, P.; Rao, C.V. Inflammatory Mediators and Gut Microbial Toxins Drive Colon Tumorigenesis by IL-23 Dependent Mechanism. Cancers 2021, 13, 5159. [Google Scholar] [CrossRef]
- Seong, J.B.; Kim, B.; Kim, S.; Kim, M.H.; Park, Y.H.; Lee, Y.; Lee, H.J.; Hong, C.W.; Lee, D.S. Macrophage peroxiredoxin 5 deficiency promotes lung cancer progression via ROS-dependent M2-like polarization. Free Radic. Biol. Med. 2021, 176, 322–334. [Google Scholar] [CrossRef]
- Song, M.; Yeku, O.O.; Rafiq, S.; Purdon, T.; Dong, X.; Zhu, L.; Zhang, T.; Wang, H.; Yu, Z.; Mai, J.; et al. Tumor derived UBR5 promotes ovarian cancer growth and metastasis through inducing immunosuppressive macrophages. Nat. Commun. 2020, 11, 6298. [Google Scholar] [CrossRef]
- Akhmetzyanova, I.; Aaron, T.; Galbo, P.; Tikhonova, A.; Dolgalev, I.; Tanaka, M.; Aifantis, I.; Zheng, D.; Zang, X.; Fooksman, D. Tissue-resident macrophages promote early dissemination of multiple myeloma via IL-6 and TNFalpha. Blood Adv. 2021, 5, 3592–3608. [Google Scholar] [CrossRef]
- Jetten, N.; Verbruggen, S.; Gijbels, M.J.; Post, M.J.; De Winther, M.P.; Donners, M.M. Anti-inflammatory M2, but not pro-inflammatory M1 macrophages promote angiogenesis in vivo. Angiogenesis 2014, 17, 109–118. [Google Scholar] [CrossRef]
- Sun, M.; Xiao, Q.; Wang, X.; Yang, C.; Chen, C.; Tian, X.; Wang, S.; Li, H.; Qiu, S.; Shu, J.; et al. Tumor-associated macrophages modulate angiogenesis and tumor growth in a xenograft mouse model of multiple myeloma. Leuk. Res. 2021, 110, 106709. [Google Scholar] [CrossRef]
- Tirier, S.M.; Mallm, J.P.; Steiger, S.; Poos, A.M.; Awwad, M.H.S.; Giesen, N.; Casiraghi, N.; Susak, H.; Bauer, K.; Baumann, A.; et al. Subclone-specific microenvironmental impact and drug response in refractory multiple myeloma revealed by single-cell transcriptomics. Nat. Commun. 2021, 12, 6960. [Google Scholar] [CrossRef]
- Zhang, D.; Huang, J.; Wang, F.; Ding, H.; Cui, Y.; Yang, Y.; Xu, J.; Luo, H.; Gao, Y.; Pan, L.; et al. BMI1 regulates multiple myeloma-associated macrophage’s pro-myeloma functions. Cell Death Dis. 2021, 12, 495. [Google Scholar] [CrossRef] [PubMed]
- De Beule, N.; De Veirman, K.; Maes, K.; De Bruyne, E.; Menu, E.; Breckpot, K.; De Raeve, H.; Van Rampelbergh, R.; Van Ginderachter, J.A.; Schots, R.; et al. Tumour-associated macrophage-mediated survival of myeloma cells through STAT3 activation. J. Pathol 2017, 241, 534–546. [Google Scholar] [CrossRef] [PubMed]
- Ullah, T.R. The role of CXCR4 in multiple myeloma: Cells’ journey from bone marrow to beyond. J. Bone Oncol. 2019, 17, 100253. [Google Scholar] [CrossRef] [PubMed]
- Stessman, H.A.; Mansoor, A.; Zhan, F.; Janz, S.; Linden, M.A.; Baughn, L.B.; Van Ness, B. Reduced CXCR4 expression is associated with extramedullary disease in a mouse model of myeloma and predicts poor survival in multiple myeloma patients treated with bortezomib. Leukemia 2013, 27, 2075–2077. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Wang, J.; Willingham, S.B.; Martin, R.; Wernig, G.; Weissman, I.L. Anti-CD47 antibodies promote phagocytosis and inhibit the growth of human myeloma cells. Leukemia 2012, 26, 2538–2545. [Google Scholar] [CrossRef] [Green Version]
- Giuliani, N.; Colla, S.; Morandi, F.; Lazzaretti, M.; Sala, R.; Bonomini, S.; Grano, M.; Colucci, S.; Svaldi, M.; Rizzoli, V. Myeloma cells block RUNX2/CBFA1 activity in human bone marrow osteoblast progenitors and inhibit osteoblast formation and differentiation. Blood 2005, 106, 2472–2483. [Google Scholar] [CrossRef]
- Roodman, G.D. Osteoblast function in myeloma. Bone 2011, 48, 135–140. [Google Scholar] [CrossRef]
- Mundy, G.R.; Raisz, L.G.; Cooper, R.A.; Schechter, G.P.; Salmon, S.E. Evidence for the secretion of an osteoclast stimulating factor in myeloma. N. Engl. J. Med. 1974, 291, 1041–1046. [Google Scholar] [CrossRef]
- Andersen, T.L.; Soe, K.; Sondergaard, T.E.; Plesner, T.; Delaisse, J.M. Myeloma cell-induced disruption of bone remodelling compartments leads to osteolytic lesions and generation of osteoclast-myeloma hybrid cells. Br. J. Haematol. 2010, 148, 551–561. [Google Scholar] [CrossRef]
- Chen, Z.; Orlowski, R.Z.; Wang, M.; Kwak, L.; McCarty, N. Osteoblastic niche supports the growth of quiescent multiple myeloma cells. Blood 2014, 123, 2204–2208. [Google Scholar] [CrossRef] [Green Version]
- Heider, U.; Hofbauer, L.C.; Zavrski, I.; Kaiser, M.; Jakob, C.; Sezer, O. Novel aspects of osteoclast activation and osteoblast inhibition in myeloma bone disease. Biochem. Biophys. Res. Commun. 2005, 338, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Sezer, O.; Heider, U.; Jakob, C.; Zavrski, I.; Eucker, J.; Possinger, K.; Sers, C.; Krenn, V. Immunocytochemistry reveals RANKL expression of myeloma cells. Blood 2002, 99, 4646–4647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, M.M.; Fairfield, H.; Falank, C.; Reagan, M.R. Adipose, Bone, and Myeloma: Contributions from the Microenvironment. Calcif Tissue Int 2017, 100, 433–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oyajobi, B.O.; Franchin, G.; Williams, P.J.; Pulkrabek, D.; Gupta, A.; Munoz, S.; Grubbs, B.; Zhao, M.; Chen, D.; Sherry, B.; et al. Dual effects of macrophage inflammatory protein-1alpha on osteolysis and tumor burden in the murine 5TGM1 model of myeloma bone disease. Blood 2003, 102, 311–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Croucher, P.I.; McDonald, M.M.; Martin, T.J. Bone metastasis: The importance of the neighbourhood. Nat. Rev. Cancer 2016, 16, 373–386. [Google Scholar] [CrossRef]
- Abe, M.; Kido, S.; Hiasa, M.; Nakano, A.; Oda, A.; Amou, H.; Matsumoto, T. BAFF and APRIL as osteoclast-derived survival factors for myeloma cells: A rationale for TACI-Fc treatment in patients with multiple myeloma. Leukemia 2006, 20, 1313–1315. [Google Scholar] [CrossRef] [Green Version]
- Lawson, M.A.; McDonald, M.M.; Kovacic, N.; Hua Khoo, W.; Terry, R.L.; Down, J.; Kaplan, W.; Paton-Hough, J.; Fellows, C.; Pettitt, J.A.; et al. Osteoclasts control reactivation of dormant myeloma cells by remodelling the endosteal niche. Nat. Commun 2015, 6, 8983. [Google Scholar] [CrossRef] [Green Version]
- Delgado-Calle, J.; Anderson, J.; Cregor, M.D.; Hiasa, M.; Chirgwin, J.M.; Carlesso, N.; Yoneda, T.; Mohammad, K.S.; Plotkin, L.I.; Roodman, G.D.; et al. Bidirectional Notch Signaling and Osteocyte-Derived Factors in the Bone Marrow Microenvironment Promote Tumor Cell Proliferation and Bone Destruction in Multiple Myeloma. Cancer Res. 2016, 76, 1089–1100. [Google Scholar] [CrossRef] [Green Version]
- Habibi, H.; Abroun, S.; Hajifathali, A.; Soleimani, M.; Kaviani, S.; Kalantari, N.; Eslahchi, S. Osteogenic inhibition in multiple myeloma. Cell J. 2013, 15, 266–271. [Google Scholar]
- Reagan, M.R.; Fairfield, H.; Rosen, C.J. Bone Marrow Adipocytes: A Link between Obesity and Bone Cancer. Cancers 2021, 13, 364. [Google Scholar] [CrossRef] [PubMed]
- Shin, E.; Koo, J.S. The Role of Adipokines and Bone Marrow Adipocytes in Breast Cancer Bone Metastasis. Int. J. Mol. Sci. 2020, 21, 4967. [Google Scholar] [CrossRef] [PubMed]
- Tabe, Y.; Konopleva, M.; Andreeff, M. Fatty Acid Metabolism, Bone Marrow Adipocytes, and AML. Front. Oncol. 2020, 10, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; He, J.; Koh, S.P.; Zhong, Y.; Liu, Z.; Wang, Z.; Zhang, Y.; Li, Z.; Tam, B.T.; Lin, P.; et al. Reprogrammed marrow adipocytes contribute to myeloma-induced bone disease. Sci. Transl. Med. 2019, 11, eaau9087. [Google Scholar] [CrossRef]
- Liu, Z.; Xu, J.; He, J.; Liu, H.; Lin, P.; Wan, X.; Navone, N.M.; Tong, Q.; Kwak, L.W.; Orlowski, R.Z.; et al. Mature adipocytes in bone marrow protect myeloma cells against chemotherapy through autophagy activation. Oncotarget 2015, 6, 34329–34341. [Google Scholar] [CrossRef] [Green Version]
- Morris, E.V.; Suchacki, K.J.; Hocking, J.; Cartwright, R.; Sowman, A.; Gamez, B.; Lea, R.; Drake, M.T.; Cawthorn, W.P.; Edwards, C.M. Myeloma Cells Down-Regulate Adiponectin in Bone Marrow Adipocytes Via TNF-Alpha. J. Bone Miner. Res. 2020, 35, 942–955. [Google Scholar] [CrossRef] [Green Version]
- Fairfield, H.; Dudakovic, A.; Khatib, C.M.; Farrell, M.; Costa, S.; Falank, C.; Hinge, M.; Murphy, C.S.; DeMambro, V.; Pettitt, J.A.; et al. Myeloma-Modified Adipocytes Exhibit Metabolic Dysfunction and a Senescence-Associated Secretory Phenotype. Cancer Res. 2021, 81, 634–647. [Google Scholar] [CrossRef]
- Asimakopoulos, F.; Hope, C.; Johnson, M.G.; Pagenkopf, A.; Gromek, K.; Nagel, B. Extracellular matrix and the myeloid-in-myeloma compartment: Balancing tolerogenic and immunogenic inflammation in the myeloma niche. J. Leukoc. Biol. 2017, 102, 265–275. [Google Scholar] [CrossRef] [Green Version]
- Glavey, S.V.; Naba, A.; Manier, S.; Clauser, K.; Tahri, S.; Park, J.; Reagan, M.R.; Moschetta, M.; Mishima, Y.; Gambella, M.; et al. Proteomic characterization of human multiple myeloma bone marrow extracellular matrix. Leukemia 2017, 31, 2426–2434. [Google Scholar] [CrossRef]
- Panchabhai, S.; Kelemen, K.; Ahmann, G.; Sebastian, S.; Mantei, J.; Fonseca, R. Tumor-associated macrophages and extracellular matrix metalloproteinase inducer in prognosis of multiple myeloma. Leukemia 2016, 30, 951–954. [Google Scholar] [CrossRef]
- Mahtouk, K.; Moreaux, J.; Hose, D.; Reme, T.; Meissner, T.; Jourdan, M.; Rossi, J.F.; Pals, S.T.; Goldschmidt, H.; Klein, B. Growth factors in multiple myeloma: A comprehensive analysis of their expression in tumor cells and bone marrow environment using Affymetrix microarrays. BMC Cancer 2010, 10, 198. [Google Scholar] [CrossRef] [PubMed]
- Klein, B.; Zhang, X.G.; Jourdan, M.; Portier, M.; Bataille, R. Interleukin-6 is a major myeloma cell growth factor in vitro and in vivo especially in patients with terminal disease. Curr. Top. Microbiol. Immunol. 1990, 166, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Ria, R.; Roccaro, A.M.; Merchionne, F.; Vacca, A.; Dammacco, F.; Ribatti, D. Vascular endothelial growth factor and its receptors in multiple myeloma. Leukemia 2003, 17, 1961–1966. [Google Scholar] [CrossRef] [Green Version]
- Iwasaki, T.; Hamano, T.; Ogata, A.; Hashimoto, N.; Kitano, M.; Kakishita, E. Clinical significance of vascular endothelial growth factor and hepatocyte growth factor in multiple myeloma. Br. J. Haematol. 2002, 116, 796–802. [Google Scholar] [CrossRef]
- Van de Donk, N.W.; Lokhorst, H.M.; Bloem, A.C. Growth factors and antiapoptotic signaling pathways in multiple myeloma. Leukemia 2005, 19, 2177–2185. [Google Scholar] [CrossRef] [Green Version]
- Kanwal, R.; Gupta, S. Epigenetic modifications in cancer. Clin. Genet. 2012, 81, 303–311. [Google Scholar] [CrossRef] [Green Version]
- Marks, D.L.; Olson, R.L.; Fernandez-Zapico, M.E. Epigenetic control of the tumor microenvironment. Epigenomics 2016, 8, 1671–1687. [Google Scholar] [CrossRef] [Green Version]
- Guo, B.; Huang, X.; Cooper, S.; Broxmeyer, H.E. Glucocorticoid hormone-induced chromatin remodeling enhances human hematopoietic stem cell homing and engraftment. Nat. Med. 2017, 23, 424–428. [Google Scholar] [CrossRef] [Green Version]
- Krasteva, V.; Crabtree, G.R.; Lessard, J.A. The BAF45a/PHF10 subunit of SWI/SNF-like chromatin remodeling complexes is essential for hematopoietic stem cell maintenance. Exp. Hematol. 2017, 48, 58–71.e15. [Google Scholar] [CrossRef]
- Xu, B.; Cai, L.; Butler, J.M.; Chen, D.; Lu, X.; Allison, D.F.; Lu, R.; Rafii, S.; Parker, J.S.; Zheng, D.; et al. The Chromatin Remodeler BPTF Activates a Stemness Gene-Expression Program Essential for the Maintenance of Adult Hematopoietic Stem Cells. Stem Cell Rep. 2018, 10, 675–683. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.; Jiang, N.; Jiang, Y.; He, Q.; He, H.; Wang, X.; Yang, L.; Li, R.; Liu, F.; Lin, X.; et al. Chromatin remodeler Znhit1 preserves hematopoietic stem cell quiescence by determining the accessibility of distal enhancers. Leukemia 2020, 34, 3348–3358. [Google Scholar] [CrossRef] [PubMed]
- Xi, R.; Xie, T. Stem cell self-renewal controlled by chromatin remodeling factors. Science 2005, 310, 1487–1489. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.A.; Huang, Y.C.; Swigut, T.; Mirick, A.L.; Garcia-Verdugo, J.M.; Wysocka, J.; Ernst, P.; Alvarez-Buylla, A. Chromatin remodelling factor Mll1 is essential for neurogenesis from postnatal neural stem cells. Nature 2009, 458, 529–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, L.; Ronan, J.L.; Wu, J.; Staahl, B.T.; Chen, L.; Kuo, A.; Lessard, J.; Nesvizhskii, A.I.; Ranish, J.; Crabtree, G.R. An embryonic stem cell chromatin remodeling complex, esBAF, is essential for embryonic stem cell self-renewal and pluripotency. Proc. Natl. Acad. Sci. USA 2009, 106, 5181–5186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Z.; Wang, Z.; Sharova, L.; Sharov, A.A.; Ling, C.; Piao, Y.; Aiba, K.; Matoba, R.; Wang, W.; Ko, M.S. BAF250B-associated SWI/SNF chromatin-remodeling complex is required to maintain undifferentiated mouse embryonic stem cells. Stem Cells 2008, 26, 1155–1165. [Google Scholar] [CrossRef] [Green Version]
- Xie, R.; Everett, L.J.; Lim, H.W.; Patel, N.A.; Schug, J.; Kroon, E.; Kelly, O.G.; Wang, A.; D’Amour, K.A.; Robins, A.J.; et al. Dynamic chromatin remodeling mediated by polycomb proteins orchestrates pancreatic differentiation of human embryonic stem cells. Cell Stem Cell 2013, 12, 224–237. [Google Scholar] [CrossRef] [Green Version]
- Mardaryev, A.N.; Gdula, M.R.; Yarker, J.L.; Emelianov, V.U.; Poterlowicz, K.; Sharov, A.A.; Sharova, T.Y.; Scarpa, J.A.; Joffe, B.; Solovei, I.; et al. p63 and Brg1 control developmentally regulated higher-order chromatin remodelling at the epidermal differentiation complex locus in epidermal progenitor cells. Development 2014, 141, 101–111. [Google Scholar] [CrossRef] [Green Version]
- Killaars, A.R.; Grim, J.C.; Walker, C.J.; Hushka, E.A.; Brown, T.E.; Anseth, K.S. Extended Exposure to Stiff Microenvironments Leads to Persistent Chromatin Remodeling in Human Mesenchymal Stem Cells. Adv. Sci. 2019, 6, 1801483. [Google Scholar] [CrossRef] [Green Version]
- Snykers, S.; Vanhaecke, T.; De Becker, A.; Papeleu, P.; Vinken, M.; Van Riet, I.; Rogiers, V. Chromatin remodeling agent trichostatin A: A key-factor in the hepatic differentiation of human mesenchymal stem cells derived of adult bone marrow. BMC Dev. Biol. 2007, 7, 24. [Google Scholar] [CrossRef] [Green Version]
- Sinha, S.; Biswas, M.; Chatterjee, S.S.; Kumar, S.; Sengupta, A. Pbrm1 Steers Mesenchymal Stromal Cell Osteolineage Differentiation by Integrating PBAF-Dependent Chromatin Remodeling and BMP/TGF-beta Signaling. Cell Rep. 2020, 31, 107570. [Google Scholar] [CrossRef]
- Napolitano, M.A.; Cipollaro, M.; Cascino, A.; Melone, M.A.; Giordano, A.; Galderisi, U. Brg1 chromatin remodeling factor is involved in cell growth arrest, apoptosis and senescence of rat mesenchymal stem cells. J. Cell Sci. 2007, 120, 2904–2911. [Google Scholar] [CrossRef] [PubMed]
- Alessio, N.; Squillaro, T.; Cipollaro, M.; Bagella, L.; Giordano, A.; Galderisi, U. The BRG1 ATPase of chromatin remodeling complexes is involved in modulation of mesenchymal stem cell senescence through RB-P53 pathways. Oncogene 2010, 29, 5452–5463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Squillaro, T.; Severino, V.; Alessio, N.; Farina, A.; Di Bernardo, G.; Cipollaro, M.; Peluso, G.; Chambery, A.; Galderisi, U. De-regulated expression of the BRG1 chromatin remodeling factor in bone marrow mesenchymal stromal cells induces senescence associated with the silencing of NANOG and changes in the levels of chromatin proteins. Cell Cycle 2015, 14, 1315–1326. [Google Scholar] [CrossRef] [PubMed]
- Colijn, S.; Gao, S.; Ingram, K.G.; Menendez, M.; Muthukumar, V.; Silasi-Mansat, R.; Chmielewska, J.J.; Hinsdale, M.; Lupu, F.; Griffin, C.T. The NuRD chromatin-remodeling complex enzyme CHD4 prevents hypoxia-induced endothelial Ripk3 transcription and murine embryonic vascular rupture. Cell Death Differ. 2020, 27, 618–631. [Google Scholar] [CrossRef] [PubMed]
- Hahner, F.; Moll, F.; Warwick, T.; Hebchen, D.M.; Buchmann, G.K.; Epah, J.; Abplanalp, W.; Schader, T.; Gunther, S.; Gilsbach, R.; et al. Nox4 promotes endothelial differentiation through chromatin remodeling. Redox. Biol. 2022, 55, 102381. [Google Scholar] [CrossRef]
- Pfister, N.T.; Fomin, V.; Regunath, K.; Zhou, J.Y.; Zhou, W.; Silwal-Pandit, L.; Freed-Pastor, W.A.; Laptenko, O.; Neo, S.P.; Bargonetti, J.; et al. Mutant p53 cooperates with the SWI/SNF chromatin remodeling complex to regulate VEGFR2 in breast cancer cells. Genes Dev. 2015, 29, 1298–1315. [Google Scholar] [CrossRef] [Green Version]
- Epelman, S.; Lavine, K.J.; Randolph, G.J. Origin and functions of tissue macrophages. Immunity 2014, 41, 21–35. [Google Scholar] [CrossRef] [Green Version]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol 2010, 11, 889–896. [Google Scholar] [CrossRef]
- Mielgo, A.; Schmid, M.C. Impact of tumour associated macrophages in pancreatic cancer. BMB Rep. 2013, 46, 131–138. [Google Scholar] [CrossRef] [Green Version]
- Karp, C.L.; Murray, P.J. Non-canonical alternatives: What a macrophage is 4. J. Exp. Med. 2012, 209, 427–431. [Google Scholar] [CrossRef] [Green Version]
- Gordon, S.; Martinez, F.O. Alternative activation of macrophages: Mechanism and functions. Immunity 2010, 32, 593–604. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A. Macrophages, innate immunity and cancer: Balance, tolerance, and diversity. Curr. Opin. Immunol. 2010, 22, 231–237. [Google Scholar] [CrossRef]
- Boutilier, A.J.; Elsawa, S.F. Macrophage Polarization States in the Tumor Microenvironment. Int. J. Mol. Sci 2021, 22, 6995. [Google Scholar] [CrossRef]
- Greten, F.R.; Eckmann, L.; Greten, T.F.; Park, J.M.; Li, Z.W.; Egan, L.J.; Kagnoff, M.F.; Karin, M. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 2004, 118, 285–296. [Google Scholar] [CrossRef] [Green Version]
- Karin, M.; Greten, F.R. NF-kappaB: Linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol. 2005, 5, 749–759. [Google Scholar] [CrossRef]
- Qian, B.; Deng, Y.; Im, J.H.; Muschel, R.J.; Zou, Y.; Li, J.; Lang, R.A.; Pollard, J.W. A distinct macrophage population mediates metastatic breast cancer cell extravasation, establishment and growth. PLoS ONE 2009, 4, e6562. [Google Scholar] [CrossRef] [Green Version]
- Ruffell, B.; Affara, N.I.; Coussens, L.M. Differential macrophage programming in the tumor microenvironment. Trends Immunol. 2012, 33, 119–126. [Google Scholar] [CrossRef] [Green Version]
- Ghisletti, S.; Natoli, G. Deciphering cis-regulatory control in inflammatory cells. Philos. Trans. R Soc. Lond. B Biol. Sci. 2013, 368, 20120370. [Google Scholar] [CrossRef] [Green Version]
- Creyghton, M.P.; Cheng, A.W.; Welstead, G.G.; Kooistra, T.; Carey, B.W.; Steine, E.J.; Hanna, J.; Lodato, M.A.; Frampton, G.M.; Sharp, P.A.; et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 2010, 107, 21931–21936. [Google Scholar] [CrossRef] [Green Version]
- Kapellos, T.S.; Iqbal, A.J. Epigenetic Control of Macrophage Polarisation and Soluble Mediator Gene Expression during Inflammation. Mediators Inflamm 2016, 2016, 6591703. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Hao, X.; Li, Z.; Feng, X.; Katz, J.; Michalek, S.M.; Jiang, H.; Zhang, P. Role of chromatin modulator Dpy30 in osteoclast differentiation and function. Bone 2022, 159, 116379. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.M.; Bronisz, A.; Hu, R.; Patel, K.; Mansky, K.C.; Sif, S.; Ostrowski, M.C. MITF and PU.1 recruit p38 MAPK and NFATc1 to target genes during osteoclast differentiation. J. Biol. Chem. 2007, 282, 15921–15929. [Google Scholar] [CrossRef] [Green Version]
- Faulkner, B.; Astleford, K.; Mansky, K.C. Regulation of Osteoclast Differentiation and Skeletal Maintenance by Histone Deacetylases. Molecules 2019, 24, 1355. [Google Scholar] [CrossRef] [Green Version]
- Busby, T.; Chen, Y.; Godfrey, T.C.; Rehan, M.; Wildman, B.J.; Smith, C.M.; Hassan, Q. Baf45a Mediated Chromatin Remodeling Promotes Transcriptional Activation for Osteogenesis and Odontogenesis. Front. Endocrinol. 2021, 12, 763392. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Zhang, Y.; Yang, T.; Qi, J.; Zhang, L.; Deng, L. HIF-1alpha disturbs osteoblasts and osteoclasts coupling in bone remodeling by up-regulating OPG expression. In Vitro Cell Dev. Biol. Anim. 2015, 51, 808–814. [Google Scholar] [CrossRef]
- Siersbaek, R.; Nielsen, R.; John, S.; Sung, M.H.; Baek, S.; Loft, A.; Hager, G.L.; Mandrup, S. Extensive chromatin remodelling and establishment of transcription factor ‘hotspots’ during early adipogenesis. EMBO J. 2011, 30, 1459–1472. [Google Scholar] [CrossRef] [Green Version]
- Tasdelen, I.; Nielsen, R.; Lelieveld, D.; Groot, M.; Koerkamp, R.B.; Holstege, F.C.; Egan, D.; Mandrup, S.; Kalkhoven, E. THE SWI/SNF protein BAF57 regulates adipogenesis. In Modulation of the Adipogenic Master Regulator PPARγ; Utrecht University Repository: Utrecht, The Netherlands, 2014; p. 61. [Google Scholar]
- Shore, A.; Karamitri, A.; Kemp, P.; Speakman, J.R.; Lomax, M.A. Role of Ucp1 enhancer methylation and chromatin remodelling in the control of Ucp1 expression in murine adipose tissue. Diabetologia 2010, 53, 1164–1173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapira, S.N.; Lim, H.W.; Rajakumari, S.; Sakers, A.P.; Ishibashi, J.; Harms, M.J.; Won, K.J.; Seale, P. EBF2 transcriptionally regulates brown adipogenesis via the histone reader DPF3 and the BAF chromatin remodeling complex. Genes Dev. 2017, 31, 660–673. [Google Scholar] [CrossRef] [Green Version]
- Xu, R.; Spencer, V.A.; Bissell, M.J. Extracellular matrix-regulated gene expression requires cooperation of SWI/SNF and transcription factors. J. Biol. Chem. 2007, 282, 14992–14999. [Google Scholar] [CrossRef] [Green Version]
- Ingram, K.G.; Curtis, C.D.; Silasi-Mansat, R.; Lupu, F.; Griffin, C.T. The NuRD chromatin-remodeling enzyme CHD4 promotes embryonic vascular integrity by transcriptionally regulating extracellular matrix proteolysis. PLoS Genet. 2013, 9, e1004031. [Google Scholar] [CrossRef] [Green Version]
- Estella, C.; Herrer, I.; Atkinson, S.P.; Quinonero, A.; Martinez, S.; Pellicer, A.; Simon, C. Inhibition of histone deacetylase activity in human endometrial stromal cells promotes extracellular matrix remodelling and limits embryo invasion. PLoS ONE 2012, 7, e30508. [Google Scholar] [CrossRef]
- Qu, J.; Ouyang, Z.; Wu, W.; Li, G.; Wang, J.; Lu, Q.; Li, Z. Functions and Clinical Significance of Super-Enhancers in Bone-Related Diseases. Front. Cell Dev. Biol. 2020, 8, 534. [Google Scholar] [CrossRef]
- Jia, Y.; Zhou, J.; Tan, T.K.; Chung, T.H.; Wong, R.W.J.; Chooi, J.Y.; Lim, J.S.L.; Sanda, T.; Ooi, M.; De Mel, S.; et al. Myeloma-specific superenhancers affect genes of biological and clinical relevance in myeloma. Blood Cancer J. 2021, 11, 32. [Google Scholar] [CrossRef]
- Alvarez-Benayas, J.; Trasanidis, N.; Katsarou, A.; Ponnusamy, K.; Chaidos, A.; May, P.C.; Xiao, X.; Bua, M.; Atta, M.; Roberts, I.A.G.; et al. Chromatin-based, in cis and in trans regulatory rewiring underpins distinct oncogenic transcriptomes in multiple myeloma. Nat. Commun. 2021, 12, 5450. [Google Scholar] [CrossRef]
- Jin, Y.; Chen, K.; De Paepe, A.; Hellqvist, E.; Krstic, A.D.; Metang, L.; Gustafsson, C.; Davis, R.E.; Levy, Y.M.; Surapaneni, R.; et al. Active enhancer and chromatin accessibility landscapes chart the regulatory network of primary multiple myeloma. Blood 2018, 131, 2138–2150. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chakraborty, C.; Mukherjee, S. Molecular Crosstalk between Chromatin Remodeling and Tumor Microenvironment in Multiple Myeloma. Curr. Oncol. 2022, 29, 9535-9549. https://doi.org/10.3390/curroncol29120749
Chakraborty C, Mukherjee S. Molecular Crosstalk between Chromatin Remodeling and Tumor Microenvironment in Multiple Myeloma. Current Oncology. 2022; 29(12):9535-9549. https://doi.org/10.3390/curroncol29120749
Chicago/Turabian StyleChakraborty, Chandraditya, and Srimoyee Mukherjee. 2022. "Molecular Crosstalk between Chromatin Remodeling and Tumor Microenvironment in Multiple Myeloma" Current Oncology 29, no. 12: 9535-9549. https://doi.org/10.3390/curroncol29120749
APA StyleChakraborty, C., & Mukherjee, S. (2022). Molecular Crosstalk between Chromatin Remodeling and Tumor Microenvironment in Multiple Myeloma. Current Oncology, 29(12), 9535-9549. https://doi.org/10.3390/curroncol29120749